

Abstract
The recent discovery of the family of Toll-like receptors has vastly expanded our understanding of the mechanisms by which the innate immune system recognizes and responds to a wide variety of microbial and endogenous pathogens. Toll-like receptors are transmembrane proteins that upon ligation with their cognate ligands trigger the production of cytokines, enzymes and other inflammatory agents. In the CNS Toll-like receptors are expressed predominantly by glial cells. In particular, the vastly abundant astrocytes are likely to be the major contributors to inflammatory responses within the CNS. Studies of the murine brain abscess model revealed that Toll-like receptor 2 plays a pivotal role in the generation of immune responses to Staphylococcus aureus. Although Toll-like receptor signaling is essential in antimicrobial defense, it may also lead to bystander injury of CNS tissue.
Keywords: astrocytes, brain abscess, bystander injury, microglia, neuroinflammation, Toll-like receptors
Toll-like receptors (TLRs)
TLRs are archetypal pattern recognition receptors (PRRs) of the innate immune system in the vertebrate and invertebrate lineages. TLRs recognize a variety of highly conserved structural motifs expressed by microbial pathogens, called pathogen-associated molecular patterns (PAMPs) (Kopp and Medzhitov 1999, 2003; Akira 2001; Kaisho and Akira 2004). Consequently, TLRs are the major sensors of invading pathogens. So far, 13 TLRs have been identified in mice and 10 in humans. Molecular phylogenetic analysis revealed the existence of six TLR families (Roach et al. 2005) with distinct specificities to recognize general classes of PAMPs (Lien and Ingalls 2002). Thus, the TLR1 family comprises TLR1, TLR2, TLR6 and TLR10, and recognizes bacterial lipoproteins. The TLR3, TLR4 and TLR5 families have only one member each, and recognize double-stranded RNA (dsRNA), lipopolysaccharide (LPS) and flagellin respectively. The TLR7 family consists of TLR7–9, and binds nucleic acids. The TLR11 family includes TLR11–13; however, the cognate PAMPs are less characterized with the exception of TLR11 that recognizes a profilin-like molecule (Yarovinsky et al. 2005). The repertoire of TLR specificities is further extended by the formation of heterodimers (Ozinsky et al. 2000) and homodimers (Bell et al. 2005) with one another as well as by association with accessory proteins (Miyake 2003). In addition to microbial PAMPs, TLRs recognize a number of host-derived molecules liberated from damaged tissues. For example, endogenous ligands for TLR4 include heat-shock proteins (Vabulas et al. 2002; Palliser et al. 2004), hyaluronan fragments (Termeer et al. 2002; Taylor et al. 2004), heparan sulfate (Johnson et al. 2002), β-defensin 2 (Biragyn et al. 2002) and fibrinogen (Smiley et al. 2001). TLR2 recognizes necrotic cells (Li et al. 2001; Paterson et al. 2003), and TLR3 may be stimulated by mammalian RNA (Kariko et al. 2004). Therefore, TLRs may also mediate the general response of the innate immune system to injury and auto-immunity.
TLRs are transmembrane proteins with a leucine-rich extracellular domain and a cytoplasmic domain that contains a conserved region called the Toll/interleukin (IL)-1 receptor (TIR) domain (Takeuchi and Akira 2001; Barton and Medzhitov 2003). Upon ligand binding TLRs dimerize and undergo conformational changes that trigger a cascade of intracellular signaling events (Akira et al. 2006) represented schematically in Fig. 1. This signaling results in the up-regulation of numerous pro-inflammatory target genes encoding cytokines, chemokines, enzymes, and other molecules essential for pathogen elimination (Takeuchi and Akira 2001; Horng et al. 2002; Barton and Medzhitov 2003). In addition, TLR3 and TLR4 also mediate a primary antiviral program by transactivating type I interferon (IFN) genes (Kawai et al. 2001; Doyle et al. 2002, 2003; Shinobu et al. 2002; Kawai and Akira 2004). The secretion of IFN instigates an autocrine/paracrine loop resulting in the activation of secondary pro-inflammatory genes (Kawai et al. 2001; Shinobu et al. 2002). Besides mounting the innate immune response, TLR-induced signaling also governs the activity of the adaptive immune system (Akira et al. 2001; Kaisho et al. 2002; Hoebe et al. 2004; Pasare and Medzhitov 2005).
Fig. 1.
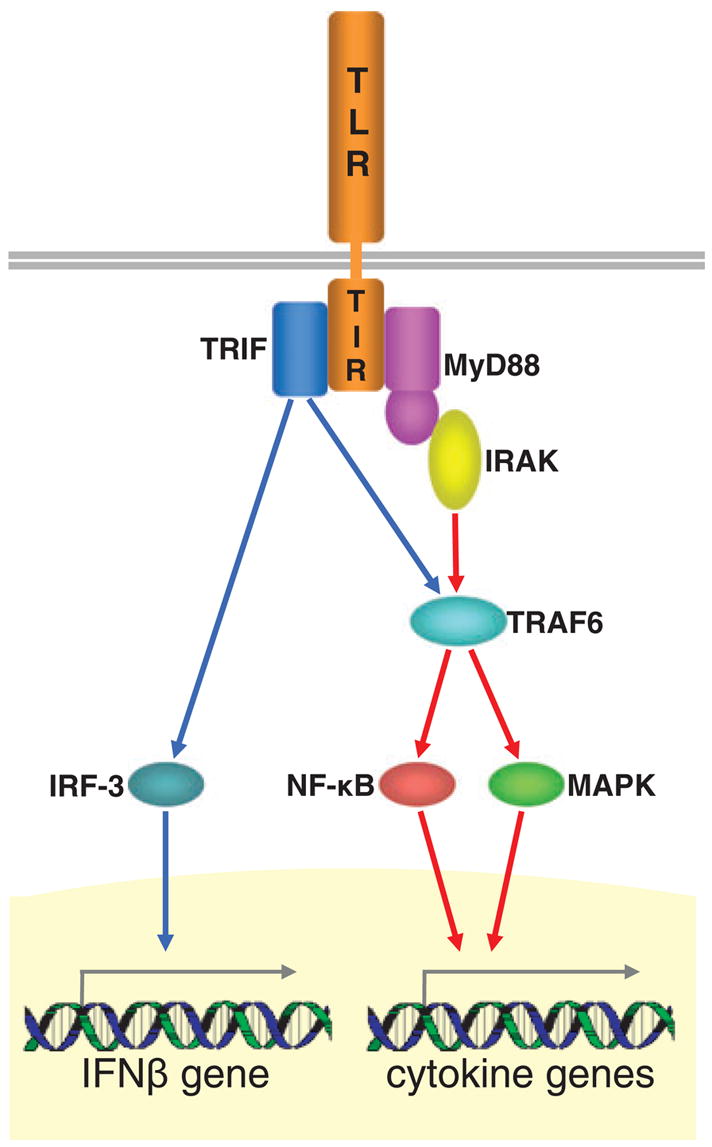
Two branches of TLR downstream signaling. All TLRs with the exception of TLR3 signal through the myeloid differentiation factor 88 (MyD88)-dependent pathway (red arrows). Upon ligand binding the cytoplasmic domain of TLRs, called the TIR domain, associates with an adaptor molecule, MyD88. The complex recruits IL-1 receptor-associated kinase (IRAK) that undergoes autophosphorylation, dissociates from the TLR signaling complex, and associates with TNF receptor-associated factor 6 (TRAF6). This association leads to the activation of NF-κB and MAPK that, in turn, up-regulate the expression of a core set of pro-inflammatory cytokine genes. In addition to this MyD88-dependent pathway, TLR3 and TLR4 signal in a MyD88-independent manner (blue arrows) through the recruitment of another adapter molecule, the TIR domain-containing adaptor inducing IFNβ (TRIF). TRIF activates the interferon regulatory factor (IRF)-3 that up-regulates the expression of the IFNβ gene. TRIF also interacts with TRAF6 leading to the activation of NF-κB and MAPK, and consequently to up-regulated expression of cytokine genes.
The expression of TLRs together with the expression of a contingent of related signaling proteins has been demonstrated in the CNS and in neural cell cultures (Kielian et al. 2002; Bsibsi et al. 2002; Dalpke et al. 2002; Rasley et al. 2002a; Bowman et al. 2003; Esen et al. 2004; Olson and Miller 2004; Carpentier et al. 2005; Farina et al. 2005; Jack et al. 2005; Nishimura and Naito 2005; Scumpia et al. 2005). All major glial cells including microglia, astrocytes and oligodendrocytes have been shown to express TLRs. Neurons may also express TLRs under certain pathological conditions (Maslinska et al. 2004). This review examines the role of TLRs in the CNS response to microbial challenge.
Astrocytes as sentinel cells for CNS pathogens
There is an increasing body of evidence that glial cells, particularly microglia and astrocytes, are pivotal in providing the first line of defense against invading microbes. Microglia are considered to be CNS-resident professional macrophages and sensor cells that function as the principal innate immune effector cells. Upon recognition of pathogens, resting microglia transform into activated microglia that migrate to and accumulate at sites of injury (Gonzalez-Scarano and Baltuch 1999; Gonzalez-Scarano and Martin-Garcia 2005). Activated microglia express a range of genes related to inflammation such as pro-inflammatory cytokines, pro-inflammatory enzymes and pro-inflammatory adhesion molecules (Gonzalez-Scarano and Baltuch 1999). The pattern of inflammatory molecule production is one that can initiate leukocyte migration through the blood–brain barrier (Persidsky 1999) and promote effector functions in these infiltrating cells. Recent studies have demonstrated the presence of mRNA and/or protein expression of TLR1 (Kielian et al. 2002), TLR2 (Kielian et al. 2002; Rasley et al. 2002a), TLR6 (Kielian et al. 2002), TLR9 (Dalpke et al. 2002), TLR3, TLR4, TLR5, TLR7 and TLR8 (Olson and Miller 2004) and the co-receptor CD14 (Kielian et al. 2002; Rasley et al. 2002b) in microglia, and have shown that such expression is increased following exposure to bacterial pathogens (Kielian et al. 2002; Rasley et al. 2002a; Olson and Miller 2004). These findings are, perhaps, not surprising considering that microglia share the same myeloid lineage as macrophages and dendritic cells – the quintessential sentinel cells.
Astrocytes are the major glial cell type in the brain and are well known to play essential roles in the development, survival and functioning of CNS neurons. However, in addition to these functions, this non-leukocytic cell type may have an additional role as an immune effector cell (Dong and Benveniste 2001). Because astrocytes vastly outnumber microglia within the CNS parenchyma, astrocytes are likely to be the major components of the CNS innate immune system. Activated astrocytes have been demonstrated to express an array of inflammatory cytokines and chemokines (Dong and Benveniste 2001). In addition to production of pro-inflammatory mediators, the stimulation of cultured astrocytes or cell lines results in expression of major histocompatibility complex (MHC) class II molecules and co-stimulatory molecules such as B7-1 and B7-2 (Soos et al. 1999). However, studies investigating the ability of astrocytes to express MHC class II following challenge in vivo have yielded equivocal results, and the functional significance of astrocyte expression of MHC antigens and co-stimulatory molecules remains controversial (Dong and Benveniste 2001). Importantly, astrocytes have been shown to become activated following challenge with clinically relevant bacterial pathogens. Borrelia burgdorferi, the Gram-negative organism responsible for Lyme neuroborreliosis, has been shown to stimulate the production of matrix metalloproteinase 9 by human and rat type I astrocytes (Perides et al. 1999), and we have recently demonstrated that this spirochete stimulates the rapid production of the inflammatory cytokine IL-6 by primary murine astrocytes (Rasley et al. 2002a). Similarly Neisseria meningitidis, an encapsulated Gram-negative organism that is an important cause of bacterial meningitis, rapidly induces IL-6 production by astrocytes (Rasley et al. 2006). Astrocytes are also highly responsive to Gram-positive bacterial species and produce the signature inflammatory cytokines IL-1β, tumor necrosis factor (TNF)-α, and IL-12 p40, following exposure to Staphylococcus aureus (Esen et al. 2004; Kielian 2004), indicating that these resident CNS cells can play an important role in the formation of brain abscess and associated damage to surrounding brain parenchyma. To date, the signals required to initiate optimal induction of the immune functions of this important glial cell type remain poorly understood and recent studies have focused on identifying the mechanisms by which astrocytes perceive bacterial pathogens.
TLR expression and immune responses to bacterial components
There is a considerable amount of circumstantial evidence for the expression of TLRs by astrocytes. LPS and Gram-positive bacterial cell wall antigens, ligands for TLR4 and TLR2 respectively (Ishii et al. 2005), have been reported to stimulate the activation of p38 mitogen-activated protein kinase (MAPK) in astrocytes (Schumann et al. 1998). In addition, LPS or bacterial DNA and synthetic oligonucleotides containing unmethylated CpG motifs (putative ligands for TLR9) can cause cultured astrocytes to express IL-1β (Lieberman et al. 1989; Gottschall et al. 1994; Kimberlin et al. 1995; Takeshita et al. 2001), TNF-α (Lieberman et al. 1989; Gottschall et al. 1994; Forloni et al. 1997; Nakamura et al. 1998), IL-6 (Lieberman et al. 1989; Benveniste et al. 1990; Gottschall et al. 1994; Nakamura et al. 1998; Takeshita et al. 2001) and chemokines (Oh et al. 1999). Furthermore, although astrocytes appear to lack membrane CD14 (Willis and Nisen 1996; Cauwels et al. 1999), these cells can respond to cell wall components of Gram-positive bacteria if soluble CD14 is present in the culture medium (Schumann et al. 1998). Taken together, these findings are consistent with the notion that astrocyte immune responses to bacterial pathogens are initiated via members of the Toll-like family of PRRs.
More recent work (Bsibsi et al. 2002; Bowman et al. 2003; Esen et al. 2004; Carpentier et al. 2005; Jack et al. 2005) has provided direct evidence for the presence of TLRs in astrocytes. Thus, cultured murine astrocytes constitutively express low levels of mRNA encoding TLR2, TLR4, TLR5 and TLR9. However, the modest expression of each TLR homolog is rapidly up-regulated following exposure to its specific bacterial ligand (Bowman et al. 2003; Carpentier et al. 2005). Interestingly, some microbial components could also up-regulate the expression of TLR homologs other than those thought to serve as their receptors (Bowman et al. 2003; Jack et al. 2005; McKimmie and Fazakerley 2005). Although it is presently unclear what mechanisms underlie such induction, it is possible that common signaling pathways control TLR homolog expression in astrocytes. Alternatively, and perhaps more intriguingly, the possibility exists that there is differential cross-talk between disparate PRR types in astrocytes. Such specific cross-talk might explain the observation that Escherichia coli-derived LPS can elicit increases in mRNA encoding both TLR4 and TLR5 in astrocytes, but not those encoding TLR2 or TLR9 (Bowman et al. 2003). Finally, it has been found that the inflammatory cytokine TNF-α and the pivotal TH1 cytokine IFNγ can also augment TLR expression in astrocytes (Carpentier et al. 2005). An ability of soluble immune mediators to regulate the expression of microbial PRRs might provide an explanation for the observation that astrocyte TLR2 expression is increased in patients with inflammatory CNS disorders including multiple sclerosis (Bsibsi et al. 2002). Together, these data suggest that exposure to microbial motifs and/or inflammatory immune mediators can potentially sensitize astrocytes by increasing expression of PRRs and lead to enhanced immune responses of these cells following CNS damage or infection.
Although an ability of microbial components to induce TLR mRNA levels implies that these receptors are functionally expressed by astrocytes, this notion is supported by our demonstration that bacterial ligands for each TLR homolog can induce the activation of nuclear factor (NF)-κB in these cells (Bowman et al. 2003). Consistent with such an effect, we demonstrated that specific TLR ligands subsequently elicit IL-6 production. This finding has since been confirmed (Carpentier et al. 2005). The TLR4 ligand LPS has also been shown to be a potent stimulus for the expression of inflammatory chemokines monocyte chemoattractant protein (MCP)-1 and regulated upon activation, normal T-cell expressed and secreted (RANTES) (Carpentier et al. 2005). Furthermore, the availability of specific antibodies to TLR4 has enabled us to demonstrate that cell surface expression of this PRR is sensitive to stimulation with LPS. We showed that LPS treatment decreased cell surface TLR4 expression on astrocytes as determined by immunocytometry (Bowman et al. 2003) in a similar manner to that reported in murine macrophages (Nomura et al. 2000). As such, these data support the notion that astrocytes express functional Toll-like PRRs for bacterial motifs either constitutively, or following microbial challenge.
In addition to results obtained utilizing bacterial LPS, commercial preparations of peptidoglycan (PGN) have been reported to elicit cytokine and chemokine production by astrocytes (Bowman et al. 2003; Esen et al. 2004) and experiments utilizing cells derived from genetically deficient animals suggest that these effects are mediated by TLR2 (Esen et al. 2004). However, a recent study has suggested that TLR2-mediated immune cell responses by other cell types may be the result of lipoprotein and/or lipoteichoic acid (LTA) contamination of commercial PGN preparations (Travassos et al. 2004). Instead, these authors suggest that responses to PGN are mediated via novel members of the nucleotide-binding oligomerization domain (NOD) family of proteins. Although the assertion that TLR2 does not recognize PGN appears to have been refuted by later studies utilizing highly purified preparations of this ligand (Dziarski and Gupta 2005), the observation that maximal immune responses by PGN-stimulated astrocytes are attenuated, but not abolished, in the absence of TLR2 (Esen et al. 2004) suggests that alternative receptors exist for the perception of this bacterial product.
Cytoplasmic NOD proteins
NOD genes encode cytoplasmic proteins with an architecture that resembles a subclass of plant disease resistance (R) molecules (reviewed by Ting and Davis 2005). These proteins possess a variable number of N-terminal domains followed by a nucleotide-binding domain and leucine-rich repeats that are similar to those thought to be responsible for bacterial ligand binding to TLRs. In some family members, including NOD1 and NOD2, the N-terminal domain consists of caspase-recruitment domains (CARDs). NOD proteins have recently been identified in both immune and non-immune cell types, and at least two members of this family of proteins appear to serve as intracellular PRRs. Although NOD1 (also designated CARD4) was initially thought to mediate LPS-induced cellular responses, it is now recognized that NOD1 detects a diaminopimelate-containing N-acetylg-lucosamine-N-acetylmuramic acid (Glc-NAc-MurNAc) tripeptide motif found in PGNs from Gram-negative bacteria (Chamaillard et al. 2003; Girardin et al. 2003a,b). In contrast, NOD2 (also designated CARD15) has been suggested to be a more general sensor of bacterial PGNs as it recognizes the minimal muramyl dipeptide (MDP) motif present in all PGNs (Girardin et al. 2003b, 2003c; Inohara et al. 2003). Both NOD1 and NOD2 have been reported to associate with Rip2 kinase (also designated RICK and CARDIAK) (Ogura et al. 2001; Chin et al. 2002; Kobayashi et al. 2002; Yoo et al. 2002) the activation of which ultimately results in the liberation of cytosolic NF-κB and nuclear translocation (reviewed by Strober et al. 2006). Hence, activation of sentinel cells via NOD receptors could underlie, at least in part, bacteria-induced immune molecule production. Interestingly, extracellular application of ligands for these intracellular receptors can initiate cellular responses (Pauleau and Murray 2003) despite the predicted inability of these molecules to cross the plasma membrane. Although it is possible that phagocytosis and/or pinocytosis/macropinocytosis could explain this effect, the precise mechanisms responsible for this uptake have yet to be determined.
The similarities between TLR and NOD signal transduction pathways have led to the suggestion that these PRRs could act in an additive or synergistic manner to evoke optimal immune responses by leukocytes (reviewed by Strober et al. 2006). Indeed, a recent report by Uehara et al. (2005) has demonstrated that NOD1- and NOD2-specific ligands can synergistically increase inflammatory chemokine production by a monocytic cell line exposed to TLR2, TLR4 or TLR9 agonists. Importantly, these investigators employed RNA interference methods to knock down the expression of mRNA encoding NOD1 and NOD2 to demonstrate that these effects were, indeed, mediated via these novel PRRs (Uehara et al. 2005). These findings are consistent with earlier studies in other cell types showing that MDP acts in a synergistic manner with LPS to elicit inflammatory cytokine production (Ribi et al. 1979; Yang et al. 2001; Wolfert et al. 2002; Li et al. 2004) and raise the intriguing possibility that NOD molecules could mediate, at least in part, the inflammatory responses of glial cells following exposure to bacterial CNS pathogens.
Astrocytes have previously been shown to express class II transactivator, a potent transcriptional activator that regulates the expression of critical genes for antigen presentation and a member of the NOD protein family (reviewed by Ting and Davis 2005). We have recently completed a series of investigations into whether these cells also express NOD1 and NOD2 (Sterka et al. 2006). Our results indicate the presence of low levels of NOD2 mRNA and protein in resting astrocytes, but these cells exhibit little or no NOD1 expression. Such a finding is consistent with the apparently preferential expression of NOD2 by professional antigen-presenting cells in contrast to the almost exclusive expression of NOD1 by most epithelial cells (reviewed by Strober et al. 2006). Interestingly, we found that NOD2 expression was rapidly up-regulated in astrocytes following exposure to the bacterial CNS pathogens B. burgdorferi and N. meningitidis (Sterka et al. 2006). Furthermore, TLR ligands also proved to be potent inducers of NOD2 expression, in agreement with the previously documented ability of LPS to induce NOD2 mRNA expression in monocytic cells (Iwanaga et al. 2003). The ability of specific TLR ligands to modulate NOD molecule expression again supports the notion that cross-talk exists between disparate PRRs in astrocytes.
Circumstantial evidence for the functional expression of NOD proteins in astrocytes comes from the finding that these cells constitutively express Rip2 kinase, an important downstream adaptor molecule for NOD-mediated activation of NF-κB (Ogura et al. 2001; Chin et al. 2002; Kobayashi et al. 2002; Yoo et al. 2002). Furthermore, levels of Rip2 kinase in astrocytes are raised following exposure to B. burgdorferi or N. meningitidis (Sterka et al. 2006). More direct evidence for the functional nature of NOD molecule expression in astrocytes comes from the demonstration that extracellular application of the NOD2 ligand MDP induces modest but significant production of IL-6 and TNF-α by these cells (Sterka et al. 2006). The limited ability of MDP alone to elicit cytokine production by astrocytes is consistent with its previously documented effects on macrophages (Pauleau and Murray 2003). However, the most compelling evidence for an important role for NOD2 in astrocyte-mediated immune responses lies in the ability of MDP to significantly augment TLR ligand-induced cytokine production in a manner that exceeds the sum of each stimulus (Sterka et al. 2006). These findings, together with the previously documented ability of NOD proteins to act in a cooperative manner with TLRs to induce immune molecule production by leukocytes (Uehara et al. 2005), leads us to suggest that NOD molecules play an important role in bacteria-induced inflammatory responses by this major CNS cell type.
Differential roles for TLR2 in CNS bacterial infection and glial activation
Brain abscess accounts for approximately one in 10 000 hospital admissions in the USA and the leading etiologic agents of disease are the streptococcal strains and S. aureus (Mathisen and Johnson 1997; Townsend and Scheld 1998). Based upon its prevalence in human CNS infection, we have used S. aureus to establish an experimental brain abscess model in the mouse that accurately reflects the course of disease progression in humans, providing an excellent system with which to identify critical molecules responsible for the establishment of CNS antibacterial immunity (Kielian et al. 2001; Baldwin and Kielian 2004; Kielian 2004).
One well characterized PAMP of S. aureus is PGN, a component of the outer bacterial cell wall (Dziarski 2003; Weber et al. 2003), and a potent TLR2 agonist (Dziarski and Gupta 2005). With regard to brain abscess, PGN is released during normal bacterial growth as well as from dying organisms within the necrotic environment that is typical of the infection. In addition, many antibiotics that are used to treat CNS Gram-positive infections enhance PGN release from the bacterial cell wall (van der Flier et al. 2003; Weber et al. 2003), liberating additional antigen to engage PRRs such as TLR2. Collectively, these findings indicate that PGN represents a PAMP of significant biological importance in brain abscess as well as other CNS Gram-positive infections. Therefore, understanding the complex interactions between various PRRs may lead to the identification of new therapeutic targets to modulate pathogenic inflammation elicited by residual PGN subsequent to pathogen elimination in the CNS.
TLR2 and S. aureus-dependent glial activation
Microglia represent the innate immune effector cells of the CNS parenchyma that exhibit S. aureus bactericidal activity (Kielian et al. 2002). With regard to brain abscess, we have recently shown using primary microglia isolated from TLR2 knockout (KO) mice that TLR2 is necessary for microglial recognition of PGN from the outer cell wall of S. aureus but plays a relatively minor role in responses to intact bacteria (Kielian et al. 2005a). Specifically, the production of several pro-inflammatory mediators, including IL-1β, TNF-β, IL-12 p40, macrophage inflammatory protein (MIP)-2 and MCP-1, were significantly attenuated in PGN-treated TLR2 KO microglia compared with wild-type (WT) cells (Kielian et al. 2005a). In contrast, although the loss of TLR2 did exert a minor effect on a select number of pro-inflammatory mediators (i.e. IL-1β and MIP-2), overall TLR2 KO microglia were still capable of responding to intact S. aureus at levels equivalent to those observed in WT cells (Kielian et al. 2005a). Microglia also express CD14, another PRR that is important for mediating cell activation in response to LPS; however, recent evidence also supports a role for CD14 in the recognition of Gram-positive PAMPs such as PGN and LTA (Cleveland et al. 1996; Gupta et al. 1996; Dziarski et al. 2000). Similar to our findings in TLR2 KO microglia, CD14 participates in PGN-dependent microglial activation whereas responses to intact S. aureus are primarily CD14-independent (Esen and Kielian 2005). Therefore, based upon its ability to augment TLR2-dependent signaling, CD14 may represent a member of a multireceptor complex responsible for the establishment of immune responses to S. aureus in brain abscess.
Although TLR2 and CD14 contribute to S. aureus-dependent microglial activation to a limited extent, collectively these findings indicate that microglia utilize additional PRRs for recognizing intact bacteria. Two potential candidates include the phagocytic scavenger receptors macrophage scavenger receptor type AI/AII (MSR) and lectin-like oxidized low-density lipoprotein receptor (LOX)-1, whose expression is significantly increased in microglia following S. aureus exposure in vitro and in brain abscesses in vivo (Kielian et al. 2005a;b). Scavenger receptors encompass a broad range of molecules involved in the non-opsonic receptor-mediated phagocytosis of selected polyanionic acids such as PGN and LTA of S. aureus in addition to intact bacteria, and are expressed on activated microglia (Husemann et al. 2002; Peiser et al. 2002). Evidence to suggest that microglia may utilize scavenger receptors for pathogen internalization is provided by our findings that bacterial phagocytosis is opsonin-independent and does not require TLRs, because microglia lacking the central adapter molecule MyD88 were still capable of phagocytizing S. aureus (T. Kielian, unpublished results). However, recent evidence suggests that TLRs may regulate phagosome formation and maturation as well as modulate the transcription of some phagocytic receptors, whereas signaling via phagocytic receptors can also influence TLR signaling, revealing the existence of receptor cross-talk between TLRs and phagocytic PRRs (Underhill and Gantner 2004). This interaction is reflected in a recent report establishing the functional cooperation between TLR2 and LOX-1 in eliciting maximal macrophage activation in response to outer membrane protein A from the cell wall of Klebsiella pneumoniae, providing evidence to support the concept that these PRRs collaborate (Jeannin et al. 2005). In addition, our recent studies have revealed that TLR2-dependent signals regulate microglial LOX-1 expression in response to S. aureus (Kielian et al. 2005a). Collectively, the cross-talk between TLR2/CD14-dependent signaling of pro-inflammatory mediators coupled with bacterial phagocytosis via MSR and/or LOX-1 would ensure the establishment of maximal antibacterial immune responses aimed at pathogen elimination from the CNS.
Astrocytes play a pivotal role in the type and extent of CNS inflammatory responses. These cells probably play an important role in the initial recruitment and activation of peripheral immune cells into the CNS during neuroinflammation through the production of several cytokines and chemokines (Benveniste 1997; Dong and Benveniste 2001). Astrocytes have recently been shown to express TLR2 (Bsibsi et al. 2002; Bowman et al. 2003; Esen et al. 2004; Carpentier et al. 2005) and, although these cells are capable of responding to the well characterized TLR2 ligand PGN (Bowman et al. 2003; Esen et al. 2004), the functional significance of this receptor was not directly demonstrated until recently. Using primary astrocytes from TLR2 KO and WT mice, we have shown that TLR2 plays a pivotal role in the recognition of S. aureus and PGN, and in subsequent cytokine and chemokine expression by astrocytes (Esen et al. 2004). Interestingly, the production of these pro-inflammatory mediators was only partially attenuated in TLR2 KO astrocytes, suggesting that, similar to microglia, alternative receptors are also involved in bacterial recognition.
The implications of TLR2-dependent glial cell activation in the context of brain abscess are probably several-fold. First, parenchymal microglia and astrocytes may be involved in the initial recruitment of professional bactericidal phagocytes, such as neutrophils and macrophages, into the CNS through their elaboration of chemokines and pro-inflammatory cytokines. Second, activated microglia have the potential to influence the type and extent of antibacterial adaptive immune responses through their up-regulation of MHC class II and co-stimulatory molecule expression. Third, if glial activation persists, the continued release of pro-inflammatory mediators could damage surrounding normal brain parenchyma through bystander injury mechanism (see below). The continued use of various PRR transgenic and KO mice for in vivo studies should facilitate our understanding of immune mechanisms contributing to brain abscess pathogenesis.
Role of TLR2 in S. aureus-induced brain abscess
TLR2 has been shown to play an important role in the host immune response to Gram-positive bacterial infections in the periphery (Takeuchi et al. 2000) and, to some extent, this receptor dictates the ensuing host antibacterial response in Streptococcus pneumoniae meningitis (Echchannaoui et al. 2002; Koedel et al. 2003). However, before our studies the functional role of TLR2 in the context of a CNS parenchymal infection, such as brain abscess, had not been examined and may differ from that of meningitis based upon the highly focal nature of lesions in the former. Therefore, we evaluated the expression of numerous pro-inflammatory mediators previously determined to be pivotal for the host immune response during the acute phase of brain abscess development to ascertain whether defects in CNS bacterial recognition were evident in TLR2-deficient animals (Kielian et al. 2001, 2004). The kinetics of pro-inflammatory mediator production, including TNF-α, IL-1β, MIP-2 and inducible nitric oxide synthase, was delayed in TLR2 KO mice compared with WT animals, with lower levels of mediators in the KO mice during the acute stage of disease (Kielian et al. 2005b). Despite these differences, TLR2 did not play a significant role in controlling the extent of infection in brain abscess, with similar bacterial titers observed between TLR2 KO and WT animals, suggesting receptor redundancy for S. aureus neutralization in the CNS (Kielian et al. 2005b). Interestingly, the inflammatory phenotype detected in TLR2 KO mice was nearly identical to that observed in CD14 KO animals (T. Kielian, unpublished observation), strongly suggesting that these two receptors may cooperate in a multireceptor complex to facilitate pathogen recognition and the subsequent shaping of the inflammatory milieu during the acute stage of infection.
Innate and adaptive immunity are linked and recent evidence demonstrates that TLR-dependent signaling leads to the initiation of adaptive immune responses (Hoebe et al. 2004; Pasare and Medzhitov 2005). Of particular interest in our brain abscess studies with TLR2 KO mice was the significant induction of the T cell-derived cytokine IL-17 in brain abscesses of KO mice compared with WT animals (Kielian et al. 2005b). Because we detected elevated IL-17 levels in TLR2 KO mice during the early stages of primary infection, attributing cytokine production to memory T cells appears unusual as animals had not been previously infected with S. aureus. It is intriguing to speculate that increased IL-17 production in TLR2 KO animals represents a compensatory mechanism to counteract the observed delay in the production of neutrophil-attracting chemokines, because IL-17 is a potent stimulus for the production of these chemoattractants (Witowski et al. 2000; Maertzdorf et al. 2002; Ruddy et al. 2004). In addition, IL-17 has been shown to be pivotal in the establishment of antimicrobial immunity because IL-17 KO mice are more susceptible to systemic bacterial infections (Ye et al. 2001; Chung et al. 2002, 2003). Currently, the biological implications of raised IL-17 levels in brain abscesses of TLR2 KO mice and how the loss of TLR2 leads to increased cytokine expression are not known, but these issues represent an area of active investigation in our laboratory.
Based upon the work presented here, in conjunction with recent studies revealing that the responses of CD14-deficient mice are nearly identical to those observed in TLR2 KO animals (T. Kielian, unpublished results), we propose that these two PRRs are involved in a multireceptor complex that regulates pro-inflammatory mediator release during the acute stage of brain abscess. However, our findings also revealed that additional PRRs are responsible for bacterial containment because S. aureus burdens were not affected in TLR2 KO mice. Logical candidates include the phagocytic PRRs MSR and LOX-1, which have recently been shown to collaborate with TLR2 to regulate innate immune responses to various pathogens (Underhill and Gantner 2004; Jeannin et al. 2005). Interestingly, similar to our findings in S. aureus-activated microglia, we have found that TLR2-dependent signals influence the extent of LOX-1 induction in the brain abscess model, suggesting possible receptor cross-talk (Kielian et al. 2005b). In addition, CD14 also regulates LOX-1 and MSR levels in brain abscesses, suggestive of TLR2–CD14 interactions (T. Kielian, unpublished observations). Collectively, these studies have highlighted an important point, namely that the development of antibacterial immune responses in the CNS parenchyma cannot be accounted for by the activity of a single receptor, a concept that has emerged over recent years (Henneke et al. 2001, 2002; Koedel et al. 2003; Mukhopadhyay et al. 2004).
TLR signaling in bystander CNS Injury
Although the innate immune defense mechanisms are critical for recovery from CNS infections, these very mechanisms may also extend tissue damage by so-called bystander or collateral injury. Bystander injury is mediated by inflammatory factors that either are themselves neurotoxic, or promote the infiltration of leukocytes into the affected area and, in turn, propagate detrimental inflammatory milieu. Consequently, the ultimate outcome of an infectious insult is determined by a dynamic balance between accelerated pathogen elimination and augmented injury of CNS tissue.
As already mentioned in the previous section, bystander injury may be an important event in the pathology of brain abscesses. For example, cell wall fragments such as PGN and LTA, as well as bacterial DNA released from dead/dying organisms within the necrotic abscess, probably persist long after the elimination of viable bacteria. These PAMPs can serve as agonists for TLR2- and TLR9-mediated activation of resident glia, i.e. microglia and astrocytes, as well as infiltrating leukocytes. In essence, the continued presence of S. aureus PAMPs would lead the CNS innate immune cells to perceive the presence of an active infection, resulting in the persistent production of pro-inflammatory mediators that are capable of exerting neurotoxic effects in chronic brain abscesses.
Viral infections of the CNS pose the serious risk of neural tissue damage and consequently to devastating neurological dysfunction. It is highly probable that bystander injury mediated through TLRs and/or other PRRs plays a decisive role in many, if not all, of these conditions. A potential mechanism entails the shedding of viral components from infected target cells. The viral components are recognized by neighboring glial cells and trigger focal inflammatory reactions that damage neural tissue. Such a mechanism has been proposed for the pathology of human immunodeficiency virus (HIV)-1-associated neurological dysfunction (neuro-AIDS) (Nathanson et al. 1994; Haughey and Mattson 2003). In this condition invading macrophages and resident microglia are the principal cells infected with HIV-1, but their number is relatively low compared with the extent of inflammation and neurodegeneration (Kure et al. 1990; Budka 1991). Moreover, neuronal loss results from bystander injury by a host of neurotoxic agents generated by glial cells (Lipton 1991; Genis et al. 1992; Wesselingh et al. 1993). Consequently, the likely scenario features the amplification of pro-inflammatory reactions by glial cells in response to viral components released from infected macrophages/microglia.
Double-stranded RNA is of a particular interest as a putative pro-inflammatory viral component, because most viruses either contain dsRNA structures within their genomes or generate dsRNA during their replication cycle (Jacobs and Langland 1996), and because dsRNA is a potent stimulant of innate immunity (Kopp and Medzhitov 1999; Akira et al. 2001). Once released, the extracellular dsRNA exerts its pro-inflammatory action through the engagement of TLR3 (Alexopoulou et al. 2001). The ligand–receptor binding triggers the up-regulation of pro-inflammatory cytokines as well as antiviral and secondary pro-inflammatory genes (Kawai et al. 2001; Doyle et al. 2002). TLR3 is expressed by microglia and astrocytes in the brain (Bsibsi et al. 2002) and in culture (Olson and Miller 2004; Carpentier et al. 2005; Farina et al. 2005; Scumpia et al. 2005). Interestingly, cultured human fetal astrocytes feature preferential expression of TLR3 over other TLRs (Farina et al. 2005). The expression of TLR3 is also profoundly up-regulated within multiple sclerosis lesions (Bsibsi et al. 2002) strongly buttressing the role of TLR3 in the pathology of this neurodegenerative disease that is believed to feature viral etiology (Fazakerley and Walker 2003; Scarisbrick and Rodriguez 2003). In the experimental setting, intraventricular administration of dsRNA induces chronic activation of glia in rat brain characterized by the expression of MHC class II in microglia, and the expression of pro-inflammatory IL-1β in astrocytes (Melton et al. 2003). Moreover, stimulation of glial cells with dsRNA up-regulates the expression of several cytokines, chemokines and co-stimulatory molecules, and the generation of nitric oxide (Olson and Miller 2004; Carpentier et al. 2005; Konat et al. 2005; Scumpia et al. 2005). In addition, secondary structures of cellular RNA are also potent activators of TLR3 (Kariko et al. 2004). Consequently, cellular RNA released from cells damaged in the course of microbial infection may be an ancillary agent to further promote bystander injury.
Conclusions
The presence of an array of TLRs and other PRRs in astrocytes provides a mechanism by which this major glial cell type can respond to disparate microbial pathogens and promote inflammation within the CNS. Furthermore, the ability of microbial motifs and/or inflammatory mediators to induce a broad up-regulation in the expression of multiple PRR types and to elicit synergistic immune responses has important implications. A scenario might be envisioned in which exposure of an astrocyte to microbial products or an inflammatory environment sensitizes that cell to further assault and enhances its ability to mount an immune response. As such, initiation of astrocyte immune functions via these PRR receptors is likely to be an important component of the pathologies of inflammatory CNS disorders such as meningitis and central nervous complications of sepsis. The studies performed to date in the mouse experimental brain abscess model have begun to elucidate the roles of TLR2 and alternative PRRs in disease pathogenesis and their effects on cytokines, which play a pivotal role in the generation of CNS antibacterial immune responses. However, there are numerous issues that remain to be resolved regarding the role of PRRs in the evolution of brain abscess. For example, the formation of a multireceptor PRR complex to facilitate efficient recognition of S. aureus in the brain has yet to be demonstrated directly. In addition, the mechanism(s) by which TLR2 influences the initiation of antibacterial adaptive immune responses in brain abscesses remain to be elucidated. Moreover, one should also be aware that PAMPs released during various stages of infection may augment inflammatory reactions and further extend CNS tissue damage through bystander injury mechanisms. Therefore, an understanding of mechanisms that govern TLR signaling in glial cells will undoubtedly facilitate the design of effective therapeutic regimens for CNS infections that would be capable of pathogen elimination without the destruction of surrounding brain parenchyma.
Acknowledgments
This work was supported by the National Institutes of Health (NS40730 and MH65297 to TK; NS051787 to GWK), the National Institute of Neurological Disorders and Stroke-supported Core Facility at University of Arkansas for Medical Sciences (P30 NS047546 to TK), the Arkansas Biosciences Institute (TK) and West Virginia University School of Medicine Research Development Grant (GWK).
Abbreviations used
- CARD
caspase-recruitment domain
- dsRNA
double-stranded RNA
- HIV
human immunodeficiency virus
- IFN
interferon
- IL
interleukin
- IRAK
IL-1 receptor-associated kinase
- IRF
IFN regulatory factor
- KO
knockout
- LOX
lectin-like oxidized low-density lipoprotein receptor
- LPS
lipopolysaccharide
- LTA
lipoteichoic acid
- MAPK
mitogen-activated protein kinase
- MCP
monocyte chemoattractant protein
- MDP
muramyl dipeptide
- MHC
major histocompatibility complex
- MIP
macrophage inflammatory protein
- MSR
macrophage scavenger receptor type AI/AII
- MyD88
myeloid differentiation factor 88
- NF-κB
nuclear factor-κB
- NOD
nucleotide-binding oligomerization domain
- PAMP
pathogen-associated molecular pattern
- PGN
peptidoglycan
- PRR
pattern recognition receptor
- TIR
Toll/IL-1 receptor
- TLR
toll-like receptor
- TNF
tumor necrosis factor
- TRAF6
TNF receptor-associated factor 6
- TRIF
TIR domain-containing adaptor inducing IFNβ
- WT
wild type
References
- Akira S. Toll-like receptors and innate immunity. Adv Immunol. 2001;78:1–56. doi: 10.1016/s0065-2776(01)78001-7. [DOI] [PubMed] [Google Scholar]
- Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- Baldwin AC, Kielian T. Persistent immune activation associated with a mouse model of Staphylococcus aureus-induced experimental brain abscess. J Neuroimmunol. 2004;151:24–32. doi: 10.1016/j.jneuroim.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Barton GM, Medzhitov R. Toll-like receptor signaling pathways. Science. 2003;300:1524–1525. doi: 10.1126/science.1085536. [DOI] [PubMed] [Google Scholar]
- Bell JK, Botos I, Hall PR, Askins J, Shiloach J, Segal DM, Davies DR. The molecular structure of the Toll-like receptor 3 ligand-binding domain. Proc Natl Acad Sci USA. 2005;102:10 976–10 980. doi: 10.1073/pnas.0505077102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benveniste EN. Cytokines: influence on glial cell gene expression and function. In: Blalock JE, editor. Neuroimmunoendorinology. Vol. 69. S Karger: Basel; 1997. pp. 31–75. [DOI] [PubMed] [Google Scholar]
- Benveniste EN, Sparacio SM, Norris JG, Grenett HE, Fuller GM. Induction and regulation of interleukin-6 gene expression in rat astrocytes. J Neuroimmunol. 1990;30:201–212. doi: 10.1016/0165-5728(90)90104-u. [DOI] [PubMed] [Google Scholar]
- Biragyn A, Ruffini PA, Leifer CA, et al. Toll-like receptor 4-dependent activation of dendritic cells by beta-defensin 2. Science. 2002;298:1025–1029. doi: 10.1126/science.1075565. [DOI] [PubMed] [Google Scholar]
- Bowman CC, Rasley A, Tranguch SL, Marriott I. Cultured astrocytes express toll-like receptors for bacterial products. Glia. 2003;43:281–291. doi: 10.1002/glia.10256. [DOI] [PubMed] [Google Scholar]
- Bsibsi M, Ravid R, Gveric D, van Noort JM. Broad expression of Toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol. 2002;61:1013–1021. doi: 10.1093/jnen/61.11.1013. [DOI] [PubMed] [Google Scholar]
- Budka H. Neuropathology of human immunodeficiency virus infection. Brain Pathol. 1991;1:163–175. doi: 10.1111/j.1750-3639.1991.tb00656.x. [DOI] [PubMed] [Google Scholar]
- Carpentier PA, Begolka WS, Olson JK, Elhofy A, Karpus WJ, Miller SD. Differential activation of astrocytes by innate and adaptive immune stimuli. Glia. 2005;49:360–374. doi: 10.1002/glia.20117. [DOI] [PubMed] [Google Scholar]
- Cauwels A, Frei K, Sansano S, Fearns C, Ulevitch R, Zimmerli W, Landmann R. The origin and function of soluble CD14 in experimental bacterial meningitis. J Immunol. 1999;162:4762–4772. [PubMed] [Google Scholar]
- Chamaillard M, Hashimoto M, Horie Y, et al. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat Immunol. 2003;4:702–707. doi: 10.1038/ni945. [DOI] [PubMed] [Google Scholar]
- Chin AI, Dempsey PW, Bruhn K, Miller JF, Xu Y, Cheng G. Involvement of receptor-interacting protein 2 in innate and adaptive immune responses. Nature. 2002;416:190–194. doi: 10.1038/416190a. [DOI] [PubMed] [Google Scholar]
- Chung DR, Chitnis T, Panzo RJ, Kasper DL, Sayegh MH, Tzianabos AO. CD4+ T cells regulate surgical and post-infectious adhesion formation. J Exp Med. 2002;195:1471–1478. doi: 10.1084/jem.20020028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung DR, Kasper DL, Panzo RJ, Chitnis T, Grusby MJ, Sayegh MH, Tzianabos AO. CD4+ T cells mediate abscess formation in intra-abdominal sepsis by an IL-17-dependent mechanism. J Immunol. 2003;170:1958–1963. doi: 10.4049/jimmunol.170.4.1958. [DOI] [PubMed] [Google Scholar]
- Cleveland MG, Gorham JD, Murphy TL, Tuomanen E, Murphy KM. Lipoteichoic acid preparations of gram-positive bacteria induce interleukin-12 through a CD14-dependent pathway. Infect Immun. 1996;64:1906–1912. doi: 10.1128/iai.64.6.1906-1912.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalpke AH, Schafer MK, Frey M, Zimmermann S, Tebbe J, Weihe E, Heeg K. Immunostimulatory CpG-DNA activates murine microglia. J Immunol. 2002;168:4854–4863. doi: 10.4049/jimmunol.168.10.4854. [DOI] [PubMed] [Google Scholar]
- Dong Y, Benveniste EN. Immune function of astrocytes. Glia. 2001;36:180–190. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- Doyle SE, Vaidya SA, O’Connell R, et al. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity. 2002;17:251–263. doi: 10.1016/s1074-7613(02)00390-4. [DOI] [PubMed] [Google Scholar]
- Doyle SE, O’Connell R, Vaidya SA, Chow EK, Yee K, Cheng G. Toll-like receptor 3 mediates a more potent antiviral response than Toll-like receptor 4. J Immunol. 2003;170:3565–3571. doi: 10.4049/jimmunol.170.7.3565. [DOI] [PubMed] [Google Scholar]
- Dziarski R. Recognition of bacterial peptidoglycan by the innate immune system. Cell Mol Life Sci. 2003;60:1793–1804. doi: 10.1007/s00018-003-3019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziarski R, Gupta D. Staphylococcus aureus peptidoglycan is a toll-like receptor 2 activator: a reevaluation. Infect Immun. 2005;73:5212–5216. doi: 10.1128/IAI.73.8.5212-5216.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziarski R, Ulmer AJ, Gupta D. Interactions of CD14 with components of gram-positive bacteria. Chem Immunol. 2000;74:83–107. doi: 10.1159/000058761. [DOI] [PubMed] [Google Scholar]
- Echchannaoui H, Frei K, Schnell C, Leib SL, Zimmerli W, Landmann R. Toll-like receptor 2-deficient mice are highly susceptible to Streptococcus pneumoniae meningitis because of reduced bacterial clearing and enhanced inflammation. J Infect Dis. 2002;186:798–806. doi: 10.1086/342845. [DOI] [PubMed] [Google Scholar]
- Esen N, Kielian T. Recognition of Staphylococcus aureus-derived peptidoglycan (PGN) but not intact bacteria is mediated by CD14 in microglia. J Neuroimmunol. 2005;170:93–104. doi: 10.1016/j.jneuroim.2005.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esen N, Tanga FY, DeLeo JA, Kielian T. Toll-like receptor 2 (TLR2) mediates astrocyte activation in response to the Gram-positive bacterium Staphylococcus aureus. J Neurochem. 2004;88:746–758. doi: 10.1046/j.1471-4159.2003.02202.x. [DOI] [PubMed] [Google Scholar]
- Farina C, Krumbholz M, Giese T, Hartmann G, Aloisi F, Meinl E. Preferential expression and function of Toll-like receptor 3 in human astrocytes. J Neuroimmunol. 2005;159:12–19. doi: 10.1016/j.jneuroim.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Fazakerley JK, Walker R. Virus demyelination. J Neurovirol. 2003;9:148–164. doi: 10.1080/13550280390194046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Flier M, Geelen SP, Kimpen JL, Hoepelman IM, Tuomanen EI. Reprogramming the host response in bacterial meningitis: how best to improve outcome? Clin Microbiol Rev. 2003;16:415–429. doi: 10.1128/CMR.16.3.415-429.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forloni G, Mangiarotti F, Angeretti N, Lucca E, De Simoni MG. Beta-amyloid fragment potentiates IL-6 and TNF-alpha secretion by LPS in astrocytes but not in microglia. Cytokine. 1997;9:759–762. doi: 10.1006/cyto.1997.0232. [DOI] [PubMed] [Google Scholar]
- Genis P, Jett M, Bernton EW, Boyle T, Gelbard HA, Dzenko K, Keane RW, Resnick L, Mizrachi Y, Volsky DJ. Cytokines and arachidonic metabolites produced during human immunodeficiency virus (HIV)-infected macrophage–astroglia interactions: implications for the neuropathogenesis of HIV disease. J Exp Med. 1992;176:1703–1718. doi: 10.1084/jem.176.6.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girardin SE, Boneca IG, Carneiro LA, et al. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science. 2003a;300:1584–1587. doi: 10.1126/science.1084677. [DOI] [PubMed] [Google Scholar]
- Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, Philpott DJ, Sansonetti PJ. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. 2003b;278:8869–8872. doi: 10.1074/jbc.C200651200. [DOI] [PubMed] [Google Scholar]
- Girardin SE, Travassos LH, Herve M, Blanot D, Boneca IG, Philpott DJ, Sansonetti PJ, Mengin-Lecreulx D. Peptidoglycan molecular requirements allowing detection by Nod1 and Nod2. J Biol Chem. 2003c;278:41 702–41 708. doi: 10.1074/jbc.M307198200. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Scarano F, Baltuch G. Microglia as mediators of inflammatory and degenerative diseases. Annu Rev Neurosci. 1999;22:219–240. doi: 10.1146/annurev.neuro.22.1.219. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Scarano F, Martin-Garcia J. The neuropathogenesis of AIDS. Nat Rev Immunol. 2005;5:69–81. doi: 10.1038/nri1527. [DOI] [PubMed] [Google Scholar]
- Gottschall PE, Tatsuno I, Arimura A. Regulation of interleukin-6 (IL-6) secretion in primary cultured rat astrocytes: synergism of interleukin-1 (IL-1) and pituitary adenylate cyclase activating polypeptide (PACAP) Brain Res. 1994;637:197–203. doi: 10.1016/0006-8993(94)91233-5. [DOI] [PubMed] [Google Scholar]
- Gupta D, Kirkland TN, Viriyakosol S, Dziarski R. CD14 is a cell-activating receptor for bacterial peptidoglycan. J Biol Chem. 1996;271:23 310–23 316. doi: 10.1074/jbc.271.38.23310. [DOI] [PubMed] [Google Scholar]
- Haughey NJ, Mattson MP. Alzheimer’s amyloid beta-peptide enhances ATP/gap junction-mediated calcium-wave propagation in astrocytes. Neuromol Med. 2003;3:173–180. doi: 10.1385/NMM:3:3:173. [DOI] [PubMed] [Google Scholar]
- Henneke P, Takeuchi O, van Strijp JA, et al. Novel engagement of CD14 and multiple toll-like receptors by group B streptococci. J Immunol. 2001;167:7069–7076. doi: 10.4049/jimmunol.167.12.7069. [DOI] [PubMed] [Google Scholar]
- Henneke P, Takeuchi O, Malley R, et al. Cellular activation, phagocytosis, and bactericidal activity against group B streptococcus involve parallel myeloid differentiation factor 88-dependent and independent signaling pathways. J Immunol. 2002;169:3970–3977. doi: 10.4049/jimmunol.169.7.3970. [DOI] [PubMed] [Google Scholar]
- Hoebe K, Janssen E, Beutler B. The interface between innate and adaptive immunity. Nat Immunol. 2004;5:971–974. doi: 10.1038/ni1004-971. [DOI] [PubMed] [Google Scholar]
- Horng T, Barton GM, Flavell RA, Medzhitov R. The adaptor molecule TIRAP provides signalling specificity for Toll-like receptors. Nature. 2002;420:329–333. doi: 10.1038/nature01180. [DOI] [PubMed] [Google Scholar]
- Husemann J, Loike JD, Anankov R, Febbraio M, Silverstein SC. Scavenger receptors in neurobiology and neuropathology: their role on microglia and other cells of the nervous system. Glia. 2002;40:195–205. doi: 10.1002/glia.10148. [DOI] [PubMed] [Google Scholar]
- Inohara N, Ogura Y, Fontalba A, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J Biol Chem. 2003;278:5509–5512. doi: 10.1074/jbc.C200673200. [DOI] [PubMed] [Google Scholar]
- Ishii KJ, Coban C, Akira S. Manifold mechanisms of Toll-like receptor–ligand recognition. J Clin Immunol. 2005;25:511–521. doi: 10.1007/s10875-005-7829-1. [DOI] [PubMed] [Google Scholar]
- Iwanaga Y, Davey MP, Martin TM, Planck SR, DePriest ML, Baugh MM, Suing CM, Rosenbaum JT. Cloning, sequencing and expression analysis of the mouse NOD2/CARD15 gene. Inflamm Res. 2003;52:272–276. doi: 10.1007/s00011-003-1170-z. [DOI] [PubMed] [Google Scholar]
- Jack CS, Arbour N, Manusow J, Montgrain V, Blain M, McCrea E, Shapiro A, Antel JP. TLR signaling tailors innate immune responses in human microglia and astrocytes. J Immunol. 2005;175:4320–4330. doi: 10.4049/jimmunol.175.7.4320. [DOI] [PubMed] [Google Scholar]
- Jacobs BL, Langland JO. When two strands are better than one: the mediators and modulators of the cellular responses to double-stranded RNA. Virology. 1996;219:339–349. doi: 10.1006/viro.1996.0259. [DOI] [PubMed] [Google Scholar]
- Jeannin P, Bottazzi B, Sironi M, et al. Complexity and complementarity of outer membrane protein A recognition by cellular and humoral innate immunity receptors. Immunity. 2005;22:551–560. doi: 10.1016/j.immuni.2005.03.008. [DOI] [PubMed] [Google Scholar]
- Johnson GB, Brunn GJ, Kodaira Y, Platt JL. Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by Toll-like receptor 4. J Immunol. 2002;168:5233–5239. doi: 10.4049/jimmunol.168.10.5233. [DOI] [PubMed] [Google Scholar]
- Kaisho T, Akira S. Pleiotropic function of Toll-like receptors. Microbes Infect. 2004;6:1388–1394. doi: 10.1016/j.micinf.2004.08.019. [DOI] [PubMed] [Google Scholar]
- Kaisho T, Hoshino K, Iwabe T, Takeuchi O, Yasui T, Akira S. Endotoxin can induce MyD88-deficient dendritic cells to support T(h)2 cell differentiation. Int Immunol. 2002;14:695–700. doi: 10.1093/intimm/dxf039. [DOI] [PubMed] [Google Scholar]
- Kariko K, Ni H, Capodici J, Lamphier M, Weissman D. mRNA is an endogenous ligand for Toll-like receptor 3. J Biol Chem. 2004;279:12 542–12 550. doi: 10.1074/jbc.M310175200. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. Toll-like receptor downstream signaling. Arthritis Res Ther. 2004;7:12–19. doi: 10.1186/ar1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Takeuchi O, Fujita T, Inoue J, Muhlradt PF, Sato S, Hoshino K, Akira S. Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J Immunol. 2001;167:5887–5894. doi: 10.4049/jimmunol.167.10.5887. [DOI] [PubMed] [Google Scholar]
- Kielian T. Immunopathogenesis of brain abscess. J Neuroinflammation. 2004;1:16. doi: 10.1186/1742-2094-1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T, Barry B, Hickey WF. CXC chemokine receptor-2 ligands are required for neutrophil-mediated host defense in experimental brain abscesses. J Immunol. 2001;166:4634–4643. doi: 10.4049/jimmunol.166.7.4634. [DOI] [PubMed] [Google Scholar]
- Kielian T, Mayes P, Kielian M. Characterization of microglial responses to Staphylococcus aureus: effects on cytokine, costimulatory molecule, and Toll-like receptor expression. J Neuroimmunol. 2002;130:86–99. doi: 10.1016/s0165-5728(02)00216-3. [DOI] [PubMed] [Google Scholar]
- Kielian T, Bearden ED, Baldwin AC, Esen N. IL-1 and TNF-alpha play a pivotal role in the host immune response in a mouse model of Staphylococcus aureus-induced experimental brain abscess. J Neuropathol Exp Neurol. 2004;63:381–396. doi: 10.1093/jnen/63.4.381. [DOI] [PubMed] [Google Scholar]
- Kielian T, Esen N, Bearden ED. Toll-like receptor 2 (TLR2) is pivotal for recognition of S. aureus peptidoglycan but not intact bacteria by microglia. Glia. 2005a;49:567–576. doi: 10.1002/glia.20144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T, Haney A, Mayes PM, Garg S, Esen N. Toll-like receptor 2 modulates the proinflammatory milieu in Staphylococcus aureus-induced brain abscess. Infect Immun. 2005b;73:7428–7435. doi: 10.1128/IAI.73.11.7428-7435.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimberlin DW, Velasco S, Paris MM, Hickey SM, McCracken GH, Jr, Nisen PD. Modulation of expression of genes involved in the inflammatory response by lipopolysaccharide and temperature in cultured human astroglial cells. Immunol Invest. 1995;24:775–785. doi: 10.3109/08820139509060705. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Inohara N, Hernandez LD, Galan JE, Nunez G, Janeway CA, Medzhitov R, Flavell RA. RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature. 2002;416:194–199. doi: 10.1038/416194a. [DOI] [PubMed] [Google Scholar]
- Koedel U, Angele B, Rupprecht T, Wagner H, Roggenkamp A, Pfister HW, Kirschning CJ. Toll-like receptor 2 participates in mediation of immune response in experimental pneumococcal meningitis. J Immunol. 2003;170:438–444. doi: 10.4049/jimmunol.170.1.438. [DOI] [PubMed] [Google Scholar]
- Konat GW, Banaszewska M, Krasowska A. Double stranded RNA triggers nitric oxide generation and cytokine expression in astrocytes. J Neurochem. 2005;94:PTW03–11. [Google Scholar]
- Kopp EB, Medzhitov R. The Toll-receptor family and control of innate immunity. Curr Opin Immunol. 1999;11:13–18. doi: 10.1016/s0952-7915(99)80003-x. [DOI] [PubMed] [Google Scholar]
- Kopp EB, Medzhitov R. Recognition of microbial infection by Toll-like receptors. Curr Opin Immunol. 2003;15:396–401. doi: 10.1016/s0952-7915(03)00080-3. [DOI] [PubMed] [Google Scholar]
- Kure K, Lyman WD, Weidenheim KM, Dickson DW. Cellular localization of an HIV-1 antigen in subacute AIDS encephalitis using an improved double-labeling immunohistochemical method. Am J Pathol. 1990;136:1085–1092. [PMC free article] [PubMed] [Google Scholar]
- Li J, Moran T, Swanson E, Julian C, Harris J, Bonen DK, Hedl M, Nicolae DL, Abraham C, Cho JH. Regulation of IL-8 and IL-1beta expression in Crohn’s disease associated NOD2/CARD15 mutations. Hum Mol Genet. 2004;13:1715–1725. doi: 10.1093/hmg/ddh182. [DOI] [PubMed] [Google Scholar]
- Li M, Carpio DF, Zheng Y, Bruzzo P, Singh V, Ouaa ZF, Medzhitov RM, Beg AA. An essential role of the NF-kappa B/Toll-like receptor pathway in induction of inflammatory and tissue-repair gene expression by necrotic cells. J Immunol. 2001;166:7128–7135. doi: 10.4049/jimmunol.166.12.7128. [DOI] [PubMed] [Google Scholar]
- Lieberman AP, Pitha PM, Shin HS, Shin ML. Production of tumor necrosis factor and other cytokines by astrocytes stimulated with lipopolysaccharide or a neurotropic virus. Proc Natl Acad Sci USA. 1989;86:6348–6352. doi: 10.1073/pnas.86.16.6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien E, Ingalls RR. Toll-like receptors. Crit Care Med. 2002;30:S1–S11. [PubMed] [Google Scholar]
- Lipton SA. HIV-related neurotoxicity. Brain Pathol. 1991;1:193–199. doi: 10.1111/j.1750-3639.1991.tb00659.x. [DOI] [PubMed] [Google Scholar]
- Maertzdorf J, Osterhaus AD, Verjans GM. IL-17 expression in human herpetic stromal keratitis: modulatory effects on chemokine production by corneal fibroblasts. J Immunol. 2002;169:5897–5903. doi: 10.4049/jimmunol.169.10.5897. [DOI] [PubMed] [Google Scholar]
- Maslinska D, Laure-Kamionowska M, Maslinski S. Toll-like receptors in rat brains injured by hypoxic–ischaemia or exposed to staphylococcal alpha-toxin. Folia Neuropathol. 2004;42:125–132. [PubMed] [Google Scholar]
- Mathisen GE, Johnson JP. Brain abscess. Clin Infect Dis. 1997;25:763–779. doi: 10.1086/515541. [DOI] [PubMed] [Google Scholar]
- McKimmie CS, Fazakerley JK. In response to pathogens, glial cells dynamically and differentially regulate Toll-like receptor gene expression. J Neuroimmunol. 2005;169:116–125. doi: 10.1016/j.jneuroim.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Melton LM, Keith AB, Davis S, Oakley AE, Edwardson JA, Morris CM. Chronic glial activation, neurodegeneration, and APP immunoreactive deposits following acute administration of double-stranded RNA. Glia. 2003;44:1–12. doi: 10.1002/glia.10276. [DOI] [PubMed] [Google Scholar]
- Miyake K. Innate recognition of lipopolysaccharide by CD14 and toll-like receptor 4-MD-2: unique roles for MD-2. Int Immunopharmacol. 2003;3:119–128. doi: 10.1016/s1567-5769(02)00258-8. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S, Herre J, Brown GD, Gordon S. The potential for Toll-like receptors to collaborate with other innate immune receptors. Immunology. 2004;112:521–530. doi: 10.1111/j.1365-2567.2004.01941.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura A, Johns EJ, Imaizumi A, Abe T, Kohsaka T. Regulation of tumour necrosis factor and interleukin-6 gene transcription by beta2-adrenoceptor in the rat astrocytes. J Neuroimmunol. 1998;88:144–153. doi: 10.1016/s0165-5728(98)00109-x. [DOI] [PubMed] [Google Scholar]
- Nathanson N, Cook DG, Kolson DL, Gonzalez-Scarano F. Pathogenesis of HIV encephalopathy. Ann N Y Acad Sci. 1994;724:87–106. doi: 10.1111/j.1749-6632.1994.tb38898.x. [DOI] [PubMed] [Google Scholar]
- Nishimura M, Naito S. Tissue-specific mRNA expression profiles of human toll-like receptors and related genes. Biol Pharm Bull. 2005;28:886–892. doi: 10.1248/bpb.28.886. [DOI] [PubMed] [Google Scholar]
- Nomura F, Akashi S, Sakao Y, et al. Cutting edge: endotoxin tolerance in mouse peritoneal macrophages correlates with down-regulation of surface toll-like receptor 4 expression. J Immunol. 2000;164:3476–3479. doi: 10.4049/jimmunol.164.7.3476. [DOI] [PubMed] [Google Scholar]
- Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Nunez G. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J Biol Chem. 2001;276:4812–4818. doi: 10.1074/jbc.M008072200. [DOI] [PubMed] [Google Scholar]
- Oh JW, Schwiebert LM, Benveniste EN. Cytokine regulation of CC and CXC chemokine expression by human astrocytes. J Neurovirol. 1999;5:82–94. doi: 10.3109/13550289909029749. [DOI] [PubMed] [Google Scholar]
- Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173:3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- Ozinsky A, Underhill DM, Fontenot JD, Hajjar AM, Smith KD, Wilson CB, Schroeder L, Aderem A. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc Natl Acad Sci USA. 2000;97:13766–13771. doi: 10.1073/pnas.250476497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palliser D, Huang Q, Hacohen N, Lamontagne SP, Guillen E, Young RA, Eisen HN. A role for Toll-like receptor 4 in dendritic cell activation and cytolytic CD8+ T cell differentiation in response to a recombinant heat shock fusion protein. J Immunol. 2004;172:2885–2893. doi: 10.4049/jimmunol.172.5.2885. [DOI] [PubMed] [Google Scholar]
- Pasare C, Medzhitov R. Toll-like receptors: linking innate and adaptive immunity. Adv Exp Med Biol. 2005;560:11–18. doi: 10.1007/0-387-24180-9_2. [DOI] [PubMed] [Google Scholar]
- Paterson HM, Murphy TJ, Purcell EJ, Shelley O, Kriynovich SJ, Lien E, Mannick JA, Lederer JA. Injury primes the innate immune system for enhanced Toll-like receptor reactivity. J Immunol. 2003;171:1473–1483. doi: 10.4049/jimmunol.171.3.1473. [DOI] [PubMed] [Google Scholar]
- Pauleau AL, Murray PJ. Role of nod2 in the response of macrophages to toll-like receptor agonists. Mol Cell Biol. 2003;23:7531–7539. doi: 10.1128/MCB.23.21.7531-7539.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiser L, Mukhopadhyay S, Gordon S. Scavenger receptors in innate immunity. Curr Opin Immunol. 2002;14:123–128. doi: 10.1016/s0952-7915(01)00307-7. [DOI] [PubMed] [Google Scholar]
- Perides G, Tanner-Brown LM, Eskildsen MA, Klempner MS. Borrelia burgdorferi induces matrix metalloproteinases by neural cultures. J Neurosci Res. 1999;58:779–790. doi: 10.1002/(sici)1097-4547(19991215)58:6<779::aid-jnr5>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Persidsky Y. Model systems for studies of leukocyte migration across the blood–brain barrier. J Neurovirol. 1999;5:579–590. doi: 10.3109/13550289909021287. [DOI] [PubMed] [Google Scholar]
- Rasley A, Anguita J, Marriott I. Borrelia burgdorferi induces inflammatory mediator production by murine microglia. J Neuroimmunol. 2002a;130:22–31. doi: 10.1016/s0165-5728(02)00187-x. [DOI] [PubMed] [Google Scholar]
- Rasley A, Bost KL, Olson JK, Miller SD, Marriott I. Expression of functional NK-1 receptors in murine microglia. Glia. 2002b;37:258–267. doi: 10.1002/glia.10034. [DOI] [PubMed] [Google Scholar]
- Rasley A, Tranguch SL, Rati DM, Marriott I. Murine microglia express the immunosuppressive cytokine, interleukin-10, following exposure to Borrelia burgdorferi or Neisseria meningitidis. Glia. 2006;53:583–592. doi: 10.1002/glia.20314. [DOI] [PubMed] [Google Scholar]
- Ribi EE, Cantrell JL, Von Eschen KB, Schwartzman SM. Enhancement of endotoxic shock by N-acetylmuramyl-L-alanyl-(1-seryl)-D-isoglutamine (muramyl dipeptide) Cancer Res. 1979;39:4756–4759. [PubMed] [Google Scholar]
- Roach JC, Glusman G, Rowen L, Kaur A, Purcell MK, Smith KD, Hood LE, Aderem A. The evolution of vertebrate Toll-like receptors. Proc Natl Acad Sci USA. 2005;102:9577–9582. doi: 10.1073/pnas.0502272102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruddy MJ, Shen F, Smith JB, Sharma A, Gaffen SL. Interleukin-17 regulates expression of the CXC chemokine LIX/CXCL5 in osteoblasts: implications for inflammation and neutrophil recruitment. J Leukoc Biol. 2004;76:135–144. doi: 10.1189/jlb.0204065. [DOI] [PubMed] [Google Scholar]
- Scarisbrick IA, Rodrigue ZM. Hit–Hit and Hit–Run: viruses in the playing field of multiple sclerosis. Curr Neurol Neurosci Report. 2003;3:265–271. doi: 10.1007/s11910-003-0087-9. [DOI] [PubMed] [Google Scholar]
- Schumann RR, Pfeil D, Freyer D, Buerger W, Lamping N, Kirschning CJ, Goebel UB, Weber JR. Lipopolysaccharide and pneumococcal cell wall components activate the mitogen activated protein kinases (MAPK) erk-1, erk-2, and p38 in astrocytes. Glia. 1998;22:295–305. doi: 10.1002/(sici)1098-1136(199803)22:3<295::aid-glia8>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Scumpia PO, Kelly KM, Reeves WH, Stevens BR. Double-stranded RNA signals antiviral and inflammatory programs and dysfunctional glutamate transport in TLR3-expressing astrocytes. Glia. 2005;52:153–162. doi: 10.1002/glia.20234. [DOI] [PubMed] [Google Scholar]
- Shinobu N, Iwamura T, Yoneyama M, Yamaguchi K, Suhara W, Fukuhara Y, Amano F, Fujita T. Involvement of TI-RAP/MAL in signaling for the activation of interferon regulatory factor 3 by lipopolysaccharide. FEBS Lett. 2002;517:251–256. doi: 10.1016/s0014-5793(02)02636-4. [DOI] [PubMed] [Google Scholar]
- Smiley ST, King JA, Hancock WW. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J Immunol. 2001;167:2887–2894. doi: 10.4049/jimmunol.167.5.2887. [DOI] [PubMed] [Google Scholar]
- Soos JM, Ashley TA, Morrow J, Patarroyo JC, Szente BE, Zamvil SS. Differential expression of B7 co-stimulatory molecules by astrocytes correlates with T cell activation and cytokine production. Int Immunol. 1999;11:1169–1179. doi: 10.1093/intimm/11.7.1169. [DOI] [PubMed] [Google Scholar]
- Sterka D, Jr, Rati DM, Marriott I. Functional expression of NOD2, a novel pattern recognition receptor for bacterial motifs, in primary murine astrocytes. Glia. 2006;53:322–330. doi: 10.1002/glia.20286. [DOI] [PubMed] [Google Scholar]
- Strober W, Murray P, Kitani A, Watanabe T. Signalling pathways and molecular interactions of NOD1 and NOD2. Nat Rev. 2006;6:9–20. doi: 10.1038/nri1747. [DOI] [PubMed] [Google Scholar]
- Takeshita S, Takeshita F, Haddad DE, Janabi N, Klinman DM. Activation of microglia and astrocytes by CpG oligodeoxynucleotides. Neuroreport. 2001;12:3029–3032. doi: 10.1097/00001756-200110080-00010. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Akira S. Toll-like receptors; their physiological role and signal transduction system. Int Immunopharmacol. 2001;1:625–635. doi: 10.1016/s1567-5769(01)00010-8. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Hoshino K, Akira S. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J Immunol. 2000;165:5392–5396. doi: 10.4049/jimmunol.165.10.5392. [DOI] [PubMed] [Google Scholar]
- Taylor KR, Trowbridge JM, Rudisill JA, Termeer CC, Simon JC, Gallo RL. Hyaluronan fragments stimulate endothelial recognition of injury through TLR4. J Biol Chem. 2004;279:17079–17084. doi: 10.1074/jbc.M310859200. [DOI] [PubMed] [Google Scholar]
- Termeer C, Benedix F, Sleeman J, Fieber C, Voith U, Ahrens T, Miyake K, Freudenberg M, Galanos C, Simon JC. Oligosaccharides of hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med. 2002;195:99–111. doi: 10.1084/jem.20001858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting JP, Davis BK. CATERPILLER: a novel gene family important in immunity, cell death, and diseases. Annu Rev Immunol. 2005;23:387–414. doi: 10.1146/annurev.immunol.23.021704.115616. [DOI] [PubMed] [Google Scholar]
- Townsend GC, Scheld WM. Infections of the central nervous system. Adv Intern Med. 1998;43:403–447. [PubMed] [Google Scholar]
- Travassos LH, Girardin SE, Philpott DJ, Blanot D, Nahori MA, Werts C, Boneca IG. Toll-like receptor 2-dependent bacterial sensing does not occur via peptidoglycan recognition. EMBO Report. 2004;5:1000–1006. doi: 10.1038/sj.embor.7400248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara A, Yang S, Fujimoto Y, Fukase K, Kusumoto S, Shibata K, Sugawara S, Takada H. Muramyldipeptide and diaminopimelic acid-containing desmuramylpeptides in combination with chemically synthesized Toll-like receptor agonists synergistically induced production of interleukin-8 in a NOD2- and NOD1-dependent manner, respectively, in human monocytic cells in culture. Cell Microbiol. 2005;7:53–61. doi: 10.1111/j.1462-5822.2004.00433.x. [DOI] [PubMed] [Google Scholar]
- Underhill DM, Gantner B. Integration of Toll-like receptor and phagocytic signaling for tailored immunity. Microbes Infect. 2004;6:1368–1373. doi: 10.1016/j.micinf.2004.08.016. [DOI] [PubMed] [Google Scholar]
- Vabulas RM, Wagner H, Schild H. Heat shock proteins as ligands of toll-like receptors. Curr Top Microbiol Immunol. 2002;270:169–184. doi: 10.1007/978-3-642-59430-4_11. [DOI] [PubMed] [Google Scholar]
- Weber JR, Moreillon P, Tuomanen EI. Innate sensors for Gram-positive bacteria. Curr Opin Immunol. 2003;15:408–415. doi: 10.1016/s0952-7915(03)00078-5. [DOI] [PubMed] [Google Scholar]
- Wesselingh SL, Power C, Glass JD, Tyor WR, McArthur JC, Farber JM, Griffin JW, Griffin DE. Intracerebral cytokine messenger RNA expression in acquired immunodeficiency syndrome dementia. Ann Neurol. 1993;33:576–582. doi: 10.1002/ana.410330604. [DOI] [PubMed] [Google Scholar]
- Willis SA, Nisen PD. Inhibition of lipopolysaccharide-induced IL-1 beta transcription by cyclic adenosine monophosphate in human astrocytic cells. J Immunol. 1996;64:519–524. [PubMed] [Google Scholar]
- Witowski J, Pawlaczyk K, Breborowicz A, et al. IL-17 stimulates intraperitoneal neutrophil infiltration through the release of GRO alpha chemokine from mesothelial cells. J Immunol. 2000;165:5814–5821. doi: 10.4049/jimmunol.165.10.5814. [DOI] [PubMed] [Google Scholar]
- Wolfert MA, Murray TF, Boons GJ, Moore JN. The origin of the synergistic effect of muramyl dipeptide with endotoxin and peptidoglycan. J Biol Chem. 2002;277:39179–39186. doi: 10.1074/jbc.M204885200. [DOI] [PubMed] [Google Scholar]
- Yang S, Tamai R, Akashi S, Takeuchi O, Akira S, Sugawara S, Takada H. Synergistic effect of muramyldipeptide with lipopolysaccharide or lipoteichoic acid to induce inflammatory cytokines in human monocytic cells in culture. Infect Immun. 2001;69:2045–2053. doi: 10.1128/IAI.69.4.2045-2053.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarovinsky F, Zhang D, Andersen JF, et al. TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science. 2005;308:1626–1629. doi: 10.1126/science.1109893. [DOI] [PubMed] [Google Scholar]
- Ye P, Rodriguez FH, Kanaly S, et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. 2001;194:519–527. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo NJ, Park WS, Kim SY, Reed JC, Son SG, Lee JY, Lee SH. Nod1, a CARD protein, enhances pro-interleukin-1beta processing through the interaction with pro-caspase-1. Biochem Biophys Res Commun. 2002;299:652–658. doi: 10.1016/s0006-291x(02)02714-6. [DOI] [PubMed] [Google Scholar]