

Abstract
Brain abscesses arise from a focal parenchymal infection by various pathogens, particularly Staphylococcus aureus. We have shown that astrocytes are activated upon exposure to S. aureus and may contribute to the excessive tissue damage characteristic of brain abscess. Therefore, modulating astrocyte activation may facilitate a reduction in brain abscess severity. Peroxisome proliferator activated receptor-γ (PPAR-γ) agonists are potent inhibitors of microglial activation; however, the effects of these compounds on S. aureus-dependent astrocyte activation have not yet been examined. Here, we demonstrate that two chemically distinct PPAR-γ agonists, 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) and ciglitazone, suppress the production of several pro-inflammatory molecules in S. aureus-stimulated astrocytes including interleukin-1β and nitric oxide (NO). Interestingly, 15d-PGJ2 attenuated Toll-like receptor 2 (TLR2) and inducible nitric oxide synthase expression, but failed to modulate macrophage inflammatory protein-2 (MIP-2/CXCL2) production, suggesting that 15d-PGJ2 is not a global inhibitor of astrocyte activation. Another novel finding of this study was the fact that both 15d-PGJ2 and ciglitazone were capable of attenuating pre-existing astrocyte activation, indicating their potential benefit in a therapeutic setting. Importantly, 15d-PGJ2 and ciglitazone were still capable of inhibiting S. aureus-induced pro-inflammatory mediator release in PPAR-γ-deficient astrocytes, supporting PPAR-γ-independent effects of these compounds. Collectively, these results suggest that 15d-PGJ2 and ciglitazone exert their anti-inflammatory actions on astrocytes primarily independent of the PPAR-γ pathway.
Keywords: astrocytes; central nervous system; 15-deoxy-Δ12,14-prostaglandin J2; peroxisome proliferator activated receptor-γ; Staphylococcus aureus; thiazolidinedione
Brain abscess represents a serious infectious disease, which is commonly caused by Staphylococcus aureus and Streptococcal species (Kielian 2004). During brain abscess evolution, a robust inflammatory response is initiated and directed towards the rapid neutralization and elimination of infectious agents (Kielian et al. 2004a). However, recent studies from our laboratory suggest that the immune response elicited upon infection is not precisely regulated and persists even beyond the infectious period (Baldwin and Kielian 2004). This dysregulated immune response is likely the cause of excessive neuronal damage and toxicity as a result of the detrimental consequences of accumulated nitric oxide (NO), cytokines and chemokines (Nguyen et al. 2002; Baldwin and Kielian 2004; Safieh-Garabedian et al. 2004). Recently, we have reported that astrocytes, the most abundant glial cell type in the CNS (Hansson and Ronnback 2003), express Toll-like receptor 2 (TLR2), a pattern recognition receptor for gram-positive bacteria and various other invariant motifs associated with a wide array of microbes (Yoshimura et al. 1999; Akira 2003; Bowman et al. 2003; Esen et al. 2004). This receptor enables S. aureus recognition by astrocytes and the subsequent production of numerous pro-inflammatory cytokines and chemokines (Esen et al. 2004), suggesting a possible role of astrocytes in the pathogenesis of S. aureus-induced brain abscess. Among the pro-inflammatory mediators produced by S. aureus-activated astrocytes include NO, tumor necrosis factor α (TNF-α), interleukin-1β (IL-1β), macrophage inflammatory protein-2 (MIP-2/CXCL2), and monocyte chemoattractant protein-1 (MCP-1/CCL2; Esen et al. 2004). With regard to their role in brain abscesses, IL-1β and TNF-α are essential for the induction of a protective antibacterial immune response, as well as regulating blood–brain barrier permeability (Mayhan 2002; Kielian et al. 2004a; Allan et al. 2005). Similar effects are postulated to occur with the free radical NO (Nau and Bruck 2002). Similarly, MIP-2 and MCP-1 are two chemoattractants released by S. aureus-activated astrocytes that are important for the extravasation of neutrophils and monocytes/macrophages into brain abscesses, respectively (Kielian et al. 2001). In this way, astrocytes participate in the early immune response to S. aureus by releasing several chemokines, which further mediate the migration of peripheral immune cells into the abscess site, thereby amplifying the inflammatory response to infection (Kielian 2004). In the mouse experimental brain abscess model developed in our laboratory using S. aureus, we have also found that astrocyte activation is a hallmark feature that continues throughout the course of infection, suggesting that these cells may contribute to the excessive tissue damage characteristic of brain abscess (Baldwin and Kielian 2004). Therefore, therapeutic regimens aimed at suppressing chronic astrocyte activation following effective bacterial neutralization may help reduce brain abscess severity.
Recently, several studies have suggested roles for ligand-activated peroxisome proliferator activated receptors (PPARs) in transducing environmental, inflammatory, developmental and metabolic signals into a defined cascade of cellular responses at the level of gene transcription (Berger and Moller 2002; Gervois et al. 2004). This superfamily of receptors includes three different isoforms, PPAR-α (NR1C1), PPAR-β/δ (NUC1; NR1C2) and PPAR-γ (NR1C3), with all three family members exhibiting a significant degree of homology in their DNA and ligand binding domains despite being encoded by three distinct genes (Lemberger et al. 1996; Vidal 2005). Among the various PPAR isoforms, PPAR-γ has been investigated most extensively, partly because some of its ligands have been approved for the treatment of diabetes as well as its suggested involvement in attenuating inflammation (Kielian and Drew 2003; Yki-Jarvinen 2004; Campbell 2005; Dumasia et al. 2005; Pegorier 2005; Staels and Fruchart 2005). PPAR-γ can be activated by several ligands, including tyrosine derivatives, fibrates, docosa-hexaenoic acid, linoleic acid, the anti-diabetic thiazolidinediones (TZDs), various lipids, eicosanoids, prostanoids including the endogenous cyclopentenone prostaglandin 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2), and certain non-steroidal anti-inflammatory drugs including ibuprofen and indomethacin (Cabrero et al. 2002; Clark 2002; Daynes and Jones 2002; Nencioni et al. 2003). Among the various PPAR-γ ligands, 15d-PGJ2 and TZDs have been shown to have potent immuno-modulatory effects on several cell types, including monocytes/macrophages, microglia, astrocytes, neutrophils and lymphocytes (Ricote et al. 1999; Asada et al. 2004; Buckingham 2005; Consoli and Devangelio 2005). Although TZDs have a higher binding affinity for PPAR-γ, several studies have revealed that 15d-PGJ2 is a more potent anti-inflammatory compound (Jiang et al. 1998; Maggi et al. 2000; Storer et al. 2005a). Furthermore, PPAR-γ agonists including TZDs have been shown to exert anti-inflammatory and neuroprotective effects in animal models of neurological disease, including multiple sclerosis (Feinstein et al. 2002), Alzheimer’s disease (Camacho et al. 2004; Heneka et al. 2005), and amyotrophic lateral sclerosis (Kiaei et al. 2005), suggesting potential therapeutic uses for these drugs in neurological conditions (Feinstein 2003).
The present study demonstrates that 15d-PGJ2 and the TZD ciglitazone are potent but selective inhibitors of pro-inflammatory mediator production by S. aureus-activated astrocytes. Importantly, this study is the first to directly demonstrate that the action of these compounds in astrocytes occurs primarily via a PPAR-γ-independent manner, as revealed by studies using PPAR-γ-deficient primary astrocytes. Finally, both 15d-PGJ2 and ciglitazone were still effective at attenuating S. aureus-dependent astrocyte activation when added as late as 6 h following bacterial exposure, indicating that these compounds are effective at modulating ongoing inflammation. Collectively, these findings demonstrate that 15d-PGJ2 and ciglitazone primarily act independently of PPAR-γ to suppress S. aureus-induced astrocyte activation. The potent anti-inflammatory activity of this class of compounds may help attenuate chronic inflammatory damage in the CNS during later stages of brain abscesses when bacterial burdens are relatively minimal.
Materials and methods
Primary astrocyte cultures
Primary astrocytes were derived from C57BL/6 pups (1–6 days of age) as previously described (Esen et al. 2004). Briefly, when mixed glial cultures reached confluency (1–2 weeks), flasks were shaken at 50 g overnight at 37°C to recover microglia. The supernatant containing microglia was collected and fresh medium was added to the flasks and incubated for another 5–7 days. This was repeated 3–4 times, whereupon flasks were trypsinized and cells re-suspended in astrocyte medium supplemented with 0.1 mM of the microglial cytotoxic agent L-leucine methyl ester (Sigma, St Louis, MO, USA). Astrocytes were treated with L-leucine methyl ester for at least 2 weeks prior to use in experiments. The purity of astrocyte cultures was evaluated by immunohistochemical staining using antibodies against glial fibrillary acidic protein (GFAP; Dako, Carpenteria, CA, USA) and CD11b (BD PharMingen, San Jose, CA, USA) to identify astrocytes and microglia, respectively. The purity of primary astrocyte cultures using this procedure was routinely greater than 95%.
PPAR-γ-deficient astrocytes were prepared as above, from cerebral cortices of post-natal day 1 mice with conditional deletion of astrocyte PPAR-γ. These mice were generated by crossing PPAR-γ f/f mice [in which PPAR-γ exon 2 is replaced with exon 2 flanked by the 19-bp flox sequence (Akiyama et al. 2002)] with transgenic mice expressing the bacterial Cre recombinase under control of the astrocyte-specific GFAP promoter (Zhuo et al. 2001). Pups were screened for presence of the Cre gene, and backcrossed to PPAR-γ f/f mice to generate mice homozygous for the floxed PPAR-γ gene and hemizygous for the Cre gene (PPAR-γ f/f Cre ±). Deletion of PPAR-γ exon 2 was confirmed by RT-PCR analysis of total astrocyte mRNA using PCR primers (PPAR-γ forward 91: 5′-GCTGATGCACTGCCTATGAG-3′; and PPAR-γ reverse 460: 5′-TGATGCTTTATCCCCACAGAC-3′), which flank PPAR-γ exon 2 and yield a 368-bp product.
Reagents
15d-PGJ2 and ciglitazone were purchased from Cayman Chemical (Ann Arbor, MI, USA). Heat-inactivated S. aureus (strain RN6390, kindly provided by Dr Ambrose Cheung, Dartmouth Medical School) was prepared as previously described (Kielian et al. 2001) and verified to have endotoxin levels < 0.03 EU/mL as determined by Limulus amebocyte lysate assay (Associates of Cape Cod, Falmouth, MA, USA). The concentrations of S. aureus and 15d-PGJ2 used were selected based on previous studies demonstrating optimal immuno-modulatory effects without any evidence of cytotoxicity (Esen et al. 2004; Storer et al. 2005a).
Nitrite assay
Nitrite (a metabolic breakdown product of NO) levels were determined by adding 50 μL of astrocyte-conditioned culture medium with 50 μL of Griess reagent (0.1% naphtyletylenediamine dihydrochloride, 1% sulfanilamide, 2.5% phosphoric acid, all from Sigma) in a 96-well plate. The absorbance at 550 nm was measured on a plate reader (Spectra Max 190; Molecular Devices, Sunnyvale, CA, USA), and nitrite concentrations calculated using a standard curve with sodium nitrite (NaNO2; Sigma; level of sensitivity 0.4 μM).
ELISA
Quantitation of cytokine and chemokine levels in astrocyte-conditioned medium was performed using standard sandwich ELISA kits according to the manufacturer’s instructions (OptEIA mouse IL-1β and TNF-α; BD PharMingen; DuoSet mouse MIP-2/CXCL2 and KC/CXCL2, R & D Systems, Minneapolis, MN, USA). The level of sensitivity for all ELISAs was 15.6 pg/mL.
Cell viability assays
The effects of 15d-PGJ2 and ciglitazone on astrocyte cell viability were evaluated using a standard MTT assay based upon the mitochondrial conversion of 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) into formazan crystals as previously described (Esen et al. 2004).
Protein extraction and Western blotting
Protein extracts were prepared from primary astrocytes cultured in 6-well plates by lysing cells in 200 μL of RIPA buffer (1% NP-40, 0.1% sodium dodecyl sulfate in phosphate-buffered saline, pH 7.4) supplemented with a Complete™ protease inhibitor cocktail tablet (Roche Molecular Biochemicals, Indianapolis, IN, USA). Lysates were allowed to incubate on ice for 30 min followed by centrifugation at 21 000 × g for 15 min at 4°C to pellet debris. Calculation of total protein in astrocyte extracts was determined using a standard protein assay [bicinchoninic acid (BCA) protein assay reagent; Bio-Rad Laboratories, Hercules, CA, USA]. PPAR-γ 2 (Affinity Bioreagents, Golden, CO, USA), TLR2 (R & D Systems) and inducible nitric oxide synthase (iNOS; Santa Cruz Biotechnology, Santa Cruz, CA, USA) expression were evaluated in 15d-PGJ2 and ciglitazone-treated astrocytes by Western blot analysis. Astrocyte protein extracts (ranging from 1 to 40 μg of total protein) were electrophoresed on 4–10% Tris HCl Ready Gels (Bio-Rad Laboratories) and transferred to a polyvinylidene difluoride membrane (Immobilon-P; Millipore, Bedford, MA, USA) using a semi-dry transfer apparatus (Bio-Rad Laboratories). Blots were probed using either PPAR-γ 2, TLR2 or iNOS antibodies followed by the appropriate IgG-horseradish peroxidase-conjugated secondary antibody (Jackson Immunoresearch, West Grove, PA, USA). Subsequently, membranes were stripped and re-probed with a rabbit anti-actin polyclonal antibody (Sigma) to verify uniformity in gel loading. Blots were developed using the Immobilon western substrate (Millipore, Billercia, MA, USA) and visualized by exposure to X-ray film.
RNA isolation and quantitative real-time RT-PCR (qRT-PCR)
Total RNA was isolated from primary astrocytes using the TriZol reagent and treated with Dnase1 (both from Invitrogen, Carlsbad, CA, USA) prior to use in qRT-PCR studies. The experimental procedure was performed as previously described (Esen et al. 2004; Kielian et al. 2005). Briefly, TLR2 and GAPDH primers and TAMRA TaqMan probes were synthesized by Applied Biosystems (Foster City, CA, USA). ABI Assays-on-Demand™ Taqman kits were utilized to examine CD36, PPAR-γ, and ATP-binding cassette (ABC) subfamily A1 transporter (ABCA1) expression. The RT reaction was carried out using the iScript cDNA synthesis kit (Bio-Rad Laboratories) according to the manufacturer’s instructions. Real-time PCR reactions were performed in a total reaction volume of 25 μL using the iCycler kit (Bio-Rad Laboratories) containing a final concentration of 400 nM of forward and reverse primers, 200 nM of Taqman probe, and 1 μL of cDNA from the RT step, and analyzed using the iCycler IQ multicolor real-time PCR detection system (Bio-Rad Laboratories). Gene expression levels following the various experimental treatments were calculated after normalizing cycle thresholds against the housekeeping gene GAPDH and are presented as the fold-induction (2−ΔΔCt) value relative to unstimulated astrocytes. For all qRT-PCR studies, we calculate changes in mRNA levels (mean ± SEM) using the mean of the fold-change in mRNA expression from three individual experiments (representing independent culture preparations). While rigorous, this approach leads to variance values that are larger than standard deviations often reported for qRT-PCR experiments which are calculated by taking the average of ‘technical replicates’, thus resulting in tighter error bars. Although we made all attempts to maintain experimental consistency throughout these studies, variations in the different cultures of primary astrocytes used (final cell numbers, degree of confluency, time from last feeding) most likely accounts for the interexperimental variations in mRNA levels. However, we consider this important to demonstrate the validity of our findings.
Statistics
Statistical differences between experimental groups were determined by the Student’s t-test at the 95% confidence interval using Sigma Stat (SPSS Science, Chicago, IL, USA).
Results
Primary mouse astrocytes express PPAR-γ
Recently, Storer et al. (2005a) reported that primary mouse astrocytes express PPAR-γ constitutively, the level of which remained unchanged even following stimulation with lipopolysaccharide (LPS). However, other reports have demonstrated alterations in PPAR-γ expression in disparate cell types, suggesting that the cell type and/or stimulus dictate the degree of receptor regulation (Ricote et al. 1998a,b; Huang et al. 1999; Tanaka et al. 1999). The effects of S. aureus stimulation on PPAR-γ expression in astrocytes, and how 15d-PGJ2 may regulate receptor levels, have not yet been investigated. As shown in Fig. 1(a), PPAR-γ mRNA expression was increased in primary astrocytes following 15d-PGJ2 treatment. In contrast, S. aureus stimulation led to a modest reduction in PPAR-γ mRNA levels; however, co-treatment with 15d-PGJ2 and S. aureus did not exert any additive or synergistic effects on astrocytic PPAR-γ mRNA expression compared with 15d-PGJ2 alone (Fig. 1a). Analysis of PPAR-γ protein levels by Western blotting demonstrated that primary astrocytes expressed PPAR-γ constitutively and receptor levels remained unchanged even upon stimulation with S. aureus or treatment with the PPAR-γ agonist 15d-PGJ2 (Fig. 1). Collectively, these results indicate that 15d-PGJ2 (and to a lesser extent S. aureus) can influence PPAR-γ mRNA steady-state levels; however, these changes were not apparent at the protein level, possibly reflecting restricted translational processes or, in part, because of the decreased sensitivity of Western blots compared with qRT-PCR. Nevertheless, expression of PPAR-γ in astrocytes is suggestive that these cells may be responsive to the immuno-modulatory effects of PPAR-γ agonists.
Fig. 1.
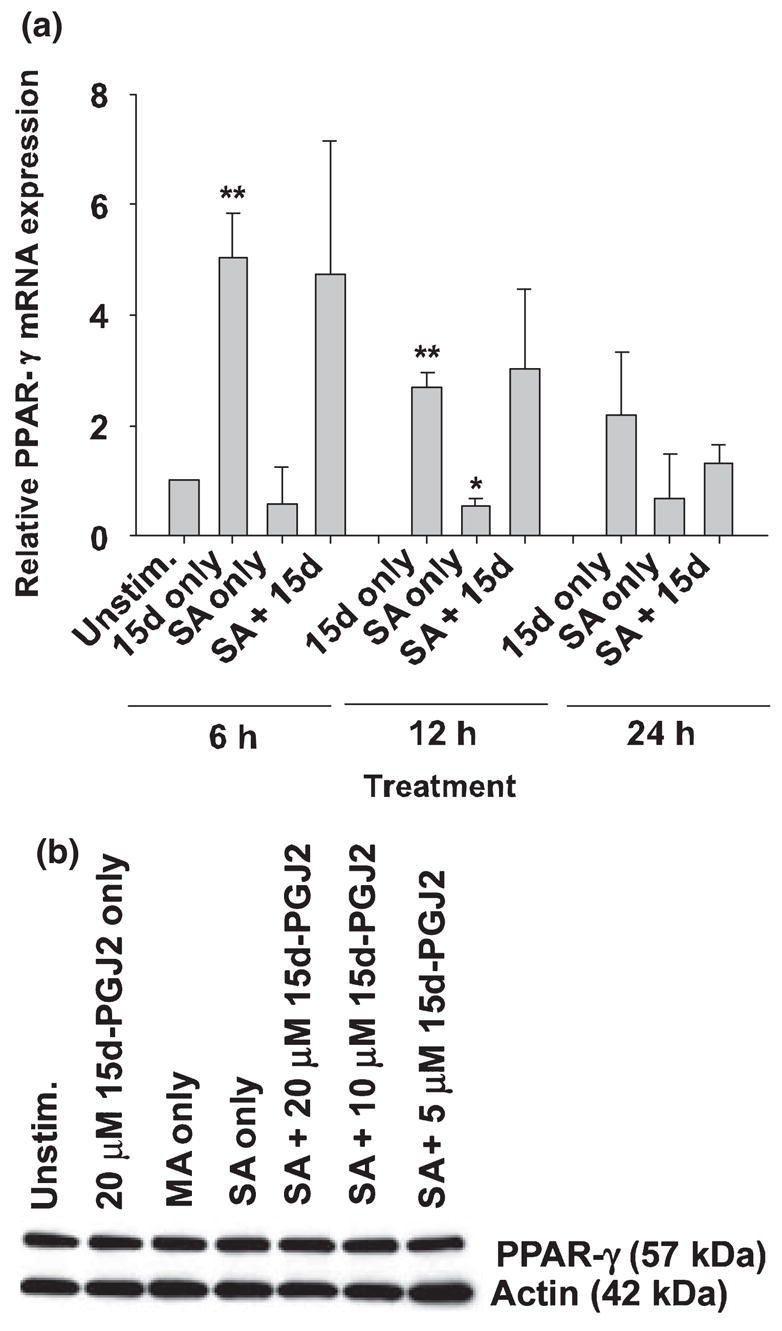
Primary mouse astrocytes express PPAR-γ. Primary astrocytes were seeded in 6-well plates at 1 × 106 cells per well. After 24 h, cells were pretreated with 15d-PGJ2 at the indicated concentrations for 1 h prior to stimulation with 108 colony forming units (cfu) of heat-inactivated S. aureus (SA). Astrocytes treated with vehicle (methyl acetate, MA) or 20 μM 15d-PGJ2 only are included as negative controls. In (a), total RNA was collected from cells at 6, 12 and 24 h following S. aureus stimulation and PPAR-γ mRNA expression was quantified by qRT-PCR as described in Materials and methods. Gene expression levels were calculated after normalizing PPAR-γ signals against the housekeeping gene GAPDH and are presented as relative mRNA expression units (mean ± SEM of three independent experiments). Significant differences between unstimulated astrocytes versus 15d-PGJ2 only or S. aureus only are denoted with asterisks (*p < 0.05, **p < 0.001). In (b), protein extracts (1 μg) from whole cell lysates were prepared at 24 h following S. aureus stimulation and assessed for PPAR-γ 2 expression by Western blotting as described in Materials and methods. Subsequently, membranes were stripped and re-probed for β-actin to verify uniformity in gel loading. Results are representative of three independent experiments.
The cyclopentenone prostaglandin 15d-PGJ2 selectively attenuates inflammatory mediator production from S. aureus -stimulated astrocytes
S. aureus-induced astrocyte activation is typified by the production of numerous pro-inflammatory mediators (Esen et al. 2004). In the current study, we investigated whether the cyclopentenone prostaglandin 15d-PGJ2 was capable of modulating the extent of pro-inflammatory mediator release from stimulated astrocytes. Pre-treatment of astrocytes with various concentrations of 15d-PGJ2 (5–20 μM) for 1 h prior to S. aureus exposure led to a dose-dependent inhibition of IL-1β expression (Fig. 2a). Cell viability assays revealed that 15d-PGJ2 was not cytotoxic at any of the concentrations examined, indicating that the anti-inflammatory effects observed were not as a result of overt cell death (Fig. 2c). In contrast to its inhibitory effects on S. aureus-induced IL-1β production, 15d-PGJ2 had no effect on the production of the CXC neutrophil chemoattractants MIP-2/CXCL2 or KC/CXCL2 (Fig. 2b and data not shown, respectively). In summary, these findings reveal that 15d-PGJ2 modulates the expression of select inflammatory mediators in S. aureus-stimulated astrocytes.
Fig. 2.
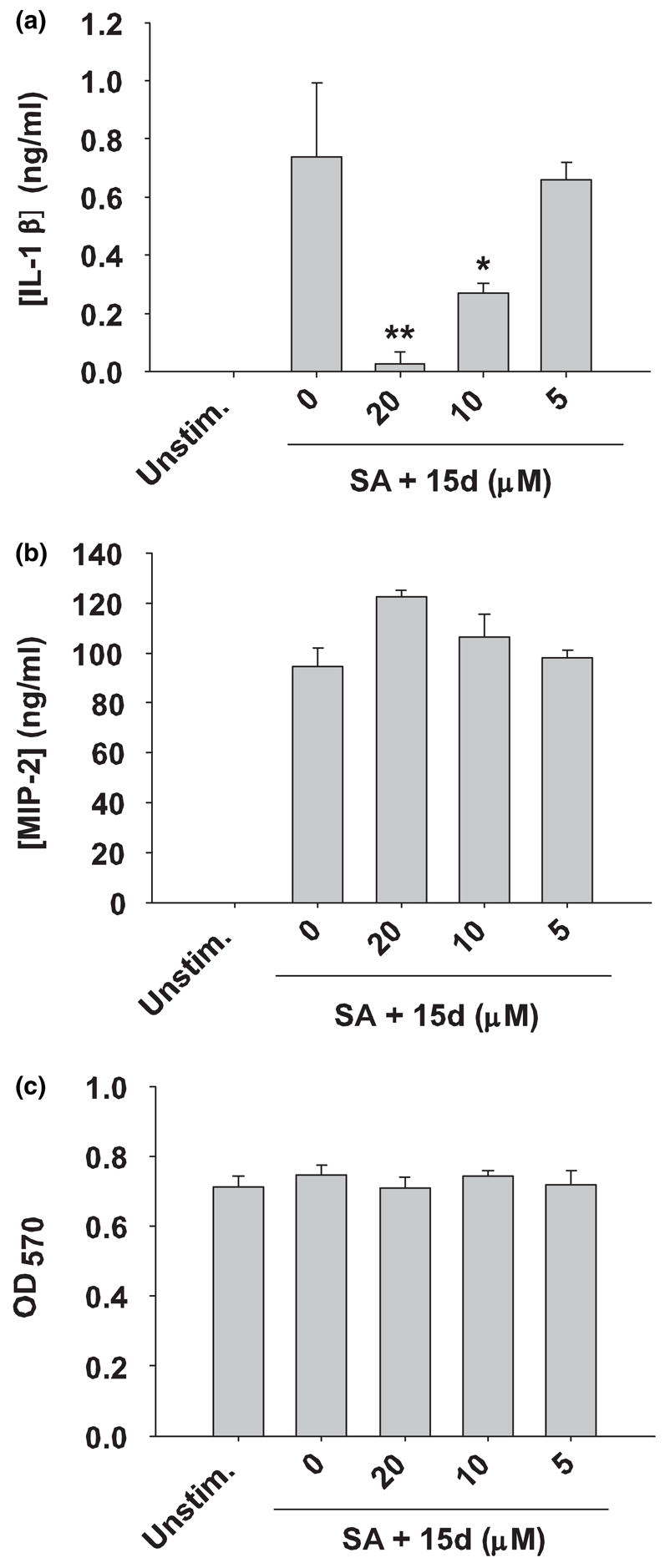
15d-PGJ2 differentially modulates pro-inflammatory cytokine and chemokine production in S. aureus-stimulated astrocytes. Primary astrocytes were seeded in 96-well plates at 1 × 105 cells per well and incubated overnight. The following day astrocytes were pre-treated for 1 h with various concentrations of 15d-PGJ2 (15d; 5–20 μM) followed by stimulation with 107 cfu heat-inactivated S. aureus (SA) for 24 h. Cell-free supernatants were collected and analyzed for IL-1β (a) and MIP-2 (b). Astrocyte viability was assessed using a standard MTT assay and the raw OD570 absorbance values are reported (c). Results are reported as the mean ± SD of three independent wells for each experimental treatment. Significant differences between astrocytes treated with S. aureus only versus cells exposed to various concentrations of 15d-PGJ2 + S. aureus are denoted with asterisks (*p < 0.05, **p < 0.001). Results are representative of five independent experiments.
S. aureus -induced iNOS expression and NO production are attenuated by 15d-PGJ2 in astrocytes
Nitric oxide is an important mediator in CNS inflammatory diseases (Bal-Price and Brown 2001, 2003; Mander and Brown 2004) and 15d-PGJ2 has previously been shown able to attenuate iNOS expression as well as NO production in astrocytes in response to the gram-negative cell wall component LPS (Giri et al. 2004; Storer et al. 2005a). However, the ability of 15d-PGJ2 to modulate astrocytic iNOS expression in response to the gram-positive CNS pathogen S. aureus has not yet been investigated. Western blot analysis demonstrated that S. aureus is a potent inducer of iNOS expression in primary astrocytes. Interestingly, pretreatment with 15d-PGJ2 attenuated S. aureus-induced iNOS expression in a dose-dependent manner (Fig. 3a). The reduction in iNOS levels directly correlated with the ability of 15d-PGJ2 to attenuate NO production in response to S. aureus (Fig. 3b). Collectively, these findings suggest that 15d-PGJ2 modulates NO production in S. aureus-stimulated astrocytes primarily by altering iNOS expression.
Fig. 3.
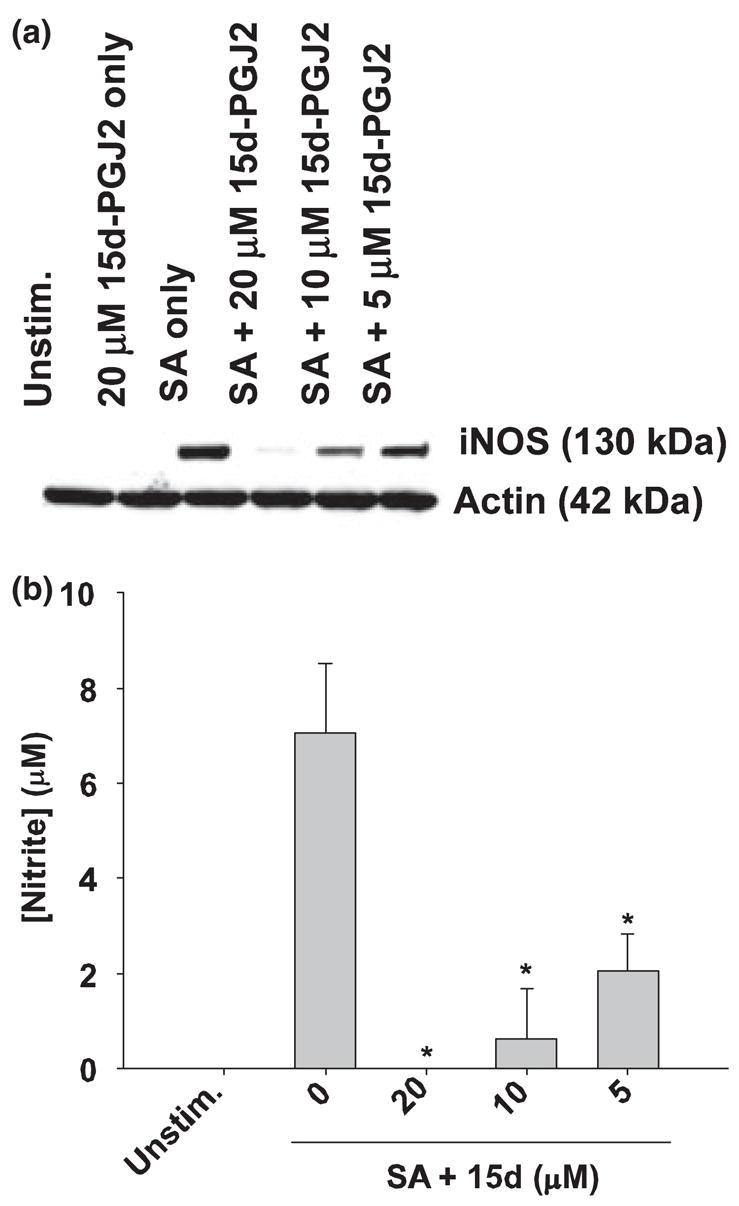
15d-PGJ2 inhibits iNOS protein expression and subsequent NO production in S. aureus-stimulated astrocytes. Astrocytes were seeded in 6-well plates at 1 × 106 cells per well. After an overnight incubation, cells were pre-treated with 20 μM of 15d-PGJ2 only, or various concentrations of 15d-PGJ2 for 1 h prior to stimulation with 108 cfu S. aureus (SA). In (a), Western blots were performed on astrocyte protein extracts (40 μg total protein) collected at 24 h following S. aureus stimulation as described in Materials and methods. Following transfer, membranes were probed with an iNOS-specific antibody and subsequently stripped and re-probed for β-actin to verify uniformity in gel loading. In (b), cell-free supernatants were analyzed for nitrite production (presented in μM). Results are reported as the mean ± SD of three independent wells for each experimental treatment. Significant differences between astrocytes treated with S. aureus versus cells exposed to the various concentrations of 15d-PGJ2 + S. aureus are denoted with asterisks (*p < 0.05). Results are representative of five independent experiments.
15d-PGJ2 modulates S. aureus -induced TLR2 expression in astrocytes
Several groups have reported that cultured astrocytes express TLR2 (Bowman et al. 2003; Esen et al. 2004; Carpentier et al. 2005; Owens 2005). In addition, recent studies from our laboratory have established that TLR2 plays a pivotal role in triggering intracellular signaling cascades involved in mediating immune responses against both S. aureus and PGN in astrocytes (Esen et al. 2004; Kielian 2006). Recently, 15d-PGJ2 has been reported to modulate TLR2-mediated dendritic cell maturation (Appel et al. 2005); however, it is not yet known whether 15d-PGJ2 can affect the expression of astrocytic TLR2 in response to S. aureus. To explore this question, TLR2 mRNA and protein levels were examined in S. aureus-stimulated astrocytes in the presence or absence of 15d-PGJ2 using qRT-PCR and Western blotting, respectively. As expected, S. aureus exposure augmented TLR2 mRNA expression, which was nearly abrogated by 15d-PGJ2 at all time points examined (Fig. 4a). Similar changes in TLR2 were observed at the protein level, where 15d-PGJ2 inhibited S. aureus-induced receptor expression in a dose-dependent manner (Fig. 4b). These findings suggest that 15d-PGJ2 is able to modulate S. aureus-induced astrocyte activation, in part, by down-regulating TLR2 expression.
Fig. 4.
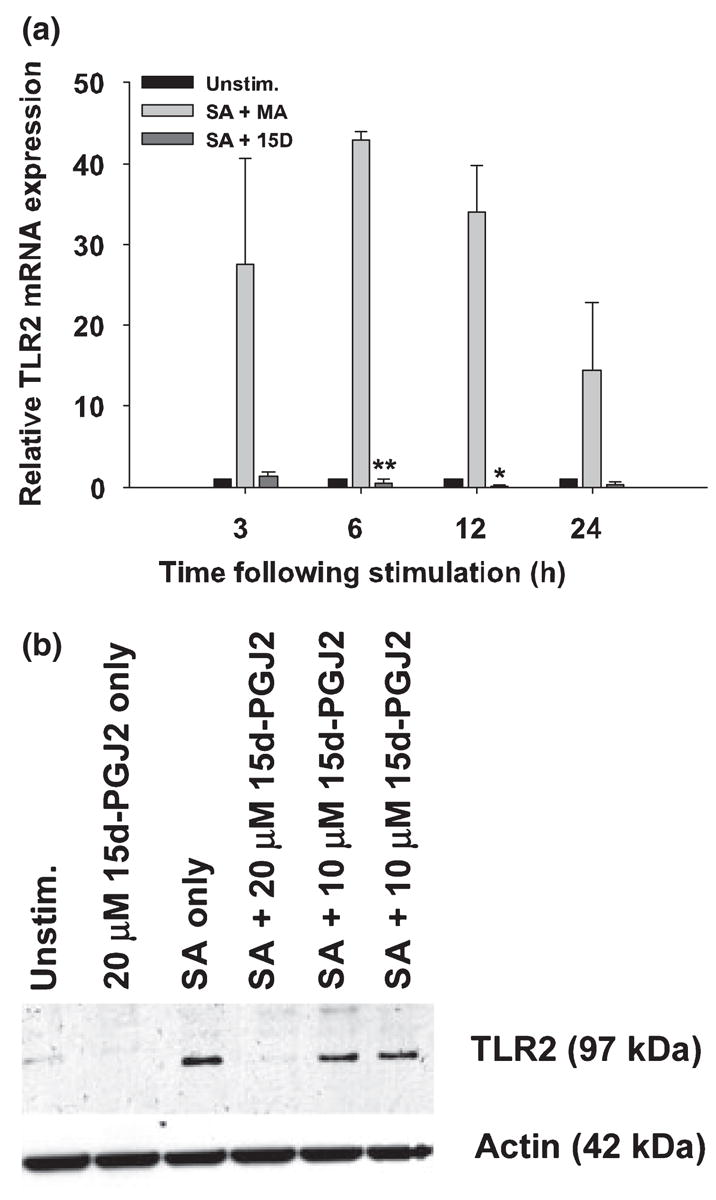
15d-PGJ2 attenuates S. aureus-induced TLR2 expression in astrocytes. Astrocytes were seeded in 6-well plates at 1 × 106 cells per well. After an overnight incubation, cells were pretreated with 20 μM of 15d-PGJ2 (15D) or vehicle only (methyl acetate, MA) for 1 h prior to stimulation with 108 cfu S. aureus (SA). In (a), total RNA was collected from cells at 3, 6, 12 and 24 h following S. aureus stimulation and TLR2 expression was quantified by qRT-PCR as described in Materials and methods. Gene expression levels were calculated after normalizing TLR2 signals against the housekeeping gene GAPDH and are presented as relative mRNA expression units (mean ± SEM of three independent experiments). Significant differences between S. aureus-stimulated astrocytes pretreated with vehicle only (methyl acetate) versus S. aureus + 20 μM of 15d-PGJ2 are denoted with asterisks (*p < 0.05, **p < 0.001). In (b) western blots were performed on astrocyte protein extracts (40 μg total protein) collected at 24 h following S. aureus stimulation as described in Materials and methods. Following transfer, membranes were probed with a TLR2-specific antibody and subsequently stripped and re-probed for β-actin to verify uniformity in gel loading. Results are representative of three independent experiments.
The synthetic PPAR-γ agonist ciglitazone attenuates pro-inflammatory mediator production from S. aureus -stimulated astrocytes
Several members of the TZD family of compounds serve as ligands for PPAR-γ and are able to exert immuno-modulatory effects on numerous classes of immune cells (Clark 2002; Daynes and Jones 2002; Zingarelli and Cook 2005). We therefore compared the ability of the TZD ciglitazone versus 15d-PGJ2 to affect inflammatory mediator production from S. aureus-stimulated astrocytes. Ciglitazone attenuated NO and IL-1β production by primary astrocytes in a dose-dependent manner (Figs 5a and b). In contrast to 15d-PGJ2, ciglitazone was capable of suppressing the production of the CXC chemoattractant MIP-2/CXCL2 (Fig. 5c). Importantly, the concentration of ciglitazone used in this study was not toxic to astrocytes at any of the doses examined (Fig. 5d). Interestingly, 15d-PGJ2 was more potent than ciglitazone in suppressing inflammation in S. aureus-stimulated astrocytes despite its lower affinity for PPAR-γ. The anti-inflammatory effects of ciglitazone occurred at concentrations much higher than its Kd value for PPAR-γ (approximately 3 μM; Willson et al. 1996), suggesting that its effects may be receptor independent. These findings indicate that, similar to 15d-PGJ2, ciglitazone also attenuates inflammatory mediator production in S. aureus-stimulated astrocytes; however, the high concentrations required suggest that PPAR-γ-independent events may be involved in these effects.
Fig. 5.
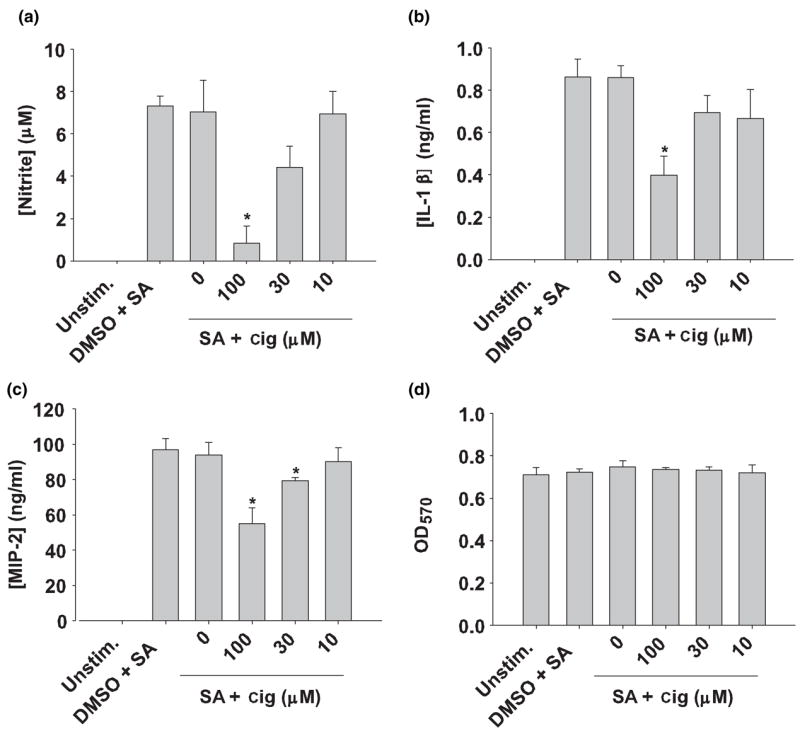
Ciglitazone modulates pro-inflammatory cytokine and chemokine production from S. aureus-stimulated astrocytes. Primary astrocytes were seeded in 96-well plates at 1 × 105 cells per well and incubated overnight. The following day, astrocytes were pre-incubated for 1 h with vehicle only (dimethylsulfoxide; DMSO), or the indicated concentrations of ciglitazone (cig; 10–100 μM) followed by treatment with 107 cfu heat-inactivated S. aureus (SA) for 24 h. Cell-free supernatants were collected and analyzed for nitrite (a), IL-1β (b) and MIP-2 (c) production. Astrocyte viability was assessed using a standard MTT assay and the raw OD570 absorbance values are reported (d). Results are reported as the mean ± SD of three independent wells for each experimental treatment. Significant differences between S. aureus-stimulated astrocytes versus S. aureus + ciglita-zone are denoted with asterisks (*p < 0.05). Results are representative of five independent experiments.
15d-PGJ2 and ciglitazone induce PPAR-responsive element (PPRE)-containing genes in astrocytes
Our data demonstrates that the PPAR-γ agonists 15d-PGJ2 and ciglitazone are capable of attenuating astrocytic expression of select pro-inflammatory mediators. However, it was important to establish whether these compounds (at the concentrations used in this study) were capable of modulating the expression of genes containing known PPREs, indicative of PPAR-γ-dependent activation. We therefore examined the ability of 15d-PGJ2 and ciglitazone to modulate expression of two PPRE-driven genes in astrocytes, namely CD36 and the ATP-binding cassette protein subtype A1 (ABCA1; Motojima et al. 1998; Akiyama et al. 2002). Both 15d-PGJ2 and ciglitazone significantly increased CD36 and ABCA1 mRNA levels in primary astrocytes, although the kinetics of induction differed between the two genes (Fig. 6). Treatment with 15d-PGJ2 augmented CD36 and ABCA1 mRNA levels in a time-dependent manner, with maximal expression observed at 24 h exposure (Figs 6a and b). In contrast, the increases as a result of treatment with ciglitazone peaked at 12 h, after which they significantly decreased (Figs 6c and d).
Fig. 6.
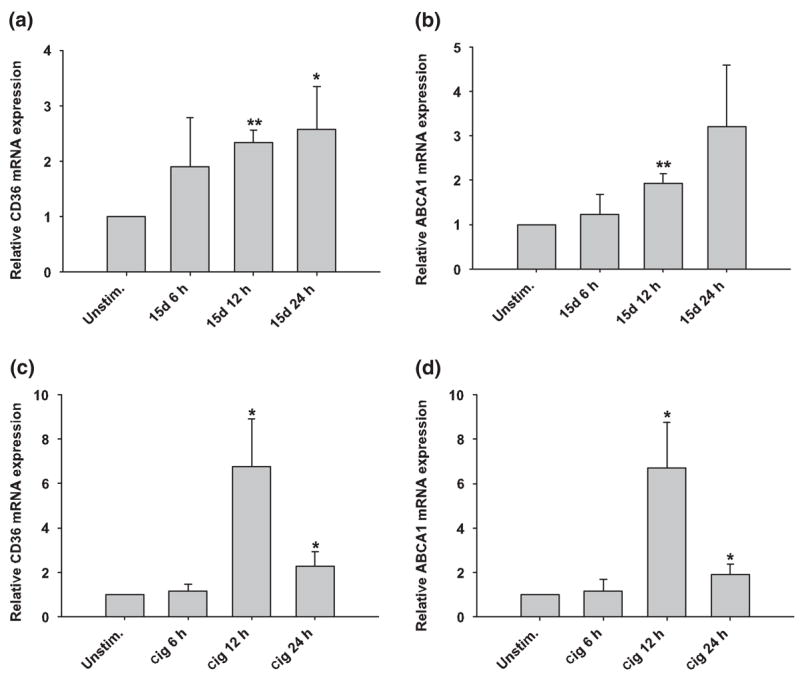
15d-PGJ2 and ciglitazone induce the expression of PPRE-responsive genes in astrocytes. Astrocytes were seeded in 6-well plates at 1 × 106 cells per well. After an overnight incubation, cells were treated with 20 μM of 15d-PGJ2 (15d) or 100 μM ciglitazone (cig) only. Total RNA was collected from cells at 6, 12 and 24 h following S. aureus stimulation, whereupon CD36 (a, c) and ABCA1 (b, d) expression was quantified by qRT-PCR as described in Materials and methods. Gene expression levels were calculated after normalizing target signals against the housekeeping gene GAPDH and are presented as relative mRNA expression units (mean ± SEM of three independent experiments). Significant differences between unstimulated versus 15d-PGJ2- or ciglitazone-treated astrocytes are denoted with asterisks (*p < 0.05, **p < 0.001).
Both 15d-PGJ2 and ciglitazone are capable of attenuating established astrocyte activation in response to S. aureus
The results presented thus far have established that both 15d-PGJ2 and ciglitazone are effective at inhibiting S. aureus-dependent astrocyte activation when administered prior to bacterial stimulation. However, it is not yet known whether these compounds can modulate pre-existing astrocyte activation. To address this clinically more relevant question, post-treatment studies were performed, where primary astrocytes were first stimulated with heat-inactivated S. aureus and subsequently treated with 15d-PGJ2 or ciglitazone at 6 h following bacterial exposure. In these experiments, S. aureus was maintained in the culture medium throughout the 24 h stimulation period, after which pro-inflammatory mediator expression was evaluated. Interestingly, this post-treatment paradigm mirrored the effects observed when PPAR-γ agonists were administered prior to S. aureus stimulation. Specifically, both 15d-PGJ2 and ciglitazone were capable of attenuating ongoing NO and IL-1β production from S. aureus-treated astrocytes, whereas MIP-2 levels were only-influenced by ciglitazone (Figs 7a–c). Similar to the pretreatment effects of 15d-PGJ2 on chemokine expression, post-stimulation treatment had no effect on the production of the CXC chemoattractant MIP-2/CXCL2 (Fig. 7c). Again, no toxic effects were observed with any of the concentrations of 15d-PGJ2 or ciglitazone tested (Fig. 7d). Collectively, these findings indicate that 15d-PGJ2 and ciglitazone are able to modulate pre-existing astrocyte activation which, to our knowledge, has not yet been reported in the literature.
Fig. 7.
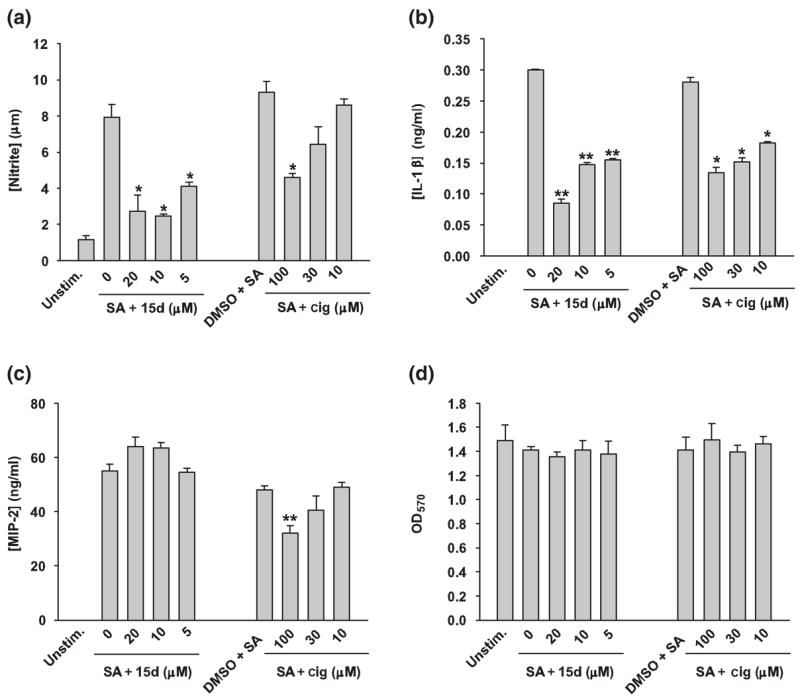
15d-PGJ2 and ciglitazone are capable of attenuating ongoing pro-inflammatory mediator expression in astrocytes. Primary astrocytes were seeded in 96-well plates at 1 × 105 cells per well and incubated overnight. The following day, astrocytes were left untreated or stimulated with 107 cfu heat-inactivated S. aureus (SA) for 6 h. Subsequently, astrocytes were treated with the indicated doses of 15d-PGJ2 (15d), ciglitazone (cig; μM, final concentration), or vehicle control (dimethylsulfoxide). At 24 h following bacterial stimulation, cell-free supernatants were collected and analyzed for nitrite (a), IL-1β (b) and MIP-2 (c) expression. Astrocyte viability was assessed using a standard MTT assay and the raw OD570 absorbance values are reported (d). Results are reported as the mean ± SD of three independent wells for each experimental treatment. Significant differences between S. aureus-stimulated astrocytes treated with vehicle only versus cells exposed to the various concentrations of 15d-PGJ2 or ciglitazone + S. aureus are denoted with asterisks (*p < 0.05, **p < 0.001). Results are representative of five independent experiments.
Both 15d-PGJ2 and ciglitazone attenuate S. aureus-dependent astrocyte activation primarily through a PPAR-γ-independent pathway
Currently, it remains unclear as to whether the anti-inflammatory effects of 15d-PGJ2 or other PPAR-γ agonists are mediated via receptor-dependent or -independent pathways (Clark 2002; Daynes and Jones 2002; Feinstein et al. 2005; Zingarelli and Cook 2005). To more definitively determine if 15d-PGJ2 and ciglitazone affect S. aureus-induced astrocyte activation independently of PPAR-γ, we investigated the ability of these compounds to modulate inflammatory responses in PPAR-γ-deficient astrocytes. Mice in which PPAR-γ was specifically deleted in astrocytes were generated using Cre-Lox technology where a human GFAP-driven Cre recombinase mouse (Zhuo et al. 2001) was crossed to a mouse containing a floxed PPAR-γ gene (Akiyama et al. 2002), generating offspring where PPAR-γ gene exon 2, necessary for activity, was deleted in astrocytes. Loss of PPAR-γ exon 2 was confirmed by PCR analysis of astrocyte mRNA using primers flanking this region (Fig. 8a). Interestingly, constitutive expression of the PPRE-driven genes CD36 and ABCA1 was not detected in PPAR-γ-deficient astrocytes, whereas both genes were constitutively expressed in wild type cells, providing additional evidence to support the loss of PPAR-γ activity in knockout (KO) astrocytes (data not shown). PPAR-γ-deficient astrocytes were still capable of producing NO and IL-1β as well as augmenting TLR2 expression in response to S. aureus, indicating that there were no global defects in the ability of these cells to recognize bacteria (Figs 8b and c, and data not shown). Importantly, both 15d-PGJ2 and ciglitazone were still able to attenuate S. aureus-dependent NO, IL-1β, and TLR2 expression (Figs 8b and c, and data not shown), consistent with the idea that these compounds can exert anti-inflammatory effects independent of PPAR-γ.
Fig. 8.

PPAR-γ null astrocytes are sensitive to the anti-inflammatory effects of 15d-PGJ2 and ciglitazone. (a) Primary astrocytes do not express full-length PPAR-γ mRNA. Primary astrocyte cultures were prepared from post-natal day 1 frontal cortices of astrocyte-conditionally null mice or wild type (WT) littermates as described in Materials and methods. After 2 weeks in culture, whole-cell RNA was isolated, converted to cDNA, and amplified using primers specific for PPAR-γ exon 2. Aliquots of the cDNA were also amplified using primers for α-tubulin to confirm RNA and cDNA integrity. The gels depict PCR products obtained from astrocyte RNA prepared from two individual WT and two PPAR-γ null (KO) mice. In (b), PPAR-γ WT and KO astrocytes were seeded in 6-well plates at 1 × 106 cells per well and incubated overnight. The following day, astrocytes were pre-incubated with 15d-PGJ2 (15d; 10–20 μM) or ciglitazone (cig; 30–100 μM) for 1 h and subsequently stimulated with 107 heat-inactivated S. aureus (SA). Cell-free supernatants were collected at 48 h following S. aureus treatment and analyzed for nitrite production (mean ± SD of four independent wells per treatment). In (c), PPAR-γ KO and WT astrocytes were pretreated with 15d-PGJ2 (20 μM) or ciglitazone (100 μM) for 1 h, followed by 107 heat-inactivated S. aureus (SA). Cell-free supernatants were collected 48 h following S. aureus treatment and IL-1β levels were quantified by ELISA. Results are reported as the mean ± SD of four independent wells for each experimental treatment. Significant differences between astrocytes treated with S. aureus only versus cells exposed to the various concentrations of 15d-PGJ2 + S. aureus or ciglitazone + S. aureus are denoted with asterisks (*p < 0.05, **p < 0.001). Results are representative of two independent experiments.
Discussion
Although the immuno-modulatory effects of PPAR-γ agonists on LPS-induced astrocyte and microglial activation have been described (Petrova et al. 1999; Giri et al. 2004; Storer et al. 2005a), whether these compounds can affect astrocytic responses to S. aureus remained unknown. This represents a question of clinical importance as S. aureus is a common CNS pathogen and we have evidence to suggest in our model of experimental brain abscess that chronic astrocytic activation may contribute to the excessive tissue damage characteristic of this disease. In the present study, we found that the PPAR-γ agonists 15d-PGJ2 and ciglitazone attenuated S. aureus-induced astrocyte activation, and that these effects were largely independent of the PPAR-γ pathway. This was demonstrated by the finding that 15d-PGJ2 and ciglitazone were still capable of attenuating S. aureus-induced NO and IL-1β release from PPAR-γ KO astrocytes. Interestingly, similar anti-inflammatory effects on S. aureus-induced astrocyte activation were observed when drugs were applied as late as 6 h following stimulation, suggesting that these compounds could be potential therapeutic agents even in conditions of pre-existing inflammation.
One intriguing finding that surfaced during the course of these studies was related to the diverse effects of 15d-PGJ2 and ciglitazone on the expression of the neutrophil chemokine MIP-2. In fact, of all of the pro-inflammatory mediators examined, MIP-2 was the only example where the actions of the two drugs differed. Specifically, 15d-PGJ2 did not attenuate MIP-2 production from S. aureus-stimulated astrocytes either in pre-treatment or post-treatment paradigms, whereas ciglitazone inhibited MIP-2 expression under both conditions. This finding is in agreement with previous reports suggesting that 15d-PGJ2 is not a global inhibitor, but rather its actions are specific for a certain subset of inflammatory mediators (Zhang et al. 2001; Kielian et al. 2004b). Because 15d-PGJ2 has been reported as a potent inhibitor of nuclear factor-κB (NF-κB) signaling (Rossi et al. 2000; Straus et al. 2000), and MIP-2 production is primarily NF-κB-mediated (Kim et al. 2003; Zampetaki et al. 2004), the inability of 15d-PGJ2 to attenuate MIP-2 expression suggests the involvement of additional NF-κB-independent mechanisms in mediating S. aureus-induced MIP-2 production in astrocytes. Moreover, in agreement with studies demonstrating that 15d-PGJ2 induces IL-8 (functional human homolog of murine MIP-2) production in human monocytic cells and T cells (Zhang et al. 2001; Harris et al. 2002), we also observed a slight increase in MIP-2 expression in murine astrocytes. Interestingly, ciglitazone attenuated MIP-2 production from S. aureus-stimulated astrocytes regardless of when the drug was administered. This finding suggests that multiple discrete signaling pathways are targeted by 15d-PGJ2 and ciglitazone to modulate S. aureus-dependent astrocyte activation; however, these differences remain to be defined.
Recent studies from our laboratory have established that the pattern recognition receptor TLR2 is pivotal for S. aureus recognition by astrocytes and subsequent pro-inflammatory mediator release (Esen et al. 2004), suggesting that this receptor plays a central role in bacterial recognition events in the CNS. S. aureus treatment led to an increase in TLR2 expression, which was reversed with 15d-PGJ2 treatment, similar to that which we have reported in primary microglia (Kielian et al. 2004b). This decrease in TLR2 expression following 15d-PGJ2 pretreatment could account for the partial non-responsiveness of astrocytes to subsequent S. aureus stimulation. However, 15d-PGJ2 was also capable of attenuating pre-existing astrocyte activation induced by S. aureus engagement of TLR2. Therefore, this finding suggests that, in addition to regulating TLR2 levels, 15d-PGJ2 may also interfere with the signaling cascade elicited following TLR2 stimulation by S. aureus. As TLR2 gene expression is driven primarily through a NF-κB pathway (Musikacharoen et al. 2001; Wang et al. 2001b), the suppression of TLR2 levels suggests that the anti-inflammatory effects of 15d-PGJ2 might involve NF-κB inhibitory actions. This possibility is in agreement with recent reports where PPAR-γ agonists were found to modulate TLR2-mediated dendritic cell maturation by interfering with the NF-κB/MAPK and PI3K/Akt pathways (Appel et al. 2005). Indeed, our findings with PPAR-γ-deficient astrocytes demonstrated that the receptor was not required for the inhibitory effects of 15d-PGJ2 on astrocytic TLR2 expression, highly suggestive that the ability of 15d-PGJ2 to modulate NF-κB and MAPK pathways is likely responsible for the observed effects.
Over the past few years, several attempts have been made to elucidate whether the anti-inflammatory effects of PPAR-γ agonists are receptor dependent or independent. Embryonic lethality of PPAR-γ homozygous KO animals prompted the development of PPAR-γ heterozygous KO mice as well as conditional KO animals (Clark 2002; Daynes and Jones 2002; Zingarelli and Cook 2005). Despite these advances, the precise molecular mechanisms underlying the anti-inflammatory effects of PPAR-γ ligands remain controversial. Several studies have suggested that the anti-inflammatory activities of PPAR-γ agonists are mediated by PPAR-γ-dependent mechanisms. For instance, it has been demonstrated that ligand-activated PPAR-γ hetrodimerizes with retinoic acid receptor (RXR) to compete with various transcription factors, such as NF-κB, activator protein-1 (AP-1), JAK-STAT – signal transducer and activator of transcription and nuclear factor of activated T cells (NFAT) for binding to limited co-activators like CBP, P300, TIF-2, AIB-1, TRAP220, DRIP205 and SRC-1, culminating in cofactor deficiency-based transcriptional inhibition (Kodera et al. 2000). However, the inability of excess cofactors to reverse this repression led to alternative explanations suggesting physical interaction-mediated antagonism between PPAR-γ and other transcription factors (De Bosscher et al. 2000). Recently, Pascual et al. (2005) demonstrated that, in response to LPS, sumoylation of ligand-activated PPAR-γ occurs, which then interacts with the NCoR-HDAC3 co-repressor complex localized on the iNOS promoter, effectively inhibiting its ubiquitination-dependent removal and eventually leading to sustained transcriptional repression. More recently, several PPAR-γ-independent actions have been suggested to account for the anti-inflammatory effects of 15d-PGJ2 and TZDs. For example, several studies have shown that 15d-PGJ2 can interfere with NF-κB and AP-1 signaling pathways (Rossi et al. 2000; Straus et al. 2000; Castrillo et al. 2001; Wang et al. 2001a), modulate extracellular-regulated kinase 1 and 2 (ERK-1 and -2), and induce IκBα expression (Guyton et al. 2003), as well as affect suppressor of cytokine signaling 1 and 3 (SOCS1 and SOCS3) expression in glial cells, which in turn inhibits the LPS-induced JAK-STAT – signal transducer and activator of transcription signaling pathway (Park et al. 2003). Importantly, the attenuation of inflammatory responses by a 15d-PGJ2 analog that possesses a cyclopentenone ring similar to 15d-PGJ2, but lacks PPAR-γ binding moieties, is also supportive of receptor-independent pathways (Guyton et al. 2001; Musiek et al. 2005; Storer et al. 2005b). With regard to TZDs, only a few reports have demonstrated anti-inflammatory effects despite being potent PPAR-γ agonists (Vaidya et al. 1999; Li et al. 2000; Thieringer et al. 2000). Moreover, the anti-inflammatory effects of TZDs are observed at concentrations significantly higher than the Kd value for binding to PPAR-γ (Buckingham 2005; Zingarelli and Cook 2005). These findings, along with the observation that PPAR-γ antagonists failed to reverse the anti-inflammatory effects of PPAR-γ agonists, also support the receptor-independent theory (Hinz et al. 2003; Grau et al. 2004).
In the current study, the most direct evidence demonstrating that the anti-inflammatory effects of 15d-PGJ2 and ciglitazone on S. aureus-induced astrocyte activation occur primarily through a PPAR-γ-independent pathway is derived from experiments using PPAR-γ KO astrocytes. Specifically, S. aureus-induced NO and IL-1β production was attenuated to nearly the same degree by 15d-PGJ2 and ciglitazone in both PPAR-γ wild type and KO astrocytes. These results clearly demonstrate a PPAR-γ-independent mechanism of action and are in agreement with a recent report by Chawla et al. (2001), demonstrating that 15d-PGJ2 and TZDs suppressed TNF-α and IL-6 production in PPAR-γ wild-type and KO macrophages to similar extents. Importantly, the use of targeted PPAR-γ gene inactivation in astrocytes using Cre-Lox technology ensures that other CNS cell types express functional PPAR-γ, which may be important from a developmental standpoint and could conceivably influence the nature by which astrocytes respond to pathologic stimuli ex vivo.
In summary, the present work demonstrates that 15d-PGJ2 and ciglitazone attenuate S. aureus-induced astrocyte activation, which is mediated primarily through a PPAR-γ-independent pathway. The inhibitory effects of 15d-PGJ2 and ciglitazone were observed when administered either prior to or subsequent to bacterial stimulation, suggesting the possible use of this class of compounds for treating pre-existing inflammatory conditions. However, future studies are warranted to obtain a better understanding of the anti-inflammatory effect of PPAR-γ agonists, which may help in the development of new treatment strategies aimed at modifying the pathophysiological responses in brain abscess and other inflammation-mediated diseases.
Acknowledgments
The authors thank Drs Nilufer Esen, Sarita Garg and Mohsin Md. Syed, in addition to Patrick Mayes, Anessa Haney, Debbie Shuffield, Shuliang Liu, and Shao Xia Lin for excellent technical and advisory support. The authors also thank Dr Paul Drew for critical review of the manuscript. This work was supported by the NIH National Institute of Neurological Disorders and Stroke (2 RO1 NS40730) to TK and the NINDS-supported Core facility at UAMS (P30 NS047546).
Abbreviations
- ABC
ATP-binding cassette
- cfu
colony forming units
- 15d-PGJ2
15-deoxy-Δ12,14-prostaglandin J2
- GFAP
glial fibrillary acidic protein
- IL-1β
interleukin-1β
- iNOS
inducible nitric oxide synthase
- KO
knockout
- LPS
lipopolysaccharide
- MTT
3-[4,5-di-methylthiazol-2-yl]-2,5-diphenyltetrazolium bromide
- NF-κB
nuclear factor-κB
- NO
nitric oxide
- PPAR-γ
peroxisome proliferator activated receptor-γ
- PPRE
PPAR-responsive element
- TNF-α
tumor necrosis factor-α
- TLR2
Toll-like receptor 2
- TZD
thiazolidinedione
References
- Akira S. Toll-like receptor signaling. J Biol Chem. 2003;278:38 105–38 108. doi: 10.1074/jbc.R300028200. [DOI] [PubMed] [Google Scholar]
- Akiyama TE, Sakai S, Lambert G, et al. Conditional disruption of the peroxisome proliferator-activated receptor γ gene in mice results in lowered expression of ABCA1, ABCG1, and apoE in macrophages and reduced cholesterol efflux. Mol Cell Biol. 2002;22:2607–2619. doi: 10.1128/MCB.22.8.2607-2619.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan SM, Tyrrell PJ, Rothwell NJ. Interleukin-1 and neuronal injury. Nat Rev Immunol. 2005;5:629–640. doi: 10.1038/nri1664. [DOI] [PubMed] [Google Scholar]
- Appel S, Mirakaj V, Bringmann A, Weck MM, Grunebach F, Brossart P. PPAR-γ agonists inhibit toll-like receptor-mediated activation of dendritic cells via the MAP kinase and NF-κB pathways. Blood. 2005;106:3888–3894. doi: 10.1182/blood-2004-12-4709. [DOI] [PubMed] [Google Scholar]
- Asada K, Sasaki S, Suda T, Chida K, Nakamura H. Anti-inflammatory roles of peroxisome proliferator-activated receptor γ in human alveolar macrophages. Am J Respir Crit Care Med. 2004;169:195–200. doi: 10.1164/rccm.200207-740OC. [DOI] [PubMed] [Google Scholar]
- Baldwin AC, Kielian T. Persistent immune activation associated with a mouse model of Staphylococcus aureus-induced experimental brain abscess. J Neuroimmunol. 2004;151:24–32. doi: 10.1016/j.jneuroim.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Bal-Price A, Brown GC. Inflammatory neurodegeneration mediated by nitric oxide from activated glia-inhibiting neuronal respiration, causing glutamate release and excitotoxicity. J Neurosci. 2001;21:6480–6491. doi: 10.1523/JNEUROSCI.21-17-06480.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med. 2002;53:409–435. doi: 10.1146/annurev.med.53.082901.104018. [DOI] [PubMed] [Google Scholar]
- Bowman CC, Rasley A, Tranguch SL, Marriott I. Cultured astrocytes express toll-like receptors for bacterial products. Glia. 2003;43:281–291. doi: 10.1002/glia.10256. [DOI] [PubMed] [Google Scholar]
- Brown GC, Bal-Price A. Inflammatory neurodegeneration mediated by nitric oxide, glutamate, and mitochondria. Mol Neurobiol. 2003;27:325–355. doi: 10.1385/MN:27:3:325. [DOI] [PubMed] [Google Scholar]
- Buckingham RE. Thiazolidinediones: pleiotropic drugs with potent anti-inflammatory properties for tissue protection. Hepatol Res. 2005;33:167–170. doi: 10.1016/j.hepres.2005.09.027. [DOI] [PubMed] [Google Scholar]
- Cabrero A, Laguna JC, Vazquez M. Peroxisome proliferator-activated receptors and the control of inflammation. Curr Drug Targets Inflamm Allergy. 2002;1:243–248. doi: 10.2174/1568010023344616. [DOI] [PubMed] [Google Scholar]
- Camacho IE, Serneels L, Spittaels K, Merchiers P, Dominguez D, De Strooper B. Peroxisome-proliferator-activated receptor γ induces a clearance mechanism for the amyloid-β peptide. J Neurosci. 2004;24:10 908–10 917. doi: 10.1523/JNEUROSCI.3987-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell IW. The clinical significance of PPARγ agonism. Curr Mol Med. 2005;5:349–363. doi: 10.2174/1566524053766068. [DOI] [PubMed] [Google Scholar]
- Carpentier PA, Begolka WS, Olson JK, Elhofy A, Karpus WJ, Miller SD. Differential activation of astrocytes by innate and adaptive immune stimuli. Glia. 2005;49:360–374. doi: 10.1002/glia.20117. [DOI] [PubMed] [Google Scholar]
- Castrillo A, Mojena M, Hortelano S, Bosca L. Peroxisome proliferator-activated receptor-γ-independent inhibition of macrophage activation by the non-thiazolidinedione agonist L-796 449. Comparison with the effects of 15-deoxy-δ12,14-prostaglandin J2. J Biol Chem. 2001;276:34 082–34 088. doi: 10.1074/jbc.M102472200. [DOI] [PubMed] [Google Scholar]
- Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evans RM. PPAR-γ-dependent and -independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat Med. 2001;7:48–52. doi: 10.1038/83336. [DOI] [PubMed] [Google Scholar]
- Clark RB. The role of PPARs in inflammation and immunity. J Leukoc Biol. 2002;71:388–400. [PubMed] [Google Scholar]
- Consoli A, Devangelio E. Thiazolidinediones and inflammation. Lupus. 2005;14:794–797. doi: 10.1191/0961203305lu2223oa. [DOI] [PubMed] [Google Scholar]
- Daynes RA, Jones DC. Emerging roles of PPARs in inflammation and immunity. Nat Rev Immunol. 2002;2:748–759. doi: 10.1038/nri912. [DOI] [PubMed] [Google Scholar]
- De Bosscher K, Vanden Berghe W, Vermeulen L, Plaisance S, Boone E, Haegeman G. Glucocorticoids repress NF-κB-driven genes by disturbing the interaction of p65 with the basal transcription machinery, irrespective of co-activator levels in the cell. Proc Natl Acad Sci USA. 2000;97:3919–3924. doi: 10.1073/pnas.97.8.3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumasia R, Eagle KA, Kline-Rogers E, May N, Cho L, Mukherjee D. Role of PPAR-γ agonist thiazolidinediones in treatment of pre-diabetic and diabetic individuals: a cardiovascular perspective. Curr Drug Targets Cardiovasc Haematol Disord. 2005;5:377–386. doi: 10.2174/156800605774370362. [DOI] [PubMed] [Google Scholar]
- Esen N, Tanga FY, DeLeo JA, Kielian T. Toll-like receptor 2 (TLR2) mediates astrocyte activation in response to the Gram-positive bacterium Staphylococcus aureus. J Neurochem. 2004;88:746–758. doi: 10.1046/j.1471-4159.2003.02202.x. [DOI] [PubMed] [Google Scholar]
- Feinstein DL. Therapeutic potential of peroxisome proliferator-activated receptor agonists for neurological disease. Diabetes Technol Ther. 2003;5:67–73. doi: 10.1089/152091503763816481. [DOI] [PubMed] [Google Scholar]
- Feinstein DL, Galea E, Gavrilyuk V, Brosnan CF, Whitacre CC, Dumitrescu-Ozimek L, Landreth GE, Pershadsingh HA, Weinberg G, Heneka MT. Peroxisome proliferator-activated receptor-γ agonists prevent experimental autoimmune encephalomyelitis. Ann Neurol. 2002;51:694–702. doi: 10.1002/ana.10206. [DOI] [PubMed] [Google Scholar]
- Feinstein DL, Spagnolo A, Akar C, Weinberg G, Murphy P, Gavrilyuk V, Russo CD. Receptor-independent actions of PPAR thiazolidinedione agonists: is mitochondrial function the key? Biochem Pharmacol. 2005;70:177–188. doi: 10.1016/j.bcp.2005.03.033. [DOI] [PubMed] [Google Scholar]
- Gervois P, Fruchart JC, Staels B. Inflammation, dyslipidaemia, diabetes and PPars: pharmacological interest of dual PPARα and PPARγ agonists. Int J Clin Pract Suppl. 2004;143:22–29. doi: 10.1111/j.1368-504x.2004.00376.x. [DOI] [PubMed] [Google Scholar]
- Giri S, Rattan R, Singh AK, Singh I. The 15-deoxy-delta12,14-prostaglandin J2 inhibits the inflammatory response in primary rat astrocytes via down-regulating multiple steps in phosphatidylinositol 3-kinase-Akt-NF-κB-p300 pathway independent of peroxisome proliferator-activated receptor γ. J Immunol. 2004;173:5196–5208. doi: 10.4049/jimmunol.173.8.5196. [DOI] [PubMed] [Google Scholar]
- Grau R, Iniguez MA, Fresno M. Inhibition of activator protein 1 activation, vascular endothelial growth factor, and cyclooxygenase-2 expression by 15-deoxy-Δ12,14-prostaglandin J2 in colon carcinoma cells: evidence for a redox-sensitive per-oxisome proliferator-activated receptor-γ-independent mechanism. Cancer Res. 2004;64:5162–5171. doi: 10.1158/0008-5472.CAN-04-0849. [DOI] [PubMed] [Google Scholar]
- Guyton K, Bond R, Reilly C, Gilkeson G, Halushka P, Cook J. Differential effects of 15-deoxy-δ12,14-prostaglandin J2 and a peroxisome proliferator-activated receptor γ agonist on macrophage activation. J Leukoc Biol. 2001;69:631–638. [PubMed] [Google Scholar]
- Guyton K, Zingarelli B, Ashton S, Teti G, Tempel G, Reilly C, Gilkeson G, Halushka P, Cook J. Peroxisome proliferator-activated receptor-γ agonists modulate macrophage activation by gram-negative and gram-positive bacterial stimuli. Shock. 2003;20:56–62. doi: 10.1097/01.shk.0000070903.21762.f8. [DOI] [PubMed] [Google Scholar]
- Hansson E, Ronnback L. Glial neuronal signaling in the central nervous system. FASEB J. 2003;17:341–348. doi: 10.1096/fj.02-0429rev. [DOI] [PubMed] [Google Scholar]
- Harris SG, Smith RS, Phipps RP. 15-deoxy-Δ12,14-PGJ2 induces IL-8 production in human T cells by a mitogen-activated protein kinase pathway. J Immunol. 2002;168:1372–1379. doi: 10.4049/jimmunol.168.3.1372. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Sastre M, Dumitrescu-Ozimek L, Hanke A, Dewachter I, Kuiperi C, O’Banion K, Klockgether T, Van Leuven F, Landreth GE. Acute treatment with the PPARγ agonist pioglitazone and ibuprofen reduces glial inflammation and Aβ1–42 levels in APPV717I transgenic mice. Brain. 2005;128:1442–1453. doi: 10.1093/brain/awh452. [DOI] [PubMed] [Google Scholar]
- Hinz B, Brune K, Pahl A. 15-Deoxy-Δ12,14-prostaglandin J2 inhibits the expression of pro-inflammatory genes in human blood monocytes via a PPAR-γ-independent mechanism. Biochem Biophys Res Commun. 2003;302:415–420. doi: 10.1016/s0006-291x(03)00195-5. [DOI] [PubMed] [Google Scholar]
- Huang JT, Welch JS, Ricote M, Binder CJ, Willson TM, Kelly C, Witztum JL, Funk CD, Conrad D, Glass CK. Interleukin-4-dependent production of PPAR-γ ligands in macrophages by 12/15-lipoxygenase. Nature. 1999;400:378–382. doi: 10.1038/22572. [DOI] [PubMed] [Google Scholar]
- Jiang C, Ting AT, Seed B. PPAR-γ agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- Kiaei M, Kipiani K, Chen J, Calingasan NY, Beal MF. Peroxisome proliferator-activated receptor-γ agonist extends survival in transgenic mouse model of amyotrophic lateral sclerosis. Exp Neurol. 2005;191:331–336. doi: 10.1016/j.expneurol.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Kielian T. Immunopathogenesis of brain abscess. J Neuroinflammation. 2004;1:16. doi: 10.1186/1742-2094-1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T. Toll-like receptors in central nervous system glial inflammation and homeostasis. J Neurosci Res. 2006;83:711–730. doi: 10.1002/jnr.20767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T, Drew PD. Effects of peroxisome proliferator-activated receptor-γ agonists on central nervous system inflammation. J Neurosci Res. 2003;71:315–325. doi: 10.1002/jnr.10501. [DOI] [PubMed] [Google Scholar]
- Kielian T, Barry B, Hickey WF. CXC chemokine receptor-2 ligands are required for neutrophil-mediated host defense in experimental brain abscesses. J Immunol. 2001;166:4634–4643. doi: 10.4049/jimmunol.166.7.4634. [DOI] [PubMed] [Google Scholar]
- Kielian T, Bearden ED, Baldwin AC, Esen N. IL-1 and TNF-α play a pivotal role in the host immune response in a mouse model of Staphylococcus aureus-induced experimental brain abscess. J Neuropathol Exp Neurol. 2004a;63:381–396. doi: 10.1093/jnen/63.4.381. [DOI] [PubMed] [Google Scholar]
- Kielian T, McMahon M, Bearden ED, Baldwin AC, Drew PD, Esen N. S. aureus-dependent microglial activation is selectively attenuated by the cyclopentenone prostaglandin 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) J Neurochem. 2004b;90:1163–1172. doi: 10.1111/j.1471-4159.2004.02579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T, Esen N, Bearden ED. Toll-like receptor 2 (TLR2) is pivotal for recognition of S. aureus peptidoglycan but not intact bacteria by microglia. Glia. 2005;49:567–576. doi: 10.1002/glia.20144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DS, Han JH, Kwon HJ. NF-κB and c-Jun-dependent regulation of macrophage inflammatory protein-2 gene expression in response to lipopolysaccharide in RAW 264.7 cells. Mol Immunol. 2003;40:633–643. doi: 10.1016/j.molimm.2003.07.001. [DOI] [PubMed] [Google Scholar]
- Kodera Y, Takeyama K, Murayama A, Suzawa M, Masuhiro Y, Kato S. Ligand type-specific interactions of peroxisome proliferator-activated receptor γ with transcriptional co-activators. J Biol Chem. 2000;275:33 201–33 204. doi: 10.1074/jbc.C000517200. [DOI] [PubMed] [Google Scholar]
- Lemberger T, Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: a nuclear receptor signaling pathway in lipid physiology. Annu Rev Cell Dev Biol. 1996;12:335–363. doi: 10.1146/annurev.cellbio.12.1.335. [DOI] [PubMed] [Google Scholar]
- Li M, Pascual G, Glass CK. Peroxisome proliferator-activated receptor γ-dependent repression of the inducible nitric oxide synthase gene. Mol Cell Biol. 2000;20:4699–4707. doi: 10.1128/mcb.20.13.4699-4707.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggi LB, Jr, Sadeghi H, Weigand C, Scarim AL, Heitmeier MR, Corbett JA. Anti-inflammatory actions of 15-deoxy-δ12,14-prostaglandin J2 and troglitazone: evidence for heat shock-dependent and -independent inhibition of cytokine-induced inducible nitric oxide synthase expression. Diabetes. 2000;49:346–355. doi: 10.2337/diabetes.49.3.346. [DOI] [PubMed] [Google Scholar]
- Mander P, Brown GC. Nitric oxide, hypoxia and brain inflammation. Biochem Soc Trans. 2004;32:1068–1069. doi: 10.1042/BST0321068. [DOI] [PubMed] [Google Scholar]
- Mayhan WG. Cellular mechanisms by which tumor necrosis factor-α produces disruption of the blood–brain barrier. Brain Res. 2002;927:144–152. doi: 10.1016/s0006-8993(01)03348-0. [DOI] [PubMed] [Google Scholar]
- Motojima K, Passilly P, Peters JM, Gonzalez FJ, Latruffe N. Expression of putative fatty acid transporter genes are regulated by peroxisome proliferator-activated receptor α and γ activators in a tissue- and inducer-specific manner. J Biol Chem. 1998;273:16 710–16 714. doi: 10.1074/jbc.273.27.16710. [DOI] [PubMed] [Google Scholar]
- Musiek ES, Gao L, Milne GL, et al. Cyclopentenone iso-prostanes inhibit the inflammatory response in macrophages. J Biol Chem. 2005;280:35 562–35 570. doi: 10.1074/jbc.M504785200. [DOI] [PubMed] [Google Scholar]
- Musikacharoen T, Matsuguchi T, Kikuchi T, Yoshikai Y. NF-κB and STAT5 play important roles in the regulation of mouse Toll-like receptor 2 gene expression. J Immunol. 2001;166:4516–4524. doi: 10.4049/jimmunol.166.7.4516. [DOI] [PubMed] [Google Scholar]
- Nau R, Bruck W. Neuronal injury in bacterial meningitis: mechanisms and implications for therapy. Trends Neurosci. 2002;25:38–45. doi: 10.1016/s0166-2236(00)02024-5. [DOI] [PubMed] [Google Scholar]
- Nencioni A, Wesselborg S, Brossart P. Role of peroxisome proliferator-activated receptor γ and its ligands in the control of immune responses. Crit Rev Immunol. 2003;23:1–13. doi: 10.1615/critrevimmunol.v23.i12.10. [DOI] [PubMed] [Google Scholar]
- Nguyen MD, Julien JP, Rivest S. Innate immunity: the missing link in neuroprotection and neurodegeneration? Nat Rev Neurosci. 2002;3:216–227. doi: 10.1038/nrn752. [DOI] [PubMed] [Google Scholar]
- Owens T. Toll-like receptors on astrocytes: patterning for immunity. J Neuroimmunol. 2005;159:1–2. doi: 10.1016/j.jneuroim.2004.10.015. [DOI] [PubMed] [Google Scholar]
- Park EJ, Park SY, Joe EH, Jou I. 15d-PGJ2 and rosiglitazone suppress Janus kinase-STAT inflammatory signaling through induction of suppressor of cytokine signaling 1 (SOCS1) and SOCS3 in glia. J Biol Chem. 2003;278:14 747–14 752. doi: 10.1074/jbc.M210819200. [DOI] [PubMed] [Google Scholar]
- Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, Glass CK. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-γ. Nature. 2005;437:759–763. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegorier JP. PPAR receptors and insulin sensitivity: new agonists in development. Ann Endocrinol (Paris) 2005;66:1S10–17. [PubMed] [Google Scholar]
- Petrova TV, Akama KT, Van Eldik LJ. Cyclopentenone prostaglandins suppress activation of microglia: down-regulation of inducible nitric-oxide synthase by 15-deoxy-Δ12,14-prostaglandin J2. Proc Natl Acad Sci USA. 1999;96:4668–4673. doi: 10.1073/pnas.96.8.4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-γ is a negative regulator of macrophage activation. Nature. 1998a;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- Ricote M, Huang J, Fajas L, Li A, Welch J, Najib J, Witztum JL, Auwerx J, Palinski W, Glass CK. Expression of the peroxisome proliferator-activated receptor γ (PPARγ) in human atherosclerosis and regulation in macrophages by colony-stimulating factors and oxidized low-density lipoprotein. Proc Natl Acad Sci USA. 1998b;95:7614–7619. doi: 10.1073/pnas.95.13.7614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricote M, Huang JT, Welch JS, Glass CK. The per-oxisome proliferator-activated receptor (PPARγ) as a regulator of monocyte/macrophage function. J Leukoc Biol. 1999;66:733–739. doi: 10.1002/jlb.66.5.733. [DOI] [PubMed] [Google Scholar]
- Rossi A, Kapahi P, Natoli G, Takahashi T, Chen Y, Karin M, Santoro MG. Anti-inflammatory cyclopentenone pro-staglandins are direct inhibitors of IκB kinase. Nature. 2000;403:103–108. doi: 10.1038/47520. [DOI] [PubMed] [Google Scholar]
- Safieh-Garabedian B, Haddad JJ, Saade NE. Cytokines in the central nervous system: targets for therapeutic intervention. Curr Drug Targets CNS Neurol Disord. 2004;3:271–280. doi: 10.2174/1568007043337300. [DOI] [PubMed] [Google Scholar]
- Staels B, Fruchart JC. Therapeutic roles of peroxisome proliferator-activated receptor agonists. Diabetes. 2005;54:2460–2470. doi: 10.2337/diabetes.54.8.2460. [DOI] [PubMed] [Google Scholar]
- Storer PD, Xu J, Chavis J, Drew PD. Peroxisome proliferator-activated receptor-γ agonists inhibit the activation of microglia and astrocytes: implications for multiple sclerosis. J Neuroimmunol. 2005a;161:113–122. doi: 10.1016/j.jneuroim.2004.12.015. [DOI] [PubMed] [Google Scholar]
- Storer PD, Xu J, Chavis JA, Drew PD. Cyclopentenone prostaglandins PGA2 and 15-deoxy-δ12,14 PGJ2 suppress activation of murine microglia and astrocytes: implications for multiple sclerosis. J Neurosci Res. 2005b;80:66–74. doi: 10.1002/jnr.20413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straus DS, Pascual G, Li M, Welch JS, Ricote M, Hsiang CH, Sengchanthalangsy LL, Ghosh G, Glass CK. 15-Deoxy-δ12,14-prostaglandin J2 inhibits multiple steps in the NF-κB signaling pathway. Proc Natl Acad Sci USA. 2000;97:4844–4849. doi: 10.1073/pnas.97.9.4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Itoh H, Doi K, et al. Down-regulation of peroxisome proliferator-activated receptor γ expression by inflammatory cytokines and its reversal by thiazolidinediones. Diabetologia. 1999;42:702–710. doi: 10.1007/s001250051218. [DOI] [PubMed] [Google Scholar]
- Thieringer R, Fenyk-Melody JE, Le Grand CB, Shelton BA, Detmers PA, Somers EP, Carbin L, Moller DE, Wright SD, Berger J. Activation of peroxisome proliferator-activated receptor γ does not inhibit IL-6 or TNF-α responses of macrophages to lipopolysaccharide in vitro or in vivo. J Immunol. 2000;164:1046–1054. doi: 10.4049/jimmunol.164.2.1046. [DOI] [PubMed] [Google Scholar]
- Vaidya S, Somers EP, Wright SD, Detmers PA, Bansal VS. 15-Deoxy-Δ12,14-prostaglandin J2 inhibits the β2 integrin-dependent oxidative burst: involvement of a mechanism distinct from peroxisome proliferator-activated receptor γ ligation. J Immunol. 1999;163:6187–6192. [PubMed] [Google Scholar]
- Vidal H. PPAR receptors: recent data. Ann Endocrinol (Paris) 2005;66:1S5–9. [PubMed] [Google Scholar]
- Wang P, Anderson PO, Chen S, Paulsson KM, Sjogren HO, Li S. Inhibition of the transcription factors AP-1 and NF-κB in CD4 T cells by peroxisome proliferator-activated receptor γ ligands. Int Immunopharmacol. 2001a;1:803–812. doi: 10.1016/s1567-5769(01)00015-7. [DOI] [PubMed] [Google Scholar]
- Wang T, Lafuse WP, Zwilling BS. NFκB and Sp1 elements are necessary for maximal transcription of toll-like receptor 2 induced by Mycobacterium avium. J Immunol. 2001b;167:6924–6932. doi: 10.4049/jimmunol.167.12.6924. [DOI] [PubMed] [Google Scholar]
- Willson TM, Cobb JE, Cowan DJ, Wiethe RW, Correa ID, Prakash SR, Beck KD, Moore LB, Kliewer SA, Lehmann JM. The structure-activity relationship between peroxisome proliferator-activated receptor γ agonism and the anti-hyperglycemic activity of thiazolidinediones. J Med Chem. 1996;39:665–668. doi: 10.1021/jm950395a. [DOI] [PubMed] [Google Scholar]
- Yki-Jarvinen H. Thiazolidinediones. N Engl J Med. 2004;351:1106–1118. doi: 10.1056/NEJMra041001. [DOI] [PubMed] [Google Scholar]
- Yoshimura A, Lien E, Ingalls RR, Tuomanen E, Dziarski R, Golenbock D. Cutting edge: recognition of Gram-positive bacterial cell wall components by the innate immune system occurs via Toll-like receptor 2. J Immunol. 1999;163:1–5. [PubMed] [Google Scholar]
- Zampetaki A, Mitsialis SA, Pfeilschifter J, Kourembanas S. Hypoxia induces macrophage inflammatory protein-2 (MIP-2) gene expression in murine macrophages via NF-κB: the prominent role of p42/p44 and PI3 kinase pathways. FASEB J. 2004;18:1090–1092. doi: 10.1096/fj.03-0991fje. [DOI] [PubMed] [Google Scholar]
- Zhang X, Wang JM, Gong WH, Mukaida N, Young HA. Differential regulation of chemokine gene expression by 15-deoxy-δ12,14 prostaglandin J2. J Immunol. 2001;166:7104–7111. doi: 10.4049/jimmunol.166.12.7104. [DOI] [PubMed] [Google Scholar]
- Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K, Messing A. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis. 2001;31:85–94. doi: 10.1002/gene.10008. [DOI] [PubMed] [Google Scholar]
- Zingarelli B, Cook JA. Peroxisome proliferator-activated receptor-γ is a new therapeutic target in sepsis and inflammation. Shock. 2005;23:393–399. doi: 10.1097/01.shk.0000160521.91363.88. [DOI] [PubMed] [Google Scholar]