

Abstract
Microglia represent one effector arm of CNS innate immunity as evident by their role in pathogen recognition. We previously reported that exposure of microglia to Staphylococcus aureus (S. aureus), a prevalent CNS pathogen, led to elevated Toll-like receptor 2 (TLR2) expression, a pattern recognition receptor capable of recognizing conserved structural motifs associated with gram-positive bacteria such as S. aureus. In this study, we demonstrate that the proinflammatory cytokine tumor necrosis factor-α (TNF-α) enhances TLR2 expression in microglia, whereas interleukin-1β has no significant effect. To determine the downstream signaling events responsible for elevated microglial TLR2 expression in response to TNF-α, a series of signal transduction inhibitors were employed. Treatment with caffeic acid phenethyl ester, an inhibitor of redox-mediated nuclear factor-kappa B activation, significantly attenuated TNF-α-induced TLR2 expression. Similar results were observed with the IKK-2 and IκB-α inhibitors SC-514 and BAY 11-7082, respectively. In contrast, no significant alterations in TLR2 expression were observed with protein kinase C or p38 mitogen-activated protein kinase inhibitors. A definitive role for TNF-α was demonstrated by the inability of S. aureus to augment TLR2 expression in microglia isolated from TNF-α knockout mice. In addition, TLR2 expression was significantly attenuated in brain abscesses of TNF-α knockout mice. Collectively, these results indicate that in response to S. aureus, TNF-α acts in an autocrine/paracrine manner to enhance TLR2 expression in microglia and that this effect is mediated, in part, by activation of the nuclear factor-kappa B pathway.
Keywords: microglia, nuclear factor-kappa B, Toll-like receptor 2, tumor necrosis factor-alpha
Microglia represent one effector arm of innate immunity in the CNS parenchyma as evident by their roles in pathogen recognition (Aloisi 2001; Hanisch 2002). As such, these cells are uniquely poised to provide an initial line of defense against invading microorganisms into the CNS prior to leukocyte infiltration. Gram-positive bacteria, such as Staphylococcus aureus, are frequent etiologic agents of CNS infectious diseases and are a common pathogen associated with brain abscess (Mathisen and Johnson 1997; Townsend and Scheld 1998). Our group has recently reported that exposure of microglia to S. aureus leads to the elaboration of a wide array of proinflammatory and bactericidal mediators and enhanced expression of surface receptors that play a pivotal role in bacterial recognition and antigen presentation (Kielian et al. 2002, 2004b, 2005a). Among the receptors modulated by S. aureus in microglia include the cytokine signaling pattern recognition receptor (PRR) Toll-like receptor 2 (TLR2) (Kielian et al. 2002, 2005a).
Toll-like receptors are a family of PRRs expressed on cells of the innate immune system that allow for the recognition of conserved structural motifs on a wide array of pathogens referred to as pathogen-associated molecular patterns (Kirk and Bazan 2005; Akira et al. 2006). To date, 13 TLRs have been identified, with TLR2 playing a pivotal role in recognizing structural components of various gram-positive bacteria, fungi, and protozoa (Kirk and Bazan 2005; Akira et al. 2006). Microglia express a large majority of these TLRs making them primed to respond immediately to infectious pathogens in the CNS parenchyma (Rivest 2003; Olson and Miller 2004; Kielian 2006). With regard to brain abscess, previous work from our laboratory has established that microglial TLR2 expression is increased following S. aureus exposure and plays an important role in bacterial recognition and subsequent activation of microglia (Kielian et al. 2002, 2005a). However, it has not yet been established whether the ability of S. aureus to augment TLR2 expression is a direct effect of bacterial stimulation or mediated indirectly through the autocrine/paracrine actions of proinflammatory cytokines produced by microglia in response to S. aureus. To evaluate whether TLR2 expression is subject to regulation by proinflammatory cytokines, we examined receptor levels in response to tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β), two major cytokine products of S. aureus-activated microglia (Kielian et al. 2005a; Esen and Kielian 2006).
In this study, we demonstrate that TNF-α is sufficient to induce TLR2 expression in primary microglia. Although recent studies have demonstrated a role for mitogen-activated protein kinase (MAPK), SP-1, and nuclear factor-kappa B (NF-κB) signaling pathways in regulating TLR2 expression in monocytes/macrophages (Musikacharoen et al. 2001; Wang et al. 2001; Haehnel et al. 2002; Wang et al. 2002), the precise downstream signaling events leading to the induction of TLR2 expression in microglia have not yet been investigated. In addition, none of these previous reports examined TLR2 protein levels, which represents another novel aspect of our study as alterations in mRNA expression do not always translate into changes at the protein level. Therefore, in the present study, we examined the role of different signal transduction pathways in regulating the TNF-α-induced increase in microglial TLR2 expression by employing pharmacological inhibitors of disparate cell signaling cascades. Caffeic acid phenethyl ester (CAPE), a non-specific inhibitor of the NF-κB pathway (Natarajan et al. 1996), significantly attenuated the TNF-α-mediated increase in TLR2 expression. In contrast, bisindolylmaleimide (BIM) and SB202190, which represent protein kinase C (PKC) and MAPK inhibitors, respectively (Toullec et al. 1991; Lee et al. 1994), failed to modulate the increase in microglial TLR2 levels following TNF-α exposure. Both BAY 11-7082 and SC-514, potent inhibitors of inhibitory-kappa B (IκB)-α phosphorylation and IκB kinase (IKK)-2, respectively (Pierce et al. 1997; Kishore et al. 2003; Karin et al. 2004; Zhi et al. 2007), attenuated the TNF-α-dependent increase in microglial TLR2 expression, suggesting an important role for the NF-κB pathway in regulating cytokine-induced TLR2 expression. Finally, a pivotal role for autocrine/paracrine TNF-α in augmenting microglial TLR2 levels in response to S. aureus was demonstrated by the inability of primary microglia isolated from TNF-α knockout (KO) mice to up-regulate TLR2 expression following bacterial exposure. Collectively, these findings demonstrate that microglial TLR2 expression can be induced by TNF-α, which signals, in part, via a NF-κB pathway.
Materials and methods
Preparation of primary mouse microglia
Primary microglia were prepared from neonatal C57BL/6 mice (postnatal day 2–4) as previously described (Kielian et al. 2004b). For studies investigating the functional importance of TNF-α in mediating the S. aureus-dependent increase in microglial TLR2 expression, primary microglia were isolated from neonatal TNF-α KO mice on a C57BL/6 background (Jackson Labs, Bar Harbor, ME, USA). Briefly, when mixed glial cultures reached confluency (7–10 days), flasks were shaken at 50 g overnight at 37°C to recover microglia. The purity of microglial cultures was evaluated by immunohistochemical staining using antibodies against CD11b (BD Pharmingen, San Diego, CA, USA) and glial fibrillary acidic protein (DAKO Corp., Carpenteria, CA, USA) to identify microglia and astrocytes, respectively, and was routinely greater than 95%.
Reagents
Recombinant mouse TNF-α and IL-1β were purchased from BD Pharmingen in a low endotoxin/no azide form. CAPE, BIM, SB202190, BAY 11-7082, and SC-514 were purchased from Calbiochem (San Diego, CA, USA). Heat-inactivated S. aureus (strain RN6390, kindly provided by Dr Ambrose Cheung, Dartmouth Medical School) was prepared as previously described (Kielian et al. 2002). All reagents and culture media were verified to have endotoxin levels <0.03 EU/mL as determined by Limulus amebocyte lysate assay (Associates of Cape Cod, Falmouth, MA, USA).
Cell viability assays
To confirm that any observed changes in microglial TLR2 expression in response to pharmacological inhibitors were not due to toxic effects, a standard MTT 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide assay based upon the mitochondrial conversion of MTT 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide into formazan crystals was performed as previously described (Kielian et al. 2004b). Results are reported as the raw OD570 readings (mean ± SD).
Quantitative real-time RT-PCR
Total RNA from microglia or brain abscess tissues was isolated using the TriZol reagent and treated with DNAse1 (both from Invitrogen, Carlsbad, CA, USA) prior to use in quantitative real-time RT-PCR (qRT-PCR) studies. The experimental procedure was performed as previously described (Kielian et al. 2005a). Briefly, TLR2 and glyceraldehyde-3-phosphate dehydrogenase primers and TAMRA Taqman probes were designed as previously described (Esen et al. 2004) and synthesized by Applied Biosystems (ABI, Foster City, CA, USA). Comparisons in gene expression were calculated after normalizing cycle thresholds against the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase and are presented as the fold-induction (2−ΔΔCt) value relative to unstimulated microglia or uninfected mice.
Protein extraction and western blotting
Protein extracts were prepared from primary microglia cultured in six-well plates by lysing cells in 200 μL of radio-immunoprecipitation assay buffer [1% Triton-X 100 and 0.1% sodium dodecyl sulfate in phosphate-buffered saline (PBS), pH 7.4] supplemented with a Complete™ protease inhibitor cocktail tablet (Roche, Indianapolis, IN, USA). Lysates were incubated on ice for 30 min followed by centrifugation at 21 000 g for 15 min at 4°C to pellet debris. Calculation of total protein in microglial extracts was determined using a standard protein assay (bicinchoninic acid protein assay reagent; Bio-Rad, Hercules, CA, USA). Changes in TLR2 protein expression in either primary microglia or brain abscess homogenates were evaluated by western blotting. Blots were probed using a goat anti-mouse TLR2 antibody (R&D Systems, Minneapolis, MN, USA) followed by a donkey anti-goat IgG-horseradish peroxidase secondary antibody (Jackson Immuno-research, West Grove, PA, USA). Blots were stripped and re-probed with a rabbit anti-actin polyclonal antibody (Sigma, St Louis, MO, USA) for normalization. Blots were developed using the Immobilon western substrate (Millipore, Billerica, MA, USA) and visualized by exposure to X-ray film.
Phagocytosis assay
The effects of TNF-α-induced TLR2 expression on microglial phagocytosis of S. aureus was performed as previously described (Esen and Kielian 2007). Briefly, C57BL/6 primary microglia were seeded onto 12 mm coverslips in 24-well plates and incubated overnight. The following day, cells were pre-treated with either medium alone or 100 ng/mL of recombinant mouse TNF-α for 24 h to successfully augment TLR2 protein levels. After the 24 h period, microglia were incubated with a heat-killed S. aureus isolate that constitutively expresses green fluorescence protein (kindly provided by Dr Ambrose Cheung) for 3 h, whereupon Hoechst 33342 (Molecular Probes, Eugene, OR, USA) was added to visualize nuclei. Cells were washed extensively with PBS and incubated with 0.05% crystal violet in 0.15 mol/L NaCl for 45 s to quench any fluorescence signal from residual extracellular bacteria. Coverslips were mounted onto glass slides using the Prolong anti-fade reagent (Molecular Probes) and sealed using nail polish. Coverslips were imaged using a Zeiss laser scanning confocal microscope (LSM 510; Carl Zeiss Microimaging Inc, Thornwood, NY, USA) with the confocal pinhole set to obtain an optical section thickness of 1.6 μm. Microglial-conditioned supernatants were also collected at 3 h following the addition of S. aureus-green fluorescence protein, centrifuged at 21 000 g for 15 min at 4°C to pellet bacteria, and stored at −80°C until analysis of proinflammatory mediator expression by ELISA.
Generation of experimental brain abscesses
Brain abscesses were induced in 6- to 8-week-old C57BL/6 wild-type (WT) or TNF-α KO mice using a stereotaxic approach as previously described (Kielian et al. 2004a, 2005b). For all studies, an equal number of age-matched males and females were used as previous work has established that both genders exhibit qualitatively similar inflammatory profiles following bacterial challenge (Kielian et al. 2001b,a, 2004a, 2005b). The animal use protocol has been approved by the University of Arkansas for Medical Sciences Institutional Animal Care and Use Committee and is in accord with the NIH guidelines for the use of rodents.
To simultaneously collect RNA and protein for analysis of TLR2 expression in vivo, brain abscesses were visualized by the stab wound created during injections, sectioned within 2–3 mm on all sides, and homogenized in PBS supplemented with protease and RNAse inhibitors. Subsequently, RNA and protein extracts were prepared as previously described (Kielian et al. 2005b).
Statistics
Statistical differences between experimental groups were determined by the Student’s t-test at the 95% confidence interval using SigmaStat (SPSS Science, Chicago, IL, USA).
Results
TNF-α is a robust inducer of TLR2 expression in microglia
We have previously demonstrated that S. aureus is a potent stimulus for the production of key proinflammatory cytokines in microglia including TNF-α and IL-1β (Kielian et al. 2002, 2004b; Esen and Kielian 2006). Another consequence of bacterial stimulation is the elevated expression of TLR2 in microglia, a PRR that is important for recognizing gram-positive pathogens such as S. aureus (Kielian et al. 2002, 2004b, 2005a). Therefore, it was plausible that S. aureus augments TLR2 expression via the autocrine/paracrine action of cytokines such as IL-1β and TNF-α elicited in response to bacterial infection. To evaluate the role of these cytokines on microglial TLR2 expression, cells were treated with various concentrations of TNF-α or IL-1β either alone or in combination, whereupon TLR2 mRNA levels were examined by qRT-PCR. Only TNF-α was capable of significantly augmenting TLR2 mRNA expression (Fig. 1). Although IL-1β treatment exerted modest increases in TLR2 mRNA levels, these changes did not reach statistical significance. In addition, the combination of TNF-α and IL-1β did not lead to further increases in TLR2 mRNA expression compared with TNF-α alone (Fig. 1). To evaluate whether the observed alterations in TNF-α-induced TLR2 mRNA expression were reflected at the protein level, western blots were performed. As was observed with qRT-PCR, TNF-α was a potent inducer of TLR2 protein expression (Fig. 2). In contrast, the highest dose of IL-1β examined (100 ng/mL) did not alter TLR2 levels compared with unstimulated microglia and the co-administration of IL-1β with TNF-α was not capable of further increasing receptor expression in comparison with cells treated with TNF-α only. We confirmed that treatment of primary microglia with the highest dose of TNF-α alone (i.e. 100 ng/mL) did not lead to any cytotoxicity (data not shown). Based on the finding that TNF-α was a potent inducer of both TLR2 mRNA and protein expression, subsequent experiments were performed using this cytokine only. Collectively, these findings indicate that TNF-α is a strong stimulus for TLR2 expression in microglia. The fact that TNF-α is a major product of S. aureus-activated microglia suggests that this cytokine may be responsible, in part, for the increase in TLR2 levels observed following S. aureus stimulation.
Fig. 1.
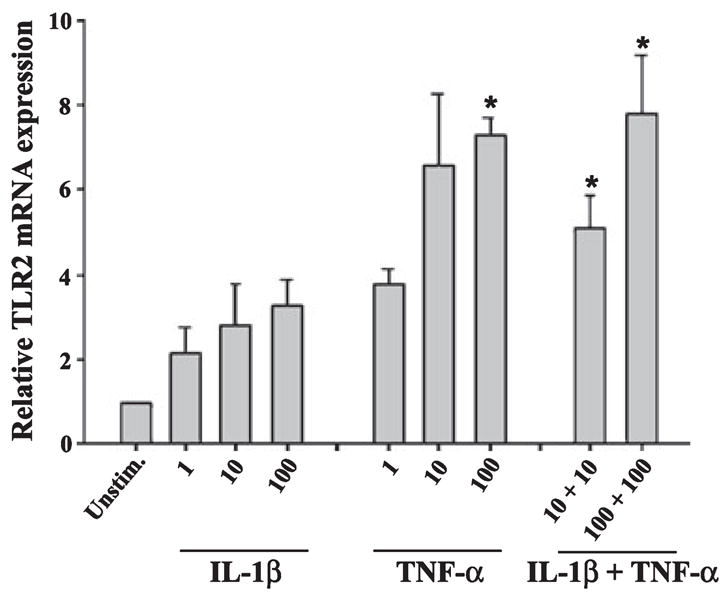
Toll-like receptor 2 (TLR2) mRNA expression in primary microglia is augmented by tumor necrosis factor-alpha (TNF-α). Microglia were seeded into six-well plates at 2 × 106 cells per well and incubated overnight. The following day, cells were treated with various concentrations of interleukin-1β (IL-1β) or TNF-α (ng/mL) either alone or in combination for 6 h, whereupon total RNA was isolated and examined for TLR2 expression by qRT-PCR. Gene expression levels were calculated after normalizing TLR2 signals against the housekeeping gene GAPDH and are presented as the fold-induction in mRNA expression of cytokine-treated microglia compared with unstimulated cells. Significant differences between unstimulated and cytokine-treated microglia are denoted with asterisks (*p < 0.05). Results represent the average of three independent experiments (mean ± SEM).
Fig. 2.

Tumor necrosis factor-alpha (TNF-α) enhances Toll-like receptor 2 (TLR2) protein expression in microglia. Microglia were seeded into six-well plates at 2 × 106 cells per well and incubated overnight. The following day, cells were treated with various concentrations of interleukin-1β (IL-1β) or TNF-α (ng/mL) either alone or in combination for 24 h, whereupon whole cell extracts (40 μg of protein per sample) were prepared and analyzed for TLR2 expression by western blotting. Blots were stripped and re-probed with an antibody specific for β-actin to verify uniformity in gel loading. Results are representative of three independent experiments.
NF-κB-dependent signaling is pivotal for the TNF-α-induced increase in microglial TLR2 expression
Recent studies have suggested the involvement of the NF-κB pathway in regulating TLR2 expression in several cell types, including macrophages (Musikacharoen et al. 2001; Wang et al. 2001, 2002; Hermoso et al. 2004); however, a role for NF-κB in dictating TLR2 expression in microglia has not yet been examined. Importantly, the mechanisms utilized for TLR2 regulation by microglia and macrophages may differ as evident by recent reports demonstrating divergent responses by both cell types (Carson et al. 1998; Schmid et al. 2002; Enose et al. 2005). One broad-scale NF-κB inhibitor is CAPE, which is known to have anti-mitogenic, anti-carcinogenic, anti-inflammatory, and immunomodulatory properties (Su et al. 1994, 1995; Natarajan et al. 1996; Watabe et al. 2004; Wei et al. 2004). In addition, CAPE has also been shown to inhibit the TNF-α-dependent activation of NF-κB (Natarajan et al. 1996). Treatment of primary microglia with CAPE was effective at attenuating TNF-α-induced TLR2 expression in dose-dependent manner (Fig. 3). As CAPE is a rather non-specific NF-κB pathway inhibitor (Epinat and Gilmore 1999), we next examined the effects of more selective inhibitors that block specific steps in the NF-κB signaling cascade. NF-κB signaling is tightly controlled by the IκB kinase complex (IKK-α/β/γ), of which IKK-β and IKK-γ are critical for cytokine induced NF-κB function, whereas IKK-α is thought to be involved in other regulatory pathways (Yamamoto and Gaynor 2004; Hayden et al. 2006; Perkins 2007). Treatment of primary microglia with SC-514, a specific inhibitor of IKK-2 (Kishore et al. 2003; Jeong et al. 2005), abrogated the increase in TLR2 expression observed following TNF-α treatment in a dose-dependent manner (Fig. 4a). Interestingly, co-administration of TNF-α with the highest dose of SC-514 examined led to significant microglial cell death (Fig. 4b), indicative of a pro-survival pathway for NF-κB in response to tumor necrosis factor receptor signaling in microglia. Importantly, lower concentrations of SC-514 were still effective at partially attenuating microglial TLR2 expression in response to TNF-α (Fig. 4a), but did not exhibit any signs of cytotoxicity (data not shown).
Fig. 3.
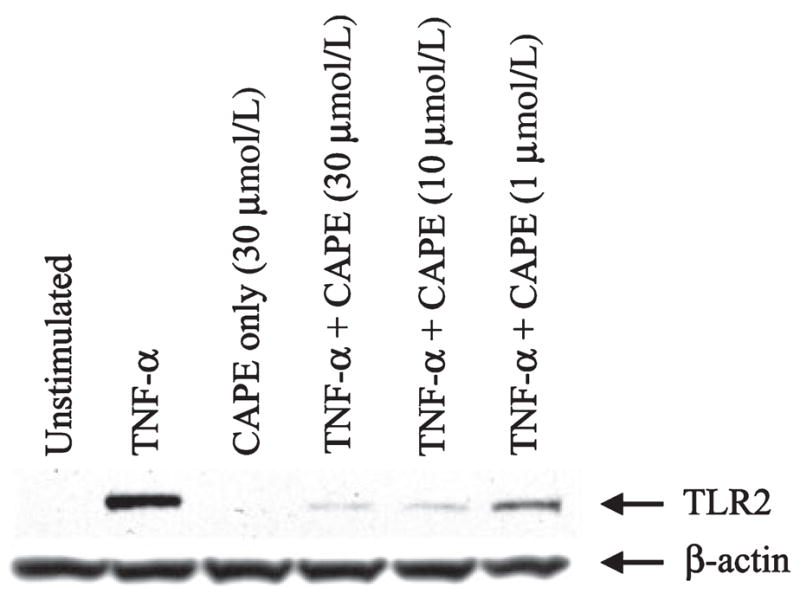
The broad-spectrum nuclear factor-kappa B inhibitor caffeic acid phenethyl ester (CAPE) attenuates tumor necrosis factor-alpha (TNF-α) -dependent Toll-like receptor 2 (TLR2) expression in a dose-dependent manner. Microglia were seeded into six-well plates at 2 × 106 cells per well and incubated overnight. The following day, cells were pre-treated with various concentrations of CAPE for 1 h prior to TNF-α stimulation (100 ng/mL) for 24 h, whereupon whole cell extracts (40 μg of protein per sample) were prepared and analyzed for TLR2 expression by western blotting. Blots were stripped and re-probed with an antibody specific for β-actin to verify uniformity in gel loading. Results are representative of three independent experiments.
Fig. 4.
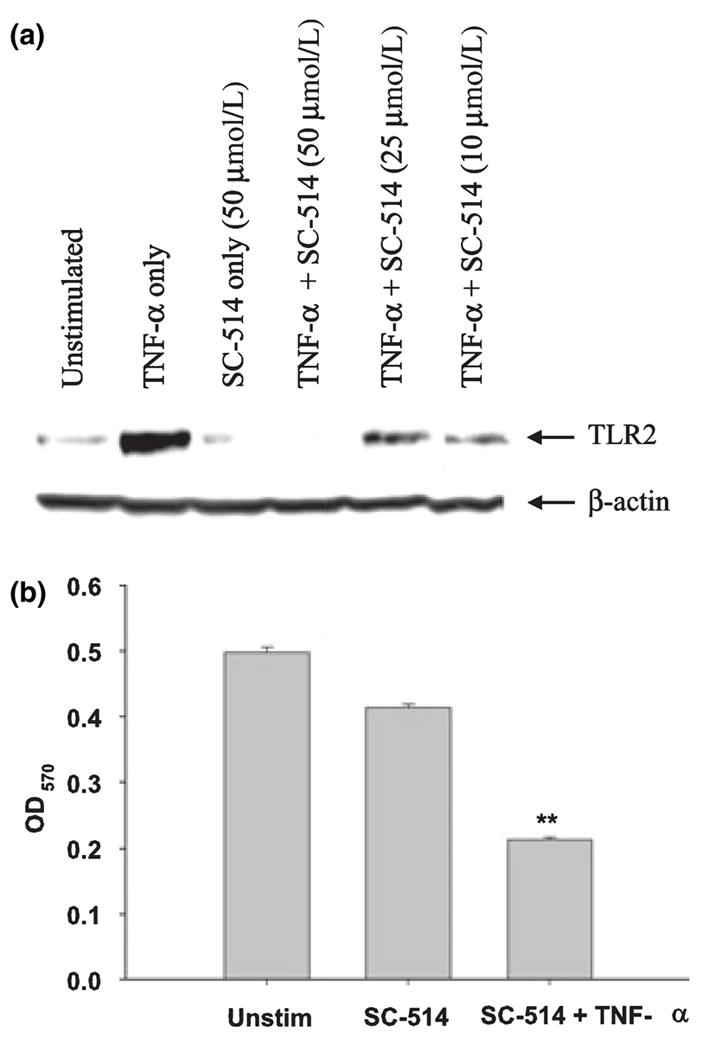
Evidence for the involvement of nuclear factor-kappa B signaling pathways in regulating the tumor necrosis factor-alpha (TNF-α) -dependent increase in Toll-like receptor 2 (TLR2) expression. Microglia were seeded into six-well plates at 2 × 106 cells per well and incubated overnight. The following day, cells were pre-treated for 1 h with various concentrations of the IKK-2 inhibitor SC-514 prior to TNF-α stimulation (100 ng/mL) for 24 h, whereupon whole cell extracts (40 μg of protein per sample) were prepared and analyzed for TLR2 expression by western blotting (a). Blots were stripped and re-probed with an antibody specific for β-actin to verify uniformity in gel loading. Results are representative of three independent experiments. In (b), microglia were pre-treated with SC-514 (50 μmol/L) for 1 h prior to TNF-α stimulation (100 ng/mL), whereupon cell viability was assessed using a standard MTT 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide assay. Raw OD570 absorbance values are reported (mean ± SD). Significant differences between unstimulated and treated microglia are denoted with asterisks (**p < 0.001).
Studies performed using another NF-κB pathway blocker, the IκB-α inhibitor BAY 11-7082 (Pierce et al. 1997), independently confirmed the involvement of NF-κB in mediating the TNF-α-dependent increase in microglial TLR2 expression (Fig. 5a). We confirmed the effectiveness of BAY 11-7082, at the concentrations used in this study, to prevent TNF-α-dependent IκB-α degradation in microglia by western blot analysis (data not shown). Importantly, co-treatment of microglia with BAY 11-7082 and TNF-α did not induce overt cell death (Fig. 5b). Collectively, these results suggest that NF-κB represents a major signaling cascade responsible for the TNF-α-mediated induction of TLR2 expression in microglia.
Fig. 5.
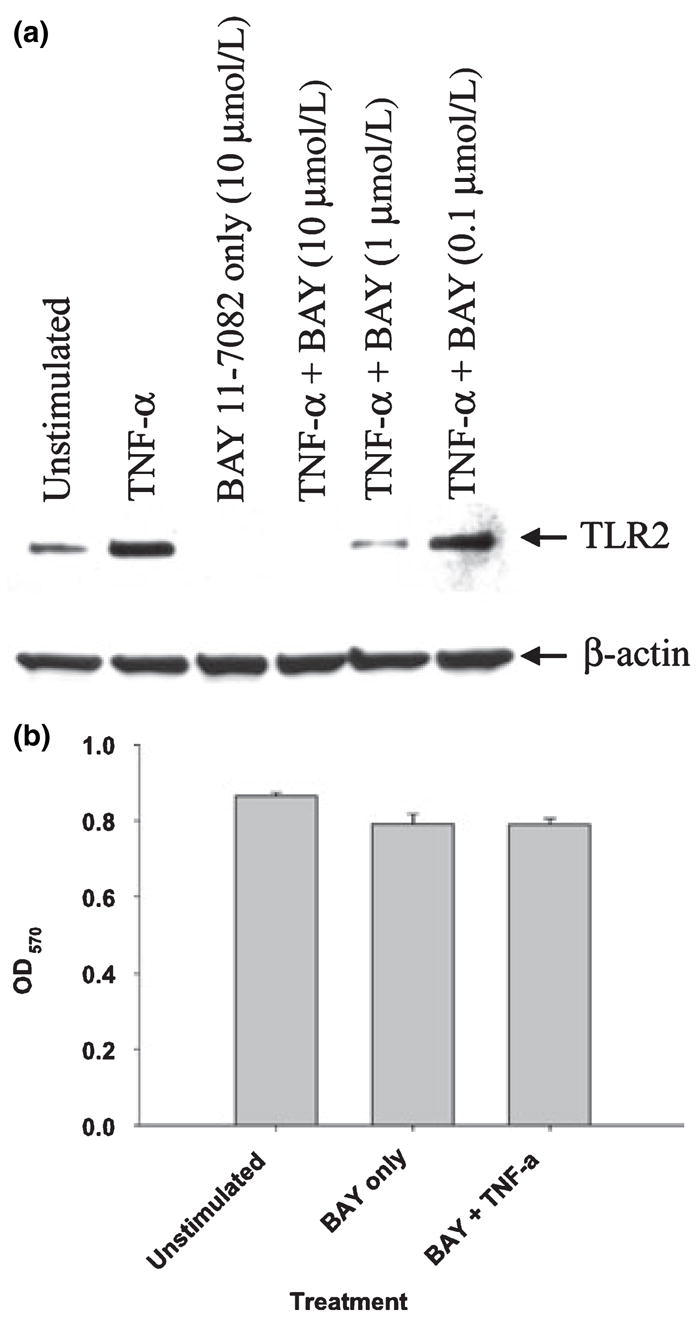
The IκB-α inhibitor BAY 11-7082 inhibits the tumor necrosis factor-alpha (TNF-α) -dependent increase in Toll-like receptor 2 (TLR2) expression without effecting cell viability. Microglia were seeded into six-well plates at 2 × 106 cells per well and incubated overnight. The following day, cells were pre-treated for 1 h with various concentrations of the IκB-α inhibitor BAY 11-7082 prior to TNF-α stimulation (100 ng/mL) for 24 h, whereupon whole cell extracts (40 μg of protein per sample) were prepared and analyzed for TLR2 expression by western blotting (a). Blots were stripped and re-probed with an antibody specific for β-actin to verify uniformity in gel loading. Results are representative of three independent experiments. In (b), microglia were pre-treated with BAY 11-7082 (10 μmol/L) for 1 h prior to TNF-α stimulation (100 ng/mL), whereupon cell viability was assessed using a standard MTT 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide assay. Raw OD570 absorbance values are reported (mean ± SD).
Neither PKC nor p38 MAPK pathways are responsible for the TNF-α-dependent induction of microglial TLR2 expression
As the increase in TLR2 expression observed following TNF-α treatment was not completely ablated with NF-κB pathway inhibitors, we broadened our analysis to examine the potential involvement of additional signaling pathways. Both PKC and p38 MAPK cascades are activated in response to TNF-α (Schutze et al. 1990; Norris et al. 1994; Brinkman et al. 1999); however, the importance of these pathways in regulating microglial TLR2 expression has not yet been examined. Neither the p38 MAPK nor PKC inhibitors SB202190 and BIM, respectively (Toullec et al. 1991; Lee et al. 1994), were capable of modulating microglial TLR2 expression in response to TNF-α (Fig. 6a and b, respectively). Interestingly, microglia treated with SB202190 alone demonstrated an increase in TLR2 expression (Fig. 6a); however, the significance of this finding is not known. Overall, these findings suggest that in response to TNF-α, microglial TLR2 expression is not influenced by either PKC-or p38 MAPK-dependent pathways.
Fig. 6.

The tumor necrosis factor-alpha (TNF-α)-dependent increase in microglial Toll-like receptor 2 (TLR2) expression is not influenced by either p38 mitogen-activated protein kinase (MAPK) or protein kinase C (PKC) pathways. Microglia were seeded into six-well plates at 2 × 106 cells per well and incubated overnight. The following day, cells were pre-treated for 1 h with various concentrations of the p38 MAPK or PKC inhibitors SB202190 (a) or bisindolylmaleimide (BIM; b), respectively. Subsequently, cells were stimulated with TNF-α (100 ng/mL) for 24 h, whereupon whole cell extracts (40 μg of protein per sample) were prepared and analyzed for TLR2 expression by western blotting. Blots were stripped and re-probed with an antibody specific for β-actin to verify uniformity in gel loading. Results are representative of three independent experiments.
The S. aureus -dependent increase in microglial TLR2 expression is due to the autocrine/paracrine action of TNF-α
Our results thus far have demonstrated that TNF-α is a potent inducer of TLR2 expression in primary microglia. To directly examine whether autocrine/paracrine TNF-α elicited upon S. aureus activation of microglia is responsible for augmenting TLR2 levels, we evaluated TLR2 expression in microglia isolated from TNF-α WT and KO mice. TLR2 expression was not detectable in TNF-α KO microglia following S. aureus stimulation, whereas receptor levels were dramatically increased in WT cells following bacterial exposure (Fig. 7). This finding established that S. aureus-induced TNF-α release is the primary mechanism leading to enhanced TLR2 expression in microglia.
Fig. 7.
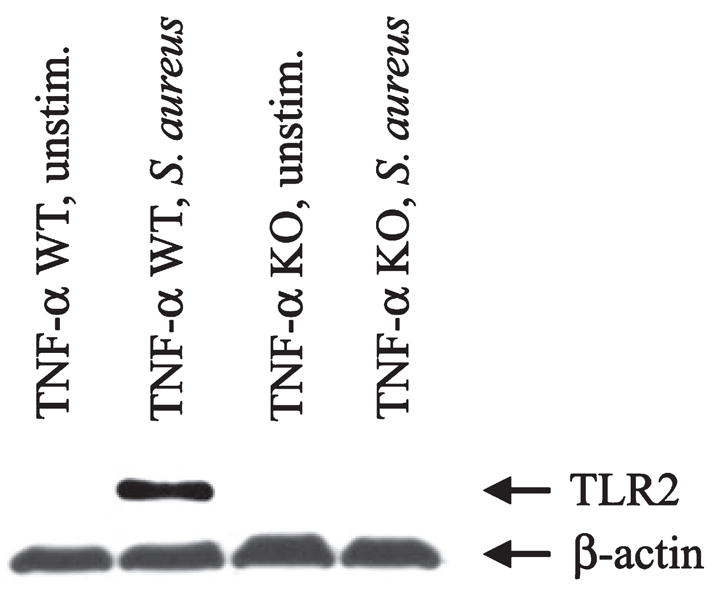
Tumor necrosis factor-alpha (TNF-α) is essential for the induction of microglial Toll-like receptor 2 (TLR2) expression in response to Staphylococcus aureus. Primary microglia isolated from TNF-α wild-type (WT) or knockout (KO) mice were seeded into six-well plates at 2 × 106 cells per well and incubated overnight. The following day, cells were stimulated with 107 colony forming units (CFU) of heat-inactivated S. aureus for 24 h, whereupon whole cell extracts were prepared (40 μg of protein per sample) and analyzed for TLR2 expression by western blotting. Blots were stripped and re-probed with an antibody specific for β-actin to verify uniformity in gel loading. Results are representative of two independent experiments.
We have previously demonstrated that TLR2 expression is increased during the course of S. aureus-induced brain abscess and that TLR2 participates in the development of select immune responses in this model (Kielian et al. 2005b). To investigate the role of TNF-α in regulating TLR2 expression in vivo, receptor levels were evaluated in brain abscesses of TNF-α WT and KO mice. TLR2 mRNA expression was significantly attenuated in brain abscesses of TNF-α KO mice throughout the course of infection (Fig. 8a). These findings were partially confirmed at the protein level by western blot analysis, where TLR2 expression was dramatically reduced in brain abscesses of TLR2 KO mice at day 3 post-infection (Fig. 8b). We were unable to detect significant TLR2 signals in brain abscesses of either TLR2 WT or KO animals at days 7 or 10 following S. aureus exposure, indicating that TLR2 expression begins to wane over time. Collectively, these results suggest that TNF-α plays a critical role in the induction of TLR2 expression following bacterial challenge.
Fig. 8.
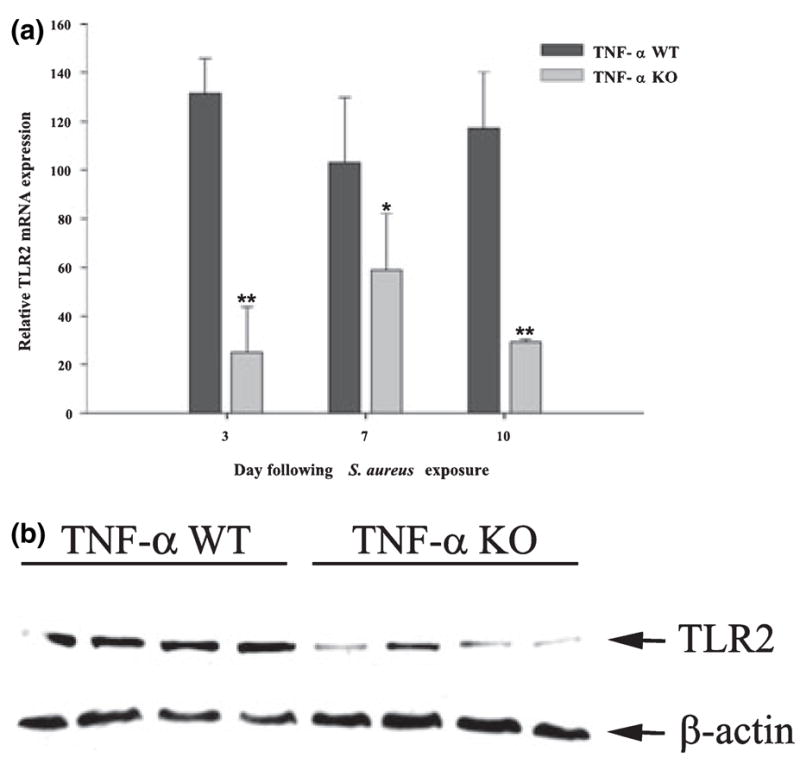
Tumor necrosis factor-alpha (TNF-α) plays a critical role in augmenting toll-like receptor 2 (TLR2) expression in vivo in a model of Staphylococcus aureus-induced brain abscess. TNF-α wild-type (WT) and knockout (KO) mice (n = 4–6 per group) received an intracerebral injection of S. aureus on day 0 and were killed at the indicated day post-infection, whereupon total RNA was isolated from brain abscesses for examination of TLR2 mRNA (a) and protein expression (b) by qRT-PCR and western blotting, respectively. For qRT-PCR, gene expression levels were calculated after normalizing TLR2 signals against the housekeeping gene GAPDH and are presented as the fold-induction in mRNA expression compared with uninfected animals. Significant differences between TNF-α WT and KO mice are denoted with asterisks (*p < 0.05, **p < 0.001). For western blots, brain abscess homogenates from four individual TNF-α WT or KO mice at day 3 post-infection were analyzed (40 μg of protein per sample). Blots were stripped and re-probed with an antibody specific for β-actin to verify uniformity in gel loading. Results are representative of two independent experiments.
Augmenting TLR2 levels by TNF-α does not impact S. aureus phagocytosis or subsequent proinflammatory mediator release
To determine whether the increase in TLR2 expression observed following TNF-α treatment resulted in heightened microglial anti-bacterial responses, the extent of S. aureus phagocytosis and proinflammatory mediator release were examined. Pre-treatment of primary WT microglia with 100 ng/mL TNF-α for a 24 h period had no significant effect on either S. aureus internalization or proinflammatory cytokine production (Fig. 9 and data not shown). Therefore, increasing TLR2 levels on microglia does not lead to overt alterations in these effector functions; however, the possibility remains that TLR2 upregulation may induce changes in alternative microglial responses not examined here.
Fig. 9.
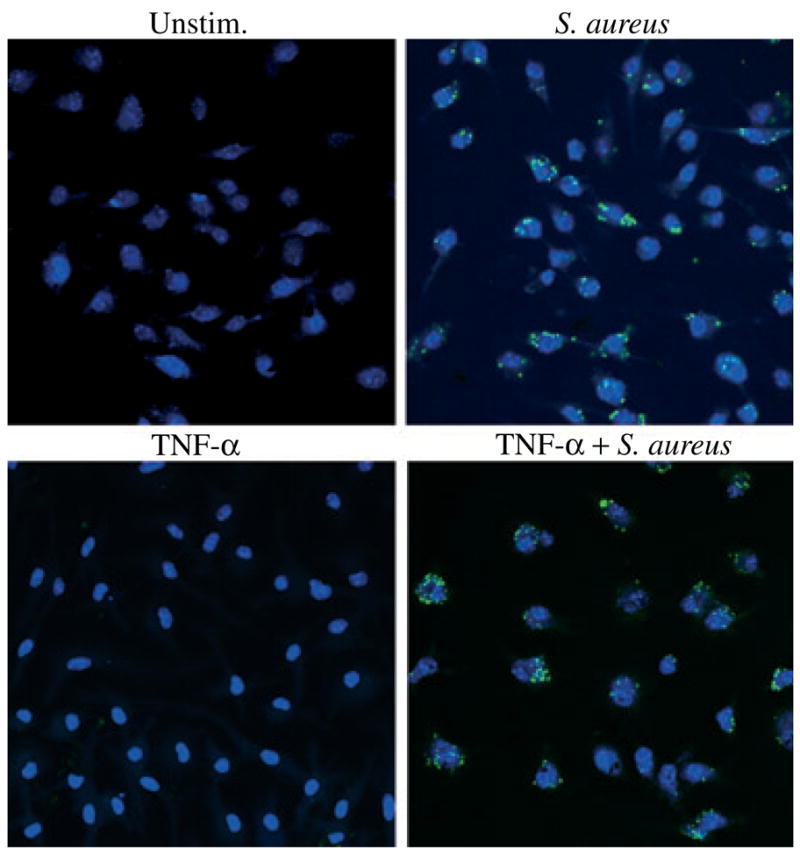
Augmenting Toll-like receptor 2 (TLR2) receptor levels with tumor necrosis factor-alpha (TNF-α) treatment has no effect on mi-croglial phagocytosis of Staphylococcus aureus. C57BL/6 primary microglia were seeded onto 12 mm coverslips at 2 × 105 cells per coverslip and incubated overnight in 24-well plates. The following day, cells were pre-treated with 100 ng/mL of recombinant mouse TNF-α for 24 h to augment TLR2 protein expression. Subsequently, microglia were incubated with 4 × 106 heat-inactivated S. aureus-green fluorescence protein (green) for 3 h and visualization of intracellular bacteria was detected using fluorescence microscopy (40×). Hoechst 33342 (blue) was used to visualize nuclei. Results are representative of three independent experiments.
Discussion
Microglia are the innate immune cells of the CNS parenchyma and express various PRRs that facilitate pathogen detection and the downstream production of various cytokines and chemokines, with the ultimate goal of efficient pathogen clearance (reviewed in Kielian 2006). One such PRR is TLR2, which is pivotal for recognition of the widest array of pathogens, including gram-positive bacteria (Kopp and Medzhitov 2003; Kirk and Bazan 2005; Akira et al. 2006). A recent study from our laboratory has shown that TLR2 is crucial for microglial responses to peptidoglycan, a cell-wall derivative of gram-positive bacteria such as S. aureus (Kielian et al. 2005a). However, it remained uncertain as to whether enhanced microglial TLR2 expression in response to S. aureus was a direct effect of pathogen exposure or indirectly mediated through the autocrine/paracrine action of cytokines released from activated cells. In response to S. aureus, microglia produce several inflammatory mediators including nitric oxide, TNF-α, IL-1β, CXCL2 (macrophage inflammatory protein-2), and CCL2 (monocyte chemoattractant protein-1) (Kielian et al. 2002, 2004b, 2005a). With regard to their role in brain abscesses, IL-1β and TNF-α are important proinflammatory mediators as IL-1β and TNF-α KO mice demonstrated delayed S. aureus clearance from the abscess site (Kielian et al. 2004a). Moreover, disease severity and mortality rates were considerably higher in TNF-α and IL-1 KO animals compared with WT mice. These findings suggest that IL-1β and TNF-α are pivotal for priming immune effector cells for rapid and efficient pathogen clearance; however the mechanisms by which these cytokines achieve these effects remain unknown. To our knowledge, this is the first report demonstrating that TNF-α produced by microglia upon bacterial challenge is responsible for augmenting TLR2 expression via an auto-crine/paracrine pathway. When comparing the abilities of TNF-α and IL-1β to regulate microglial TLR2 levels, only the former was found to exert significant effects. Moreover, the inability of TNF-α KO microglia to up-regulate TLR2 following S. aureus stimulation is strong evidence to implicate TNF-α as a pivotal mediator for augmenting TLR2 expression during inflammatory conditions. Direct in vivo evidence to support this concept was provided by the finding that TLR2 expression was significantly attenuated during brain abscess development in TNF-α KO mice.
The mouse TLR2 promoter contains numerous cis-acting binding sites for several transcription factors including two NF-κB, two CCAAT/enhancer-binding protein (C/EBP), a cAMP response element-binding protein, and one STAT site (Musikacharoen et al. 2001). The present study demonstrates that the NF-κB pathway is an important signaling cascade responsible for the TNF-α-dependent increase in microglial TLR2 expression. Our findings are in agreement with an earlier report where mutation of both NF-κB binding sites in the mouse TLR2 promoter abrogated TLR2 induction by TNF-α; however, unlike our study, TLR2 protein expression was not examined (Musikacharoen et al. 2001). We utilized a series of diverse pharmacological inhibitors of NF-κB activity in the current study as primary microglia are notoriously difficult to transfect, precluding the use of NF-κB-driven reporter constructs. However, our use of numerous inhibitors acting at distinct steps along the NF-κB pathway enabled independent confirmation of the importance of NF-κB in regulating TLR2 gene expression in primary microglia, strengthening the validity of our findings.
Treatment with the IKK-2 inhibitor SC-514 attenuated TLR2 expression in a dose-dependent manner in TNF-α-stimulated microglia. However, this combination also resulted in significant cytotoxicity as evident from cell viability assays. This finding is in agreement with previous reports demonstrating an anti-apoptotic role for NF-κB against TNF-α-mediated cell death in various cell types (Beg and Baltimore 1996; Van Antwerp et al. 1996; Li et al. 1999; Aggarwal 2003; Grivennikov et al. 2006). Importantly, this cytotoxic effect of NF-κB inhibition and TNF-α administration was not observed at lower concentrations of SC-514 that were still effective at negating the TNF-α-dependent increase in microglial TLR2 expression. Interestingly, no cytotoxicity was observed when microglia were treated with a combination of S. aureus and SC-514 (data not shown). As TNF-α is not the only mediator produced by S. aureus-activated microglia, this finding suggests that bacteria stimulate alternative signaling cascades that are capable of sustaining cell survival, even when NF-κB activity is impaired. Additional evidence to implicate NF-κB as a key regulatory pathway in dictating microglial TLR2 expression was demonstrated by the ability of a second NF-κB inhibitor, BAY 11-7082, to attenuate the TNF-α-dependent increase in TLR2 expression. Unlike SC-514, no cytotoxicity was observed upon simultaneous treatment of microglia with BAY 11-7082 and TNF-α. The differential effects of SC-514 and BAY on microglial survival could be explained by the fact that the IKK complex exerts a more central role in the NF-κB pathway, whereas the target of BAY 11-7082, IκB-α, does not (Karin et al. 2004; Scheidereit 2006; Perkins 2007). The central inhibitory effect of SC-514 via IKK blockade may lead to more widespread inhibition of alternative forms of NF-κB (other than p50/p65 heterodimers), resulting in a comprehensive impact on cell survival pathways. In contrast, NF-κB can be activated via IκB-α-independent mechanisms, which may be sufficient to afford microglial survival in response to TNF-α. Moreover, recent studies have shown that IKK itself can translocate to the nucleus and influence the expression of NF-κB-dependent as well as NF-κB-independent genes (Anest et al. 2003; Yamamoto et al. 2003; Gloire et al. 2006), suggesting that the IKK complex may have an independent role in cell survival.
To investigate the functional significance of augmenting microglial TLR2 expression in response to TNF-α, we evaluated whether cytokine treatment led to changes in S. aureus phagocytosis and/or subsequent proinflammatory mediator release by microglia. Our original prediction was that increased TLR2 expression would enable microglia to more efficiently recognize bacteria and enhance antimicrobial immune responses, as TLR2 is a potent cytokine signaling receptor. In contrast, our data suggests that prior TNF-α exposure does not significantly alter the degree of microglial IL-1β or CXCL2 release following S. aureus challenge. However, it is important to acknowledge that the treatment paradigm utilized during our in vitro studies is much less complex compared with what occurs during CNS infections such as brain abscess. Namely, it is unlikely that microglia would be exposed to TNF-α only prior to bacterial challenge as typically pathogen recognition by PRRs leads to the induction of TNF-α release in addition to an intricate array of proinflammatory molecules. It remains possible that enhanced TLR2 expression may significantly impact the host immune response during brain abscess development; however, this possibility remains speculative. It was predicted that increasing TLR2 levels with TNF-α pre-treatment would not have any consequences on microglial phagocytosis of S. aureus as TLR2 itself is not a phagocytic receptor (Underhill et al. 1999; Henneke et al. 2002; Watanabe et al. 2007), a finding which we confirmed in the current study.
When attempting to extrapolate the findings obtained with exogenous TNF-α treatment on microglial TLR2 expression with changes that may occur during S. aureus-induced brain abscess development, several factors must be considered. First, the concentration of TNF-α used in the majority of our experiments (100 ng/mL) is higher compared with levels we have previously reported associated with brain abscesses in vivo (100 pg/mL). However, it is important to note that we homogenize brain abscess tissues in a sizeable volume of PBS supplemented with protease and RNAse inhibitors (i.e. 500 μL). In doing so, we are likely significantly underestimating actual cytokine concentrations achieved locally within the site of inflammation due to this dilutional effect. However, this volume is required in order to obtain sufficient material for downstream cytokine/chemokine testing as well as quantitation of bacterial burdens associated with brain abscesses. Second, abscess homogenization disrupts local inflammatory microdomains where cytokine levels may be significantly higher compared with other areas. An example would be that cytokine expression may be highest within the necrotic center of the developing abscess where the majority of bacteria localize. In contrast, towards the abscess margins, cytokine levels may be diminished due to a reduction in triggering stimuli (i.e. bacteria). Finally, of note, significant increases in microglial TLR2 expression were also observed with lower concentrations of TNF-α, which represent levels approximating those detected in experimental brain abscesses (Kielian et al. 2005b, 2007). Relative to its impact during CNS inflammatory responses, when expressed at high levels or for prolonged periods of time, TNF-α has been associated with neuron and oligodendrocyte toxicity (Tamatani et al. 1999; Jurewicz et al. 2003; Li et al. 2004; Hovelmeyer et al. 2005; McCoy et al. 2006; Nakazawa et al. 2006). Therefore, administering high levels of exogenous TNF-α during the course of brain abscess development to augment TLR2 expression would not likely represent a viable treatment strategy due to the toxicity associated with elevated cytokine levels.
In summary, the present work demonstrates that TNF-α is a potent stimulus of TLR2 expression in microglia. In addition, using primary microglia from TNF-α KO mice, we demonstrate for the first time that the autocrine/paracrine actions of TNF-α are pivotal for augmenting microglial TLR2 expression in response to bacterial stimulation. An important role for TNF-α in increasing TLR2 levels in vivo was demonstrated by the failure to significantly augment TLR2 expression in brain abscesses of TNF-α KO mice.
Acknowledgments
The authors thank Maria Burkovetskaya, Aaron Baldwin and Anessa Haney for their excellent technical assistance. This work was supported by the NIH National Institutes of Mental Health (RO1 MH65297) and Neurological Disorders and Stroke (NS055385) to TK and the NINDS supported Core Facility at UAMS (P30 NS047546). Support for the Digital and Confocal Microscopy Laboratory at the University of Arkansas for Medical Sciences is provided by NIH/INBRE P20 RR6460 and NIH/NCRR S10 RR19395.
Abbreviations used
- BIM
bisindolylmaleimide
- CAPE
caffeic acid phenethyl ester
- IκB
inhibitory-kappa B
- IKK
IκB kinase
- IL-1β
interleukin-1β
- KO
knockout
- MAPK
mitogen-activated protein kinase
- NF-κB
nuclear factor-kappa B
- PBS
phosphate-buffered saline
- PKC
protein kinase C
- PRR
pattern recognition receptor
- TLR
Toll-like receptor
- TNF-α
tumor necrosis factor-alpha
- WT
wild-type
References
- Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol. 2003;3:745–756. doi: 10.1038/nri1184. [DOI] [PubMed] [Google Scholar]
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Aloisi F. Immune function of microglia. Glia. 2001;36:165–179. doi: 10.1002/glia.1106. [DOI] [PubMed] [Google Scholar]
- Anest V, Hanson JL, Cogswell PC, Steinbrecher KA, Strahl BD, Baldwin AS. A nucleosomal function for IkappaB kinase-alpha in NF-kappaB-dependent gene expression. Nature. 2003;423:659–663. doi: 10.1038/nature01648. [DOI] [PubMed] [Google Scholar]
- Beg AA, Baltimore D. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- Brinkman BM, Telliez JB, Schievella AR, Lin LL, Goldfeld AE. Engagement of tumor necrosis factor (TNF) receptor 1 leads to ATF-2- and p38 mitogen-activated protein kinase-dependent TNF-alpha gene expression. J Biol Chem. 1999;274:30882–30886. doi: 10.1074/jbc.274.43.30882. [DOI] [PubMed] [Google Scholar]
- Carson MJ, Reilly CR, Sutcliffe JG, Lo D. Mature microglia resemble immature antigen-presenting cells. Glia. 1998;22:72–85. doi: 10.1002/(sici)1098-1136(199801)22:1<72::aid-glia7>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Enose Y, Destache CJ, Mack AL, Anderson JR, Ullrich F, Ciborowski PS, Gendelman HE. Proteomic finger-prints distinguish microglia, bone marrow, and spleen macrophage populations. Glia. 2005;51:161–172. doi: 10.1002/glia.20193. [DOI] [PubMed] [Google Scholar]
- Epinat JC, Gilmore TD. Diverse agents act at multiple levels to inhibit the Rel/NF-kappaB signal transduction pathway. Oncogene. 1999;18:6896–6909. doi: 10.1038/sj.onc.1203218. [DOI] [PubMed] [Google Scholar]
- Esen N, Kielian T. Central role for MyD88 in the responses of microglia to pathogen-associated molecular patterns. J Immunol. 2006;176:6802–6811. doi: 10.4049/jimmunol.176.11.6802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esen N, Kielian T. Effects of low dose GM-CSF on microglial inflammatory profiles to diverse pathogen-associated molecular patterns (PAMPs) J Neuroinflammation. 2007;4:10. doi: 10.1186/1742-2094-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esen N, Tanga FY, DeLeo JA, Kielian T. Toll-like receptor 2 (TLR2) mediates astrocyte activation in response to the Gram-positive bacterium Staphylococcus aureus. J Neurochem. 2004;88:746–758. doi: 10.1046/j.1471-4159.2003.02202.x. [DOI] [PubMed] [Google Scholar]
- Gloire G, Dejardin E, Piette J. Extending the nuclear roles of IkappaB kinase subunits. Biochem Pharmacol. 2006;72:1081–1089. doi: 10.1016/j.bcp.2006.06.017. [DOI] [PubMed] [Google Scholar]
- Grivennikov SI, Kuprash DV, Liu ZG, Nedospasov SA. Intracellular signals and events activated by cytokines of the tumor necrosis factor superfamily: from simple paradigms to complex mechanisms. Int Rev Cytol. 2006;252:129–161. doi: 10.1016/S0074-7696(06)52002-9. [DOI] [PubMed] [Google Scholar]
- Haehnel V, Schwarzfischer L, Fenton MJ, Rehli M. Transcriptional regulation of the human toll-like receptor 2 gene in monocytes and macrophages. J Immunol. 2002;168:5629–5637. doi: 10.4049/jimmunol.168.11.5629. [DOI] [PubMed] [Google Scholar]
- Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40:140–155. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- Hayden MS, West AP, Ghosh S. NF-kappaB and the immune response. Oncogene. 2006;25:6758–6780. doi: 10.1038/sj.onc.1209943. [DOI] [PubMed] [Google Scholar]
- Henneke P, Takeuchi O, Malley R, et al. Cellular activation, phagocytosis, and bactericidal activity against group B streptococcus involve parallel myeloid differentiation factor 88-dependent and independent signaling pathways. J Immunol. 2002;169:3970–3977. doi: 10.4049/jimmunol.169.7.3970. [DOI] [PubMed] [Google Scholar]
- Hermoso MA, Matsuguchi T, Smoak K, Cidlowski JA. Glucocorticoids and tumor necrosis factor alpha cooperatively regulate toll-like receptor 2 gene expression. Mol Cell Biol. 2004;24:4743–4756. doi: 10.1128/MCB.24.11.4743-4756.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovelmeyer N, Hao Z, Kranidioti K, et al. Apoptosis of oligodendrocytes via Fas and TNF-R1 is a key event in the induction of experimental autoimmune encephalomyelitis. J Immunol. 2005;175:5875–5884. doi: 10.4049/jimmunol.175.9.5875. [DOI] [PubMed] [Google Scholar]
- Jeong SJ, Pise-Masison CA, Radonovich MF, Park HU, Brady JN. A novel NF-kappaB pathway involving IKKbeta and p65/RelA Ser-536 phosphorylation results in p53 inhibition in the absence of NF-kappaB transcriptional activity. J Biol Chem. 2005;280:10326–10332. doi: 10.1074/jbc.M412643200. [DOI] [PubMed] [Google Scholar]
- Jurewicz A, Matysiak M, Tybor K, Selmaj K. TNF-induced death of adult human oligodendrocytes is mediated by c-jun NH2-terminal kinase-3. Brain. 2003;126:1358–1370. doi: 10.1093/brain/awg146. [DOI] [PubMed] [Google Scholar]
- Karin M, Yamamoto Y, Wang QM. The IKK NF-kappa B system: a treasure trove for drug development. Nat Rev Drug Discov. 2004;3:17–26. doi: 10.1038/nrd1279. [DOI] [PubMed] [Google Scholar]
- Kielian T. Toll-like receptors in central nervous system glial inflammation and homeostasis. J Neurosci Res. 2006;83:711–730. doi: 10.1002/jnr.20767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T, Cheung A, Hickey WF. Diminished virulence of an alpha-toxin mutant of Staphylococcus aureus in experimental brain abscesses. Infect Immun. 2001a;69:6902–6911. doi: 10.1128/IAI.69.11.6902-6911.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T, Barry B, Hickey WF. CXC chemokine receptor-2 ligands are required for neutrophil-mediated host defense in experimental brain abscesses. J Immunol. 2001b;166:4634–4643. doi: 10.4049/jimmunol.166.7.4634. [DOI] [PubMed] [Google Scholar]
- Kielian T, Mayes P, Kielian M. Characterization of microglial responses to Staphylococcus aureus: effects on cytokine, costimulatory molecule, and Toll-like receptor expression. J Neuroimmunol. 2002;130:86–99. doi: 10.1016/s0165-5728(02)00216-3. [DOI] [PubMed] [Google Scholar]
- Kielian T, Bearden ED, Baldwin AC, Esen N. IL-1 and TNF-alpha play a pivotal role in the host immune response in a mouse model of Staphylococcus aureus-induced experimental brain abscess. J Neuropathol Exp Neurol. 2004a;63:381–396. doi: 10.1093/jnen/63.4.381. [DOI] [PubMed] [Google Scholar]
- Kielian T, McMahon M, Bearden ED, Baldwin AC, Drew PD, Esen N. Staphylococcus aureus-dependent microglial activation is selectively attenuated by the cyclopentenone prostaglandin 15-deoxy-Delta12,14-prostaglandin J2 (15d-PGJ2) J Neurochem. 2004b;90:1163–1172. doi: 10.1111/j.1471-4159.2004.02579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T, Esen N, Bearden ED. Toll-like receptor 2 (TLR2) is pivotal for recognition of Staphylococcus aureus peptidoglycan but not intact bacteria by microglia. Glia. 2005a;49:567–576. doi: 10.1002/glia.20144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T, Haney A, Mayes PM, Garg S, Esen N. Toll-like receptor 2 modulates the proinflammatory milieu in Staphylococcus aureus-induced brain abscess. Infect Immun. 2005b;73:7428–7435. doi: 10.1128/IAI.73.11.7428-7435.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T, Phulwani NK, Esen N, Syed MM, Haney AC, McCastlain K, Johnson J. MyD88-dependent signals are essential for the host immune response in experimental brain abscess. J Immunol. 2007;178:4528–4537. doi: 10.4049/jimmunol.178.7.4528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk P, Bazan JF. Pathogen recognition: TLRs throw us a curve. Immunity. 2005;23:347–350. doi: 10.1016/j.immuni.2005.09.008. [DOI] [PubMed] [Google Scholar]
- Kishore N, Sommers C, Mathialagan S, et al. A selective IKK-2 inhibitor blocks NF-kappa B-dependent gene expression in inter-leukin-1 beta-stimulated synovial fibroblasts. J Biol Chem. 2003;278:32861–32871. doi: 10.1074/jbc.M211439200. [DOI] [PubMed] [Google Scholar]
- Kopp E, Medzhitov R. Recognition of microbial infection by Toll-like receptors. Curr Opin Immunol. 2003;15:396–401. doi: 10.1016/s0952-7915(03)00080-3. [DOI] [PubMed] [Google Scholar]
- Lee JC, Laydon JT, McDonnell PC, et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- Li R, Yang L, Lindholm K, Konishi Y, Yue X, Hampel H, Zhang D, Shen Y. Tumor necrosis factor death receptor signaling cascade is required for amyloid-beta protein-induced neuron death. J Neurosci. 2004;24:1760–1771. doi: 10.1523/JNEUROSCI.4580-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ZW, Chu W, Hu Y, Delhase M, Deerinck T, Ellisman M, Johnson R, Karin M. The IKKbeta subunit of IkappaB kinase (IKK) is essential for nuclear factor kappaB activation and prevention of apoptosis. J Exp Med. 1999;189:1839–1845. doi: 10.1084/jem.189.11.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathisen GE, Johnson JP. Brain abscess. Clin Infect Dis. 1997;25:763–779. doi: 10.1086/515541. quiz 780–761. [DOI] [PubMed] [Google Scholar]
- McCoy MK, Martinez TN, Ruhn KA, Szymkowski DE, Smith CG, Botterman BR, Tansey KE, Tansey MG. Blocking soluble tumor necrosis factor signaling with dominant-negative tumor necrosis factor inhibitor attenuates loss of dopa-minergic neurons in models of Parkinson’s disease. J Neurosci. 2006;26:9365–9375. doi: 10.1523/JNEUROSCI.1504-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musikacharoen T, Matsuguchi T, Kikuchi T, Yoshikai Y. NF-kappa B and STAT5 play important roles in the regulation of mouse Toll-like receptor 2 gene expression. J Immunol. 2001;166:4516–4524. doi: 10.4049/jimmunol.166.7.4516. [DOI] [PubMed] [Google Scholar]
- Nakazawa T, Nakazawa C, Matsubara A, Noda K, Hisatomi T, She H, Michaud N, Hafezi-Moghadam A, Miller JW, Benowitz LI. Tumor necrosis factor-alpha mediates oligodendrocyte death and delayed retinal ganglion cell loss in a mouse model of glaucoma. J Neurosci. 2006;26:12633–12641. doi: 10.1523/JNEUROSCI.2801-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan K, Singh S, Burke TR, Jr, Grunberger D, Aggarwal BB. Caffeic acid phenethyl ester is a potent and specific inhibitor of activation of nuclear transcription factor NF-kappa B. Proc Natl Acad Sci USA. 1996;93:9090–9095. doi: 10.1073/pnas.93.17.9090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris JG, Tang LP, Sparacio SM, Benveniste EN. Signal transduction pathways mediating astrocyte IL-6 induction by IL-1 beta and tumor necrosis factor-alpha. J Immunol. 1994;152:841–850. [PubMed] [Google Scholar]
- Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173:3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- Pierce JW, Schoenleber R, Jesmok G, Best J, Moore SA, Collins T, Gerritsen ME. Novel inhibitors of cytokine-induced IkappaBalpha phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. J Biol Chem. 1997;272:21096–21103. doi: 10.1074/jbc.272.34.21096. [DOI] [PubMed] [Google Scholar]
- Rivest S. Molecular insights on the cerebral innate immune system. Brain Behav Immun. 2003;17:13–19. doi: 10.1016/s0889-1591(02)00055-7. [DOI] [PubMed] [Google Scholar]
- Scheidereit C. IkappaB kinase complexes: gateways to NF-kappaB activation and transcription. Oncogene. 2006;25:6685–6705. doi: 10.1038/sj.onc.1209934. [DOI] [PubMed] [Google Scholar]
- Schmid CD, Sautkulis LN, Danielson PE, Cooper J, Hasel KW, Hilbush BS, Sutcliffe JG, Carson MJ. Heterogeneous expression of the triggering receptor expressed on myeloid cells-2 on adult murine microglia. J Neurochem. 2002;83:1309–1320. doi: 10.1046/j.1471-4159.2002.01243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutze S, Nottrott S, Pfizenmaier K, Kronke M. Tumor necrosis factor signal transduction. Cell-type-specific activation and translocation of protein kinase C. J Immunol. 1990;144:2604–2608. [PubMed] [Google Scholar]
- Su ZZ, Lin J, Grunberger D, Fisher PB. Growth suppression and toxicity induced by caffeic acid phenethyl ester (CAPE) in type 5 adenovirus-transformed rat embryo cells correlate directly with transformation progression. Cancer Res. 1994;54:1865–1870. [PubMed] [Google Scholar]
- Su ZZ, Lin J, Prewett M, Goldstein NI, Fisher PB. Apoptosis mediates the selective toxicity of caffeic acid phenethyl ester (CAPE) toward oncogene-transformed rat embryo fibroblast cells. Anticancer Res. 1995;15:1841–1848. [PubMed] [Google Scholar]
- Tamatani M, Che YH, Matsuzaki H, Ogawa S, Okado H, Miyake S, Mizuno T, Tohyama M. Tumor necrosis factor induces Bcl-2 and Bcl-x expression through NFkappaB activation in primary hippocampal neurons. J Biol Chem. 1999;274:8531–8538. doi: 10.1074/jbc.274.13.8531. [DOI] [PubMed] [Google Scholar]
- Toullec D, Pianetti P, Coste H, et al. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J Biol Chem. 1991;266:15771–15781. [PubMed] [Google Scholar]
- Townsend GC, Scheld WM. Infections of the central nervous system. Adv Intern Med. 1998;43:403–447. [PubMed] [Google Scholar]
- Underhill D, Ozinsky A, Hajjar AM, Stevens A, Wilson CB, Bassetti M, Aderem A. The Toll-like receptor 2 is recruited to macrophage phagosomes and discriminates between pathogens. Nature. 1999;401:811–815. doi: 10.1038/44605. [DOI] [PubMed] [Google Scholar]
- Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- Wang T, Lafuse WP, Zwilling BS. NFkappaB and Sp1 elements are necessary for maximal transcription of toll-like receptor 2 induced by Mycobacterium avium. J Immunol. 2001;167:6924–6932. doi: 10.4049/jimmunol.167.12.6924. [DOI] [PubMed] [Google Scholar]
- Wang T, Lafuse WP, Takeda K, Akira S, Zwilling BS. Rapid chromatin remodeling of Toll-like receptor 2 promoter during infection of macrophages with Mycobacterium avium. J Immunol. 2002;169:795–801. doi: 10.4049/jimmunol.169.2.795. [DOI] [PubMed] [Google Scholar]
- Watabe M, Hishikawa K, Takayanagi A, Shimizu N, Nakaki T. Caffeic acid phenethyl ester induces apoptosis by inhibition of NFkappaB and activation of Fas in human breast cancer MCF-7 cells. J Biol Chem. 2004;279:6017–6026. doi: 10.1074/jbc.M306040200. [DOI] [PubMed] [Google Scholar]
- Watanabe I, Ichiki M, Shiratsuchi A, Nakanishi Y. TLR2-mediated survival of Staphylococcus aureus in macrophages: a novel bacterial strategy against host innate immunity. J Immunol. 2007;178:4917–4925. doi: 10.4049/jimmunol.178.8.4917. [DOI] [PubMed] [Google Scholar]
- Wei X, Zhao L, Ma Z, Holtzman DM, Yan C, Dodel RC, Hampel H, Oertel W, Farlow MR, Du Y. Caffeic acid phenethyl ester prevents neonatal hypoxic-ischaemic brain injury. Brain. 2004;127:2629–2635. doi: 10.1093/brain/awh316. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Gaynor RB. IkappaB kinases: key regulators of the NF-kappaB pathway. Trends Biochem Sci. 2004;29:72–79. doi: 10.1016/j.tibs.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Verma UN, Prajapati S, Kwak YT, Gaynor RB. Histone H3 phosphorylation by IKK-alpha is critical for cytokine-induced gene expression. Nature. 2003;423:655–659. doi: 10.1038/nature01576. [DOI] [PubMed] [Google Scholar]
- Zhi L, Ang AD, Zhang H, Moore PK, Bhatia M. Hydrogen sulfide induces the synthesis of proinflammatory cyto-kines in human monocyte cell line U937 via the ERK-NF-κB pathway. J Leukoc Biol. 2007;81:1322–1332. doi: 10.1189/jlb.1006599. [DOI] [PubMed] [Google Scholar]