

Abstract
Library samples containing 2,5-disubstituted oxadiazoles were identified as potent hits in a high throughput screen (HTS) of the NIH Molecular Libraries Small Molecule Repository (MLSMR) directed at discovering inhibitors of cathepsin L. However, when synthesized in pure form, the putative actives were found to be devoid of biological activity. Analyses by LC-MS of original library samples indicated the presence of a number of impurities, in addition to the oxadiazoles. Synthesis and bioassay of the probable impurities led to the identification of a thiocarbazate that likely originated via ring opening of the oxadiazole. Previously unknown, thiocarbazates (−)-11 and (−)-12 were independently synthesized as single enantiomers and found to inhibit cathepsin L in the low nanomolar range.
The cathepsins comprise a family of lysosomal protease enzymes whose primary function (i.e., protein degradation) plays a critical role in normal cellular homeostasis.1 Over expression of cathepsin L and/or abnormal activity has been implicated in a number of disease states.2 For example, cathepsin L is responsible for bone resorption through degradation of collagen type I; this disregulation is believed to lead to osteo- and rheumatoid arthritis.3 In addition, several infective organisms, such as SARS and Ebola viruses, utilize cathepsin L-like proteins for replication in human cells.4 The large number of disease states associated with cathepsin L calls for an understanding of the biological function.2
Recently, the Penn Center for Molecular Discovery (PCMD),5 completed a high throughput screening (HTS) campaign of the NIH Molecular Libraries Small Molecule Repository (MLSMR) to identify inhibitors of members of the papain-like cysteine protease family, including cathepsins B, L, and S.6 In this Letter, we detail our continuing efforts to create a comprehensive, publicly available profile of small-molecule inhibitors of the cysteine protease class, and herein describe the identification of a novel class of potent Cathepsin L inhibitors.
Previously reported inhibitors of cathepsin L include the peptides, leupeptin and aprotinin, and the fluoromethyl ketone, Z-LLL-FMK.3,7 The few known, potent small molecule inhibitors are either peptidic and therefore likely to suffer from physiological instability and poor permeability, or are non-selective for cathepsin L.3,8,9 The identification of potent, selective, stable, and cell permeable small molecule inhibitors would therefore be a valuable tool to interrogate cathepsin L and cathepsin L-like function, as well as to provide potential starting points for drug discovery and development.10–15
Initial HTS results of our Cathepsin L screen, indicated that several structurally related oxadiazoles exhibited potent inhibitory activity (Table 1).16–18
Table 1.
Cathepsin L inhibitory activity of oxadiazole-containing library samplesa
 | |||
|---|---|---|---|
| PubChem SID | R1 | R2 | IC50 (μM)b |
| 861540 (1) | 2-ethylbenzene | H | 0.13 ± 0.01 |
| 861087 (2) | 2,4-dimethylbenzene | H | 0.16 ± 0.05 |
| 861840 (3) | Me | Me | 0.17 ± 0.02 |
| 861542 (4) | 2,3-dimethylbenzene | H | 0.30 ± 0.04 |
| 861992 (5) | Et | Et | 0.51 ± 0.02 |
Library samples containing the parent oxadiazole with impurities.
IC50 values are reported as mean ± standard deviation (number of determinations = 3).
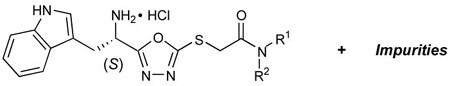
The most potent hit, cataloged in the MLSMR as disubstituted oxadiazole 1, exhibited an IC50 of 0.13 μM.17 Wells containing related analogs (2–5), differing only in the amide nitrogen substitution, displayed a range of activities (IC50 = 0.16–0.51 μM), suggesting a nascent structure activity relationship. As is our practice, library samples containing the putative active oxadiazoles were evaluated for both purity and integrity by LC-MS analysis. This analysis indicated that the primary constituent of each sample was indeed the expected oxadiazole, in up to 60% purity, however numerous impurities were also present. To confirm the biological activity attributed to 1, a synthetic sequence was developed to generate the oxadiazoles in pure form.
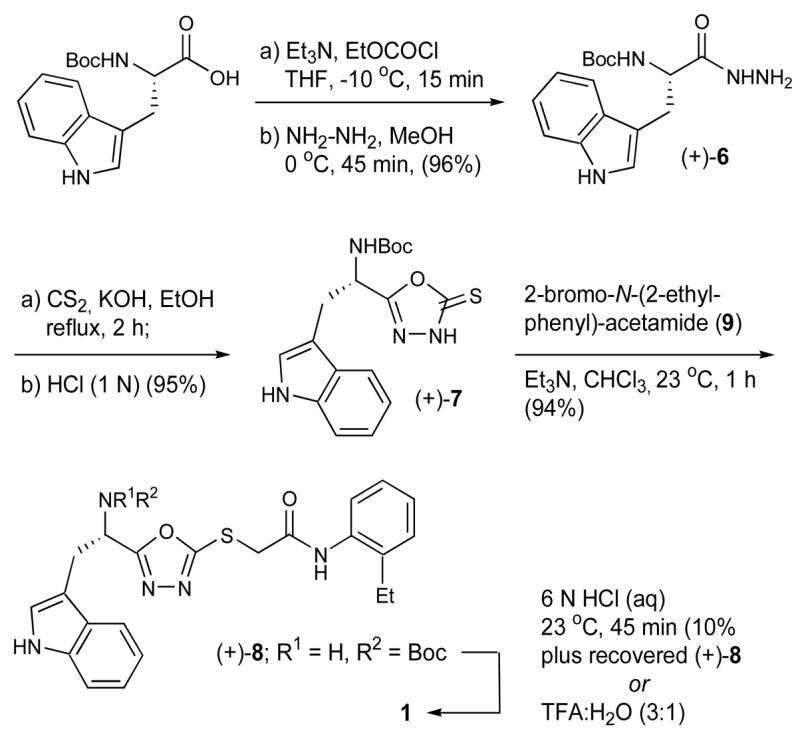
Although little literature precedence exists for the construction of compounds such as 1, we realized that modification of the Woodward-Confalone19 approach to α-amino acid substituted oxadiazolethiones could provide an entry to this class of sulfur-based 2,5-disubstituted oxadiazoles. Toward this end, commercially available L-Boc-Trp-OH was converted in high yield to the corresponding hydrazide (+)-6, on a 10 gram scale (Scheme 1).20
Scheme 1.
Synthesis of 2,5-disubstituted oxadiazoles.
Cyclization with carbon disulfide in ethanol at reflux afforded the expected substituted thione (+)-7 as a stable solid in excellent yield, which upon chemoselective alkylation with 2-bromo-N-(2-ethyl-phenyl)-acetamide21 (9) efficiently generated (+)-8 (94% yield). Further study indicated that isolation of (+)-7 was unnecessary, and (+)-8 could be prepared directly from (+)-6, in a single flask. Both sequences furnished (+)-8 in high yield. In similar fashion, the antipode, (−)-8, was prepared starting from D-Boc-Trp-OH. High enantiomeric purities (>99%) were demonstrated via chiral supercritical fluid chromatography.
Initial attempts to remove the Boc-group utilizing HCl in dioxane (4 N) or HCl in water (6 N) were compromised by the poor solubility of (+)-8. However, small quantities of the HCl salt of 1 could be obtained by treatment with 6 N HCl over 45 minutes. Upon LC-MS analysis of synthetic 1, we recognized that the impurities formed during the acid promoted Boc-deprotection step were identical to those found in the library screening samples (vide infra). Optimal conditions to remove the Boc-group were eventually developed. Specifically, treatment of (+)-8 with 25% water in TFA for 15 minutes, followed in turn by adjustment of the pH to 8.0 with NaHCO3 and aqueous extraction with methylene chloride furnished the free-base of 1 in 89% yield and 99% after column chromatography. In the end, oxadiazole 1 was prepared on gram scale from L-Boc-Trp-OH; the overall yield was 76%.
To our surprise, the free base of 1 was found to be completely devoid of activity when assayed against cathepsin L.18 This result suggested that an impurity present in the original library sample was responsible for the observed activity. Based on LC-MS analyses of the biologically active samples, we hypothesized that the active component was likely the ring-opened product 10, formed via acid-promoted addition of H2O to (+)-8 (Figure 1).
Figure 1.
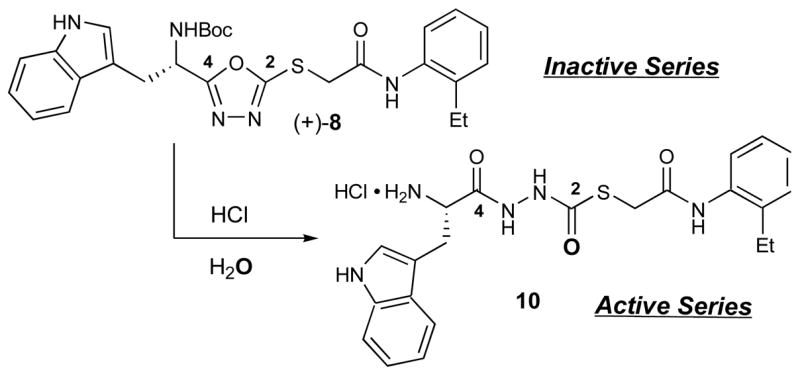
Conversion of oxadiazole (+)-8 to thiocarbazate 10.
The presence of a molecular [M+1] ion equal to 440 amu in the LC-MS analysis of impure samples was consistent with the presence of 10, thereby providing strong support for the hypothesis.
Thiocarbazates such as 10 have not been described previously in the literature. However, these compounds do bear structural resemblance to aza-peptides22–24 (e.g. A; Figure 2),25 examples of which have been reported to exhibit cysteine protease inhibitory activity through a mechanism involving attack by the active site cysteine on the carbamate carbonyl.26
Figure 2.
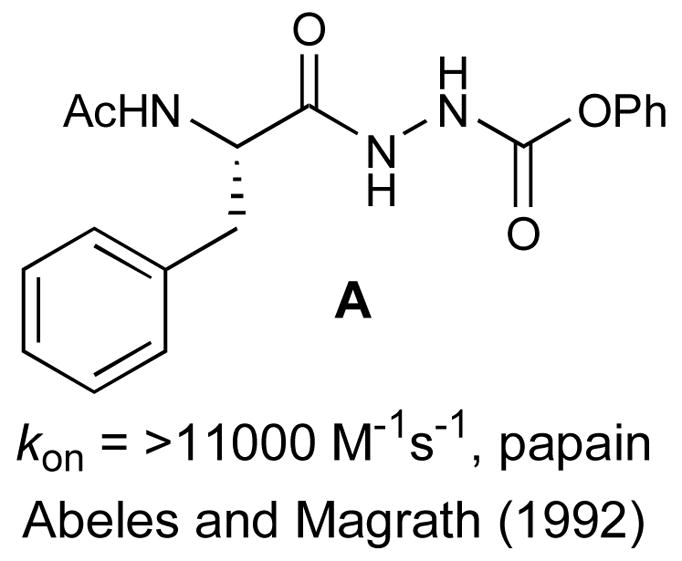
Known aza-peptide cathepsin inhibitor A.
To test the hypothesis that 10 was indeed the active species, we devised an expedient synthesis of this structural class. We envisioned that introduction of the C2 carbonyl unit of 10 could be achieved via chemistry in parallel to that used to prepare 1, beginning with hydrazide (+)-6. We began the synthesis by converting tryptophan hydrazide (+)-6 to an intermediate thiosemicarbazide (not shown) employing carbonyl sulfide (S=C=O) gas dissolved in ethanol (Scheme 2).27
Scheme 2.
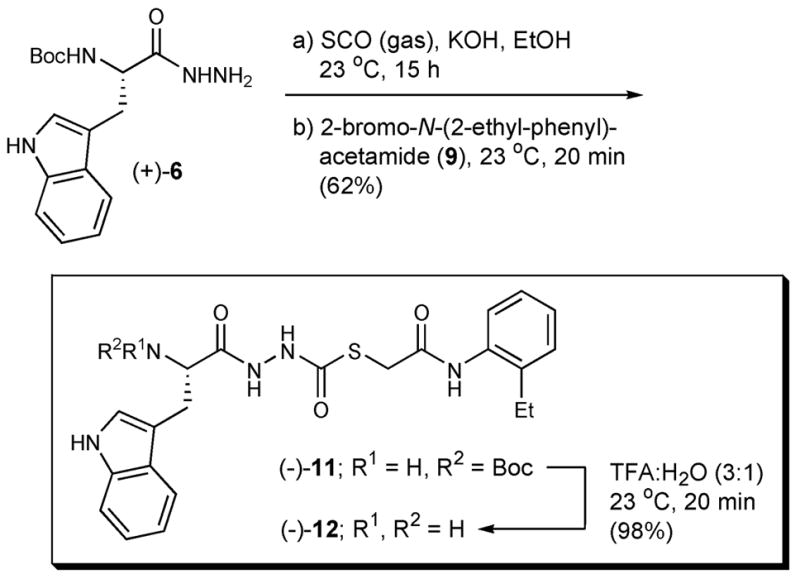
Synthetic procedure to prepare thiocarbazates.
The intermediate thiosemicarbazide did not precipitate from solution, therefore 2-bromo-N-(2-ethyl-phenyl)-acetamide21 (9) was added to the reaction flask to generate thiocarbazate (−)-11.28 Pleasingly, the yield for the two-step, one flask operation was 62%. Deprotection of (−)-11, employing our previously developed conditions (25% water in TFA), generated the free base (−)-12 in 98% yield with >95% purity. This 3-step sequence permitted construction of (−)-12 on gram scale, starting from L-Boc-Trp-OH; the overall yield was 58%.
As anticipated, both (−)-11(S) and (−)-12(S) exhibited potent inhibitory activity against cathepsin L with IC50 values of 56 nM and 133 nM, respectively.29 The R-enantiomer, (+)-11(R),30 was only modestly active against cathepsin L (IC50 = 34 μM).29 Some variability in IC50 values determined for (−)-12 prompted us to explore the stability of both (−)-11 and (−)-12 in solvents relevant to the bioassay. While (−)-11 was found to be completely stable in DMSO, as well as in the buffer employed in the Cathepsin L assay [e.g., NaOAc (20 mM), pH 5.5; EDTA (1 mM); cysteine (5 mM)], the free-base (−)-12 proved unstable in DMSO, generating decomposition products 13, 14, and 15 after only 1 hour at room temperature (Figure 3).
Figure 3.
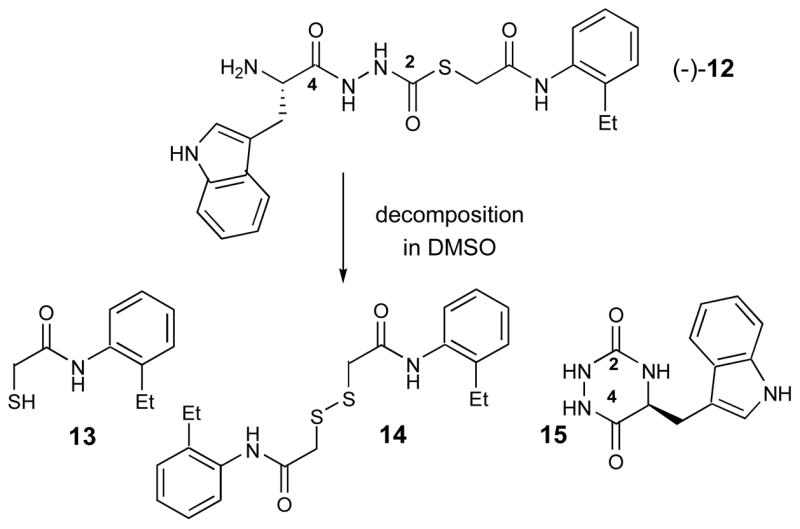
Decomposition products of (−)-12 in DMSO.
Presumably 13–15 are formed from (−)-12 via intramolecular attack of the primary amine on the C2 thiocarbazate moiety, and release of 13. These products, prepared either via synthesis (13 and 14) or by HPLC purification of decomposed material (15), were assayed for cathepsin L activity and found to be inactive. Due to the instability of (−)-12 under the assay conditions, IC50 values vary somewhat, rendering interpretation of the bioassay data difficult. We are however, confident in the accuracy of the bioassay results for (−)-11, as this compound is stable under all conditions evaluated. Thus, future efforts will be focused on the more stable thiocarbazate (−)-11 and related congeners.
As most cysteine protease inhibitors contain an electrophilic “warhead,” and work through a mechanism involving reaction with the active site cysteine,7,26 we strongly believe these novel thiocarbazates behave similarly, and are active by virtue of their electrophilic carbonyl moiety. The formation of 15 supports this mechanism, and is indicative of the electrophilicity of the C2 carbonyl moiety. Reactivity at the anilide carbonyl is also a possibility, and cannot be ruled out. However, we observe no evidence of side-products resulting from reaction at this position (cf. Figure 3). A complete biochemical characterization of this novel chemotype is underway.31
In summary, samples from the NIH MLSMR revealed promising cathepsin L inhibitory activity attributed to a series of 2,5-disubstituted oxadiazoles (1–5). Analytical analyses (LC-MS) of the library samples, however, indicated numerous impurities, thus requiring the development of an efficient synthesis for the putative active 2,5-disubstituted oxadiazoles. Synthetic samples of pure 2,5-disubstituted oxadiazoles were found to be completely devoid of cathepsin L inhibitory activity. Careful LC-MS investigation of both the library and synthetic samples revealed a thiocarbazate to be the active component. Bioassay of the synthetic thiocarbazates confirmed the hypothesis: (−)-11 and (−)-12 display potent inhibitory activity against cathepsin L, with IC50 values of 56 nM and 133 nM, respectively, however instability of (−)-12 was also noted. The design, syntheses, and biological evaluation of analogs of the highly potent lead thiocarbazate class are now ongoing in our laboratories, and will be reported in due course.
Acknowledgments
Financial support for this work was provided by the NIH (5U54HG003915-02 & 5U54HG003915-03). We thank Professor Barry S. Cooperman (University of Pennsylvania) and Dr. Mary Pat Beavers (University of Pennsylvania) for helpful discussions. We also thank Christina Kraml (Lotus Separations) for chiral SFC analysis. Finally we thank Dr. G. T. Furst (NMR Facility) and Dr. R. Kohli (Mass Spectrometry Facility) at the University of Pennsylvania for assistance in obtaining NMR and high-resolution mass spectra.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.McGrath ME. Annu Rev Biophys Biomol Struct. 1999;28:181. doi: 10.1146/annurev.biophys.28.1.181. [DOI] [PubMed] [Google Scholar]
- 2.Vasiljeva O, Reinheckel T, Peters C, Turk D, Turk V, Turk B. Curr Pharm Des. 2007;13:385. doi: 10.2174/138161207780162962. [DOI] [PubMed] [Google Scholar]
- 3.Esser RE, Angelo RA, Murphey MD, Watts LM, Thornburg LP, Palmer JT, Talhouk JW, Smith RE. Arthritis Rheum. 1994;37:236. doi: 10.1002/art.1780370213. [DOI] [PubMed] [Google Scholar]
- 4.Simmons G, Gosalia DN, Rennekamp AJ, Reeves JD, Diamond SL, Bates P. Proc Natl Acad Sci USA. 2005;102:11876. doi: 10.1073/pnas.0505577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Penn Center for Molecular Discovery: http://www.seas.upenn.edu/~pcmd/Molecular Library Screening Center Network: http://nihroadmap.nih.gov/molecularlibraries/
- 6.Myers MC, Napper AD, Motlekar N, Shah PP, Chiu CH, Beavers MP, Diamond SL, Huryn DM, Smith AB., III Bioorg Med Chem Lett. 2007;17:4761. doi: 10.1016/j.bmcl.2007.06.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Otto HH, Schirmeister T. Chem Rev. 1997;97:133. doi: 10.1021/cr950025u. [DOI] [PubMed] [Google Scholar]
- 8.Montaser M, Lalmanach G, Mach L. Biol Chem. 2002;383:1305. doi: 10.1515/BC.2002.147. [DOI] [PubMed] [Google Scholar]
- 9.Fujishima A, Imai Y, Nomura T, Fujisawa Y, Yamamoto Y, Sugawara T. FEBS Lett. 1997;407:47. doi: 10.1016/s0014-5793(97)00216-0. [DOI] [PubMed] [Google Scholar]
- 10.Schirmeister T, Kaeppler U. Mini-Rev Med Chem. 2003;3:361. doi: 10.2174/1389557033488079. [DOI] [PubMed] [Google Scholar]
- 11.Hernandez AA, Roush WR. Curr Opin Chem Bio. 2002;6:459. doi: 10.1016/s1367-5931(02)00345-9. [DOI] [PubMed] [Google Scholar]
- 12.Marquis RW, James I, Zeng J, Trout REL, Thompson S, Rahman A, Yamashita DS, Xie R, Gress CJ, Blake S, Lark MA, Hwang SM, Tomaszek T, Offen P, Head MS, Cummings MD, Veber DF. J Med Chem. 2005;48:6870. doi: 10.1021/jm0502079. [DOI] [PubMed] [Google Scholar]
- 13.Zhou NE, Guo D, Thomas G, Reddy AVN, Kaleta J, Purisima E, Menard R, Micetich RG, Singh R. Bioorg Med Chem Lett. 2003;13:139. doi: 10.1016/s0960-894x(02)00831-4. [DOI] [PubMed] [Google Scholar]
- 14.Chiva C, Barthe P, Codina A, Gairí M, Molina F, Granier C, Pugnière M, Inui T, Nishio H, Nishiuchi Y, Kimura T, Sakakibara S, Albericio F, Giralt E. J Am Chem Soc. 2003;125:1508. doi: 10.1021/ja0207908. [DOI] [PubMed] [Google Scholar]
- 15.Huang L, Ellman JA. Bioorg Med Chem Lett. 2002;12:2993. doi: 10.1016/s0960-894x(02)00619-4. [DOI] [PubMed] [Google Scholar]
- 16.PubChem: http://pubchem.ncbi.nlm.nih.gov/
- 17.PubChem web address for PCMD cathepsin L hits: http://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=460
- 18.HTS and initial followup (cherry picks) studies were conducted with the following assay buffer: 20 mM sodium acetate, 1 mM EDTA, and 5 mM DTT, pH5.5. Confirmatory results were obtained utilizing the following assay conditions, replacing DTT with cysteine in the assay buffer: Compounds were serially diluted in DMSO and transferred into a 96-well Corning 3686 assay microplate to give 16 dilutions ranging from 50 μM to 1.5 nM. Human liver cathepsin L (Calbiochem 219402) was activated by incubating with assay buffer for 30 min. Assay buffer consisted of 20 mM sodium acetate, 1 mM EDTA, and 5 mM cysteine, pH 5.5. Upon activation, cathepsin L (300 pM) was incubated with 1 μM Z-Phe-Arg-AMC substrate and test compound in 100 μL of assay buffer for 1 h at room temperature. Fluorescence of AMC released by enzyme-catalyzed hydrolysis of Z-Phe-Arg-AMC was read on a PerkinElmer Envision microplate reader (excitation 355 nm, emission 460 nm). Data was scaled using internal controls and fit to a four parameter logistic model (IDBS XLfit equation 205) to obtain IC50 values in triplicate.
- 19.Confalone PN, Woodward RB. J Am Chem Soc. 1983;105:902. [Google Scholar]
- 20.Verardo G, Geatti P, Lesa B. Synthesis. 2005:559. [Google Scholar]
- 21.For a general procedure for the preparation of α-bromo anilides see: von Geldern TW, Tasker AS, Sorensen BK, Winn M, Szczepankiewicz BG, Dixon DB, Chiou WJ, Wang L, Wessale JL, Adler A, Marsh KC, Nguyen B, Opgenorth TJ. J Med Chem. 1999;42:3668. doi: 10.1021/jm990170q.
- 22.Baggio R, Shi Y-Q, Wu Y-q, Abeles RH. Biochemistry. 1996;35:3351. doi: 10.1021/bi952879e. [DOI] [PubMed] [Google Scholar]
- 23.Thompson SK, Halbert SM, Bossard MJ, Tomaszek TA, Levy MA, Zhao B, Smith WW, Abdel-Meguid SS, Janson CA, D’Alessio KJ, McQueney MS, Amegadzie BY, Hanning CR, DesJarlais RL, Briand J, Sarkar SK, Huddleston MJ, Ijames CF, Carr SA, Garnes KT, Shu A, Heys JR, Bradbeer J, Zembryki D, Lee-Rykaczewski L, James IE, Lark MW, Drake FH, Gowen M, Gleason JG, Veber DF. Proc Natl Acad Sci USA. 1997;94:14249. doi: 10.1073/pnas.94.26.14249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xing R, Hanzlik RP. J Med Chem. 1998;41:1344. doi: 10.1021/jm970802d. [DOI] [PubMed] [Google Scholar]
- 25.Magrath J, Abeles RH. J Med Chem. 1992;35:4279. doi: 10.1021/jm00101a004. [DOI] [PubMed] [Google Scholar]
- 26.For a review on irreversible inhibitors of serine, cysteine, and threonine proteases see: Powers JC, Asglan JL, Ekici OD, James KE. Chem Rev. 2002;102:4639–4750. doi: 10.1021/cr010182v.
- 27.Chande MS, Singh-Jathar K. Indian J Chem. 1998;37B:352. [Google Scholar]
- 28.Characterization data for (−)-11 (PubChem SID26681509) mp = 131 °C; [α]D23 = −14.8 (c 0.5, AcOH); IR (KBr) 3412, 3343, 2971, 1680, 1529, 1167 cm−1; 1H NMR (500 MHz, DMSO-d6, VT-350K)δ 10.61 (br s, 1H), 10.04, (br s, 1H), 9.88 (br s, 1H), 9.14 (br s, 1H), 7.61 (d, J = 7.6 Hz, 1H), 7.43 (d, J = 7.4 Hz, 1H), 7.32 (d, J = 8.2 Hz, 1H), 7.21 (d, J = 7.3 Hz, 1H), 7.16–7.11 (m, 3H), 7.05 (t, J = 7.1 Hz, 1H), 6.97 (t, J = 7.4 Hz, 1H), 6.47 (br s, 1H), 4.31 (br s, 1H), 3.73 (br s, 2H), 3.17 (dd, J = 14.7, 4.1 Hz, 2H), 2.96 (m, 2H), 2.59 (q, J = 7.5 Hz, 2H), 1.29 (br s, 9H), 1.13 (t, J = 7.5 Hz, 3H); 13C NMR (125 MHz, DMSO-d6, major rotamer) δ 172.7, 171.6, 166.9, 155.2, 137.3, 136.0, 135.4, 128.5, 127.1, 125.7, 125.5, 125.2, 124.0, 120.9, 118.5, 118.1, 111.3, 109.9, 78.0, 53.6, 33.1, 28.1, 27.7, 23.6, 14.2; high resolution mass spectrum (ES+) m/z 562.2126 [(M+Na)+; calcd for C27H33N5O5SNa: 562.2100]. [α]D23= −14.8
- 29.IC50 values and mean ± standard deviations: (−)-11(S) 56 nM ± 4 nM; (−)-12(S) 133 nM ± 3 nM; (+)-11(R) 34 μM ± 2 μM.
- 30.Optical roation for (+)-11: [α]D23 = +12.8. The enantiomeric purity of both (−)-11 and (+)-11 were assayed using an OD-RH chiral column with the following LC parameters: 1.0 mL/min with a linear gradient of 90% water in acetonitrile to 10% water in acetonitrile over 15 min. Using this method, baseline separation was obtained for the enantiomers and retention times for (−)-11 and (+)-11 were 14.01 min and 13.02 min, respectively. The synthesis, outlined in Scheme 2., produced both enantiomers in >99% enantiomeric purity.
- 31.Shah PP, Myers MC, Beavers MP, Purvis JE, Huryn DM, Smith AB, III, Diamond SL. (Manuscript in preparation) [Google Scholar]