

Abstract
Purpose
The goals of this study were to determine if single-nucleotide polymorphisms in DNA damage repair genes and cell cycle regulating genes affect clinical response to combined gemcitabine radiation therapy and the overall survival (OS) of patients with pancreatic cancer.
Experimental Design
We evaluated six single-nucleotide polymorphisms of the ATM, ATM and Rad3-related (ATR), CHEK1, and CHEK2 genes in 119 patients with potentially resectable pancreatic cancer who were enrolled in clinical trials at The University of Texas M. D. Anderson Cancer Center from February1999 toJanuary 2006, with follow-up until February 2007. Patients received neoadjuvant concurrent gemcitabine and radiation therapy with or without gemcitabine-cisplatin induction therapy. Genotypes were determined and tested for associations with OS by Kaplan-Meier estimation, the log-rank test, and Cox regression analysis. P values of ≤0.05 were considered significant.
Results
The ATM G60A and CHEK1 G35A genotypes were significant (P < 0.05), and the ATR C340T genotype borderline significantly (P = 0.079) associated with OS. The hazard ratio of CHEK1 35AA was 2.01 (95% confidence interval, 1.20–3.37; P = 0.007) compared with CHEK1 35GG/GA with adjustments for race, sex, diabetes status, CA19-9 level, and success of tumor resection. A significant combined genotype effect was observed between ATM 60GA/GG, ATR 340CT/CC, and CHEK1 35AA with median OS times of 31.0, 16.2, and 10.5 months for patients carrying ≤1, 2, and 3 deleterious alleles, respectively (P = 0.004).
Conclusions
These observations suggest that polymorphic variations of DNA damage response genes affect clinical response to gemcitabine radiation therapy and OS of patients with resectable pancreatic cancer.
Pancreatic cancer is the fourth leading cause of cancer death in the United States, with an estimated 33,370 deaths in 2007, and it is one of the most aggressive human cancers with a 5-year survival rate of <5% (1). Pancreatic cancer is usually diagnosed at a late stage of the disease because it lacks early disease-specific signs and symptoms and progresses rapidly. Therefore, surgical resection is not an option for most patients, although it is the only potentially curative treatment. Neoadjuvant or adjuvant chemotherapy and radiation therapy are commonly used in the attempt to improve the surgical outcome and prolong overall survival (OS) in patients with resectable or nonresectable tumors. Gemcitabine has been the current standard chemotherapy for pancreatic cancer since Burris et al. (2) reported that it conferred a better clinical responses and longer OS than 5-fluorouracil. Recent studies have shown that the median OS for patients with pancreatic cancer treated with gemcitabine is ~6 months and 1-year survival rates are ~20% (3). Preoperative gemcitabine combined with radiation therapy has recently been attempted for not only unresectable but also borderline resectable locally advanced tumors, providing the opportunity for down-staging and rarely allowing patients to undergo resection (4–6).
The treatment efficacy of cytotoxic drugs is determined not only by the amount of therapy-induced DNA damage but also by the capacity of tumor cells to repair the damaged DNA or initiate apoptosis (7). Damage to DNA can induce several cellular responses, including DNA repair, transcriptional response, cell cycle arrest, and apoptosis (8). Host genetic variability that modulates DNA repair efficiency and cell viability may affect individual responses to cytotoxic therapies and thus OS.
Gemcitabine incorporates into DNA and causes a masked chain termination and an accumulation of cells in the S phase of the cell cycle. Gemcitabine also induces DNA replication stress by inhibiting ribonucleotide reductase, which results in an imbalanced nucleotide pool for DNA synthesis. DNA double-strand breaks are the most lethal DNA damage induced by radiation. DNA double-strand breaks and DNA replication stress activate the ATM and ATM and Rad3-related (ATR) signaling pathways, respectively, which transduce the signal to downstream genes, thereby inducing DNA repair, cell cycle arrest, or apoptosis. A previous study showed that cells lacking RAD9A, CHEK1, or ATR were more sensitive to gemcitabine, and this sensitization is p53 independent (9). Furthermore, ATM depletion also sensitized cells to gemcitabine and ionizing radiation. Gemcitabine activates the ATR/CHEK1 pathway (9). In response to ionizing radiation–induced DNA double-strand breaks, the ATM/CHEK2 pathway prevents DNA synthesis and cell cycle progression (10). To date, the role of the ATM/CHEK2 pathway in cells treated with gemcitabine is unknown (9).
Previous studies have shown that single-nucleotide polymorphisms (SNP) of DNA repair genes affect not only a patient’s risk for breast and lung cancers but also on the OS of such patients receiving cytotoxic therapies (7, 11–13). Because of the known associations between gemcitabine and radiation therapy–induced DNA damage and activation of the ATR/CHEK1 and ATM/CHEK2 pathways, we hypothesized that genetic variations in these pathways may affect sensitivity to such therapies and thus overall prognosis. We tested this hypothesis in a relatively homogeneous population of 119 patients with potentially resectable pancreatic cancer who had undergone neoadjuvant gemcitabine-based chemotherapy plus radiation therapy. We evaluated six SNPs of the ATM, ATR, CHEK1, and CHEK2 genes in this exploratory investigation.
Materials and Methods
Patient recruitment and data collection
The study involved 119 patients who, at the time of diagnosis, had potentially resectable adenocarcinoma of the head of the pancreas and had not received any treatment for pancreatic cancer. All patients were enrolled onto one of two sequential phase II clinical trials (ID98-020 and ID01-341) of preoperative (neoadjuvant) combined chemotherapy-radiation therapy for pancreatic cancer at The University of Texas M. D. Anderson Cancer Center conducted sequentially from February 1999 to 2006 and were observed through February 2007. These 119 patients represent the subset of patients enrolled in these clinical trials who consented to blood donation. The study was approved by the institutional review board of M. D. Anderson Cancer Center. Patients in the ID98-020 trial (n = 54) had received gemcitabine-based chemoradiotherapy consisting of weekly gemcitabine (400 mg/m2) for 4 wk and radiation (30 Gy in 10 fractions) for 2 wk. Patients in the ID01-341 trial (n = 65) had received induction therapy of gemcitabine (750 mg/m2/d) and cisplatin (30 mg/m2/d) every 2 wk for 4 wk and radiation (30 Gy in 10 fractions) for 2 wk. The same eligibility criteria for patient recruitment were applied for both protocols, and no significant difference in any clinical feature was observed between the two patient populations. Clinical information was collected from the patients’ medical records. Serum CA19-9 levels were measured at the time of cancer diagnosis. Tumor size was estimated from measurements made by endoscopic ultrasonography or radiologic imaging at the time of cancer diagnosis or extrapolated from actual measurements of the resected tumors if pretreatment measurements were not available. Tumor differentiation was evaluated in most surgically resected tumors and a few biopsy samples. The preoperative treatment effect was evaluated histologically in resected tumors according to previously published criteria (14), i.e., tumors with >90% viable cancer cells were defined as treatment effect grade I, 51% to 90% viable cells as grade IIA, 10% to 50% viable cells as grade IIB, and <10% viable cells as grade III. Postsurgical treatment or treatment received after tumor recurrence was not considered in this study.
DNA extraction and genotyping
Whole blood was collected from patients at the time of enrollment, and DNA was extracted from peripheral lymphocytes using a DNA isolation kit (Qiagen). Polymorphisms were detected using the TaqMan genotyping assays provided by Applied Biosystems.
Approximately, 5% of the samples were analyzed in duplicate, and discrepancies were seen in <1% of all duplicates. Patients whose samples yielded discordant results in two analyses were excluded from the final data analysis. The genes, chromosome locations, nucleotide substitutions, amino acid changes, reference SNP identification numbers, and reported allele frequencies of the six SNPs evaluated in this study are summarized in Table 1. The ATM G60A SNP was selected because of its prior association with increased risk of cancer (15). The three ATR SNPs selected are all nonsynonymous SNPs with a minor allele frequency of >10%. The single intronic CHEK1 and CHEK2 SNP was selected for explorative purpose because neither prior association nor nonsynonymous SNP with a minor allele frequency of >10% was identified for these two genes when we initialized the study.
Table 1.
SNPs evaluated
| Gene | Chromosome | SNP | Reference SNP ID no. | Minor allele frequency (all races)* | Minor allele frequency (Caucasian)* |
|---|---|---|---|---|---|
| ATM | 11q22-q23 | IVS61 +60, G>A | 664143 | 0.33 | 0.34 |
| ATR | 3q22-q24 | Ex4 +340C>T, T211M | 2227928 | 0.31 | 0.38 |
| Ex43 -76G>A, R2425Q | 2229032 | 0.16 | 0.17 | ||
| Ex8 +44A>T, G592G | 2227930 | 0.32 | 0.40 | ||
| CHEK1 | 11q24-q24 | IVS11 +35, G>A | 521102 | 0.41 | 0.43 |
| CHEK2 | 22q12.1 | IVS8 -200, T>C | 2267130 | 0.35 | 0.45 |
Allele frequencies came from the National Cancer Institute SNP500 cancer database.
Survival measurements
Dates of death were obtained and cross-checked using at least one of the following sources: Social Security Death Index,4 in-patient medical records, or the M. D. Anderson Cancer Center tumor registry. OS times were calculated from the date of pathologic diagnosis to the date of death or last follow-up. Data for patients who were alive at the last day of follow-up evaluation were censored at that time.
Statistical methods
The genotype distribution was tested for Hardy-Weinberg equilibrium with the goodness-of-fit χ2 test. Median follow-up times were computed with censored observations only, whereas median survival times (MST) were calculated for all patients. Hazard ratios and 95% confidence intervals (95% CI) were estimated using univariable or multivariate Cox proportional hazard models. In addition to age, sex, and ethnicity, known or potential clinical prognostic factors (such as serum CA19-9 values at diagnosis) and tumor resection status (done/not done) were included in the multivariate model when appropriate. All statistical testing was conducted with SPSS software, version 12.0 (SPSS), and statistical significance was defined as P ≤ 0.05. We estimated the false-positive report probability for the observed statistically significant association using the methods described by Wacholder et al. (16). We considered that a prior probability of 25% might be appropriate when there is biological plausibility and availability of previous epidemiologic evidence for such an association. The false-positive report probability value for noteworthiness was set as 0.2. Multiplicity adjusted P value was also calculated (17).
Results
Patient characteristics and clinical predictors
The patient characteristics and clinical features of their tumors are summarized in Table 2. The median age of the 119 patients was 65 years (range, 38–83 years). There were 84 (71%) deaths, and the MST was 28.7 months (95% CI, 24.4–32.9). The median follow-up time was 51.7 months (95% CI, 43.1–60.4) for the patients who were still alive at the end of the follow up. There was no significant difference in OS by age, race, tumor size, and treatment effects. Women had longer OS times than men, but the difference was not statistically significant (P = 0.09). Diabetes, a higher serum level of CA 19-9 at diagnosis, tumor not resected, poor tumor differentiation, and node-positive resection were significantly associated with reduced OS (Table 2).
Table 2.
Patients characteristics (n = 119)
| Variable | No. patients | No. deaths | MST (mo) | Log-rank P value |
|---|---|---|---|---|
| Age (y) | 0.86 | |||
| ≤50 | 15 | 13 | 20.3 | |
| 51–60 | 32 | 21 | 22.5 | |
| 61–70 | 43 | 29 | 21.2 | |
| >70 | 29 | 21 | 21.2 | |
| Sex | 0.09 | |||
| Male | 69 | 52 | 16.5 | |
| Female | 50 | 32 | 24.5 | |
| Race | 0.85 | |||
| White | 108 | 77 | 21.2 | |
| Hispanic | 5 | 4 | 18.2 | |
| African American | 5 | 3 | 33.6 | |
| Other | 1 | 0 | NA | |
| Diabetes status | 0.04 | |||
| Negative | 88 | 57 | 24.5 | |
| Positive | 31 | 27 | 16.9 | |
| Tumor size (cm) | 0.36 | |||
| ≤2 | 35 | 22 | 21.2 | |
| >2 | 83 | 61 | 20.9 | |
| Tumor differentiation | 0.001 | |||
| Well to moderate | 64 | 38 | 29.0 | |
| Poor | 23 | 21 | 15.5 | |
| CA19-9 (units/mL) | 0.03 | |||
| ≤47 | 30 | 17 | 51.3 | |
| 48–500 | 58 | 41 | 23.9 | |
| 501–1,000 | 13 | 11 | 18.9 | |
| >1,000 | 18 | 15 | 14.4 | |
| Tumor resection status | <0.001 | |||
| Negative | 35 | 35 | 10.5 | |
| Positive | 84 | 49 | 33.6 | |
| Node status | 0.01 | |||
| Negative | 40 | 20 | 53.8 | |
| Positive | 44 | 29 | 23.9 | |
| Treatment effect* | 0.76 | |||
| G1/G2A (≥50%) | 34 | 21 | 31.0 | |
| G2B/G3 (<50%) | 49 | 23 | 33.9 |
Abbreviations: G1, grade I; G2A, grade IIA; G2b, grade IIB; G3, grade III.
On viable cells in resected tumors.
Genotype frequency and effect on OS
The six SNPs of interest were successfully amplified in 95.6% to 99.1% of the samples. Genotype frequencies of the nine SNPs were found to be in Hardy-Weinberg equilibrium (χ2 = 0.001–0.77; all P > 0.1). The ATM G60A SNP was in linkage disequilibrium with the ATM T-77C SNP that was examined in our previous study (ref. 18; D′ = 0.85). The ATR C340T SNP was in linkage disequilibrium with the ATR G-76A (D′ = 1) and A44T (D′ = 0.98) SNP. No significant racial difference in genotype frequency was observed (data not shown). Of the six SNPs, two showed a significant effect on OS. The genotype frequencies, MSTs, and hazard ratios (95% CI) are shown in Table 3. A significantly reduced OS was associated with the presence of heterozygous variant alleles of ATM G60A and homozygous variant alleles of CHEK1 G35A. The MST associated with ATM G60A SNPs was 16.6 months for the GA genotype compared with 23.9 and 36.0 months for the GG and AA genotypes, respectively (log-rank P = 0.048; Fig. 1). The MST associated with CHEK1 SNPs was 14.3 months for the AA genotype compared with 26.4 and 21.4 months for the GA and GG genotypes, respectively (log-rank P = 0.039; Fig. 1). The ATM 60GA and CHEK1 35AA genotype remained a significant independent predictor of survival after adjusting for sex, race, diabetes, CA19-9, and tumor resection (P = 0.007; Table 3). Assuming there were six independent tests, the Bonferroni adjusted P value (0.007 × 6 = 0.042) remained significant for both SNPs. The ATR C340T CC and CHEK2 T-200C CT/TT genotypes were nonsignificantly associated with reduced OS (log-rank P = 0.079 and 0.122, respectively).
Table 3.
OS by genotype
| Genotype | No. cases | No. deaths | MST ± SE (mo) | P (log-rank) | Hazard ratio* (95% CI) | P |
|---|---|---|---|---|---|---|
| ATM G60A | ||||||
| AA | 23 | 13 | 36.0 ± 8.3 | 1.0 | ||
| GA | 51 | 38 | 16.6 ± 2.3 | 2.35 (1.24–4.46) | 0.007 | |
| GG | 44 | 32 | 23.9 ± 3.7 | 0.048 | 1.34 (0.70–2.60) | 0.379 |
| AA versus GA/GG | 0.048 | 1.77 (0.97–3.23) | 0.059 | |||
| ATR C340T | ||||||
| TT | 39 | 26 | 31.0 ± 7.9 | 1.0 | ||
| CT | 54 | 35 | 18.4 ± 2.3 | 1.03 (0.61–1.73) | 0.925 | |
| CC | 21 | 18 | 21.4 ± 6.7 | 0.188 | 0.88 (0.46–1.69) | 0.438 |
| TT/CT versus CC | 0.079 | 0.84 (0.47–1.51) | 0.565 | |||
| ATR G-76A | ||||||
| GG | 89 | 62 | 20.9 ± 1.7 | 1.0 | ||
| GA | 27 | 20 | 24.3 ± 3.4 | 1.26 (0.75–2.13) | 0.379 | |
| AA | 2 | 1 | 12.9 | 0.840 | 0.62 (0.08–4.78) | 0.555 |
| ATR A44T | ||||||
| AA | 41 | 29 | 26.4 ± 7.2 | 1.0 | ||
| TA | 56 | 37 | 18.7 ± 2.6 | 1.02 (0.62–1.67) | 0.854 | |
| TT | 21 | 17 | 18.9 ± 6.2 | 0.294 | 0.81 (0.43–1.55) | 0.511 |
| AA versus TA/TT | 0.445 | 0.95 (0.60–1.51) | 0.863 | |||
| CHEK1 G35A | ||||||
| GG | 29 | 19 | 21.4 ± 3.5 | 1.0 | ||
| GA | 60 | 41 | 26.4 ± 3.8 | 0.88 (0.50–1.55) | 0.638 | |
| AA | 29 | 23 | 14.3 ± 2.6 | 0.039 | 1.85 (0.97–3.52) | 0.061 |
| GG/GA versus AA | 0.012 | 2.01 (1.20–3.37) | 0.007 | |||
| CHEK2 T-200C | ||||||
| CC | 20 | 12 | 35.7 ± 5.1 | 1.0 | ||
| CT | 62 | 46 | 18.9 ± 1.9 | 1.42 (0.74–2.75) | 0.297 | |
| TT | 35 | 24 | 21.2 ± 4.3 | 0.291 | 1.52 (0.72–3.19) | 0.273 |
| CC versus CT/TT | 0.122 | 1.45 (0.76–2.74) | 0.257 | |||
Hazard ratio adjusted for sex, race, diabetes, CA19-9, and tumor resection.
Fig. 1. Kaplan-Meier plot of OS by genotype.
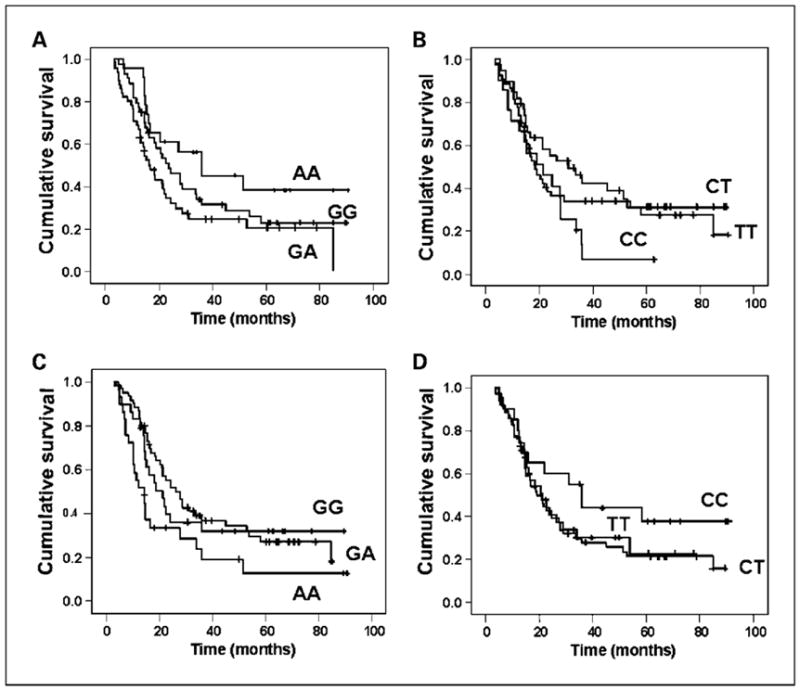
A, ATM G60A; B, ATR C340T; C, CHEK1 G35A; and D, CHEK2 C-200T.
Furthermore, the ATR C340T SNP showed a borderline significant association with the fraction of patients who actually achieved tumor resection after the preoperative chemoradiation. Eighty-two percent (32 of 39) of the patients carrying the TT genotype had their tumors resected compared with 72% (39 of 54) and 52% (11 of 21) of those carrying the TC and CC genotypes, respectively (P = 0.051, χ2 test). Patients who did not achieve tumor resection were mostly due to early disease progression.
Combined genotype effects
Because ATM, ATR, and CHEK1 are all involved in the same DNA damage response pathway, we explored the effect of combined genotypes of three deleterious alleles on OS of the patients. The deleterious alleles included ATM 60 GA/GG, ATR 340CT/CC, and CHEK1 35AA, all of which were related to a shorter OS. As the number of deleterious alleles increased, OS decreases (Table 4). The MSTs were 31.0, 18.2, and 10.5 months for patients carrying ≤1, 2, and 3 deleterious alleles, respectively (log-rank P = 0.004). When all three deleterious alleles were included in the Cox regression model, both ATM (AA versus AG/GG) and CHEK1 (GG/GA versus AA) genotypes remained as significant predictors for survival (Table 5). The multiplicity-adjusted P value was significant for CHEK1 but not ATM SNP. We estimated the false-positive report probability of the ATM and CHEK1 SNP to be 0.135 and 0.038, respectively, given a prior probability of 25%. Both are below the threshold of 0.20 indicating noteworthiness. The effects of tumor resection and diabetes on OS remained highly significant (P < 0.001 and P = 0.001, respectively), whereas CA19-9 level became marginally significant or nonsignificant (P = 0.091, 0.058, and 0.148 for CA19-9 level of 48–500, 501–1,000, and >1,000 units/mL, respectively).
Table 4.
OS by combined genotype
| No. deleterious alleles | No. cases | No. deaths | MST (mo)* | Hazard ratio (95% CI) | P |
|---|---|---|---|---|---|
| ≤1 | 49 | 31 | 31.0 | Reference | |
| 2 | 57 | 42 | 18.2 | 1.57 (0.95–2.58) | 0.078 |
| 3 | 13 | 11 | 10.5 | 3.04 (1.46–6.32) | 0.003 |
Log-rank P = 0.004.
Table 5.
Effects of genotype on survival as analyzed in a multivariate Cox regression (n = 112)
| Variable | Hazard ratio (95% CI) | P |
|---|---|---|
| ATM (AA versus AG/GG) | 1.92 (1.03–3.59) | 0.041 |
| ATR (TT versus CT/CC) | 1.06 (0.64–1.77) | 0.822 |
| CHEK1 (GG/GA versus AA) | 2.84 (1.59–5.09) | <0.001 |
| Sex (male versus female) | 0.87 (0.53–1.41) | 0.568 |
| Race (White, Hispanic, African American, others) | 0.70 (0.39–1.26) | 0.232 |
| CA19-9 (reference, V47) | 1.76 (0.97–1.55) | 0.091 |
| 48–500 | 2.47 (0.97–6.31) | 0.058 |
| 501–1,000 | 1.78 (0.82–3.90) | 0.148 |
| >1,000 | 2.61 (1.48–4.59) | 0.001 |
| Diabetes (no versus yes) Tumor resection (no versus yes) | 0.13 (0.08–0.24) | <0.001 |
NOTE: Tumor grade was not included in this model because information was missing from 27% of the 119 patients.
Discussion
In this study, we evaluated the associations between ATM, ATR, CHEK1, and CHEK2 gene SNPs and the clinical outcomes of patients with resectable pancreatic cancer. Our findings suggest that the ATM G60A and CHEK1 G35A genotype significantly affected OS. A significant combined genotype effect of ATM 60GA/GG, ATR 340CT/CC, and CHEK1 35AA on OS was detected. The CHEK1 G35A genotype remained as a significant predictor for survival after adjusting for all other clinical and genetic factors and after adjusting for multiple testing. These data are the first to show that there is an important role for DNA damage response genes in the OS of patients with resectable pancreatic cancer.
The ATM gene encodes a protein kinase that is activated by DNA damage caused by ionizing radiation or reactive oxygen intermediates to induce the transactivation of various proteins that function in cell-cycle arrest, apoptosis, DNA repair, and centrosome duplication (19). Several studies have shown that the ATM genotype/haplotype alters the cellular sensitivity to ionizing radiation (10, 11, 20–22). However, the findings of investigations into the effect of ATM G60A genotype on patient survival or cancer risk have been inconsistent. Two studies found no significant association between the ATM G60A genotype and relapse-free survival in prostate cancer (23) or between this genotype and lung cancer response to chemotherapy alone, chemotherapy plus radiation therapy, or radiotherapy alone (7). On the other hand, one study showed that the ATM GA heterozygote was associated with a significantly higher risk of lung cancer than the AA and GG homozygotes (15). This observation is consistent with our current finding on the GA heterozygote and our previous finding on the ATM -77TC heterozygote (18), both were associated with shorter OS compared with their respective homozygotes. The ATM -77TC heterozygote was previously associated with a reduced radiosensitivity among patients with breast cancer compared with the TT homozygote (24). These observations suggest that these two ATM SNPs might be dominant-negative mutations. To date, no functional studies have been reported linking an altered protein function or cellular phenotype with the presence of these SNPs. It has been postulated that the G60A site exists in protein-binding motifs that have potential as binding sites of intronic splicing enhancers or repressors. Therefore, G60A is a possible site that might be related to the splicing process of exon 61, leading to inaccurate splicing (23).
The ATR gene encodes a protein kinase that responds to a wide range of DNA lesions and seems to be particularly important in maintaining the integrity of the DNA replication apparatus following damage that arrests the progression of this complex (25). ATR phosphorylates CHEK1 (on Ser345) in response to agents that arrest DNA replication (26). A previous study showed that the ATR T340C genotype was associated with a decreased risk of non–small cell lung cancer (12). When we tested this genotype association in pancreatic cancer, we found a marginally significant difference in OS and the success rate of tumor resection (P = 0.079 and 0.051, respectively). These observations could be by chance alone. Because gemcitabine induces DNA replication arrest and an in vitro study showed that ATR-deficient cells are more sensitive to gemcitabine (10), the association between ATR genotypes and clinical response to gemcitabine needs further investigation in a larger patient population.
The CHEK1 gene encodes a cell cycle checkpoint kinase that is phosphorylated through ATR-mediated regulation in response to agents that arrest DNA replication (26). CHEK1 is essential for cell viability, and it plays a crucial role in replication origin firing and S-phase delay (27). It is known that gemcitabine-mediated radiosensitization requires cell accumulation in the S phase (28), and gemcitabine stimulates CHEK1 to initiate a G2-M cell cycle checkpoint (10). CHEK1 negatively regulates cell entry into mitosis in response to gemcitabine and acts to coordinate the cell cycle with DNA synthesis, thus preventing premature mitotic entry in gemcitabine-treated cells (10). A recent in vitro study has shown that CHEK1 kinase inhibitor substantially enhanced gemcitabine-induced cell killing (29). The significant association between the homozygous variant genotype of CHEK1 and reduced OS observed in our study could be attributed to a poorer response to preoperative gemcitabine-based chemoradiation therapy. The observed association between CHEK1-genotype and survival has some potential clinical implications. Patients carrying the genotypes that confer a higher activity of CHEK1 may have a poor response to gemcitabine alone but a better response to combined therapy of CHEK1 inhibitor and gemcitabine. On the other hand, patients carrying the genotypes that confer a lower activity of CHEK1 may less likely respond to CHEK1 inhibitor-initiated gemcitabine sensitization. Because it is difficult to obtain tissue specimens from pancreatic cancers, determining a patient’s genotype could be a valuable surrogate for tumor testing. Further investigations into the genotype-phenotype associations of CHEK1 and their associations with clinical outcome of patients treated with gemcitabine are warranted.
The CHEK2 gene encodes a cell cycle checkpoint kinase that is activated through the ATM-mediated signaling pathway. Previous studies found no evidence of polymorphic variants of the CHEK2 gene associated with risk of breast cancer (13). Our results also do not support a role for the CHEK2 polymorphic variant in cellular response to gemcitabine and radiation therapy in pancreatic cancer. This is consistent with the fact CHEK2 has no given role in cellular response to gemcitabine and radiation therapy (10). Indeed, one previous study reported that effect of ATM on survival is mediated by an ATM substrate other than CHEK2 (9).
Because the four genes selected in this study are all involved in the same cellular response to the DNA damage signaling pathway, it is not surprising that a strong combined genotype effect on survival was detected, which underscores the importance of a candidate gene and pathway-based approach in genotyping investigation. Although each gene/SNP may have a subtle effect on drug response or tumor progression, the consequence of the combined effect of several critical genes in the same pathway could be prominent. The ultimate goal of this research is to identify the genetic profiles that can be used in the clinic as predictors for response to therapy or prognostic factors for survival. Our results need to be confirmed in other study populations. If confirmed, analysis of these genotypes may eventually be applied in the clinic to determine treatment modalities.
Acknowledgments
Grant support: NIH RO1 grant CA098380 (D. Li), Specialized Programs of Research Excellence P20 grant CA101936 (J.L. Abbruzzese), NIH Cancer Center Core grant CA16672, and Lockton Research Funds research grant (D. Li).
Footnotes
References
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 2.Burris HA, III, Moore MJ, Andersen J, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15:2403–13. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 3.Hochster HS, Haller DG, de Gramont A, et al. Consensus report of the international society of gastrointestinal oncology on therapeutic progress in advanced pancreatic cancer. Cancer. 2006;107:676–85. doi: 10.1002/cncr.22036. [DOI] [PubMed] [Google Scholar]
- 4.Varadhachary GR, Tamm EP, Abbruzzase JL, et al. Borderline resectable pancreatic cancer: definitions, management, and role of preoperative therapy. Ann Surg Oncol. 2006;13:1035–46. doi: 10.1245/ASO.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 5.Kim R, Saif MW. Is there an optimal neoadjuvant therapy for locally advanced pancreatic cancer? JOP. 2007;8:279–88. [PubMed] [Google Scholar]
- 6.Massucco P, Capussotti L, Magnino A, et al. Pancreatic resections after chemotherapy for locally advanced ductal adenocarcinoma: analysis of perioperative outcome and survival. Ann Surg Oncol. 2006;13:1202–8. doi: 10.1245/s10434-006-9032-x. [DOI] [PubMed] [Google Scholar]
- 7.Su D, Ma S, Liu P, et al. Genetic polymorphisms and treatment response in advanced non-small cell lung cancer. Lung Cancer. 2007;56:281 –8. doi: 10.1016/j.lungcan.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 8.Sancar A, Lindsey-Boltz LA, «nsal-Kac̊maz KU, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 9.Karnitz LM, Flatten KS, Wagner JM, et al. Gemcitabine-induced activation of checkpoint signaling pathways that affect tumor cell survival. Mol Pharmacol. 2005;68:1636–44. doi: 10.1124/mol.105.012716. [DOI] [PubMed] [Google Scholar]
- 10.Morgan MA, Parsels LA, Parsels JD, Mesiwala AK, Maybaum J, Lawrence TS. Role of checkpoint kinase1 in preventing premature mitosis in response to gemcitabine. Cancer Res. 2005;65:6835–42. doi: 10.1158/0008-5472.CAN-04-2246. [DOI] [PubMed] [Google Scholar]
- 11.Ange’ le S, Romestaing P, Moullan N, et al. ATM haplotypes and cellular response to DNA damage: association with breast cancer risk and clinical radiosensitivity. Cancer Res. 2003;63:8717–25. [PubMed] [Google Scholar]
- 12.Zienolddiny S, Campa D, Lind H, et al. Polymorphisms of DNA repair genes and risk of non-small cell lung cancer. Carcinogenesis. 2006;27:560–7. doi: 10.1093/carcin/bgi232. [DOI] [PubMed] [Google Scholar]
- 13.Goode EL, Dunning AM, Kuschel B, et al. Effect of germ-line genetic variation on breast cancer survival in a population-based study. Cancer Res. 2002;62:3052–7. [PubMed] [Google Scholar]
- 14.Evans DB, Rich TA, Byrd DR, et al. Preoperative chemoradiation and pancreaticoduodenectomy for adenocarcinoma of the pancreas. Arch Surg. 1992;127:1335–9. doi: 10.1001/archsurg.1992.01420110083017. [DOI] [PubMed] [Google Scholar]
- 15.Kim JH, Kim H, Lee KY, et al. Genetic polymorphisms of ataxia telangiectasia mutated affect lung cancer risk. Hum Mol Genet. 2006;15:1181–6. doi: 10.1093/hmg/ddl033. [DOI] [PubMed] [Google Scholar]
- 16.Wacholder S, Chanock S, Garcia-Closas M, El Ghormli L, Rothman N. Assessing the probability that a positive report is false: an approach for molecular epidemiology studies. J Natl Cancer Inst. 2004;96:434–42. doi: 10.1093/jnci/djh075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bland JM, Altman DG. Multiple significance tests: the Bonferroni method. BMJ. 1995;310:170. doi: 10.1136/bmj.310.6973.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li D, Frazier M, Evans DB, et al. Single nucleotide polymorphisms of Rec Q1, RAD54L, and ATM genes are associated with reduced survival of pancreatic cancer. J Clin Oncol. 2006;24:1720–8. doi: 10.1200/JCO.2005.04.4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shiloh Y. ATMandrelatedproteinkinases:safeguard-inggenomeintegrity. Nat Rev Cancer. 2003;3:155–68. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- 20.Falck J, Mailand N, Sylju3sen RG, Bartek J, Lukas J. The ATM-Chk2 – 25A checkpoint pathway guards against radioresistant DNA synthesis. Nature. 2001;410:842–7. doi: 10.1038/35071124. [DOI] [PubMed] [Google Scholar]
- 21.Gutierrez-Enriquez S, Fernet M, Do «rk T, et al. Functional consequences of ATM sequence variants for chromosomal radiosensitivity. Genes Chromosomes Cancer. 2004;40:109–19. doi: 10.1002/gcc.20025. [DOI] [PubMed] [Google Scholar]
- 22.Alsbeih G, El-Sebaie M, Al-Harbi N, Al-Buhairi M, Al-Hadyan K, Al-Rajhi N. Radiosensitivity of human fibroblasts is associated with amino acid substitution variants in susceptible genes and correlates with the number of risk alleles. Int J Radiat Oncol Biol Phys. 2007;67:1320–3. doi: 10.1016/j.ijrobp.2006.12.050. [DOI] [PubMed] [Google Scholar]
- 23.Browning REL, IV, Li H, Shinohara ET, et al. ATM polymorphism IVS62+60G>A is not associated with disease aggressiveness in prostate cancer. Urology. 2006;67:1320–3. doi: 10.1016/j.urology.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 24.Angele S, Romestaing P, Moullan N, et al. ATM haplotypes and cellular response to DNA damage: association with breast cancer risk and clinical radiosensitivity. Cancer Res. 2003;63:8717–25. [PubMed] [Google Scholar]
- 25.Zhou B-BS, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–9. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 26.Liu Q, Guntuku S, Cui XS, et al. Chk1is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–59. [PMC free article] [PubMed] [Google Scholar]
- 27.Feijoo C, Hall-Jackson C, Wu R, et al. Activation of mammalian Chk1during DNA replication arrest: a role for Chk1 in the intra-S phase checkpoint monitoring replication origin firing. J Cell Biol. 2001;154:913–23. doi: 10.1083/jcb.200104099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ostruszka LJ, Shewach DS. The Role of Cell Cycle Progression in Radiosensitization by 2′,2prime;-Difluoro-2prime;-deoxycytidine. Cancer Res. 2000;60:6080–8. [PubMed] [Google Scholar]
- 29.Matthews DJ, Yakes FM, Chen J, et al. Pharmacological abrogation of S-phase checkpoint enhances the anti-tumor activity of gemcitabine in vivo. Cell Cycle. 2007;6:104–10. doi: 10.4161/cc.6.1.3699. [DOI] [PubMed] [Google Scholar]