

Abstract
The majority of cell adhesion molecules are N-glycosylated but the role of N-glycans in intercellular adhesion in epithelia remains ill-defined. Reducing N-glycan branching of cellular glycoproteins by swainsonine, the inhibitor of N-glycan processing, tightens and stabilizes cell-cell junctions as detected by a 3-fold decrease in the paracellular permeability and a 2- to 3-fold increase in the resistance of the adherens junction proteins to extraction by non-ionic detergent. In addition, exposure of cells to swainsonine inhibits motility of MDCK cells. Mutagenic removal of N-glycosylation sites from the Na,K-ATPase β1 subunit impairs cell-cell adhesion and decreases the effect of swainsonine on the paracellular permeability of the cell monolayer and also on detergent resistance of adherens junction proteins, indicating that the extent of N-glycan branching of this subunit is important for intercellular adhesion. The N-glycans of the Na,K-ATPase β1 subunit and E-cadherin are less complex in tight renal epithelia than in the leakier intestinal epithelium. The complexity of the N-glycans linked to these proteins gradually decreases upon the formation of a tight monolayer from dispersed MDCK cells. This correlates with a cell-cell adhesion-induced increase in expression of GnT-III (stops N-glycan branching) and a decrease in expression of GnTs IVC and V (promote N-glycan branching) as detected by real-time quantitative PCR. Consistent with these results, partial silencing of the gene encoding GnT-III increases branching of N-glycans linked to the Na,K-ATPase β1 subunit and other glycoproteins and results in a 2-fold increase in the paracellular permeability of MDCK cell monolayers. These results suggest epithelial cells can regulate tightness of cell junctions via remodeling of N-glycans, including those linked to the Na,K-ATPase β1-subunit.
The generation of a polarized epithelium depends on the establishment of cell-cell contacts that induce the formation of the adherens junctions followed by assembly of the tight junctions (1,2). Adherens junctions mechanically link adjacent cells, whereas tight junctions seal intercellular contacts and limit paracellular movement of fluid and solutes. These tight junctions can vary in tightness in different organs and even in different parts of the same organ. For example, renal epithelia are less permeable than intestine epithelia and the renal collecting duct is less permeable than the proximal tubule (3,4). Also, the paracellular permeability of an epithelium can be regulated in response to different physiological conditions (5,6).
Several membrane-inserted proteins which are involved in formation and maintenance of intercellular junctions have been identified. Occludin, JAM-1 and claudins are cell adhesion molecules of the tight junctions (1,2), while cadherins and nectins are membrane components of the adherens junctions (7). In addition, the Na,K-ATPase, an essential membrane transport enzyme, has been found to be important for intercellular adhesion (8–12).
The Na,K-ATPase is a P2-type cation pump that catalyzes outward active transport of 3 Na+ in exchange for exoplasmic 2 K+. It is a heterodimer with an α subunit and a glycosylated β subunit. The former subunit contains the catalytic site, whereas the latter subunit is necessary for normal pump maturation and trafficking (13,14). Additional tissue-specific regulatory subunits of the Na,K-ATPase that belong to the FXYD family of small membrane proteins have also been identified (15,16). This enzyme generates ion gradients important for the regulation of cell osmolality, membrane potential, cell volume, intracellular pH and intracellular Ca2+ concentration. Further, by producing a transmembrane Na+ gradient, this pump drives vectorial transport of a number of solutes across epithelia. Finally, it plays a role in intracellular signaling by a mechanism which is independent of ion transport, but similar to ion transport by this pump is sensitive to ouabain (17).
An additional role for this enzyme, also independent of its ion-pumping function, is to facilitate adhesion between adjoining cells of various epithelia. Indeed, the β2 subunit of the Na,K-ATPase was first described as an adhesion protein in glial cells (AMOG) in the rat brain (12). Specific isoforms of the Na,K-ATPase play a crucial role in intercellular junction formation in a Drosophila tracheal epithelium (9,10,18). The β1 subunit of the Na,K-ATPase is necessary for normal intercellular adhesion in MDCK cells, a cell line with characteristics of the distal renal tubule (11,19). N-glycosylation of this subunit is important for normal cell-cell adhesion, since MDCK cells expressing the unglycosylated mutant of the Na,K-ATPase β1 subunit display a significantly lower rate of formation of cell-cell contacts than non-transfected MDCK cells (11).
Recent studies indicate that as the mature monolayer develops, the degree of N-glycosylation of adhesion molecules is altered. The complexity of N-glycans linked to E-cadherin is decreased in densely populated MDCK cell cultures when compared with sparsely populated MDCK cell cultures (20). Consistent with these observations, the relative molecular weight of N-glycans linked to the N-cadherin decreases as non confluent RPE cells become confluent (21). Also, during development of cell-cell adhesion of epithelial GE11 cells, GnT-III, the enzyme that stops branching of N-glycans is up-regulated (22).
These data suggest that changes in the structure of the N-glycans linked to adhesion molecules might be expected to affect intercellular adhesion. Since N-glycans linked to the Na,K-ATPase β1 subunit are required for the initial steps of formation of intercellular contacts (11), it is possible that complexity of N-glycans of this subunit also changes with development of cell-cell junctions and a specific N-glycan structure is necessary for normal cell to cell adhesion.
To examine these issues, we altered the extent of N-glycan branching either by exposure of MDCK cells in culture to swainsonine, an inhibitor of N-glycan branching, or by post-transcriptional silencing the gene encoding GnT-III, a critical N-glycan stop-branching enzyme, using RNA interference and assessed the impact of these perturbations on cell-cell adhesion. The results of these studies demonstrate that prevention of N-glycan branching by swainsonine tightens and stabilizes cell-cell junctions. Conversely, increased N-glycan branching due to partial silencing of the gene encoding GnT-III, loosens cell-cell contacts. Consistent with these observations, the normal development of cell-cell adhesion is associated with reduced complexity of N-glycans of the Na,K-ATPase β1 subunit and other adhesion proteins. These changes correlate with increased expression of GnT-III, the enzyme that reduces branching, and decreased expression of GnT-IVC and GnT-V, enzymes that promote branching. These results suggest tightness of cell junctions can be modulated through modification of N-glycans.
The tightening and stabilizing effects of swainsonine are attenuated in a cell line expressing the unglycosylated mutant of the Na,K-ATPase β1 subunit; findings indicating that the structure of the N-glycans linked to this subunit is important for normal cell-cell adhesion.
Experimental Procedures
Construction of MDCK stable cell lines
Stable cell lines expressing a fusion protein with YFP linked to the N-terminus of the Na,K-ATPase β1 subunit (YFP-β1)1 and mutated YFP-β1 fusion protein lacking three N-glycosylation sites (N123) were obtained as described previously (11,23). Non-transfected MDCK cells and cells expressing YFP-β1 and N123 were grown in Corning Costar polyester transwell inserts (Corning Incorporated) in 6-well plates in DMEM medium (Cellgro).
Primary Antibodies
The following monoclonal antibodies were used for Western blot analysis: against E-cadherin (Alexis Biochemicals), against the Na,K-ATPase β1 subunit, clone M17-P5-F11 (Affinity Bioreagents), against GFP, clones 7.1 and 13.1, that also recognized YFP (Roche Molecular Biochemicals), against β1-integrin (BD Transduction Laboratories), and against the Na+/Ca2+ exchanger (Novus Biologicals, Inc). Also, a polyclonal antibody against polycystin-2 (Chemicon International) was used.
Cell culture
Cells were grown in DMEM medium (Cellgro Mediatech) containing 4.5 g/L glucose, 2 mM L-glutamine, 8 mg/L phenol red, 100 units/mL penicillin, 0.1 mg/mL streptomycin, and 10% FBS unless specified otherwise.
Isolation of basolateral or total plasma membrane proteins of MDCK cells using surface specific biotinylation
Cells were maintained for 6 days after becoming confluent in transwell inserts. Biotinylation of surface proteins was performed according to previously described procedures (24,25). Cell monolayers were biotinylated with EZ-Link™ Sulfo-NHS-SS-biotin (Pierce) that was added either into the well only (basolateral surface of the tight cell monolayers) or into both insert and well (total surface of the dispersed cells). After quenching the biotinylation reaction, cells were washed and then lysed by incubation with 200 L of 0.15M NaCl in 15 mM Tris pH 8.0 with 1% Triton X-100 and 4 mM EGTA. Cell extracts were clarified by centrifugation (15,000 g, 10 min) at 4°C. To isolate surface biotinylated proteins, the cell extract was incubated with 100 L of streptavidin-agarose beads (Sigma-Aldrich) in a total volume of 1 mL of 0.15M NaCl in 15 mM Tris pH 8.0 with 0.5% Triton X-100 and 4 mM EGTA at 4°C with continuous rotation for 60 min. The bead-adherent complexes were washed three times on the beads, and then proteins were eluted from the beads by incubation in 40 L of SDS-PAGE sample buffer (4% SDS, 0.05% bromophenol blue, 20% glycerol, 1% β-mercaptoethanol in 0.1 M Tris pH 6.8) for 5 min at 80°C.
Preparation of crude microsomal membranes from kidney and intestine
Rabbit kidney and small intestine were homogenized with a tight Dounce homogenizer (Wheaton, Millwille, NY). The homogenate was collected, layered onto a 42% sucrose solution and spun in a Beckman SW28 swinging bucket rotor at 25,000 rpm for 1 hr at 4°C. The fraction at the interface of buffer/sucrose was collected and diluted to a total volume of 15 ml in buffer A. This membrane-enriched fraction was collected by centrifugation in a Beckman 75Ti rotor (35,000 rpm, 4°C, 1 hr). The pellet was resuspended in 10 mM PIPES/TRIS buffer containing 2 mM EGTA and 2 mM EDTA, pH 7.0 by homogenization with a 2 ml Teflon homogenizer (Wheaton, Millwille, NY). Protein concentration was determined by the modified Lowry protein Assay Reagent (Pierce). The typical protein concentration was 5–10 g/l. The membranes were aliquoted, flash frozen and stored at −80°C.
Confocal microscopy
Confocal microscopic images were acquired using the Zeiss LSM 510 laser scanning confocal microscope and LSM 510 software, version 3.2.
Western blot analysis of the total and plasma-membrane-resident proteins of MDCK cells and microsomal membranes from kidney and small intestine
To equalize the amounts of proteins in cell extracts, the confluent cells from one well insert and the dispersed cells from three well inserts were collected in 200 μL of the lysis buffer. Samples containing 20 L of the MDCK cell extract mixed with 15 μL of SDS-PAGE sample buffer, or 1 g of microsomal membrane proteins in 20 μL of SDS-PAGE sample buffer, or biotinylated proteins eluted from the agarose-streptavidin beads were loaded onto 4–12% gradient SDS-PAGE gels (Invitrogen). Proteins were separated by SDS-PAGE using MES/SDS running buffer (0.05 M MES, 0.05 M Tris base, 0.1% SDS and 1 mM ADTA free acid, pH 7.3), transferred onto a nitrocellulose membrane and detected by Western blot analysis using the appropriate primary antibody and the anti-mouse, anti-rabbit or anti-rat IgG conjugated to alkaline phosphatase (Promega) as a secondary antibody. Alkaline phosphatase was detected using nitro blue tetrazolium and 5-bromo-4-chloro-3-indolyl-phosphate in alkaline phosphatase buffer (150 mM NaCl, 1mM MgCl2 in 10mM Tris-HCl, pH 9.0). Immunoblots were quantified by densitometry using Kodak 1D 3.6 software.
Analysis of N-glycans of the E-cadherin and the Na,K-ATPase β1 subunit using endoglycosidases
MDCK cells, either dispersed or organized into tight monolayers, were biotinylated as described above. The biotinylated proteins adherent to agarose-streptavidin beads obtained from a single well insert (tight monolayers) or three well inserts (dispersed cells) were treated with the following glycosidases: 30 mU/mL EndoH (Prozyme) or 30000 U/mL PNGase F (New England Biolabs). The reaction mixtures containing bead-adherent biotinylated proteins of MDCK cells, the appropriate buffers and detergents in the total volume of 50 μL were incubated for 3 hours at 37 °C according to the manufacturer’s instructions. Then the reaction mixtures were diluted with 30 μL of the SDS-PAGE sample buffer and incubated for 5 min at 80°C. The aliquots were loaded onto SDS-PAGE gels followed by Western blot analysis using the appropriate antibodies.
Treatment of MDCK cells by tunicamycin, swainsonine and sialidase
The MDCK cell monolayers, grown on the transwell inserts for 4 days after the cells became confluent, were incubated with 1 μg/mL tunicamycin C2 homolog (Sigma) for 48 hours, or 2 μg/μL swainsonine (Sigma) for 48 hours, or 5 U/μL sialidase (Prozyme) for 18 hours.
Paracellular permeability of cell monolayers
MDCK cell monolayers grown on transwell porous inserts were incubated in DMEM medium without phenol red and without FBS (Cellgro Mediatech) that was added into the well (lower chamber) and insert (upper chamber). The fluorescent membrane-impermeable dye, BCECF free acid (10 μM), was added into the lower chamber. Accumulation of the dye in the upper chamber was determined by taking 50 μL aliquots, diluting them in 3 mL of PBS, pH 7.2 and measuring the fluorescence intensity every 30 min during cell incubation at room temperature for 2 hours. Accumulation of BCECF in the upper chamber reflects paracellular flux of the dye through the monolayer since this dye is membrane-impermeable and, therefore, can penetrate the monolayer only between the cells. The slope of the linear regression of the fluorescence intensity plotted versus time was used as a measure of the paracellular permeability. Typically, the paracellular permeability of the tight monolayer is about 100-fold less than the paracellular permeability of sub-confluent cells or cells incubated in Ca2+-free buffer where cell junctions are fully disrupted (Suppl. Fig. 1).
RT real-time quantitative polymerase chain reaction
Total RNA from MDCK cells, either sparsely plated, or organized into the tight monolayers, was isolated using RNAqueous (Ambion, USA). Typical RNA concentrations were 100 200 ng/μL. The RNA was measured for purity and stability with a Bioanalyser 2100 (Agilent Technologies). 2000 ng of total RNA in total 40 μL volume were converted to cDNA by use of Omniscript RT Kit (Qiagen) and an oligo(dT) 12 18 primer (Invitrogen), according to the to the manufacturer’s protocol. One μL of RT product was amplified by real-time PCR using the SYBR Premix ExTaq Perfect Real-Time PCR Kit (TaKaRa, Japan), according to the manufacturer’s protocol. The primers used are shown in Suppl. Table 1. Real-time PCR was performed in eight-well strips using a DNA Engine Opticon 2 (MJ Research). The cycle of threshold (Ct) was determined for each primer set. The efficiency of amplification was determined by generating a standard curve for each primer pair using a corresponding PCR product as a template. The PCR products were purified by using the MinElute Gel Extraction Kit (Qiagen). The range of PCR product concentrations was chosen for each primer set so that the standard curve included the values of Ct observed for the RNA samples isolated from both tight monolayers and dispersed cells. The resulting values of Ct were plotted against the logarithm of the PCR product copy number (Suppl. Fig. 2). Quantification of the mRNA levels in the tight monolayers relative to that in the dispersed cells was performed as described previously (26) using the Na,K-ATPase β1 subunit mRNA as a reference. The cycle of threshold for the Na,K-ATPase β1 subunit (Ct) was 13.3 ± 0.38 and 13.2 ± 0.25 in the dispersed cells and tight monolayers, respectively. All reactions were carried out in duplicates and three separate mRNA isolations were performed.
GnT-III and GnT-IVC gene silencing using siRNA
MDCK cells were grown on porous well inserts for two days after becoming confluent in the regular DMEM with 10% FBS. Two Then cells were rinsed with the medium without FBS and antibiotics and transfected with the dicer-substrate siRNA (IDT). The sequences of siRNAs are shown in Suppl. Table 2. 100 pmole siRNA were added to 250 μL serum-free DMEM. In a separate tube, 6 μL Lipofectamine 2000 (Invitrogen) were added to 250 μL serum-free DMEM. Diluted siRNA and Lipofectamine were incubated at room temperature for 5 min, then mixed with each other and incubated for additional 20 min. The mixture was diluted with 1.5 mL serum-containing DMEM and added to the cells (1.5 mL to the lower chamber and 0.5 mL to the upper chamber). The efficiency of transfection was close to 100% as detected by using a fluorescently labeled transfection control (IDT) (Suppl. Fig. 3). The next day, the medium was changed to regular serum-containing DMEM. The transfection procedure was repeated two days later. The measurement of the paracellular permeability was performed on the day 6 after cells became confluent. A parallel sample of siRNA-treated cells was used for a Western blot analysis of the glycosylation status of the Na,K-ATPase β1 subunit. Another parallel sample was used to isolate RNA for determination of the levels of mRNA of GnT-III and GnT-IVC using the RT real-time quantitative PCR. Three independent transfection experiments for each siRNA were performed.
Statistical analysis was performed using Student’s t-test (GraphPad Prism 4 software and Microsoft Excel). Statistical significance is specified in the figure legends.
RESULTS
Branching of N-glycans is reduced with development of cell-cell junctions
MDCK cells sparsely plated on the porous well inserts (Fig. 1A, Dispersed cells) spread over the surface, replicate and migrate. Cell migration is abruptly inhibited after formation of the confluent monolayer (Fig. 1A, Day 0). However, cells of the confluent monolayer continue to divide over about a week until they reach a critical density and stop replicating. They increase in number 6-fold, decrease in horizontal dimensions 2.4-fold (Fig. 1A, Day 6) and increase in height about 6-fold (not shown). In parallel, cells of the monolayer developed competent tight junctions as detected by a gradual decrease in paracellular permeability to the membrane-impermeable dye, BCECF free acid, which reaches its minimum on day 3 and is maintained until day 6 (Fig. 1B). The maturation process is accompanied by stabilization of adherens junctions as seen by the gradual increase in the fractions of the E-cadherin and the Na,K-ATPase β1 subunit that were retained in the cells after treatment of cells with non-ionic detergent (Fig. 1C). This was associated with a gradual increase in gel mobility of the Na,K-ATPase β1 subunit (Fig. 1C). In agreement with previously published results (20), gel mobility of E-cadherin was also increased in the tight cell monolayers as compared to the dispersed cells (Fig. 1C).
Fig. 1. Progression of the dispersed MDCK cells into the tight cell monolayer is associated with a gradual decrease in the paracellular permeability, increase in resistance of E-cadherin and the Na,K-ATPase to the non-ionic detergent, and increase in electrophoretic gel mobility of both proteins.
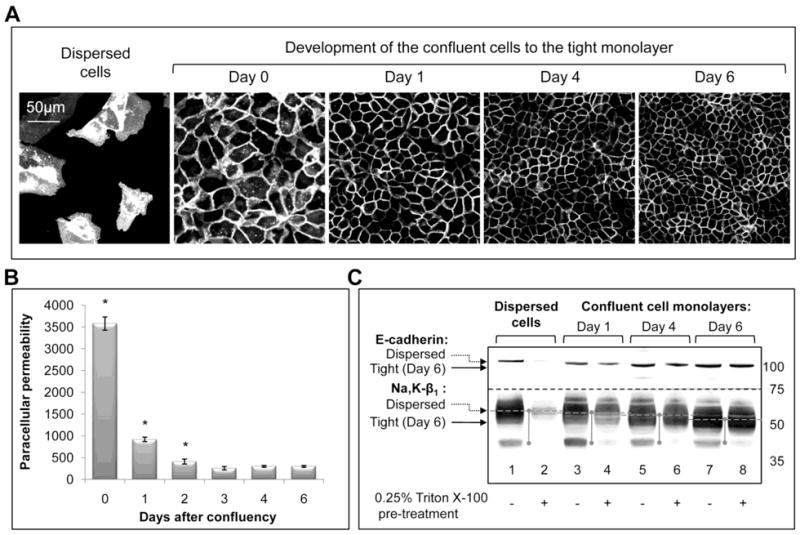
MDCK cells expressing YFP-β1 were sparsely plated on the porous well inserts, grown until confluent and then were kept on the inserts for 6 days.
A, Confocal microscopy images of the cells showing development of the tight cell monolayer from dispersed cells.
B, Paracellular permeability of the MDCK cell monolayer for the membrane impermeable dye, BCECF, free acid, gradually decreases during maturation of the confluent cells for 3 days and then is maintained at the same level until day 6.
C, As detected by a Western blot analysis, the gel mobility of E-cadherin and the Na,K-ATPase β1 subunit gradually increases during the development of the tight MDCK cell monolayer from the dispersed cells (lanes 1, 3, 5 and 7) as does resistance to detergent extraction (lanes 2, 4, 6 and 8). Where indicated, the cells were treated with 0.25% Triton X-100 and washed with PBS before cell lysis.
Error bars, ± s.d. (n=4). * - significant difference with “Day 6”, P<0.01, Student’s t-test.
Also, gel mobility of E-cadherin and the Na,K-ATPase β1 subunit was greater in rabbit kidney that contains relatively tight epithelia as compared to the leaky epithelium of small intestine (Fig. 2, lanes 1 and 2). These results suggest that there is a correlation between the tightness of epithelia in both cultured cells and animal tissues and gel mobility of E-cadherin and the Na,K-ATPase β1 subunit. A similar correlation was detected for several other cellular glycoproteins, β1-integrin (Fig. 2, lanes 3 and 4), Na+/Ca2+ exchanger and polycystin-2 (not shown), suggesting that the N-glycosylation status of cellular glycoproteins is different in tight and leaky epithelia. Consistent with this interpretation, deglycosylation of the Na,K-ATPase β1 subunit from the dispersed cells and tight monolayers using PNGase F resulted in the products having the same gel mobility (Fig. 3, lanes 1 and 6, bottom pane, dashed arrow). Similarly, deglycosylated E-cadherin from the dispersed cells and tight monolayers migrated to the same position on SDS-PAGE (Fig. 3, lanes 1 and 6, top panel, dashed arrow). Also, deglycosylated renal and intestinal E-cadherin and Na,K-ATPase β1 subunit had the same gel mobility (not shown).
Fig. 2. Electrophoretic gel mobility of E-cadherin, β1-integrin and the Na,K-ATPase β1 subunit isolated from kidney is greater than that of the proteins isolated from small intestine.

Microsomal membrane proteins isolated from kidney and small intestine were analyzed by SDS-PAGE followed by Western blot analysis. The intestinal Na,K-ATPase β1 subunit, E-cadherin and β1-integrin migrate more slowly (lanes 1 and 3, dotted arrows) than the corresponding proteins of the kidney (lanes 2, and 4, solid arrows).
Fig. 3. The complex-type N-glycans of E-cadherin and the Na,K-ATPase β1 subunit have fewer branches or shorter branches in the tight MDCK cell monolayers as compared to the dispersed MDCK cells.
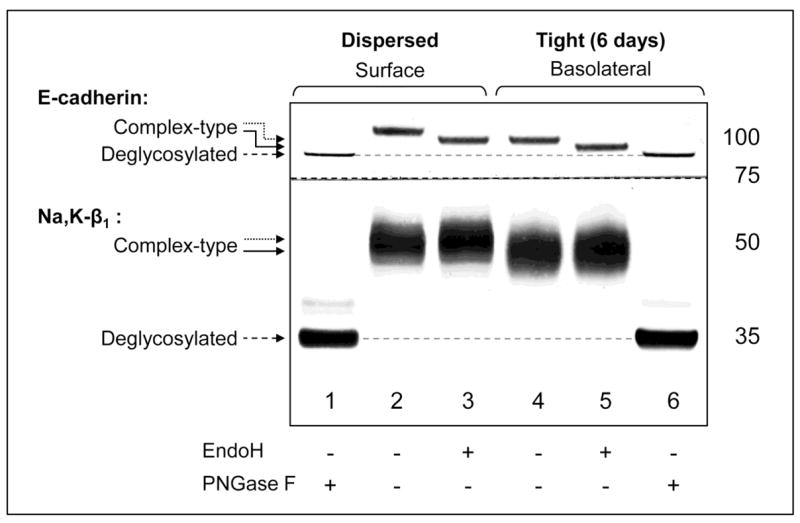
Plasma membrane proteins isolated from the dispersed cells or tight cell monolayers were treated with glycosidases, EndoH or PNGase F, followed by Western blot analysis. In the dispersed cells, surface E-cadherin is susceptible to EndoH (compare lanes 2 and 3, top panel), indicating that E-cadherin contains at least one hybrid or high-mannose N-glycan. The product of EndoH cleavage migrates more slowly (lane 3, dotted arrow) than the PNGase F product (lane 1, dashed arrow), indicating that E-cadherin also contains at least one complex N-glycan. The plasma membrane fraction of the Na,K-ATPase β1 subunit of the dispersed cells is resistant to EndoH (compare lanes 2 and 3, bottom panel), indicating that the glycoprotein contains only complex N-glycans. In the membrane fractions isolated from the tight monolayers (lanes 4–6), susceptibility to glycosidases of E-cadherin and the Na,K-ATPase β1 subunit is the same as in dispersed cells (lanes 1–3), showing that the type of N-glycosylation of both E-cadherin and the Na,K-ATPase β1 subunit does not change during formation of the tight monolayer from the dispersed cells. The EndoH products of both E-cadherin and Na,K-ATPase β1 subunit of dispersed cells migrate more slowly (lane 3, dotted arrows) than the corresponding proteins of the tight monolayers (lane 5, solid arrows) indicating that the composition of complex-type N-glycans becomes less complex during formation of the tight monolayer from the dispersed cells.
It is known that virtually all types of N-linked glycans are removed from glycoproteins by PNGase F, while glycoproteins containing complex N-glycans are resistant to EndoH cleavage. The analytical cleavage of surface proteins of MDCK cells by these glycosidases showed that the Na,K-ATPase β1 subunit contains only complex N-glycans (Fig. 3) in agreement with previously published results (27), while E-cadherin contains at least one hybrid or high-mannose N-glycan and at least one complex-type N-glycan (Fig. 3) consistent with recently published data on N-glycan composition of the human E-cadherin expressed in CHO cells (20). Importantly, susceptibility of E-cadherin and the Na,K-ATPase β1 subunit to glycosidases is the same in the samples isolated from the tight monolayers (Fig. 3, lanes 4–6) and from the dispersed cells (Fig. 3, lanes 1–3), indicating the type of glycosylation does not change upon the monolayer formation in contrast to the model suggested previously (20). Since the products of EndoH cleavage contain only complex-type N-glycans, the difference in gel mobility of the EndoH-treated glycoproteins in the dispersed cells and tight monolayers (Fig. 3, lanes 3 and 5) reflects the difference in the structure and/or monosaccharide composition of the complex N-glycans attached to each protein. Either the number of branches or the length of branches decreases in individual complex-type N-glycans during the development of the tight cell monolayer from the dispersed cells.
Various glycosyltransferases generate highly diverse N-glycans. Four of them, Golgi N-acetylglucosamine-glycosyltransferases (GnT) III to VI are known to be responsible for variations in the number of branches in N-glycans (Fig. 4A). GnTs IV, V and VI promote branching of N-glycans, while GnT-III stops branching. The latter enzyme can add the bisecting GlcNAc to hybrid or complex N-glycans and prevent the downstream action of the branching enzymes. For example, the action of this enzyme on the product of the glycosyltransferase II results in the formation of bi-antennary complex N-glycans (Fig. 4A, bottom).
Fig. 4. The decreased complexity of N-glycans in tight MDCK cell monolayers correlates with up-regulation of the genes encoding the Golgi enzymes that stop branching of N-glycans and down-regulation of the genes encoding enzymes that facilitate branching of N-glycans.
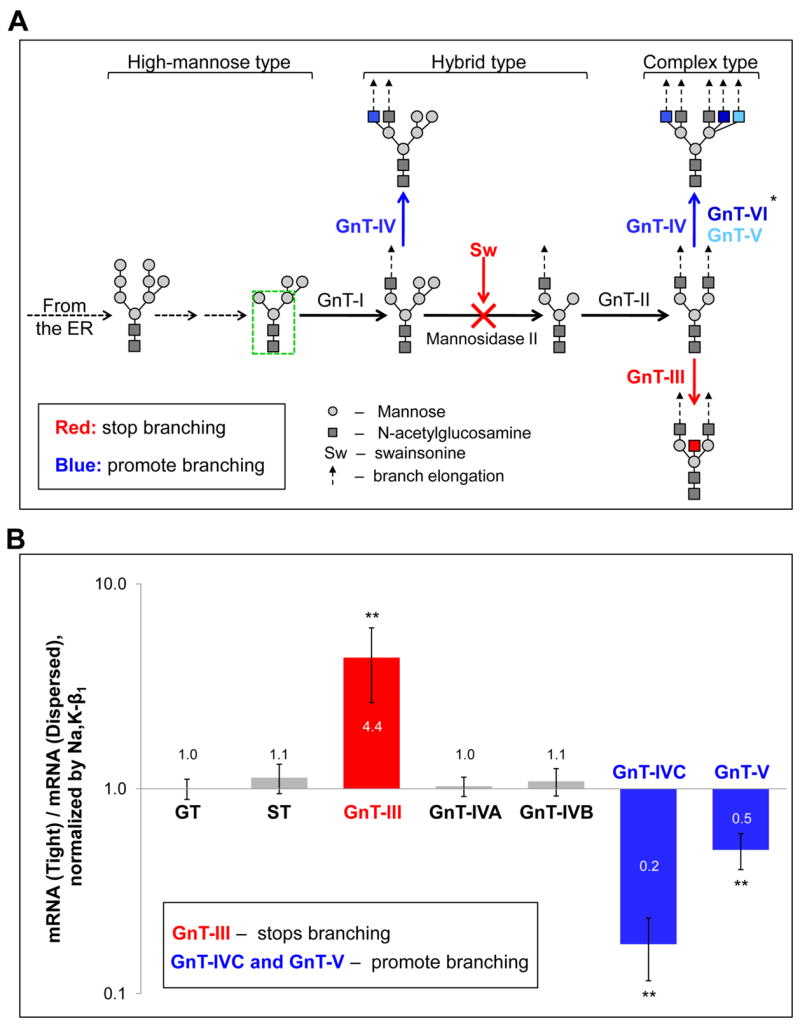
A, A simplified scheme showing the role of N-acetylglucosamine-glycosyltransferases (GnT) in N-glycan branching. Six different GnTs (I – VI) can add N-acetylglucosamine (GlcNAc) residues to the three-mannosyl core of N-glycans (green rectangle) and start the formation of diverse structures. Addition of GlcNAc residues, except for those added by GnT-III, allows elongation of the chains, referred as antennae, with additional monosaccharide linkages due to the action of other glycosyltransferases (dashed arrows). GnT-I is a key regulatory enzyme that initiates formation of hybrid and complex type N-glycans, while GnT-II initiates formation of the complex-type N-glycans. GnTs IV, V and VI promote branching of N-glycans by adding GlcNAc into the positions shown by different shades of blue. The individual N-glycans can be modified by one, two or all three of these enzymes resulting in two- to five-antennary N-glycans. In contrast, GnT-III (red) stops branching by adding a bisecting GlcNAc to any hybrid or complex N-glycan structures shown on the scheme that prevent downstream action of GnTs II, IV, V and VI. A similar stop-branching effect can be achieved by incubation of cells with the Golgi mannosidase II inhibitor, swainsonine (Sw), that prevents mannose trimming and consequent action of GnT-II, resulting in preservation of the hybrid type structure of N-glycans.
B, Formation of the tight monolayer from dispersed cells is accompanied by an increase in the levels of expression of the genes encoding GnT-III and a decrease in the levels of expression of GnT-IVC and GnT-V as determined by RT real-time PCR of RNA samples isolated from dispersed cells and from tight cell monolayers grown for 6 days after confluency. Quantification of the results was performed as described in Experimental procedures and demonstrated in Supplemental Fig. 2.
Abbreviations: GT - UDP-Gal:betaGlcNAc beta-1,4-galactosyltransferase 1, membrane-bound form; ST – beta-galactoside-alpha-2,3-sialyltransferase, GnT – N-acetylglucosamine-glycosyltransferase. *-The expression of GnT-VI has not been tested because the sequence of this enzyme in dogs is unknown. Error bars, ± s.d. (n=3). ** P<0.001, Student’s t-test.
All chains where the original mannose residues are substituted by GlcNAc residues, except for those formed by GnT-III, can be elongated due to the action of other various glycosyltransferases (Fig. 4A dashed arrows) that compete with each other for the N-glycan substrate. As a result, individual branches vary in the length and carbohydrate composition. The most common extension of individual branches occurs due to the multiple linkages of sialic acid residues by sialyltransferases. Individual branches can be also significantly elongated as a result of polylactosamine extensions. The key enzyme that contributes to polylactosamine synthesis is beta-1,4-galactosyltransferase.
To test which of these glycosyltransferases contribute to the changes in N-glycosylation observed upon intercellular adhesion, we compared the levels of mRNA encoding the enzymes in the tight monolayers to those in dispersed cells using real-time PCR. The level of the Na,K-ATPase β1 subunit mRNA was the same in tight monolayers and dispersed cells, providing an internal control. Similarly, the levels of 1,4-galactosyltransferase and sialyltransferase, the enzymes responsible for elongation of N-glycan branches, did not change upon cell-cell adhesion (Fig. 4B).
In contrast, the levels of mRNA encoding the enzymes that promote branching, GnT-IVC and GnT-V, were reduced by (83 ± 6)% and (50 ± 10)%, respectively, while the level of the mRNA encoding an enzyme that stops branching, GnT-III, was increased (4.4 ± 1.7)-fold at the day 6 of the tight monolayer development as compared to the dispersed cells (Fig. 4B). The increase in the mRNA level of GnT-III and decrease in GnT-IVC developed gradually, starting from the day when cells became confluent, while a significant decrease in GnT-V was observed only at the day 6 (Suppl Fig. 4). These changes in mRNA and corresponding changes in the amount of the three enzymes, GnT-III, GnT-IVC and GnT-V, would result in an increase in gel mobility of glycoproteins upon formation of the tight cell monolayer from dispersed cells due to a reduction in the number of branches in N-glycans. Therefore, the data on the differential expression levels of glycosyltransferases in the dispersed cells and the tight cell monolayers are consistent with the observed differences in N-glycosylation of the E-cadherin and the Na,K-ATPase β1 subunit in the dispersed cells and the tight cell monolayers (Fig. 3, lanes 2 and 4).
Reduced branching of N-glycans promotes cell-cell adhesion in epithelia
The following approaches to modify N-glycosylation of cellular proteins were employed. To prevent N-glycosylation of newly synthesized proteins, cells were treated with tunicamycin, an inhibitor of N-glycosylation which prevents the synthesis of the glycosylated lipid precursor of N-glycans. As expected, the glycosylated Na,K-ATPase β1 subunit was replaced by non-glycosylated subunit in the inhibitor-treated cells as seen from the gel mobility shift (Fig. 5A, lane 1). To reduce branching of N-glycans, cells were treated with an inhibitor of N-glycan processing, swainsonine. Swainsonine (Sw) blocks mannose trimming from N-glycans and hence the consequent action of GnT-II, preventing further branching of N-glycans (Fig. 4A). As a result, swainsonine preserves the hybrid type structure of newly synthesized glycoproteins, which is evident from the increase in gel mobility of the Na,K-ATPase β1 subunit in the inhibitor-treated cells (Fig. 5A, lane 3).
Fig. 5. Prevention of branching of N-glycans by swainsonine increases tightness of the MDCK cell monolayer, while silencing of the gene encoding a stop-branching enzyme, GnT-III, loosens cell-cell contacts.
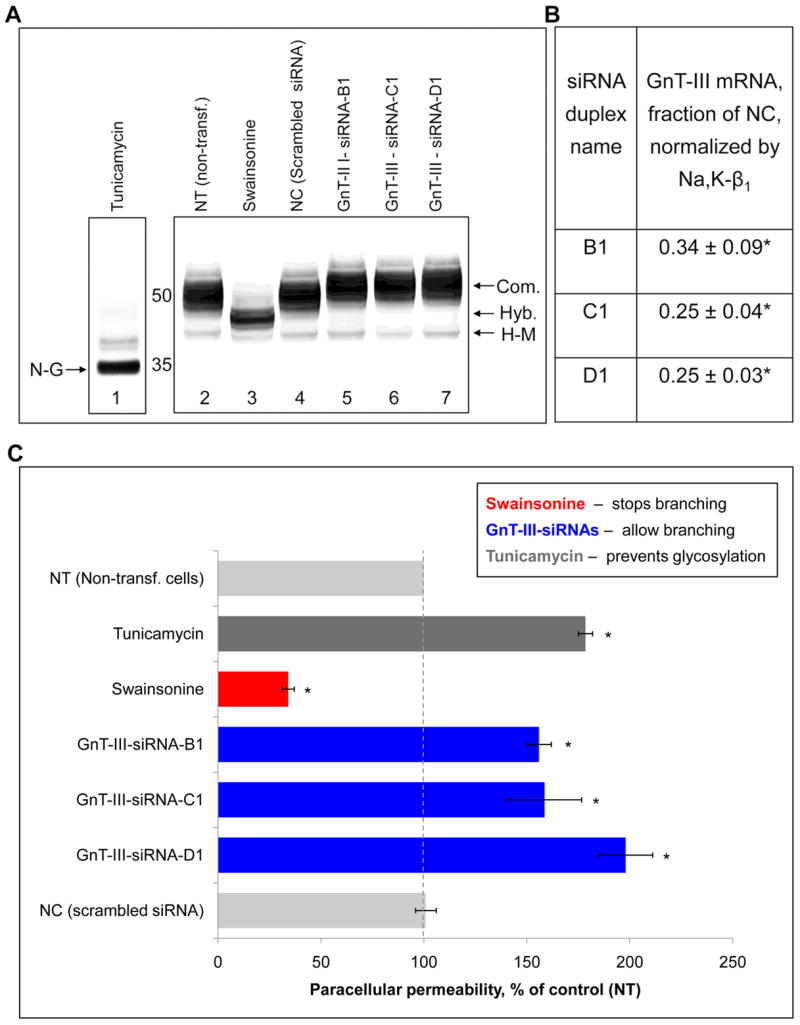
Tight monolayers of MDCK cells were grown on porous transwell inserts without inhibitors for 4 days after the cells became confluent and then for 2 days in the absence or presence of 2 μg/mL swainsonine or 1 μg/mL tunicamycin. As indicated, other cells were transfected with either scrambled siRNA (negative control) or with siRNA against GnT-III. Transfection was performed as described in Experimental procedures.
A, Electrophoretic mobility of the Na,K-ATPase β1 subunit was increased in the cells treated with tunicamycin and swainsonine and decreased in the cells transfected with siRNAs against GnT-III as detected by Western blot analysis.
B, The decrease in the mRNA level of GnT-III by siRNAs as detected by RT real-time PCR.
C, Treatment of cells with tunicamycin significantly increased, while swainsonine decreased the paracellular permeability of the cell monolayers. Transfection of cells with three different siRNAs against GnT-III significantly increased the paracellular permeability.
Abbreviations: Com. - complex, Hyb. - hybrid, H-M –high-mannose, and D-G – deglycosylated. Error bars and errors, ± s.d. (n=3). * P<0.01, Student’s t-test.
We also tried to reduce the level of N-glycan branching using siRNA specific to GnT-IVC, the enzyme that promotes N-glycan branching. However, none of the six tested siRNAs affected the level of expression of the enzyme and gel mobility of the Na,K-ATPase β1 subunit (Suppl. Table 2). As expected, none of the six GnT-IVC-specific siRNAs affected paracellular permeability of the MDCK cell monolayer (Suppl. Table 2).
To increase the degree of N-glycan branching, we used siRNA specific to GnT-III to inhibit the expression of this stop-branching enzyme. Three of the six tested GnT-III-specific siRNAs significantly decreased GnT-III mRNA concentration by (66 ± 9)%, (75 ± 4)% and (75 ± 3)%, respectively (Fig. 5B and Suppl. Table 2). The decrease in gel mobility of the Na,K-ATPase β1 subunit (Fig. 5A, lanes 5, 6 and 7) suggests that branching of N-glycans was increased as a result of silencing of GnT-III.
Exposure of cells to tunicamycin increased the paracellular permeability of the cell monolayers by (79 ± 3)%, indicating that N-glycans are important for integrity of the tight junctions. In contrast, the paracellular permeability was decreased by (66 ± 3)% as a result of pre-treatment of MDCK cells with swainsonine (Fig. 5C). Conversely, three GnT-III-specific siRNAs increased the paracellular permeability by (56 ± 6), (58 ± 18) and (98 ± 13)% (Fig. 5C). Therefore, reducing N-glycan branching tightens intercellular junctions, while increasing N-glycan branching loosens cell-cell contacts.
Both E-cadherin and the Na,K-ATPase β1 subunit were resistant to treatment of the tight MDCK cell monolayers by 0.25% Triton X-100 (Fig. 6, lane 2) but were partially extracted by 0.5% detergent (Fig. 6, lane 3) and largely removed by 0.75% Triton X-100 (Fig. 6, lane 4). In the swainsonine-treated cells, the Na,K-ATPase β1 subunit was more resistant to extraction by increasing detergent concentrations, while E-cadherin was completely resistant to detergent extraction at concentrations up to 0.75% (Fig. 6, lanes 5-8). The relative resistance of cellular proteins to detergent extraction is probably due to their association with the cytoskeleton. Therefore, the increased detergent resistance of the E-cadherin and the Na,K-ATPase β1 subunit in the swainsonine-treated cells suggests that reduced branching of N-glycans increases stability of association of the adherens junctional complex with the cytoskeleton.
Fig. 6. Swainsonine increases detergent resistance of the E-cadherin and the Na,K-ATPase β1 subunit in the tight monolayers of MDCK cells.

A, Tight monolayers of MDCK cells were grown in the absence (lanes 1–4) or presence of swainsonine (lanes 5–8) as described in the legend to Fig. 5. Cell monolayers were biotinylated from the basolateral side as described in Experimental procedures. As indicated, right before lysis, cells were incubated either with PBS (lanes 1 and 5), or with 0.25% (lanes 2 and 6), or with 0.5% (lanes 3 and 7) or with 0.75% Triton X-100 (lanes 4 and 8) in PBS for 15 min and rinsed with PBS. The gel mobility and detergent resistance of both E-cadherin and the Na,K-ATPase β1 subunit increases in the presence of swainsonine as detected by a Western blot analysis.
B–C, Quantification of the results showing that swainsonine significantly increases the resistance of the Na,K-ATPase β1 subunit (B) and E-cadherin (C) to Triton X-100 extraction.
Error bars, ± s.d. (n=3). Significant difference with untreated cells: * P<0.05, ** - P<0.001, Student’s t-test.
To test whether the effect of swainsonine on cell-cell adhesion of MDCK cells is due to the reduced content of the sialic acid residues in the hybrid type N-glycans as compared to the complex-type N-glycans, MDCK cells grown and polarized on porous well inserts were incubated in the presence or absence of sialidase overnight. Sialidase treatment increased gel mobility of the Na,K-ATPase β1 subunit and E-cadherin indicating that some sialic acid residues were cleaved from the surface N-glycans, but this did not affect the detergent resistance of the Na,K-ATPase β1 subunit and E-cadherin and the paracellular permeability of the MDCK cell monolayer (data not shown). Therefore, the effect of swainsonine on cell-cell adhesion of MDCK cells does not appear to be a result of the reduced content of sialic acid residues in glycoproteins of the inhibitor-treated cells.
To test whether swainsonine affects cell motility, tight monolayers of the MDCK cells were damaged to provoke cell migration into the wound area (Fig. 7). Cell migration was inhibited by 46% in swainsonine-treated cells as quantified by measuring the area of the wound covered by migrated cells during “wound healing” process.
Fig. 7. Swainsonine inhibits migration of MDCK cells as detected by the “wound healing” assay.

Tight monolayers of the MDCK cells expressing YFP-β1 were grown in the absence (top panels) or presence of swainsonine (bottom panels) as described in the legend to Fig. 5. The cell monolayers were damaged using a scalpel (left panels). The “wound healing” process was assessed by examination of serial confocal microscopic images of the same field every 2 hrs while incubated in regular medium. The “wound healing” process was inhibited by swainsonine as quantified by measuring the area of the wound covered by migrated cells after 8 hrs of epithelization of the defect.
Errors, ± s.d. (n=5). * P=0.00039, Student’s t-test.
The degree of branching of N-glycans of the Na,K-ATPase β1 subunit modulates the efficacy of cell-cell adhesion
Remodeling of N-glycan branching affects stability and integrity of the cell-cell junctions, suggesting that the structure of N-glycans linked to particular cellular glycoproteins is important for intercellular adhesion. It is possible that one of these glycoproteins is the Na,K-ATPase β1 subunit, since N-glycans of this subunit were found important for cell-cell contact formation (11). To test if the structure of N-glycans of this subunit is important for cell-cell adhesion, we used the cell line, in which 55% of the endogenous Na,K-ATPase β1 subunit is replaced by the unglycosylated mutant of this subunit, N123, while the abundance of the of the Na,K-ATPase α-subunits on the lateral membrane is not changed (11).
The unglycosylated mutant of the Na,K-ATPase β1 subunit was less resistant to extraction by Triton X-100 from the basolateral membrane than the normally glycosylated β1 subunit (Fig. 8A, lanes 1–4 and Fig. 6A, lanes 1–4) consistent with previously published results (11). Remarkably, E-cadherin was also more easily removed from the basolateral plasma membrane by Triton X-100 in the cell line expressing the N123 mutant compared to non-transfected cells (Fig. 8A, lanes 1–4 and Fig. 6A, lanes 1–4). Thus, E-cadherin was fully resistant to 0.25% Triton X-100 in non-transfected cells, while 20% of the protein was removed by detergent in the mutant-expressing cells. Similarly, the detergent resistance of E-cadherin was decreased in tunicamycin-treated cells (not shown). These results suggest that the N-glycans of the Na,K-ATPase β1 subunit are important for stabilization of adherens junctions by cytoskeletal elements. Consistent with this interpretation, both over-expression of unglycosylated mutant of the N,K-ATPase β1 subunit and exposure of cells to tunicamycin significantly distorted intercellular junctions in the middle section of the cells and slightly distorted them in the sup-apical region (Fig. 9).
Fig. 8. Prevention of glycosylation of the Na,K-ATPase β1 subunit decreases detergent resistance of E-cadherin and abolishes a swainsonine-induced increase in detergent resistance of E-cadherin.

A, Tight monolayers of MDCK cells expressing unglycosylated mutant of the Na,K-ATPase β1 subunit were grown in the absence (lanes 1–4) or presence of swainsonine (lanes 5–8) and treated with Triton X-100 as described in the legend to Fig. 6. As expected, swainsonine did not change gel mobility of N123, but slightly increased gel mobility of E-cadherin. Swainsonine did not affect detergent stability of either E-cadherin or the mutated Na,K-ATPase β1 subunit.
B–C, Quantification of the results shows that both unglycosylated mutant of the Na,K-ATPase β1 subunit, N123 (B), and E-cadherin (C) resident in the basolateral plasma membrane are less resistant to extraction by Triton X-100 in the cell line expressing N123 mutant as compared to non-transfected MDCK cells (Fig. 6). Swainsonine did not change detergent resistance of the unglycosylated Na,K-ATPase β1 subunit (B) and E-cadherin (C) in the mutant-expressing cell line, in contrast to the protective effect of the inhibitor in non-transfected cells (Fig. 6).
Error bars, ± s.d. (n=3). Significant difference with non-transfected MDCK cells: * P<0.05, Student’s t-test. ** - P<0.001, Student’s t-test.
Fig. 9. Prevention of glycosylation of the Na,K-ATPase β1 subunit and exposure of cell to tunicamycin results in the distortion of cell-cell junctions.

Confocal microscopy images of MDCK cells expressing YFP-β1, untreated (a, d and g) or treated with tunicamycin (c, f and i), and confocal microscopy images of MDCK cells expressing the unglycosylated mutant of the Na,K-ATPase β1 subunit, N123 (b, e and h). Lateral contacts in the middle section show a beaded appearance in the control and the presence of many dissociated regions both in the mutant-expressing cells and tunicamycin-treated cultures. Images were taken on day 6 after cell confluency.
As expected, detergent resistance of the unglycosylated form of the Na,K-ATPase β1 subunit was not affected by swainsonine (Fig. 8). Susceptibility of E-cadherin to detergent also was not significantly changed in the cells exposed to swainsonine, suggesting that the presence and degree of branching of N-glycans of the Na,K-ATPase β1 subunit are important for stability of E-cadherin to detergent extraction.
The paracellular permeability of the monolayer formed by the cells expressing the unglycosylated mutant of the Na,K-ATPase β1 subunit was significantly increased compared to that in non-transfected cells (Fig. 10), suggesting that N-glycans of this subunit are also important for the integrity of the tight junctions. Swainsonine decreased the paracellular permeability in both non-transfected and the mutant-expressing cell lines (Fig. 10A). However, the swainsonine-induced decrease in the permeability was significantly less in the transfected cell line than in non-transfected cells (Fig. 10A, table). Therefore, the tightening effect of swainsonine in non-transfected cells, at least in part, is related to reduction of branching of N-glycans linked to the Na,K-ATPase β1 subunit.
Fig. 10. Prevention of glycosylation of the Na,K-ATPase β1 subunit increases the paracellular permeability of the MDCK cell monolayer and decreases the tightening effect of swainsonine.
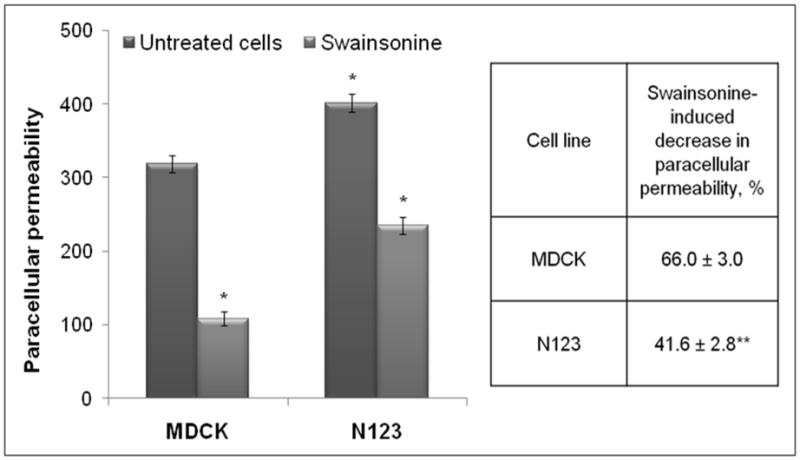
Tight monolayers of non-transfected MDCK cells and cells transfected with the unglycosylated mutant of the Na,K-ATPase β1 subunit were grown in the absence or presence of swainsonine as described in the legend to Fig. 5. The paracellular permeability of the monolayer formed by the mutant-transfected cells (N123) was increased by 26% compared to non-transfected cells (MDCK). Swainsonine treatment of non-transfected MDCK cells decreased paracellular permeability in both cell lines. However the inhibitor effect was significantly less prominent in the mutant-expressing cells (table).
Error bars and errors, ± s.d. (n=4). * - significant difference with non-transfected MDCK cells. P<0.001, Student’s t-test. ** - Significant difference with swainsonine-induced decrease in the paracellular permeability in non-transfected cells. P<0.001, Student’s t-test.
The lack of N-glycans on the β1 subunit does not affect the activity of the Na,K-ATPase (28). Detergent resistance, the paracellular permeability and sensitivity to swainsonine of the MDCK cell line expressing the wild type YFP-β1-was the same as in the non-transfected cells (not shown). Therefore, the decreased sensitivity to swainsonine in the mutant cells is related to the lack of N-glycans in the β1 subunit and is not due to the non-specific effects of transfection or impaired Na,K-ATPase activity.
DISCUSSION
The results of the present study show that the structure of N-glycans linked to adhesion proteins affect the process of intercellular adhesion and elaboration of a tight epithelium. Exposure of MDCK cell monolayers to the inhibitor of N-glycosylation, tunicamycin, significantly increases the paracellular permeability of the monolayers, indicating that N-glycans are important for integrity of the mature epithelium. In contrast, paracellular permeability is significantly reduced when normal branching of N-glycans is prevented by exposure of the cell monolayer to swainsonine, suggesting that less branched N-glycans are preferable for tighter intercellular contacts.
The effects of tunicamycin and swainsonine on paracellular permeability indicate that the lack or modification of N-glycans affect the tight junctions. In parallel, both inhibitors affect the adherens junctions. Tunicamycin distorts the structure of intercellular contacts below the tight junction region and decreases detergent resistance of adherens junction proteins, while swainsonine increases stability of adherens junction proteins to detergent extraction. The constituents of the tight junctions are not glycosylated. In contrast, cell adhesion molecules of the adherens junctions, E-cadherin and nectin, are glycoproteins. Also, the Na,K-ATPase β1 subunit that is important for stability of adherens junctions is glycosylated. Therefore, the lack or modification of N-glycans probably affects the tight junctions through changes in the adherens junctions.
This interpretation is consistent with numerous reports on cross-talk between the adherens and tight junctions. The assembly of the tight junctions is always preceded by the formation of the adherens junctions and can be inhibited by an E-cadherin-blocking antibody (30–32). Conversely, disruption or weakening of the adherens junctions by depletion of E-cadherin and catenins or by chelating Ca2+ in the culture medium causes partial or complete disassembly of the tight junctions (33,34). Therefore, the integrity of the tight junctions depends on the intactness and stability of the adherens junctions. The integrity of the tight junctions is maintained and regulated by a variety of signaling pathways that are initiated by the cytoplasmic components of the adherens junctions such as β-catenin and afadin (7,29).
Although swainsonine should modify all cellular glycoproteins, the increase in both tightness of the monolayer and stability of E-cadherin to detergent extraction observed after exposure to swainsonine is attenuated in the cell line expressing the unglycosylated Na,K-ATPase β1 subunit, observations confirming the tightening and stabilizing effects of the inhibitor in non-transfected cells are related, at least in part, to the action of swainsonine on the N-glycan branching of the Na,K-ATPase β1 subunit.
During the progression of MDCK cells from individually attached dispersed cells to a mature cell monolayer, the complexity of N-glycans linked to E-cadherin and Na-K-ATPase is reduced. This reduction appears to reflect the decrease in N-glycan branching due to changes in expression of the enzymes that determine the degree of N-glycan branching, GnT-III, GnT-IVC and GnT-V. These changes occur in parallel with a gradual decrease in the paracellular permeability of the epithelium. Furthermore, inhibiting the expression of a stop-branching enzyme, GnT-III, by RNA interference causes an increase in paracellular permeability of the mature cell monolayer, indicating that reduced branching of N-glycans is an important element of the complex process of formation and maintenance of the tight junctions.
These findings suggest that modification of N-glycans could serve as a mechanism whereby inherent paracellular permeability of epithelia of different tissues is modified. In support of this possibility, the N-glycans of the Na,K-ATPase β1 subunit, E-cadherin and other glycoproteins isolated from the renal collecting duct, epithelia known to have a high electrical resistance, are less complex than those of the glycoproteins obtained from the relatively leaky intestinal epithelium.
Reducing N-glycan branching by treatment with swainsonine suppresses motility of MDCK cells as detected by the “wound healing” assay. This observation is consistent with the suppression of fibroblastic cell motility and invasiveness of malignant cells induced by measures that reduce N-glycan branching (35–38).
Formation of a tight cell monolayer from dispersed MDCK cells is accompanied by changes in cell motility. Dispersed MDCK cells that are more motile compared to the cells comprising the tight monolayer manifest glycoproteins with a relatively high degree of N-glycan branching. This is consistent with the data on correlation between high motility of fibroblastic and malignant cells and increased branching of N-glycans (39–43). Formation of cell-cell contacts between dispersed cells induces changes in the expression levels of GnTs III, IVC and V and results in a decrease in N-glycan branching. Reduced branching would inhibit cell migration. In addition, reducing N-glycan branching facilitates intercellular adhesion by tightening and stabilizing cell-cell junctions, which, in turn, causes additional inhibition of cell motility.
Therefore, reducing N-glycan branching by modulating activities of glycosyltransferases may contribute to the contact inhibition of locomotion, an important property of normal epithelial cells. Cancer cells may lack the ability to regulate expression of glycosyltransferases upon cell-cell contact and fail to reduce branching of N-glycans when they make contact. As a consequence, tumor cells would stay motile even when they reach confluency and would not form stable intercellular junctions.
The findings that exposure of MDCK cells in culture to swainsonine tightens cell monolayers could have important implications for the treatment of various disorders characterized by disruption of cell adhesion. The healing of the disrupted skin epithelia after injury or tubular integrity with acute kidney injury might be facilitated by treatment with swainsonine. This possibility remains to be examined.
Thus, the correlation between N-glycan branching, cell-cell adhesion and cell motility in epithelia may explain the biological requirement for heterogeneity of complex N-glycans in vertebrates. Highly branched N-glycans are needed to promote cell motility, while less branched N-glycans facilitate intercellular adhesion. The proposed mechanism of modulation of intercellular adhesion via remodeling of N-glycans, in particular those of the Na,K-ATPase β1 subunit, suggests a regulatory role of specific glycosyltransferases. The precise signaling pathways involved in relationship between cell motility, cell-cell adhesion and expression of glycosyltransferases remain to be elucidated.
Supplementary Material
Footnotes
Supported in part by NIH grants DK077149, DK46917, DK58333, D53642, and USVA
We thank Dr. Jeff Kraut for careful reading of the manuscript and helpful suggestions.
The abbreviations used are: YFP-β1 - the fusion protein between the yellow fluorescent protein and the Na,K-ATPase β1 subunit; GnT - N-acetylglucosamine-glycosyltransferase.
References
- 1.Farquhar MG, Palade GE. J Cell Biol. 1963;17:375–412. doi: 10.1083/jcb.17.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shin K, Fogg VC, Margolis B. Annu Rev Cell Dev Biol. 2006;22:207–235. doi: 10.1146/annurev.cellbio.22.010305.104219. [DOI] [PubMed] [Google Scholar]
- 3.Turner JR. Am J Pathol. 2006;169:1901–1909. doi: 10.2353/ajpath.2006.060681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kapus A, Szaszi K. Biochem Cell Biol. 2006;84:870–880. doi: 10.1139/o06-202. [DOI] [PubMed] [Google Scholar]
- 5.Diamond JM. Physiologist. 1977;20:10–18. [PubMed] [Google Scholar]
- 6.Van Itallie CM, Anderson JM. Annu Rev Physiol. 2006;68:403–429. doi: 10.1146/annurev.physiol.68.040104.131404. [DOI] [PubMed] [Google Scholar]
- 7.Miyoshi J, Takai Y. Adv Drug Deliv Rev. 2005;57:815–855. doi: 10.1016/j.addr.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 8.Rajasekaran SA, Palmer LG, Quan K, Harper JF, Ball WJ, Jr, Bander NH, Peralta Soler A, Rajasekaran AK. Mol Biol Cell. 2001;12:279–295. doi: 10.1091/mbc.12.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Genova JL, Fehon RG. J Cell Biol. 2003;161:979–989. doi: 10.1083/jcb.200212054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paul SM, Ternet M, Salvaterra PM, Beitel GJ. Development. 2003;130:4963–4974. doi: 10.1242/dev.00691. [DOI] [PubMed] [Google Scholar]
- 11.Vagin O, Tokhtaeva E, Sachs G. J Biol Chem. 2006;281:39573–39587. doi: 10.1074/jbc.M606507200. [DOI] [PubMed] [Google Scholar]
- 12.Gloor S, Antonicek H, Sweadner KJ, Pagliusi S, Frank R, Moos M, Schachner M. J Cell Biol. 1990;110:165–174. doi: 10.1083/jcb.110.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blanco G, Mercer RW. Am J Physiol. 1998;275:F633–650. doi: 10.1152/ajprenal.1998.275.5.F633. [DOI] [PubMed] [Google Scholar]
- 14.Crambert G, Hasler U, Beggah AT, Yu C, Modyanov NN, Horisberger JD, Lelievre L, Geering K. J Biol Chem. 2000;275:1976–1986. doi: 10.1074/jbc.275.3.1976. [DOI] [PubMed] [Google Scholar]
- 15.Sweadner KJ, Rael E. Genomics. 2000;68:41–56. doi: 10.1006/geno.2000.6274. [DOI] [PubMed] [Google Scholar]
- 16.Geering K. Am J Physiol Renal Physiol. 2006;290:F241–250. doi: 10.1152/ajprenal.00126.2005. [DOI] [PubMed] [Google Scholar]
- 17.Pierre SV, Xie Z. Cell Biochem Biophys. 2006;46:303–316. doi: 10.1385/cbb:46:3:303. [DOI] [PubMed] [Google Scholar]
- 18.Paul SM, Palladino MJ, Beitel GJ. Development. 2007;134:147–155. doi: 10.1242/dev.02710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barwe SP, Rajasekaran SA, Rajasekaran AK. Cell Mol Biol (Noisy-le-grand) 2006;52:41–47. [PubMed] [Google Scholar]
- 20.Liwosz A, Lei T, Kukuruzinska MA. J Biol Chem. 2006;281:23138–23149. doi: 10.1074/jbc.M512621200. [DOI] [PubMed] [Google Scholar]
- 21.Youn YH, Hong J, Burke JM. Invest Ophthalmol Vis Sci. 2006;47:2675–2685. doi: 10.1167/iovs.05-1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iijima J, Zhao Y, Isaji T, Kameyama A, Nakaya S, Wang X, Ihara H, Cheng X, Nakagawa T, Miyoshi E, Kondo A, Narimatsu H, Taniguchi N, Gu J. J Biol Chem. 2006;281:13038–13046. doi: 10.1074/jbc.M601961200. [DOI] [PubMed] [Google Scholar]
- 23.Vagin O, Turdikulova S, Sachs G. J Biol Chem. 2005;280:43159–43167. doi: 10.1074/jbc.M508262200. [DOI] [PubMed] [Google Scholar]
- 24.Gottardi CJ, Dunbar LA, Caplan MJ. Am J Physiol. 1995;268:F285–295. doi: 10.1152/ajprenal.1995.268.2.F285. [DOI] [PubMed] [Google Scholar]
- 25.Kroepfl JF, Gardinier MV. J Neurochem. 2001;77:1301–1309. doi: 10.1046/j.1471-4159.2001.00343.x. [DOI] [PubMed] [Google Scholar]
- 26.Pfaffl MW. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Treuheit MJ, Costello CE, Kirley TL. J Biol Chem. 1993;268:13914–13919. [PubMed] [Google Scholar]
- 28.Laughery MD, Todd ML, Kaplan JH. J Biol Chem. 2003;278:34794–34803. doi: 10.1074/jbc.M302899200. [DOI] [PubMed] [Google Scholar]
- 29.Rajasekaran AK, Hojo M, Huima T, Rodriguez-Boulan E. J Cell Biol. 1996;132:451–463. doi: 10.1083/jcb.132.3.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yap AS, Brieher WM, Gumbiner BM. Annu Rev Cell Dev Biol. 1997;13:119–146. doi: 10.1146/annurev.cellbio.13.1.119. [DOI] [PubMed] [Google Scholar]
- 31.Gumbiner B, Simons K. J Cell Biol. 1986;102:457–468. doi: 10.1083/jcb.102.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gumbiner B, Stevenson B, Grimaldi A. J Cell Biol. 1988;107:1575–1587. doi: 10.1083/jcb.107.4.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rothen-Rutishauser B, Riesen FK, Braun A, Gunthert M, Wunderli-Allenspach H. J Membr Biol. 2002;188:151–162. doi: 10.1007/s00232-001-0182-2. [DOI] [PubMed] [Google Scholar]
- 34.Capaldo CT, Macara IG. Mol Biol Cell. 2007;18:189–200. doi: 10.1091/mbc.E06-05-0471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Granovsky M, Fata J, Pawling J, Muller WJ, Khokha R, Dennis JW. Nat Med. 2000;6:306–312. doi: 10.1038/73163. [DOI] [PubMed] [Google Scholar]
- 36.Guo HB, Lee I, Bryan BT, Pierce M. J Biol Chem. 2005;280:8332–8342. doi: 10.1074/jbc.M413532200. [DOI] [PubMed] [Google Scholar]
- 37.Isaji T, Gu J, Nishiuchi R, Zhao Y, Takahashi M, Miyoshi E, Honke K, Sekiguchi K, Taniguchi N. J Biol Chem. 2004;279:19747–19754. doi: 10.1074/jbc.M311627200. [DOI] [PubMed] [Google Scholar]
- 38.Kino T, Inamura N, Nakahara K, Kiyoto S, Goto T, Terano H, Kohsaka M, Aoki H, Imanaka H. J Antibiot (Tokyo) 1985;38:936–940. doi: 10.7164/antibiotics.38.936. [DOI] [PubMed] [Google Scholar]
- 39.Handerson T, Pawelek JM. Cancer Res. 2003;63:5363–5369. [PubMed] [Google Scholar]
- 40.Ishibashi Y, Dosaka-Akita H, Miyoshi E, Shindoh M, Miyamoto M, Kinoshita I, Miyazaki H, Itoh T, Kondo S, Nishimura M, Taniguchi N. Oncology. 2005;69:301–310. doi: 10.1159/000089680. [DOI] [PubMed] [Google Scholar]
- 41.Dosaka-Akita H, Miyoshi E, Suzuki O, Itoh T, Katoh H, Taniguchi N. Clin Cancer Res. 2004;10:1773–1779. doi: 10.1158/1078-0432.ccr-1047-3. [DOI] [PubMed] [Google Scholar]
- 42.Murata K, Miyoshi E, Kameyama M, Ishikawa O, Kabuto T, Sasaki Y, Hiratsuka M, Ohigashi H, Ishiguro S, Ito S, Honda H, Takemura F, Taniguchi N, Imaoka S. Clin Cancer Res. 2000;6:1772–1777. [PubMed] [Google Scholar]
- 43.Takahashi T, Hagisawa S, Yoshikawa K, Tezuka F, Kaku M, Ohyama C. J Urol. 2006;175:90–93. doi: 10.1016/S0022-5347(05)00044-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.