

Abstract
A facile synthesis of oligosaccharide-thiazoline derivatives of N-glycans as a novel class of inhibitors for endo-β-N-acetylglucosaminidases was described. It was found that the external sugar residues on the N-glycan core could enhance the inhibitory potency. While the Manβ1,4GlcNAc- and Man3GlcNAc-thiazolines were only moderate inhibitors, the large Man9GlcNAc-thiazoline demonstrated potent inhibitory activity, with an IC50 of 0.22 and 0.42 μM against the Arthrobacter enzyme (Endo-A) and the human endo-β-N-acetylglycosaminidase (hENGase), respectively. It was also observed that the oligosaccharide thiazolines could differentially inhibit endo-β-N-acetylglucosaminidases from different sources. These oligosaccharide thiazolines represent the first class of endo-β-N-acetylglucosaminidase inhibitors.
1. Introduction
Endo-β-N-acetylglucosaminidases (EC 3.2.1.96) (ENGases) are a class of endoglycosidases that hydrolyze the β-1,4-glycosidic bond in the N,N′-diacetylchitobiose core of N-glycans in glycoproteins. The enzymes are widely distributed in nature and are implicated to play a role in glycoprotein metabolism.1–3 For example, ENGases have been found in bacteria, including Endo-H from Streptomyces plicatus4, Endo-A (from Arthrobacter protophormiae),5,6 Endo-Fsp from Flavobacterium sp,7 and Endo-F1,2,3 from Flavobacterium meningosepticum;8 in fungi, such as Endo-M from Mucor hiemalis;9 in animals, such as Endo-HO (from hen oviduct);10 and in Caenorhabditis elegans (Endo-CE).2 ENGases were also found in human tissues and are implicated to play a role in the processing of N-glycans in the cytosol.3,11 ENGases from different sources have demonstrated varied substrate specificity. For example, Endo-H, Endo-A, Endo-Fsp, Endo-F1, and Endo-CE are specific for high-mannose type N-glycans; Endo-F2 and Endo-F3 are specific for bi-and tri-antennary complex type N-glycans, respectively; whereas Endo-M can hydrolyze both high-mannose and complex type N-glycans. Among them, the Endo-H and Endo-F2 have been frequently used as tools for structural characterization and functional analysis of N-glycans in glycoproteins. In addition to hydrolytic activity, some ENGases such as Endo-A and Endo-M possess transglycosylation activity and have been found valuable applications for the synthesis of homogeneous glycopeptides. For further structural and functional studies, we are interested in developing effective inhibitors against this important class of endoglycosidases, for which no inhibitors have ever been available.
In analogy to some family 18 chitinases12 and family 20 N-acetylhexosaminidases,13 the ENGase-catalyzed hydrolysis of N-glycans was also implicated to proceed in a substrate-assisted mechanism through the 2-acetamido neighboring group participation to form an oxazolinium ion intermediate.14–16 More direct evidence comes from the fact that Endo-A and Endo-M could take synthetic sugar oxazolines as substrates in transglycosylation.17–19 Based on the findings that N-acetylglucosamine-derived thiazoline (GlcNAc-thiazoline) (1) was a mechanism-based inhibitor for N-acetylhexosaminidases,20–22 we reasoned that an oligosaccharide thiazoline, in which the anomeric oxygen of the corresponding N-glycan oxazoline is replaced with a sulfur atom, would behave as an inhibitor against the endoglycosidase. We describe in this paper the first synthesis of such oligosaccharide thiazolines corresponding to the N-glycans of glycoproteins, including the di-, tetra-, and deca-saccharide thiazoline derivatives (2–4) (Figure 1). The inhibitory activities of the synthetic oligosaccharide thiazolines against an array of ENGases were examined.
Figure 1.
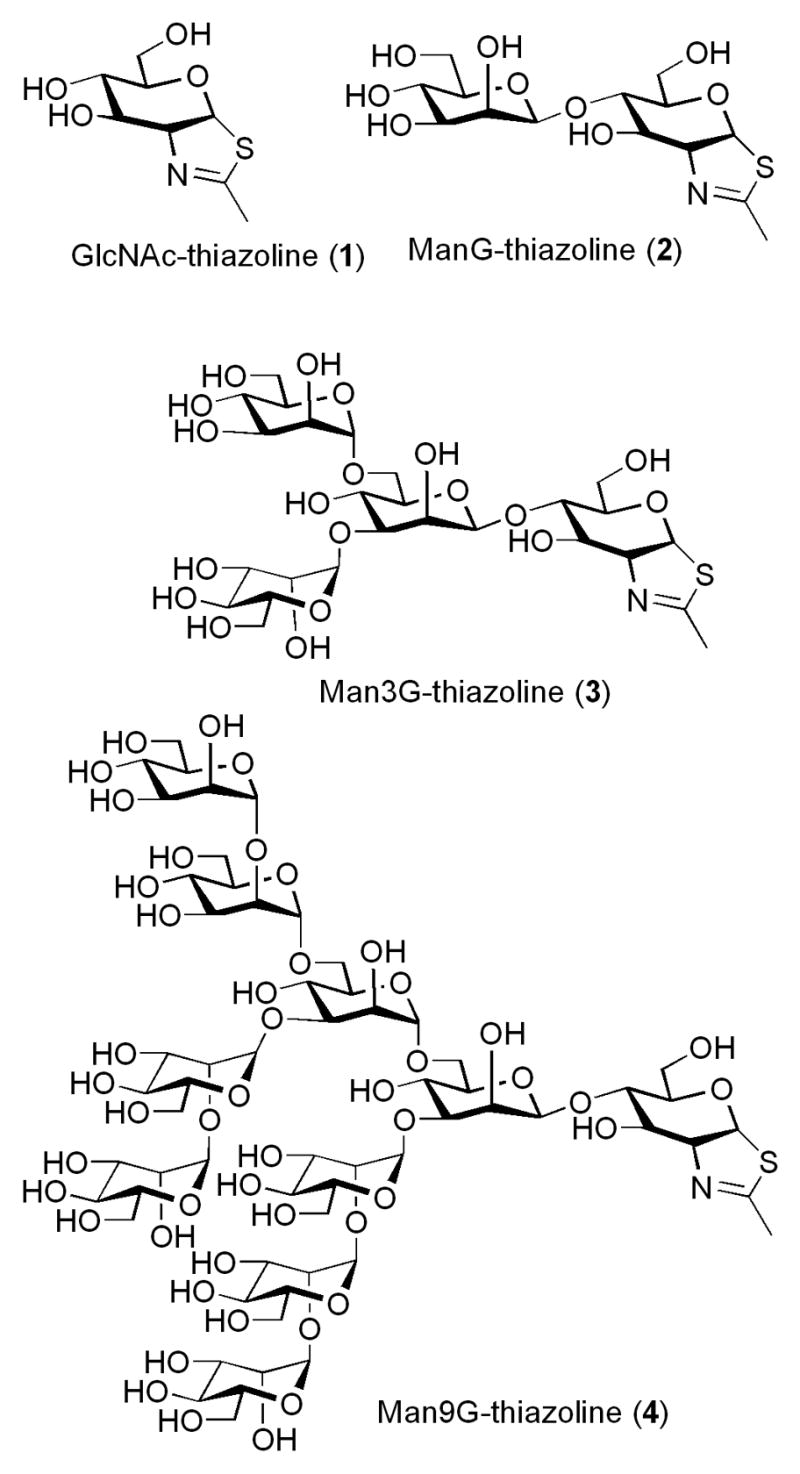
Structures of GlcNAc-thiazoline and oligosaccharide thiazolines
2. Results and discussion
2.1. Synthesis
The synthesis of the disaccharide thiazoline 2 started from the peracetylated Manβ1,4GlcNAc derivative 5 that we have prepared previously.18,19 Treatment of an α/βmixture of 5 (α:β ca. 3:1) with Lawesson’s reagent [2,4-bis(4-methoxyphenyl)-1,3-dithia-2,4-diphosphetane-2,4-disulfide]23 in toluene at 80°C gave the thioacetamide α-isomer 6 as the major product, together with the expected thiazoline derivative 7 as the minor product. Apparently the thioacetamide α-isomer (6) could not be cyclized to form the thiazoline derivative under the reaction condition. This result is consistent with previous observations with monosaccharide derivatives that the α-anomers of peracetylated GlcNAc and GalNAc were resistant to further SN2-type cyclization because of the unfavorable configuration of the 1-α-OAc leaving group.20,24 To convert 6 to 7, we have examined several Lewis acid catalysts. It was found that the combination of TMS-Cl and BF3.Et2O was the best catalyst that could transform the thioacetamide 6 to the thiazoline 7 in an excellent yield. Other catalysts, such as BF3.Et2O or TMS-Br would either lead to a low yield or result in decomposition of the thiazoline product. De-O-acetylation of 7 gave the disaccharide thiazoline 2 (Scheme 1). The preparation of the tetrasaccharide thiazoline 3 followed a similar synthetic pathway. Thus, reaction of the tetrasaccharide derivative 818 with Lawesson’s reagent, followed by treatment with TMS-Cl and BF3.Et2O in the presence of 2,4,6-collidine gave the thiazoline derivative 9 in 76% yield in two steps. Compound 9 was de-O-acetylated to give the tetrasaccharide thiazoline 3 (Scheme 2). The large oligosaccharide thiazoline 4 was synthesized using the high-mannose type oligosaccharide Man9GlcNAc 10 as the starting material, which was prepared from soybean agglutinin following our published procedure.25 Compound 10 was first acetylated with Ac2O/Py to give the peracetylated compound 11. Then compound 11 was treated with Lawesson’s reagent and subsequently with TMS-Cl/BF3.Et2O/collidine to give the thiazoline derivative 12. The yield of 12 from 10 was 73% in two steps. Finally, de-O-acetylation of 12 afforded the Man9G-thiazoline 4(Scheme 3).
Scheme 1.
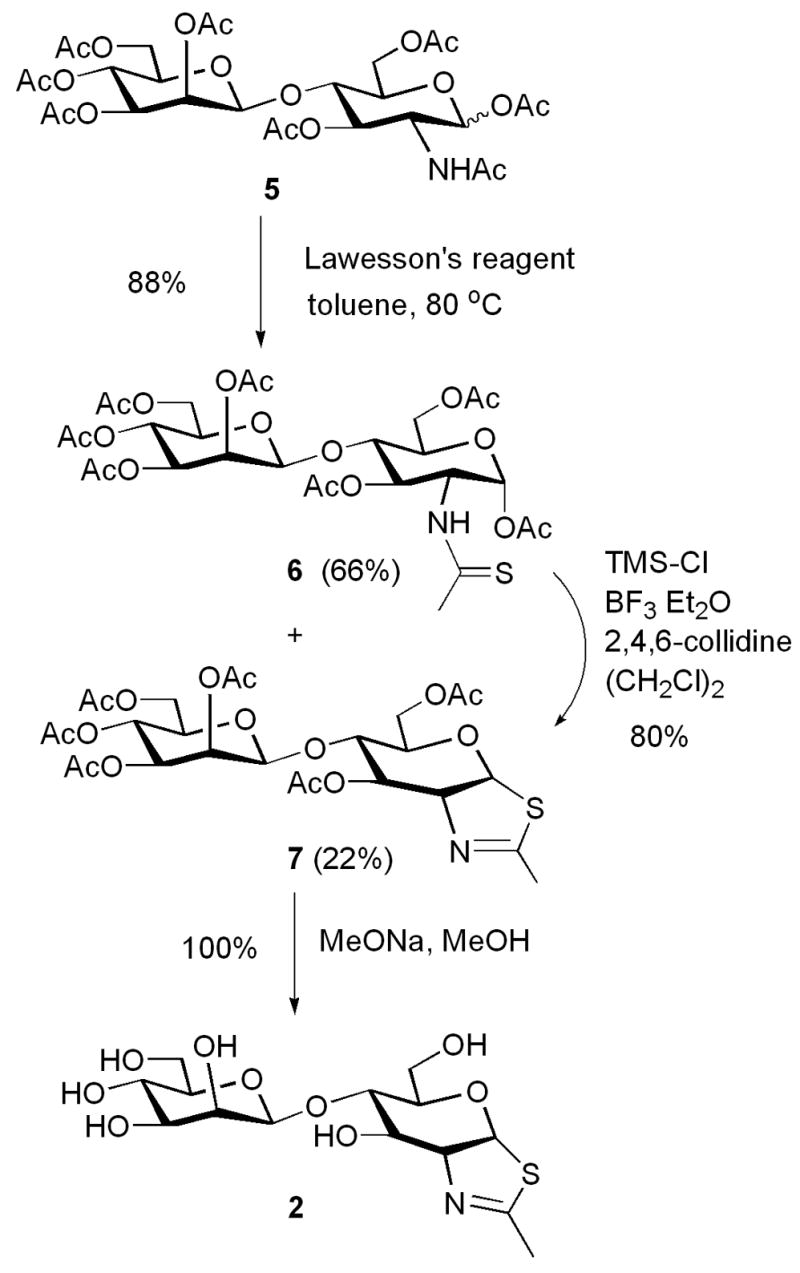
Synthesis of ManGlcNAc-thiazoline
Scheme 2.
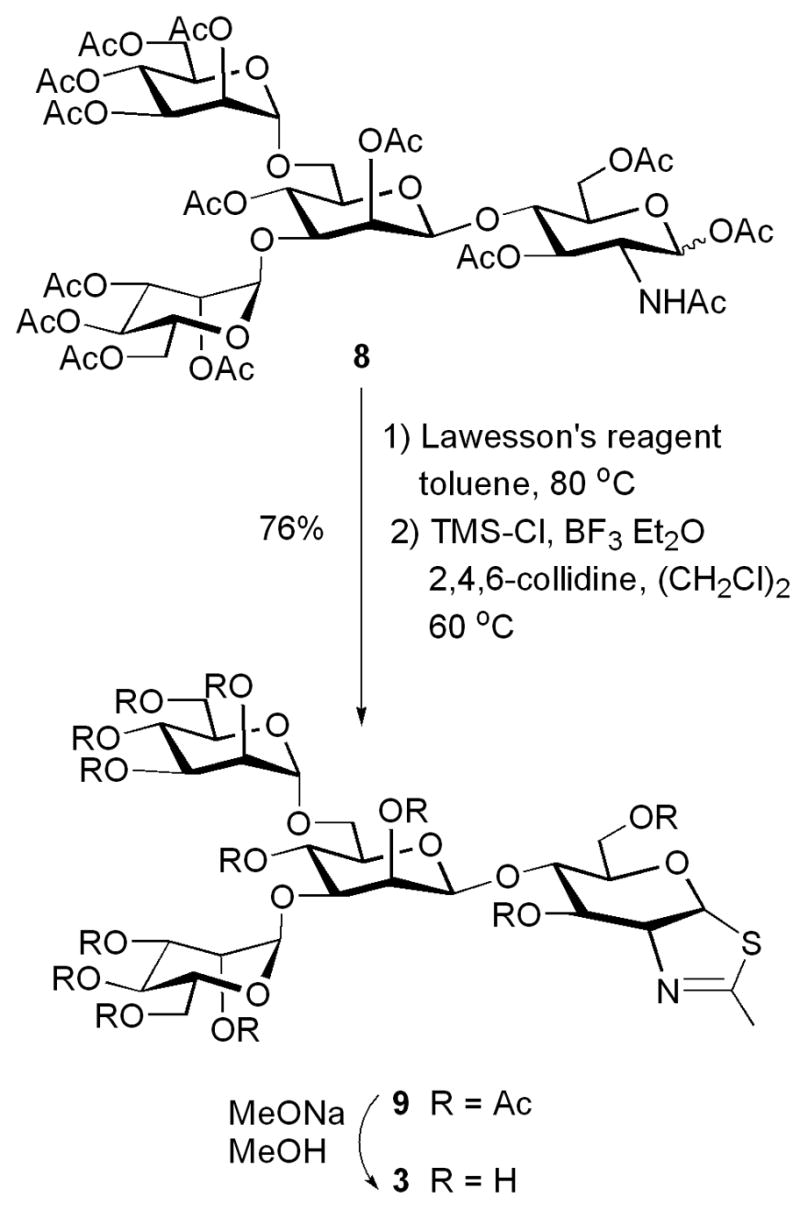
Synthesis of Man3GlcNAc-thiazoline
Scheme 3.
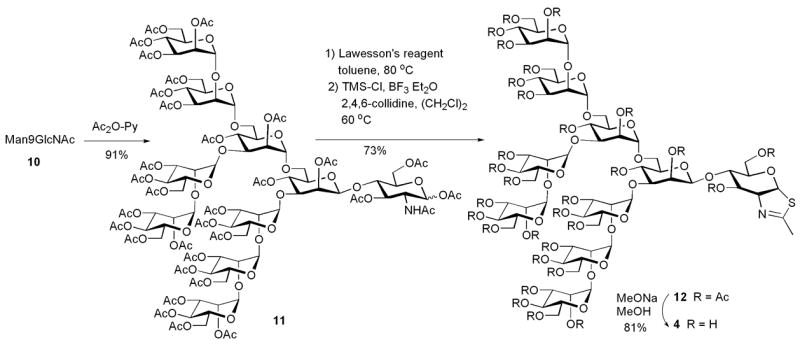
Synthesis of Man9GlcNAc-thiazoline
2.2. Inhibitory activities
The inhibitory activities of the synthetic thiazoline compounds were examined against an array of endo-β-N-acetylglucosaminidases. For Endo-A and Endo-M, Man9GlcNAc2Asn was used as the substrate and the hydrolytic activity of the enzymes in the presence or absence of the respective inhibitors were measured by quantifying the hydrolytic product Man9GlcNAc using the HPAEC-PED method.25 It was found that all the three oligosaccharide thiazolines (2–4) showed inhibitory activities against Endo-A. But the larger oligosaccharide derivative 4 was much more potent in inhibiting Endo-A activity than the tetrasaccharide and disaccharide derivatives 2 and 3, respectively (Figure 2A). The IC50 (concentrations for 50% inhibition) of compounds 2, 3, and 4 against Endo-A were estimated to be 38, 13, and 0.22 μM, respectively. The oligosaccharide thiazolines 2–4 could also inhibit the fungus enzyme Endo-M (Figure 2B). The IC50 of compounds 2, 3, and 4 against Endo-M were 80, 64, and 6 μM, respectively. In both cases, the inhibitory potency of the synthetic compounds is in the following order: the decasaccharide thiazoline 4 ≫ the tetrasaccharide thiazoline 3 > the di-saccharide thiazoline 2. In comparison between the enzymes, the respective oligosaccharide thiazolines are less active in inhibiting Endo-M than Endo-A. If the IC50 data was taken as an indication for inhibition potency, the Man9GlcNAc-thiazoline 4 would be about 27-fold more active against Endo-A than Endo-M. Next, we examined the oligosaccharide thiazolines against another bacterial enzyme, Endo-Fsp from Flavobacterium sp7. It was found that no inhibitory activities against this enzyme were observed for compounds 2 and 3, even at a 100 μM concentration. But, interestingly, the Man9GlcNAc-thiazoline 4 was found to be a strong inhibitor against this enzyme (75% inhibition at 1 μM of 4) (data not shown). We also tested the large oligosaccharide thiazoline 4 against a human endo-β-N-acetylglucosaminidase (hENGase), which was proposed to involve in the metabolism of N-glycans in cytosol.3 It was found that the Man9GlcNAc-thiazoline 4 was a potent inhibitor against hENGase with an IC50 of 0.42 μM (Figure 2C). While the oligosaccharide thiazolines are inhibitors against the endoglycosidases, the monosaccharide thiazoline 1 did not exhibit any inhibitory activity at up to 500 μM against Endo-A or Endo-M (data not shown). These results suggest that the disaccharide Manβ1,4GlcNAc-thiazoline moiety might be the minimal recognition unit for the endoglycosidases. But additional sugar residues extended from the disaccharide core are essential for a potent inhibitory activity, probably through an enhanced binding to the enzymes. On the other hand, the significant difference in inhibitory activity of the same oligosaccharide thiazoline against different ENGases indicates that the enzymes from different sources could respond to the oligosaccharide thiazoline inhibitors differentially, making it possible for selective inhibition. A potent and specific inhibitor for ENGases should be highly valuable for studying the biological functions of ENGases in vivo, such as their role in the metabolism of N-glycans and glycoproteins.3 One potential problem is the cell membrane permeability of the synthetic inhibitors when they are used in vivo for inhibiting intracellular ENGases. The free oligosaccharide thiazolines may not be permeable. If this is the case, corresponding per-O-acetylated derivatives (7, 9, and 12) of the oligosaccharide thiazolines may be directly used for in vivo studies. The acetylated derivatives are expected to have better membrane permeability and the O-acetyl groups should be removable in situ to restore the original free oligosaccharides by cellular esterases (deacetylases), as exemplified by the use of per-O-acetylated mannosamine derivatives for cell surface glyco-engineering in cell culture and in living animals.26,27
Figure 2. Inhibition of endoglycosidases by the synthetic oligosaccharide thiazolines.
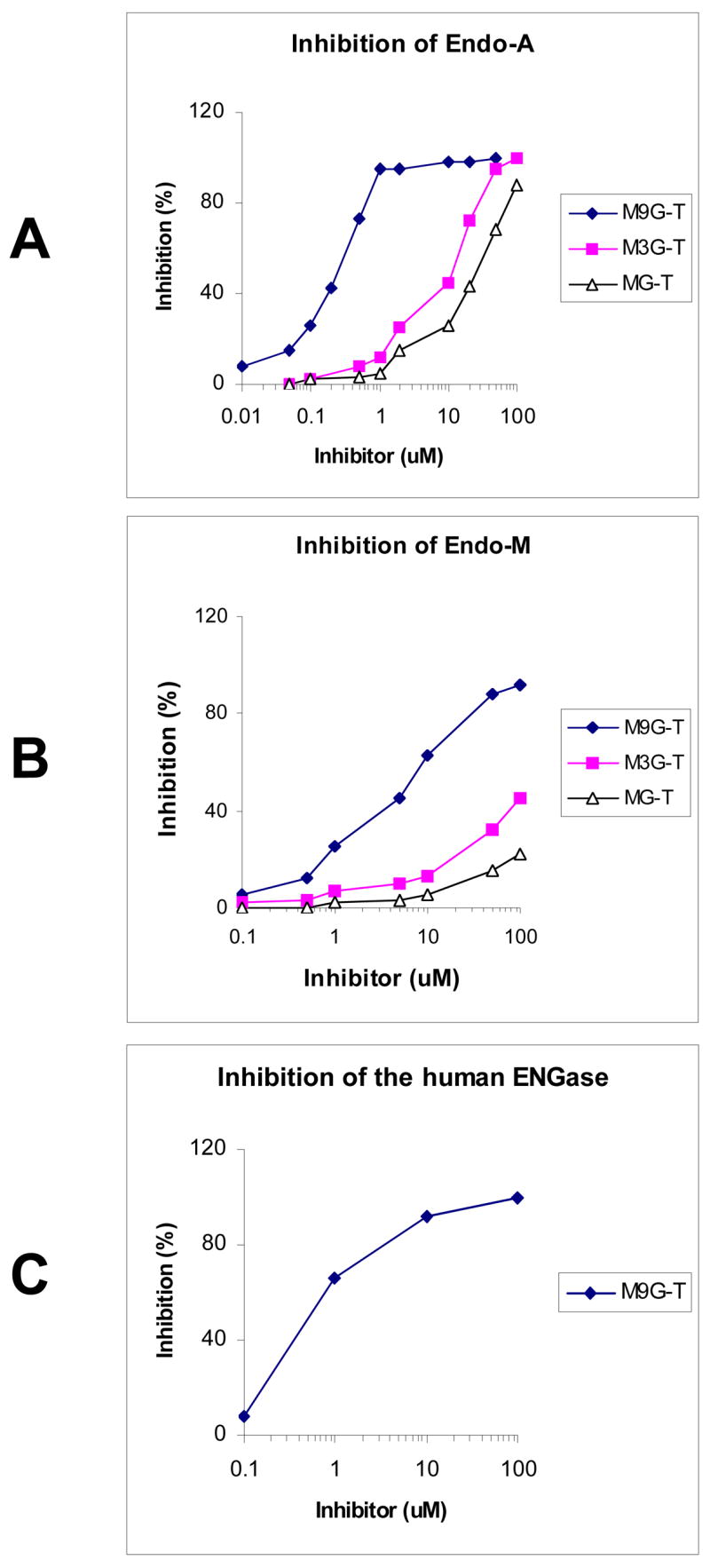
Man9GlcNAc2-Asn and Man9GlcNAc2-PA were used as the substrates for the inhibitory assays. The substrates were incubated with the respective enzyme in the presence of a serial dilutions of the synthetic oligosaccharide thaizoline inhibitors.
3. Conclusion
An array of oligosaccharide thiazolines corresponding to N-glycans was synthesized and their inhibitory activities against endo-β-N-acetylglucosaminidases (ENGases) were examined. It was found that the largest oligosaccharide thiazoline Man9GlcNAc-thiazoline (4) is the most potent inhibitor against all the ENGases tested. ENGases from different sources respond to the inhibitors differentially, implicating the possibility to design specific inhibitors through structural modification. This is the first class of mechanism-based inhibitors against ENGases. These nonhydrolyzable transition state analogs may be useful for X-ray structural studies through co-crystallization. In addition, a potent and specific inhibitor for ENGases will be highly valuable for studying the biological functions of this class of endoglycosidases in vivo, such as their role in N-glycan metabolism.
4. Experimental
4.1. Synthesis of oligosaccharide thiazolines
4.1.1. Materials and methods
All chemicals, biochemicals and reagents were purchased from Sigma/Aldrich Chemical Company (St. Louis, MO). TLC was performed on glass plates coated with silica gel 60 F254 (E. Merck). Flash column chromatography was performed on silica gel 60 (EM Science, 230–400 mesh). 1H and 13C NMR spectra were recorded on an Inova 500 NMR machine. The ESI-MS spectra were measured on a micromass ZQ-4000 single quadruple mass spectrometer.
4.1.2. O-(2,3,4,6-tetra-O-acetyl-β-D-mannopyranosyl)-(1→4)-(3,6-di-O-acetyl-1,2-dideoxy-α-D-glucopyrano)-[2,1-d]-2-thiazoline (7)
To a solution of 5 (800 mg, 0.12 mmol) in toluene (8 mL) was added Lawesson’s reagent (53 mg, 0.13 mmol) and the mixture was heated at 80°C for 1.5 h. Then the mixture was cooled and the solvent was evaporated. The residue was subject to flash silica gel column chromatography (EtOAc/CH2Cl2, 3/7) to afford a mixture of the thioacetamide 6 and the thiazoline derivative 7 (72 mg, 88%, 6:7 = 3:1) as a yellow syrup. Without further purification, the mixture (39 mg) was dissolved in anhydrous ClCH2CH2Cl (2 ml), to which TMSCl (35 μl, 277 μmol), BF3.Et2O (36 μl, 284 μmol) and 2, 4, 6-collidine (37 μl, 279 μmol) were added. The resulting solution was stirred at r.t. for 3 h. The reaction mixture was diluted with CH2Cl2. The organic layer was separated and washed with aq. NaHCO3 and brine.The organic layer was dried over Na2SO4 and filtered. The filtrate was concentrated in vacuo and the residue was subject to flash silica gel column chromatography (3:7 EtOAc: CH2Cl2) to give the peracetylated thiazoline derivative 7 (30 mg, 80%) as a yellow foam. 1H NMR (CDCl3, 500 MHz): δ 6.24 (d, J = 7.5 Hz, 1H, H-1), 5.88 (s, 1H, H-3), 5.47 (d, J = 2.5 Hz, 1H, H-2′), 5.32 (t, J = 10 Hz, 1H, H-4′), 5.11 (dd, J = 9.5, 3 Hz, 1H, H-3′), 4.89 (s, 1H. H-1′), 4.50 (m, 1H, H-2),4.33–4.18 (m, 4H), 3.78–3.75 (m, 2H), 3.45–3.40 (m, 1H), 2.33 (d, J = 1.5 Hz, 3H, CH3C(=N)-), 2.25 (s, 3H, CH3CO2-), 2.17 (s, 3H, CH3CO2-), 2.15 (s, 6H, 2 x CH3CO2-), 2.10 (s, 3H, CH3CO2-), 2.05 (s, 3H, CH3CO2-); 13C NMR (CDCl3,125 MHz): δ 170.5, 170.4, 170.3, 169.6, 169.3, 167.8, 100.4, 89.9, 77.4, 76.6, 72.7, 70.8, 70.3, 69.2, 68.2, 65.3, 61.5, 62.0, 20.8; ESI-MS: calcd for C26H35NO15S, M = 633.6; Found, 634.1 (M+H)+.
4.1.3.O-(β-D-mannopyranosyl)-(1→4)-(1,2-dideoxy-α-D-glucopyrano)-[2,1-d]-2-thiazoline (2)
To a solution of compound 7 (17 mg, 27 μmol) in MeOH (2 mL) was added MeONa/MeOH (0.5 M, 60 μl). The solution was stirred at r.t. for 2 h and then neutralized with Dowex 50w-x8 (H+ form). The mixture was filtrated and the filtrate was concentrated. The residue was dissolved in water and lyophilized to give the thiazoline 2 (10 mg, quantitative) as a yellow solid. 1H NMR (D2O, 500 MHz): δ 6.24 (d, J = 6.5 Hz, 1H, H-1), 4.66 (s, 1H, H-1′), 4.45 (s, 1H, H-2′), 3.91–3.87(m, 2H), 3.72–3.65(m, 4H), 3.56–3.46 (m, 4H), 3.32–3.28 (m, 1H), 2.23 (s, 3H, CH3C(=N)-); 13C NMR(D2O, 125 MHz): δ 172.0, 101.4, 87.6, 78.6, 77.6, 76.4, 72.8, 72.0, 70.4, 69.6, 66.7, 61.8, 61.0, and 19.4; ESI-MS: Calcd for C14H23NO9S, M = 381.4; Found, 382.1 (M +H)+.
4.1.4. O-(2,3,4,6-tetra-O-acetyl-α-D-mannopyranosyl)-(1→6)-[(2,3,4,6-tetra-O-acetyl-αD-mannopyranosyl)-(1→3)]-2,4-di-O-acetyl--β-D-mannopyranosyl-(1→4)-(3,6-di-O-acetyl-1,2-dideoxy-α-D-glucopyrano)-[2,1-d]-2-thiazoline (9)
A solution of 8 (31 mg, 25 μmol) in toluene (3 mL) was treated with Lawesson’s reagent (10 mg, 25μmol) at 80°C for 1.5 h. The solvent was then evaporated and the residue was subject to flash silica gel column chromatography (3:7 EtOAc: CH2Cl2) to afford a yellow syrup which contains both the thioacetamide and the thiazoline derivative. The mixture was then dissolved inanhydrous ClCH2CH2Cl (2 ml). To the solution were added TMSCl (16 μl, 130 μmol), BF3.Et2O (16 μl, 130 μmol), and 2, 4, 6-collidine (17 μl, 130 μmol). The mixture was stirred at r.t. for 3 h. The reaction mixture was diluted with CH2Cl2 (3 mL). The organic layer was washed with NaHCO3 and brine, dried over Na2SO4 and filtered. The filtrate was concentrated in vacuo and the residue was subject to flash silica gel column chromatography (EtOAc/CH2Cl2, 3/7) to give the peracetylated thiazoline derivative 9 (23 mg, 76%) as a yellow foam. 1H NMR (CDCl3, 500 MHz): δ 6.26 (d, J = 7 Hz, 1H, H-1), 5.97 (s, 1H, H-3), 5.46 (s, 1H, H-2′), 5.35–5.29 (m, 5H), 5.23–5.18 (m, 2H), 5.06–5.04 (m, 2H), 4.91 (s, 1H), 4.85 (s, 1H, H-1′), 4.53 (m, 1H, H-2), 4.41 (dd, J = 12.5, 4 Hz, 1H), 4.34–4.13 (m, 8H), 3.96–3.94 (m, 2H), 3.76–3.71 (m, 2H), 2.33 (s, 3H, CH3C(=N)-), 2.27 (s, 3H, CH3CO2-), 2.20 (s, 3H, CH3CO2-), 2.18(s, 9H, 3 x CH3CO2-), 2.17 (s, 3H, CH3CO2-), 2.16 (s, 3H, CH3CO2-), 2.14 (s, 3H, CH3CO2-), 2.13 (s, 3H, CH3CO2-), 2.09 (s, 3H, CH3CO2-), 2.03 (s, 3H, CH3CO2-), 2.00 (s, 3H, CH3CO2-); 13C NMR (CDCl3, 125 MHz): δ 170.9, 170.8, 170.3, 170.2, 170.1, 170.0, 169.8, 169.7, 169.6, 169.5, 167.4, 99.4, 99.1, 97.7, 88.9, 73.1, 70.2, 70.1, 70.0, 69.5, 69.4, 69.1, 69.0, 68.9, 68.6, 68.3, 67.6, 66.0, 65.9, 63.6, 62.2, 20.9, 20.8, 20.7; ESI-MS: Calcd for C50H67NO31S, M = 1210.12; Found, 1211.3 (M+H)+.
4.1.5. O-(α-D-mannopyranosyl)-(1→6)-[(α-D-mannopyranosyl)-(1→3)]-β-D-mannopyranosyl-(1→4)-(1,2-dideoxy-α-D-glucopyrano)-[2,1-d]-2-thiazoline (3)
To a solution of compound 9 (12 mg, 10 μmol) in MeOH (2 mL) was added MeONa/MeOH (0.5 M, 20 μ) and the mixture was stirred at r.t. for 2 h. The reaction solution was neutralized with Dowex 50w-x8 (H+ form) and filtered. The filtrate was concentrated and the residue was dissolved in water and lyophilized to afford the thiazoline 3 (7 mg, quantitative) as a yellow solid. 1H NMR (CD3OD, 500 MHz): δ6.39 (d, J = 6.5 Hz, 1H, H-1), 5.12 (s, 1H, H-1′ ′), 4.93 (s, 1H, H-1′ ′ ′), 4.68 (s, 1H, H-1′), 4.56 (s, 1H. H-3), 4.41 (s, 1H, H-2′), 4.28 (s, 1H, H-2′ ′ ′), 4.27 (s, 1H, H-2′ ′), 4.14 (m, 1H, H-2), 4.03–3.62 (m, 18H), 3.49 (m, 1H), 2.32 (s, 3H, CH3C(=N)-); 13C NMR(CD3OD,125 MHz): δ 171.1, 103.1, 102.5, 100.4, 89.9, 80.6, 79.0, 77.8, 74.4, 73.4, 72.7, 70.9, 70.5, 70.3, 70.2, 69.9, 69.0, 66.8, 66.7, 65.8, 65.6, 65.1, 61.7, 61.1, 60.9, 19.4; ESI MS: Calcd for C26H43NO19S, M = 705.7; Found, 706.1 (M+H)+.
4.1.6. Preparation of the peracetylated Man9GlcNAc-thiazoline (12)
The oligosaccharide Man9GlcNAc (90 mg, 53 μmol), prepared from soybean flour by the previously described procedure 25, was acetylated by treatment with pyridine (5 mL) and acetic anhydride (5 mL) at r.t. for 20 h. The reaction mixture was concentrated under vacuum to dryness and the residue was subject to flash silica gel column chromatography (CH2Cl2/MeOH, 20/1) to give the peracetylated Man9GlcNAc 11 (145 mg, 91%) as a pale-yellow syrup: ESI-MS: calculated for C124H167NO82, M = 2983.6; found, 2984.3 (M+H)+ and 1492.7 (M+2H)2+. The conversion of 11(26 mg, 9 μmol) into the peracetylated Man9GlcNAc-thiazoline 12 was performed in the same way as for the preparation of thiazoline derivative 9, including treatment with Lawesson’s reagent and then with TMS-Cl/BF3.Et2O/collidine, to give 12 (16 mg, 63% in two steps) as a yellow foam. 1H NMR (CDCl3, 500 MHz): δ 6.26 (d, J = 7.0 Hz, 1H, H-1), 5.86 (s, 1H,), 5.52 –4.82 (m, 31H), 4.35–4.00 (m, 31H), 3.88–3.84 (m, 4H), 3.59–3.57 (m, 2H), 2.37 (s, 3H, CH3C(=N)-), 2.30–2.06 (m, 90H, 30xCH3CO2-); 13C NMR (CDCl3, 500MHz): 171.5, 171.3, 171.2, 171.1, 171.0, 169.5, 169.4, 169.2, 168.9, 132.1, 131.5, 128.9, 101.5, 100.9, 100.8, 100.7, 100.5, 100.4, 100.2, 99.8, 97.5, 88.9, 73.5, 69.5, 69.3, 69.1, 68.9, 20.5. ESI-MS: Calcd for C122H163NO79S, M = 2939.6; Found, 2940.2 (M+H)+.
4.1.7. Preparation of the Man9GlcNAc-thiazoline (4)
The peracetylated thiazoline 12 (15 mg, 5 μmol) was dissolved in MeOH (2 mL) containing MeONa (5 μmol), and the solution was stirred at r.t. for 2 h. After neutralization with Dowex 50w-x8 (H+ form), the solvent was evaporated. The residue was dissolved in water and lyophilized to give the Man9GlcNAc-thiazoline 4 (9 mg, quantitative) as a yellow solid. 1H NMR (D2O, 500 MHz): δ 6.26 (d, J = 7.0 Hz, 1H, H-1), 5.34 (s, 2H), 5.27 (s, 2H), 5.24 (s, 2H), 5.09 (s, 3H), 4.98 (s, 7H), 4.87 (s, 1H), 4.46 (s, 4H), 4.12 –3.51(m, 43H), 3.37–3.29 (m, 5H), 2.24 (s, 3H, CH3C(=N)); 13C NMR (D2O, 125MHz): 168.1, 132.4, 131.2, 128.7, 102.9, 102.5, 100.3, 89.9, 71.2, 70.8, 63.5, 62.0, 19.3. ESI MS: Calculated for C62H103NO49S, M = 1678.5; Found, 1679.5 (M +H)+.
4.2. Inhibition assays
4.2.1. Inhibition of Endo-A, Endo-M, and Endo-Fsp
The inhibition assays against Endo-A and Endo-M were performed using Man9GlcNAc2Asn as the substrate. For Endo-Fsp, Man5GlcNAc2Asn was used as the substrate. Briefly, a solution of Man9GlcNAc2Asn (500 μM) in a phosphate buffer (50 μL, 50 mM, pH 6.5) was incubated with limited amount of respective ENGase in the presence or absence of serial dilutions of the thiazoline derivative. The mixture was incubated at 30°C for 5 min and the reaction was stopped by heating at 100°C for 3 min. The reaction mixture was analyzed by HPAEC-PED and the hydrolytic product Man9GlcNAc was quantitated following our previously described procedure.25
4.2.2. Preparation and Assay for human ENGase
pcDNA-hENGase3 was transfected into Hek293 cells using lipofectamine 2000 (Invitrogen) according to the manufacture’s protocol. A protein extract was prepared 48 h after the transfection. For cells from a 10 cm-dish (~107 cells), 300 μl of buffers (20 mM Hepes-KOH buffer (pH 7.0) containing 5 mM MgCl2, 150 mM potassium acetate, 250 mM sorbitol, 5 mM DTT, 1 mM AEMSF (Roche), and 1 X Complete protease inhibitor cocktails (Roche) was added. After homogenization, soluble fraction was obtained by 100,000 × g for 1 h and used for enzyme fraction. Enzyme assay was carried out in 10 μl solution of 40 mM of Mes-NaOH buffer (pH 6.1) containing 1 μM of Man9GlcNAc2-PA (Takara BioInc., Kusatsu, Japan), 2 μl of enzyme fraction, and various concentration of Man9GlcNAc-thiazoline (4) (10 mM – 0.1 μM). Incubation was performed for 3 min at 37°C and the reaction was stopped by boiling the sample for 5 min. The enzyme activity was quantitated by HPLC using C18 column (TSK gel ODS-80TM; 7.5 × 75 mm; Tosoh Corp, Tokyo, Japan). Elution was performed in 0.1 M ammonium acetate buffer (pH 4.0) at a flow rate of 1.0 ml/min with a linear gradient of 1-butanol from 0.025% to 0.5% for 55 min. Column was washed for 20 min with the starting buffer between the analyses.
Supplementary Material
Acknowledgments
The work was supported by the National Institutes of Health (R01 GM073717 and R01 GM080374).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Tarentino AL, Plummer TH., Jr Methods Enzymol. 1994;230:44–57. doi: 10.1016/0076-6879(94)30006-2. [DOI] [PubMed] [Google Scholar]
- 2.Kato T, Fujita K, Takeuchi M, Kobayashi K, Natsuka S, Ikura K, Kumagai H, Yamamoto K. Glycobiology. 2002;12:581–7. doi: 10.1093/glycob/cwf073. [DOI] [PubMed] [Google Scholar]
- 3.Suzuki T, Yano K, Sugimoto S, Kitajima K, Lennarz WJ, Inoue S, Inoue Y, Emori Y. Proc Natl Acad Sci USA. 2002;99:9691–6. doi: 10.1073/pnas.152333599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tarentino AL, Trimble RB, Maley F. Methods Enzymol. 1978;50:574–80. doi: 10.1016/0076-6879(78)50065-7. [DOI] [PubMed] [Google Scholar]
- 5.Takegawa K, Nakoshi M, Iwahara S, Yamamoto K, Tochikura T. Appl Environ Microbiol. 1989;55:3107–3112. doi: 10.1128/aem.55.12.3107-3112.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takegawa K, Yamaguchi S, Kondo A, Iwamoto H, Nakoshi M, Kato I, Iwahara S. Biochem Int. 1991;24:849–55. [PubMed] [Google Scholar]
- 7.Takegawa K, Mikami B, Iwahara S, Morita Y, Yamamoto K, Tochikura T. Eur J Biochem. 1991;202:175–80. doi: 10.1111/j.1432-1033.1991.tb16359.x. [DOI] [PubMed] [Google Scholar]
- 8.Trimble RB, Tarentino AL. J Biol Chem. 1991;266:1646–51. [PubMed] [Google Scholar]
- 9.Yamamoto K, Kadowaki S, Watanabe J, Kumagai H. Biochem Biophys Res Commun. 1994;203:244–52. doi: 10.1006/bbrc.1994.2174. [DOI] [PubMed] [Google Scholar]
- 10.Kato T, Hatanaka K, Mega T, Hase S. J Biochem (Tokyo) 1997;122:1167–73. doi: 10.1093/oxfordjournals.jbchem.a021877. [DOI] [PubMed] [Google Scholar]
- 11.Overdijk B, van der Kroef WM, Lisman JJ, Pierce RJ, Montreuil J, Spik G. FEBS Lett. 1981;128:364–6. doi: 10.1016/0014-5793(81)80118-4. [DOI] [PubMed] [Google Scholar]
- 12.Brameld KA, Shrader WD, Imperiali B, Goddard WA., 3rd J Mol Biol. 1998;280:913–23. doi: 10.1006/jmbi.1998.1890. [DOI] [PubMed] [Google Scholar]
- 13.Mark BL, Vocadlo DJ, Knapp S, Triggs-Raine BL, Withers SG, James MN. J Biol Chem. 2001;276:10330–7. doi: 10.1074/jbc.M011067200. [DOI] [PubMed] [Google Scholar]
- 14.Rao V, Guan C, Van Roey P. Structure. 1995;3:449–57. doi: 10.1016/s0969-2126(01)00178-2. [DOI] [PubMed] [Google Scholar]
- 15.Van Roey P, Silva GH, Rao V, Plummer TH, Jr, Tarentino AL, Guan C. J Mol Biol. 1994;237:157–9. doi: 10.1006/jmbi.1994.1214. [DOI] [PubMed] [Google Scholar]
- 16.Waddling CA, Plummer TH, Jr, Tarentino AL, Van Roey P. Biochemistry. 2000;39:7878–85. doi: 10.1021/bi0001731. [DOI] [PubMed] [Google Scholar]
- 17.Fujita M, Shoda S, Haneda K, Inazu T, Takegawa K, Yamamoto K. Biochim Biophys Acta. 2001;1528:9–14. doi: 10.1016/s0304-4165(01)00164-7. [DOI] [PubMed] [Google Scholar]
- 18.Li B, Zeng Y, Hauser S, Song H, Wang LX. J Am Chem Soc. 2005;127:9692–3. doi: 10.1021/ja051715a. [DOI] [PubMed] [Google Scholar]
- 19.Zeng Y, Wang J, Li B, Hauser S, Li H, Wang LX. Chem Eur J. 2006;12:3355–3364. doi: 10.1002/chem.200501196. [DOI] [PubMed] [Google Scholar]
- 20.Knapp S, Vocadlo D, Gao Z, Kirk B, Lou J, Withers SG. J Am Chem Soc. 1996;118:6804–6805. [Google Scholar]
- 21.Reid CW, Blackburn NT, Legaree BA, Auzanneau FI, Clarke AJ. FEBS Lett. 2004;574:73–9. doi: 10.1016/j.febslet.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 22.Macauley MS, Whitworth GE, Debowski AW, Chin D, Vocadlo DJ. J Biol Chem. 2005;280:25313–22. doi: 10.1074/jbc.M413819200. [DOI] [PubMed] [Google Scholar]
- 23.Cava MP, Levinson MI. Tetrahedron. 1985;41:5061–5087. [Google Scholar]
- 24.Ritter TK, Wong CH. Tetrahedron Lett. 2001;42:615–618. [Google Scholar]
- 25.Wang LX, Ni J, Singh S, Li H. Chem Biol. 2004;11:127–34. doi: 10.1016/j.chembiol.2003.12.020. [DOI] [PubMed] [Google Scholar]
- 26.Yarema KJ, Mahal LK, Bruehl RE, Rodriguez EC, Bertozzi CR. J Biol Chem. 1998;273:31168–79. doi: 10.1074/jbc.273.47.31168. [DOI] [PubMed] [Google Scholar]
- 27.Prescher JA, Dube DH, Bertozzi CR. Nature. 2004;430:873–7. doi: 10.1038/nature02791. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.