

The [FeFe] hydrogenase enzymes are the most efficient catalysts known for the reduction of protons to H2.[1] The active site exists in two functional states (Scheme 1), Hred, which is S =0, and Hox, which is S = 1/2.[2] Research in this area is aimed at elucidating the mechanism of the enzymatic catalysis and at using this information to develop protein-free bioinspired synthetic catalysts.[3] A specific research goal is the preparation of molecules that resemble the functional states of the active site with the expectation that function will follow form. Most studies on diiron dithiolato carbonyl complexes rely on organic ligands (e.g. phosphanes) in place of the naturally occurring cyanide and μ-SR[Fe4S4] ligands,[4] which have complicated acid–base behavior that is often difficult to control outside of the protein. Another barrier to modeling has been the rarity of mixed-valence diiron dithiolate compounds with the appropriate structures, stability, and reactivity.
Scheme 1.
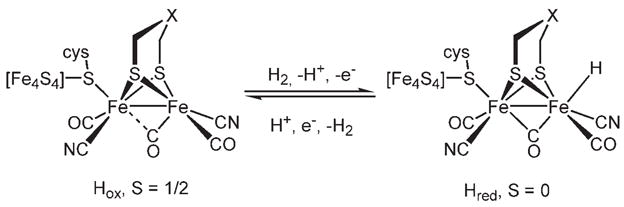
Two functional states of the active site of the [FeFe] hydrogenases.
The first evidence for mixed valency in diiron dithiolate models was obtained in the one-electron oxidation of [Fe2{(SCH2)2CMeCH2SMe}(CN)2(CO)4] 2−, which afforded a thermally sensitive mixed-valence derivative with IR and EPR spectroscopy signatures resembling those for the CO-inhibited enzyme.[5] In very recent work, the oxidation of [Fe2(S2C3H6)(CO)4(PMe3)L1] (L1 =1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene) was shown to give a mixed-valence derivative with structural and spectroscopic features resembling Hox.[6]
Our recently reported species [Fe2(S2C2H4)(CO)3(PMe3)-(dppv)] (1, dppv =cis-1,2-C2H2(PPh2)2) is attractive, because, like the active site, this diiron framework bears three donor ligands and three CO ligands.[7] We previously showed that oxidation of 1 in MeCN gives [1(NCMe)]2+. In MeCN solution, one equivalent of oxidizing agent gives an approximately 1:1 mixture of unreacted starting material and [1(NCMe)]2+. Oxidation of 1 in the weakly coordinating solvent CH2Cl2, however, proceeds quite differently.
Addition of one equivalent of FcBF4 (Fc+=[Fe(C5H5)2]+) to a CH2Cl2 solution of 1 at −45 °C resulted in complete consumption of the diiron complex, as indicated by in situ IR spectroscopy. We probed the oxidation of 1 by cyclic voltammetry on a solution of 1 in 0.1m Bu4NPF6/CH2Cl2. The voltammetry revealed a reversible couple at 20 mV vs. Ag/AgCl (ca. 300 mV vs. NHE). No other redox events were observed between 0.6 and −1 V.
The purple-colored compound 1-BF4 is stable in solution for several minutes at room temperature and is sufficiently stable that we were able to grow single crystals for X-ray diffraction (see below). The X-band EPR spectrum of polycrystalline 1-BF4 (15 K) displays g values at 2.14, 2.03, 2.01 as well as a signal for an apparent impurity at g =2.05; this rhombic pattern is similar to that seen for Hox from D. desulfuricans (g =2.10, 2.04, 1.99).[1,8]
Crystals of 1-BF4 were obtained from CH2Cl2/hexanes at −20 °C. Crystallographic analysis revealed an unsolvated 1:1 salt. In general, the [Fe2(SR)2L6] framework approaches that proposed for the Hox state of the active site.[2,9] Of specific interest, the oxidation has caused the {Fe(CO)(dppv)} center to rotate about the Fe–Fe axis. The apical site on the {Fe(dppv)} center is vacant, and the [BF4]− anion is approximately 5 Å away from this “distal” Fe center. The “proximal” {Fe(CO)2(PMe3)} moiety remains relatively unperturbed, and overall the bond lengths match those in 1 within 0.02 Å.[7] The unique CO ligand is only partially bridging, with a relatively long Feprox–C separation of 2.62 Å compared to the Fedist–CO bond length of 1.781(3) Å (∢(Fedist-C-O) = 170.2(3)°; Figure 1). We have recently described [Fe2-(S2C2H4)(CO)(μ-COAX3)(dppv)2] (AX3 = AlBr3, B(C6F5)3), which is not mixed-valence but also adopts the rotated structure.[10]
Figure 1.
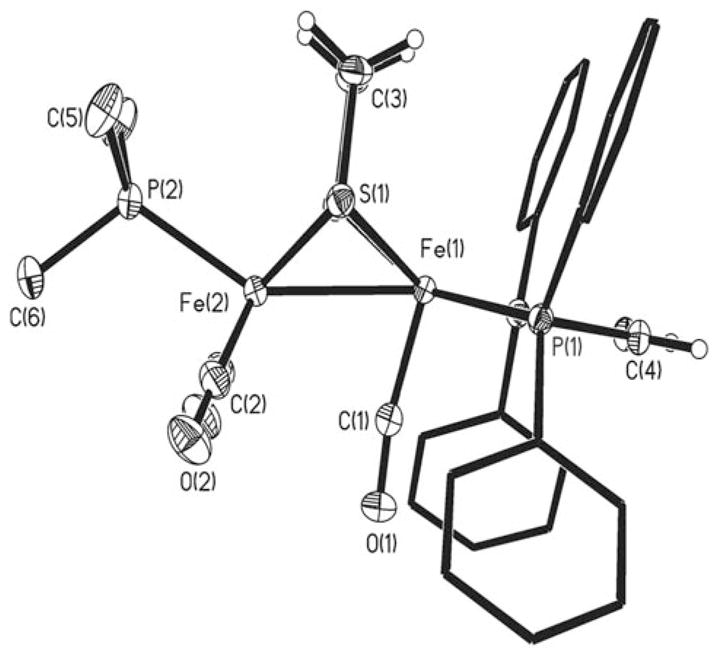
Structure of the cation in [Fe2(S2C2H4)(CO)3(PMe3)(dppv)]BF4 with thermal ellipsoids set at the 30% probability level. For clarity, methyl and phenyl hydrogen atoms are omitted. Selected distances [Å] and angles [°]: Fe(1)-Fe(2) 2.5598(5), Fe(1)-S(1) 2.2742(5), Fe(2)-S(1) 2.2474(6), Fe(1)-C(1), 1.781(3), Fe(2)-C(2), 1.793(2); Fe(1)-Fe(2)-C(1) 72.02(9), Fe(1)-C(1)-O(1) 170.1(3).
The unsaturated character of 1-BF4 is indicated by the speed of its reaction with CO, which occurs in seconds at − 45 °C. The product is proposed to be the adduct [1(CO)]BF4 (Scheme 2). The carbonylation is indicated by a shift of the ν̃CO bands (2021, 1970, 1883 cm−1 to 2030, 1987, 1783 cm− 1; see Table 1); the band for the bridging CO ligand (ν̃μ-CO) shifts by 100 cm−1. This dramatic shift for the bridging CO group is possibly due to the ligand moving into a bridging position from a bent-semibridging position. The spectrum of [1(CO)]BF4 matches that seen by Pickett upon oxidation of [Fe2{(SCH2)2CMeCH2SMe}(CN)2(CO)4]2−.[5] The observation of only three ν̃CO bands for this tetracarbonyl species results from accidental degeneracy: the 1987 cm−1 band arises from both the {Fe(dppv)(CO)terminal} site and the asymmetric component of the {Fe(CO)2(PMe3)} site. Carbonylation of 1-BF4 with 13CO resulted in the appearance of one new band in the IR spectrum (Figure 2), which is consistent with regioselective labeling. This new band is proposed to arise from the splitting of the 1987 cm−1 band in 1+ (see above). Significantly, ν̃μ-CO is barely affected (3 cm−1). In general, the ν̃CO bands for the terminal CO ligands in both 1-BF4 and [1(CO)]BF4 are higher in energy than those for Hox.[1,2] The IR spectra for 1-BF4 and [1(CO)]BF4 bear a resemblance to the patterns seen for the enzyme from D. desulfuricans.[11]
Scheme 2.
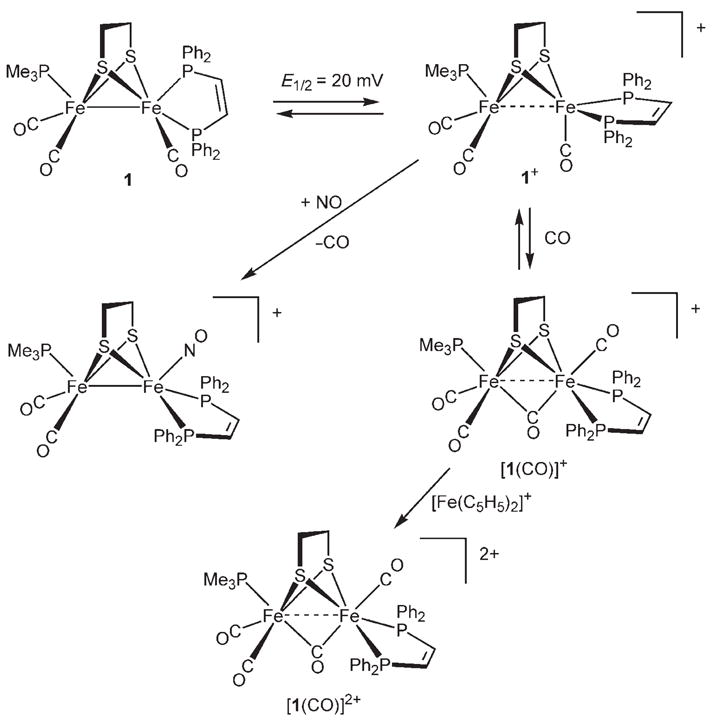
Synthesis and reactions of [Fe2(S2C2H4)(CO)3(PMe3)(dppv)]+.
Table 1.
IR spectroscopic data for Hox models and the enzyme.
| Complex | ν̃CO [cm−1] |
|---|---|
| [Fe2(S2C2H4)(CO)3(PMe3)(dppv)]+ (1+) | 2021, 1970, 1883 |
| [Fe2(S2C2H4)(μ-CO)(CO)3(PMe3)(dppv)]+ ([1(CO)]+) | 2030, 1987, 1783 |
| [Fe2(S2C2H4)(μ-CO)(CO)3(PMe3)(dppv)]2+ ([1(CO)]2+) | 2063, 2028, 1939 |
| Hox (D. desulfuricans)[11] | 1965, 1940, 1802 |
| HoxCO (D. desulfuricans)[11] | 2016, 1972, 1963, 1811 |
Figure 2.
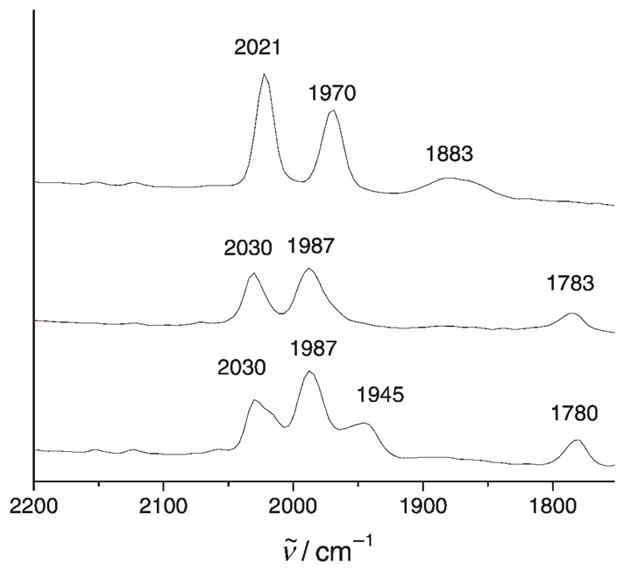
IR spectra (−40 °C, CH2Cl2 solution) of [Fe2(S2C2H4)(CO)3-(PMe3)(dppv)]BF4 before CO treatment (top) and after treatment with 12CO (middle) and 13CO (bottom).
An N2 purge slowly (0 °C, 30 min) displaced CO from [1(CO)]BF4 to give 1-BF4 with only moderate decomposition. A stable tetracarbonyl compound was generated by treating [1(CO)]BF4 with FcBF4. This reaction afforded the diamagnetic dication [1(CO)]2+, which was isolated in analytical purity as its tetrafluoroborate salt. NMR spectroscopy data indicate that [1(CO)]2+ has Cs symmetry (Scheme 2). Under an atmosphere of CO, [1(CO)]2+ is stable for hours at room temperature.
The radical character of 1-BF4 was demonstrated by its reactivity toward NO. At −45 °C, 1-BF4 rapidly and irreversibly binds NO. The product, which forms with loss of CO, is the diamagnetic dicarbonyl nitrosyl [Fe2(S2C2H4)(CO)2(NO)(PMe3)(dppv)]BF4 (ν̃NO = 1772 cm−1), which was isolated in analytical purity. 1H and 31P NMR spectra show that this {FeI2} species is Cs-symmetric, which uniquely requires that the NO ligand occupies the apical site on the {Fe(dppv)} center (Scheme 2).
In summary, the mixed-valence salt [Fe2(S2C2H4)(CO)3(PMe3)(dppv)]BF4 displays the structure and reactivity proposed for Hox, one of the two functional states of the [FeFe] hydrogenases.[1] Future reports will describe further spectroscopic characterization of this new generation of model complexes.
Experimental Section
[Fe2(S2C2H4)(CO)3(PMe3)(dppv)]BF4, 1-BF4: A solution of 1 (0.400 g, 0.526 mmol) in CH2Cl2 (5 mL) at −45 °C was treated with a solution of FcBF4 (0.143 g, 0.526 mmol) in CH2Cl2 (15 mL). The resulting purple solution was transferred into hexanes (200 mL) cooled to −45 °C to precipitate the product. Crystals were grown via slow diffusion of hexanes into a methylene chloride solution at −20°C. Yield 0.15 g (34%). The low yield is attributed to mechanical losses due to the large volume flask required for the precipitation. The method was optimized for the removal of the ferrocene coproduct, not yield. In situ IR measurements indicate that the conversion is virtually quantitative. IR (CH2Cl2): ν̃CO = 2021 (vs), 1970 (s), 1883 cm−1 (m, br). Elemental analysis calcd for C34H35BF4Fe2O3P3S2 : C 48.20, H 4.16; found: C 47.33, H 4.27.
Carbonylation of [Fe2(S2C2H4)(CO)3(PMe3)(dppv)]BF4 : A solution of 1 (0.050 g, 0.066 mmol) in CH2Cl2 (5 mL) at −45 °C was treated with a solution of FcBF4 (0.018 g, 0.066 mmol) in CH2Cl2 (2 mL). In situ IR spectra confirmed the formation of [Fe2(S2C2H4)(CO)3(PMe3)(dppv)]BF4. Maintaining the solution at −45°C, the solution was treated with a stream of CO (ca. 0.1 mLs−1 for 1 min) with no precautions to exclude ambient light. IR (CH2Cl2): ν̃CO = 2030 (s), 1987 (vs), 1783 cm−1 (m, br). The 13CO labeling experiment was conducted similarly.
[Fe2(S2C2H4)(CO)4(PMe3)(dppv)](BF4)2 : A solution of 1 (1.0 g, 1.31 mmol) in CH2Cl2 (10 mL) at −45 °C was treated with a solution of FcBF4 (0.71 g, 2.63 mmol) in CH2Cl2 (10 mL) under a CO atmosphere. The solution was allowed to warm to room temperature under a CO atmosphere and concentrated to 5 mL. The brown solid precipitated upon addition of 40 mL of hexanes. Yield: 0.98 g (91%). 1H NMR (500 MHz, CD2Cl2): δ = 8.8−7.3 (m, 20H; C6H5), 4.2 (s, 2H; PCH), 3.5 (m, 2H; SCH2), 2.0 (d, JPH = 12 Hz, 9H; PCH3), 1.4 ppm (m, 2H; SCH2). 31P NMR (202 MHz, CD2Cl2): δ = 68.0 (d, JPP =3 Hz), 38.2 ppm (t, JPP = 3 Hz). IR (CH2Cl2): ν̃CO = 2063, 2028, 1939 cm−1. Elemental analysis calcd for C35H35B2F8Fe2O4P3S2 : C 43.70, H 3.67; found: C 43.82, H 3.81.
[Fe2(S2C2H4)(CO)2(NO)(PMe3)(dppv)]BF4: A solution of [Fe2(S2C2H4)(CO)3(PMe3)(dppv)] (0.050 g, 0.066 mmol) in 5 mL of CH2Cl2 at −45 °C was treated with a solution of FcBF4 (0.018 g, 0.066 mmol) in CH2Cl2 (2 mL). An in situ IR spectrum of the reaction mixture showed [Fe2(S2C2H4)(CO)3(PMe3)(dppv)]+ in solution. The reaction mixture was treated with a stream of NO and after 1 min the IR spectrum showed complete conversion to [Fe2(S2C2H4)(CO)3(NO)(PMe3)(dppv)]+, which was spectroscopically identical to a sample prepared from the reaction of 1 and NOBF4. 1H NMR (500 MHz, CD2Cl2): δ =8.8−7.3 ppm (m, 20H; C6H5).31P NMR (202 MHz, CD2Cl2): δ =74.1, 25.0 ppm. IR (CH2Cl2): ν̃CO=2002, 1955 cm−1; ν̃NO = 1772 cm−1 ESI-MS: m/z 762.2 ([Fe2(S2C2H4)(CO)2(NO)(PMe3)(dppv)]+). Elemental analysis calcd for C33H35BF4Fe2NO3P3S2: C 46.67, H 4.15, N 1.65; found: C 46.66, H 4.37, N 1.62.
Footnotes
This research was supported by the US National Institutes of Health. We thank Teresa Prussak-Wieckowska for assistance on X-ray crystallography.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.Peters JW, Lanzilotta WN, Lemon BJ, Seefeldt LC. Science. 1998;282:1853. doi: 10.1126/science.282.5395.1853. [DOI] [PubMed] [Google Scholar]; Nicolet Y, Piras C, Legrand P, Hatchikian CE, Fontecilla-Camps JC. Structure. 1999;7:13. doi: 10.1016/s0969-2126(99)80005-7. [DOI] [PubMed] [Google Scholar]; Frey M. Chem Bio Chem. 2002;3:153. [Google Scholar]
- 2.Nicolet Y, Lemon BJ, Fontecilla-Camps JC, Peters JW. Trends Biochem Sci. 2000;25:138. doi: 10.1016/s0968-0004(99)01536-4. [DOI] [PubMed] [Google Scholar]
- 3.Ezzaher S, Capon JF, Gloaguen F, Pétillon FY, Schollhammer P, Talarmin J, Pichon R, Kervarec N. Inorg Chem. 2007;46:3426. doi: 10.1021/ic0703124. [DOI] [PubMed] [Google Scholar]; Ekström J, Abrahamsson M, Olson C, Bergquist J, Kaynak FB, Eriksson L, Sun L, Becker HC, Åkermark B, Hammarström L, Ott S. Dalton Trans. 2006:4599. doi: 10.1039/b606659c. [DOI] [PubMed] [Google Scholar]; Tye JW, Hall MB, Darensbourg MY. Proc Natl Acad Sci USA. 2005;102:16911. doi: 10.1073/pnas.0508740102. [DOI] [PMC free article] [PubMed] [Google Scholar]; Georgakaki IP, Darensbourg MY. Comp Coord Chem II. 2004;8:549. [Google Scholar]; Borg SJ, Behrsing T, Best SP, Razavet M, Liu X, Pickett CJ. J Am Chem Soc. 2004;126:16988. doi: 10.1021/ja045281f. [DOI] [PubMed] [Google Scholar]; Evans DJ, Pickett CJ. Chem Soc Rev. 2003;32:268. doi: 10.1039/b201317g. [DOI] [PubMed] [Google Scholar]
- 4.van der Vlugt JI, Rauchfuss TB, Whaley CM, Wilson SR. J Am Chem Soc. 2005;127:16012. doi: 10.1021/ja055475a. [DOI] [PubMed] [Google Scholar]; Schwartz L, Eilers G, Eriksson L, Gogoll A, Lomoth R, Ott S. Chem Commun. 2006:520. doi: 10.1039/b514280f. [DOI] [PubMed] [Google Scholar]; Dong W, Wang M, Liu X, Jin K, Li G, Wang F, Sun L. Chem Commun. 2006:305. doi: 10.1039/b513270c. [DOI] [PubMed] [Google Scholar]
- 5.Razavet M, Borg SJ, George SJ, Best SP, Fairhurst SA, Pickett CJ. Chem Commun. 2002:700. doi: 10.1039/b111613b. [DOI] [PubMed] [Google Scholar]
- 6.Liu T, Darensbourg MY. J Am Chem Soc. 2007;129:7008. doi: 10.1021/ja071851a. [DOI] [PubMed] [Google Scholar]
- 7.Justice AK, Zampella G, De Gioia L, Rauchfuss TB, van der Vlugt JI, Wilson SR. Inorg Chem. 2007;46:1655. doi: 10.1021/ic0618706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Albracht SPJ, Roseboom W, Hatchikian EC. J Biol Inorg Chem. 2005;11:88. doi: 10.1007/s00775-005-0039-8. [DOI] [PubMed] [Google Scholar]
- 9.Nicolet Y, de Lacey AL, Vernede X, Fernandez VM, Hatchikian EC, Fontecilla-Camps JC. J Am Chem Soc. 2001;123:1596. doi: 10.1021/ja0020963. [DOI] [PubMed] [Google Scholar]
- 10.Justice AK, Zampella G, De Gioia L, Rauchfuss TB. Chem Commun. 2007:2019. doi: 10.1039/b700754j. [DOI] [PubMed] [Google Scholar]
- 11.De Lacey AL, Stadler C, Cavazza C, Hatchikian EC, Fernandez VM. J Am Chem Soc. 2000;122:11232. [Google Scholar]