

Summary
SN1-type alkylating agents that produce cytotoxic O6-methyl-G (O6-meG) DNA adducts induce cell cycle arrest and apoptosis in a manner requiring the DNA mismatch repair (MMR) proteins MutSα and MutLα. Here, we show that checkpoint signaling in response to DNA methylation occurs during S phase and requires DNA replication that gives rise to O6-meG/T mispairs. DNA binding studies reveal that MutSα specifically recognizes O6-meG/T mispairs, but not O6-meG/C. In an in vitro assay, ATR-ATRIP, but not RPA, is preferentially recruited to O6-meG/T mismatches in a MutSαand MutLα-dependent manner. Furthermore, ATR kinase is activated to phosphorylate Chk1 in the presence of O6-meG/T mispairs and MMR proteins. These results suggest that MMR proteins can act as direct sensors of methylation damage and help recruit ATR-ATRIP to sites of cytotoxic O6-meG adducts to initiate ATR checkpoint signaling.
Introduction
Cells are continually exposed to exogenous and endogenous sources of DNA damage and respond via a series of DNA repair pathways. DNA damage can also activate checkpoint signaling to halt the progression of the cell cycle or induce apoptosis. DNA damage signaling involves members of the phosphoinositide 3-kinase-related kinase (PIKK) family, including DNA-dependent protein kinase (DNA-PK), ATM, and ATR (Abraham, 2004). ATM and DNA-PK respond to DNA double-strand breaks (DSBs), whereas ATR has been implicated in the response to a broad range of DNA lesions resulting from UV exposure, alkylating agents, ionizing radiation, and hypoxia as well as stalled replication forks. ATR also safeguards chromosome integrity during replicative stress in undamaged cells and regulates origin firing in normal S phase (Shechter et al., 2004). These damage transducers function together with mediator and adaptor proteins as part of a sophisticated network of interacting proteins to phosphorylate a large number of downstream effector proteins, e.g., Chk1, Nbs1, SMC1, and p53, ultimately leading to repair, transcriptional activation, cell cycle arrest, and apoptosis.
Although ATR is clearly implicated in the damage response, the molecular mechanism underlying the initial DNA damage sensor step is not well understood. In human cells, ATR forms a complex with ATR-interacting protein (ATRIP) and both proteins are required for the DNA damage checkpoint pathway (Cortez et al., 2001). A single-strand DNA binding protein, RPA, facilitates the recruitment of ATR-ATRIP to regions of single-strand DNA (ssDNA) and is required for the formation of ionizing radiation-induced ATR foci (You et al., 2002; Zou and Elledge, 2003) . Thus, RPA-coated ssDNA is proposed to be a DNA intermediate that activates ATR. Binding of ATR to ssDNA may not always require RPA; however, ATR by itself can bind ssDNA directly, and RPA-independent ssDNA binding perhaps mediated by other proteins has been reported for ATR-ATRIP (Bomgarden et al., 2004; Unsal-Kacmaz and Sancar, 2004). The role of RPA in damage checkpoint signaling is less clear. A requirement for RPA has been reported for the phosphorylation of Chk1 by ATR in response to DNA damage (Wang and Qin, 2003; Zou and Elledge, 2003), However, others report that Chk1 phosphorylation in response to UV, hydroxyurea, or ionizing radiation is not inhibited in cells depleted of RPA1 or RPA2 (Dodson et al., 2004). In a similar vein, phosphorylation of SMC1 in response to alkylation damage also occurs in RPA-depleted cells (Wang and Qin, 2003), and mutant human and Xenopus ATRIP proteins lacking an N-terminal region required for interaction with RPA and stable DNA binding nevertheless support ATR-mediated phosphorylation of Chk1 in response to DNA damage (Ball et al., 2005; Kim et al., 2005). Finally, topoisomerase IIβ binding protein 1 (TopBP1) strongly stimulates the kinase activity of ATR via a specific protein-protein interaction and is postulated to serve as a direct activator of ATR (Kumagai et al., 2006).
SN1-type alkylating agents such as N-methyl-N'-nitro-N-nitrosoguanidine (MNNG) produce several covalent DNA modifications, including cytotoxic O6-meG, and induce ATR-dependent signaling activation, cell cycle arrest, and apoptosis. Alkylation damage signaling and apoptosis induction require the MMR proteins MutSα and MutLα (Karran, 2001; Stojic et al., 2004a), but not ExoI (Wang and Qin, 2003; Cejka et al., 2005). Human MutSα binds to O6-meG mispairs in vitro (Duckett et al., 1996; Berardini et al., 2000). A large number of downstream targets, including p53, Chk1, Chk2, CDC25A, and SMC1, are activated in this MMR-protein-dependent signaling cascade (Duckett et al., 1999; Stojic et al., 2004b; Wang and Qin, 2003), placing MMR proteins upstream of ATR kinase or in the initiating steps of DNA damage signaling.
MMR contributes to the fidelity of DNA replication by targeting mismatches in newly synthesized DNA for correction (Schofield and Hsieh, 2003; Kunkel and Erie, 2005). MutSα, a heterodimer of MSH2 and MSH6 subunits, recognizes base-base mispairs and loops of a single unpaired base, and together with MutLα, a heterodimer of MLH1 and PMS2, targets the newly synthesized strand containing the error for excision and subsequent gap repair. In addition to recognizing mismatches involving normal DNA bases, MutSα also recognizes base pairs involving modified or damaged bases, including alkylated bases, cisplatin adducts, UV-induced adducts, oxidized bases, and fluoropyrimi-dines.
A fundamental question regarding the role of MMR in damage signaling is whether processing of damaged or modified sites by MMR is a prerequisite for checkpoint signal transduction. The finding that loss of MMR conferred tolerance to DNA alkylating agents led to the proposal that repeated attempts or futile cycles of MMR triggered by O6-meG mispairs resulted ultimately in cell death (reviewed in Karran [2001]). At low concentrations, alkylating agents induce replication inhibition in the second S phase posttreatment, resulting in G2 arrest (Stojic et al., 2004b; Zhukovskaya et al., 1994). In MMR-proficient 293T cells, ATM and Chk2 phosphorylation as well as Chk1 phosphorylation peak are similarly delayed, and ATR and RPA foci are observed 24−48 hr postdamage (Stojic et al., 2004b). The appearance of chromosomal aberrations and sister chromatid exchanges induced by MNNG are also delayed (Kaina, 2004). These results have led to the proposal that an intermediate that accumulates as a result of MMR-provoked excision of O6-meG mispairs triggers replication fork arrest in the second cell cycle that is rescued by homologous recombination. However, even in cases where MNNG induces delayed arrest in the second cell cycle posttreatment, ATR- and MMR-dependent phosphorylation of Chk1 is observed in the first cell cycle within 12 hr postdamage (Stojic et al., 2004b). Thus, MMR-dependent activation of ATR kinase activity occurs fairly rapidly and raises the possibility of an alternate signaling pathway.
The notion that MutSα has a direct role as a DNA damage sensor binding to O6-meG mispairs and communicating directly with the checkpoint machinery has been proposed previously (Kat et al., 1993; Fishel, 1999; Li, 1999). MMR and the corresponding antimutator role can be separated functionally from the DNA damage response role of MMR proteins. For example, cells having reduced levels of MSH2 or MLH1 protein are competent for MMR but fail to carry out checkpoint activation or undergo apoptosis in response to DNA alkylation (Lettieri et al., 1999; Cejka et al., 2003; Claij and te Riele, 2004). Even more intriguing has been recent reports of two separation of function alleles in murine Msh2 and Msh6 that encode the two subunits of MutSα. These mutations in or near the nucleotide binding site of MSH2 and MSH6 disable mismatch repair but leave intact the apoptotic response to DNA damaging agents, including MNNG (Yang et al., 2004; Lin et al., 2004). These separation of function alleles argue strongly that MMR and damage signaling involve distinct pathways. This notion is supported by protein-protein interactions between Msh6p and Mek1p, the ATR homolog in budding yeast (Gavin et al., 2006), and between human Msh2 and the checkpoint proteins Chk1 and Chk2 (Adamson et al., 2005; Brown et al., 2003).
Here, we investigate the early steps of DNA alkylation damage signaling involving MMR proteins and ATR and ask (1) when in the cell cycle is the checkpoint machinery engaged, (2) are MMR proteins and ATR recruited to O6-meG mispairs in vitro, and (3) does recognition of O6-meG by MMR proteins activate ATR kinase activity?
Results
Activation of Signaling in S Phase
As a starting point for in vitro studies, we confirmed that HeLa cells possess a damage signaling response to DNA alkylation by the SN1-type alkylating agent MNNG that is dependent on the MMR protein MSH2 and ATR, but not ATM (Figures S1A–S1D available in the Supplemental Data with this article online). Damage signaling was monitored by phosphorylation at S966 of SMC1 or S317 of Chk1 after treatment of HeLa cells with 10 μM MNNG. Lowering the dose of MNNG to 2 μM also elicited phosphorylation of SMC1 and Chk1 but only in the presence of O6-benzylguanine, an inhibitor of O6-methylgua-nine-DNA methyltransferase (MGMT) (Figure S1). Thus, in these HeLa cells, MMR-dependent phosphorylation of Chk1 after MNNG treatment is attributable to O6-meG adducts.
HeLaS3 cells exhibit checkpoint signaling after MNNG treatment during S phase as expected. Synchronized cultures of HeLaS3 cells released from a double-thymi-dine block were treated with 10 μM MNNG at various points in the cell cycle (Figure 1A). Phosphorylation of downstream targets Chk1 and SMC1 in MNNG-treated cells coincided with the relative abundance of cells in S phase peaking 2 hr after release from the thymidine block (Figure 1A). As the cells progressed through G2/M and G1 of the second cell cycle, levels of phosphorylated Chk1 and SMC1 decreased, rising again in the following S phase at 24 hr. Triggering of checkpoint signaling specifically during S phase was also observed at a lower dose of 2 μM MNNG but only in the presence of O6-benzylguanine, confirming again that damage signaling induced by MNNG is due to the presence of O6-meG adducts (Figure 1B).
Figure 1.
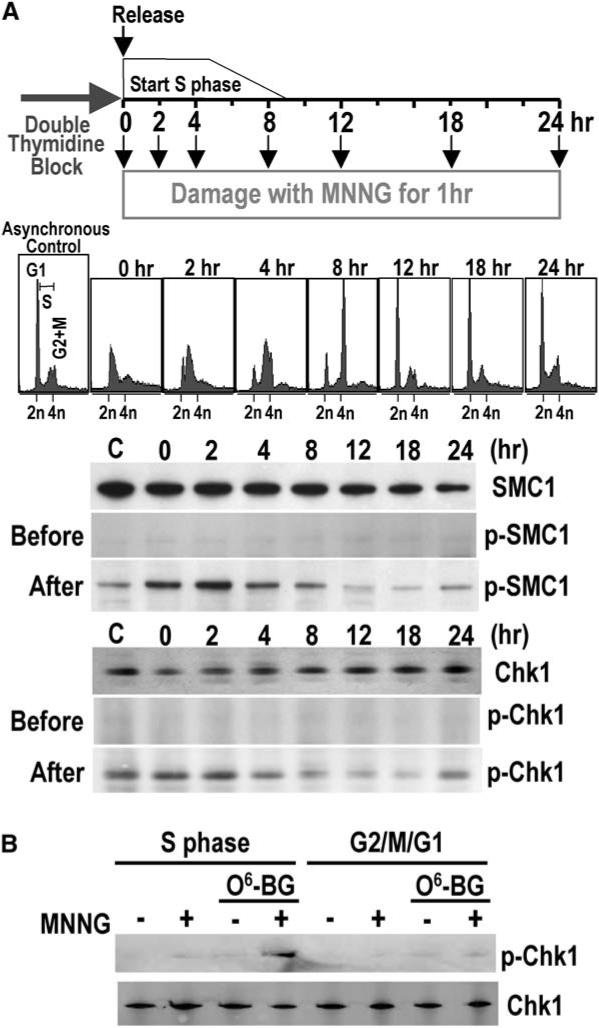
MNNG Damage Signaling Coincides with S Phase (A) Cell cycle dependence of MNNG damage signaling. Synchronized HeLaS3 cells were released from a double thymidine block and cultured for varying times in complete medium followed by incubation for 1 hr in serum-free medium in the presence or absence of 10 μM MNNG prior to harvesting. Synchronization was monitored by FACS prior to MNNG treatment. Phosphorylated and nonphosphorylated SMC1 and Chk1 were detected by immunoblotting. “C” is an asynchronous cell population. (B) DNA damage signaling in response to O6-meG adducts. HeLaS3 cells synchronized in S phase (2 hr after release from a double thymidine block) or G2-M-G1 phases (12 hr after release) were incubated in the presence or absence of O6-benzyl-guanine followed by incubation for 2 hr in serum-free medium containing 2 μM MNNG. Lysates were analyzed by immunoblotting.
The confinement of the damage response largely to S phase suggested that DNA replication might be required to elicit ATR signaling. Asynchronous cultures of HeLa cells or HeLa cells 30 min postrelease from a double-thymidine block were pretreated with aphidicolin prior to exposure to 10 μM MNNG for 1 or 2 hr (Figure 2). Aphidicolin treatment itself induced phosphorylation of SMC1 due to replication arrest, as expected. However, it is evident that aphidicolin treatment partially quenched phosphorylation of SMC1 in MNNG-treated cells. Thus, inhibiting replication can inhibit ATR checkpoint signaling in response to alkylation damage.
Figure 2.
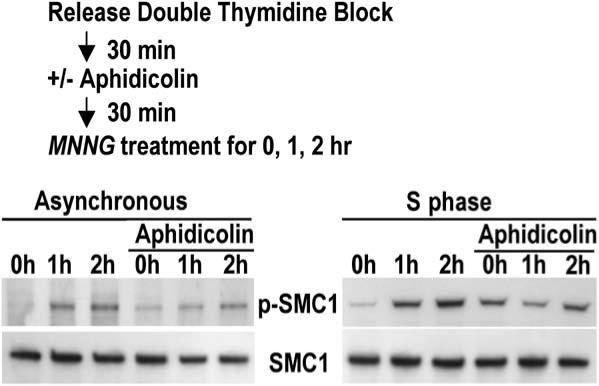
DNA Damage Signaling Triggered by MNNG Modification Depends on DNA Replication HeLaS3 cells, from either unsynchronized or synchronized cultures, were incubated in the presence or absence of aphidicolin followed by the addition of 10 μM MNNG for 1 or 2 hr in serum-free medium. Lysates were analyzed by immunoblotting to monitor phosphorylation of SMC1.
MutSα Binds Preferentially to O6-meG/T Mismatches
Whereas O6-meG/C exists in unreplicated DNA that has been subjected to MNNG modification, O6-meG/T mismatches are produced after replication in S phase because DNA polymerase preferentially incorporates dTMP opposite O6-meG (Dosanjh et al., 1991). Because checkpoint signaling peaks in S phase cells (Figure 1), the question arises as to whether MutSα preferentially recognizes O6-meG/T versus O6-meG/C mispairs.
Electrophoretic mobility shift assays were performed with purified recombinant MutSα (Figure S2) and DNA duplexes containing a single O6-meG/T, O6-meG/C, or G/T mismatch or a corresponding G/C base pair control (Figure 3). MutSα bound O6-meG/T mismatches as well as G/T mismatches (half-maximal binding at MutSα concentrations of 10 nM). In contrast, O6-meG/C was only bound nonspecifically with a relative affinity comparable to that for a G/C homoduplex control (half-maximal binding >100 nM). Because mismatch binding by MutS and MutS homologs exhibits sequence context effects, we examined binding in two other sequence contexts: one derived from the pBC-KS+ plasmid used in cross-linking experiments (see below) and one previously described in binding studies of yeast MSH2-MSH6 (Cejka et al., 2005). Preferential binding to a G/T mismatch and to an O6-meG/T mismatch in the pBC-KS+ sequence context was observed (Figure S3) though the relative affinity of the O6-meG/T mismatch was reduced in this sequence context compared that seen in Figure 3 (half-maximal binding at 8 nM and 34 nM MutSα for G/T and O6-meG/T, respectively). In a third sequence context, the O6-meG/T mismatch was not recognized any better than a G/C homoduplex (half-maximal binding at >100nM MutSα). Thus, although this sequence context elicits strong binding of an O6-meG/T mismatch by yeast MSH2-MSH6, murine MutSα fails to bind specifically to O6-meG/T in this sequence context despite retaining recognition of a G/T mismatch. The basis for the distinction by MutSα between G/T and O6-meG/T mismatches is not clear at present but may be relevant to the differing biological responses triggered by these two mismatches: repair and damage signaling.
Figure 3.
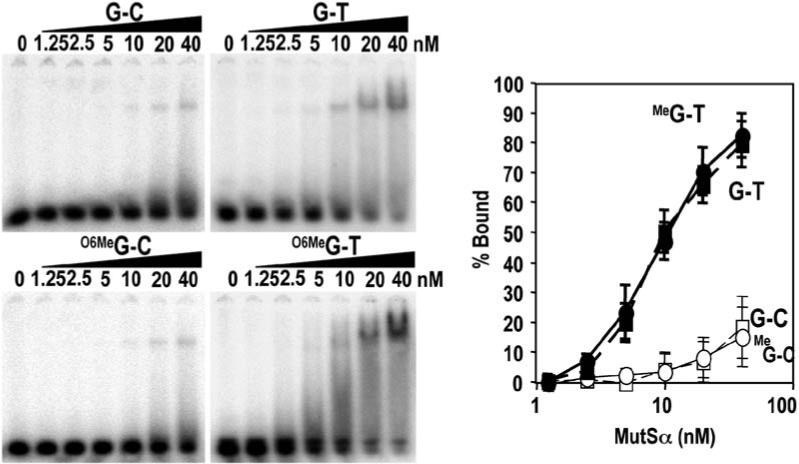
MutSα Binds to O6-meG/T Mispairs Gel mobility shift assays were performed with 41 bp DNA duplexes containing G/C, G/T, O6-meG/C, or O6-meG/T base pairs. 32P 5′ labeled DNA was incubated with the indicated concentration of purified recombinant MutSα and subjected to native gel electrophoresis. Concentrations of MutSα that gave half-maximal binding were 10 ± 0.9 nM for O6-meG/T, 10 ± 1.1 nM for G/T, and >100 nM for O6-meG/C and G/C. Error bars represent the SDs of three independent experiments.
Recruitment of ATR-ATRIP and MMR Proteins to O6-meG/T Mismatches
The preferential recognition of O6-meG/T mispairs, but not O6-meG/C mispairs, by MutSα and the requirement for DNA synthesis in S phase for DNA damage signaling raised the question of whether ATR might be recruited to sites of O6-meG/T mispairs to activate the ATR kinase. To determine whether MutSα, MutLα, or ATR-ATRIP is localized to O6-meG adducts, we utilized an in vitro crosslinking and immunoprecipitation assay (CrIP) (Figure 4A). Covalently closed, circular DNA substrates based on the pBC-KS+ plasmid containing a single O6-meG/T, O6-meG/C, or G/T mismatch or a control G/C base pair at site 1 were incubated with HeLaS3 nuclear extracts followed by crosslinking and shearing to prepare protein-DNA complexes. These complexes were subjected to immunoprecipitation with several different antisera, and after crosslink reversal and deproteinization, the substrates were subjected to paired PCR reactions to amplify site 1, containing the defined mismatches, and site 2 that served as an internal control for nonspecific DNA binding. Preferential association of a protein to mismatched DNA would be expected to yield enhanced recovery of site 1 over site 2, whereas nonspecific DNA binding should yield similar recoveries in both sites. Control experiments indicated that sites 1 and 2 were amplified with similar efficiencies (Figure 4B).
Figure 4.
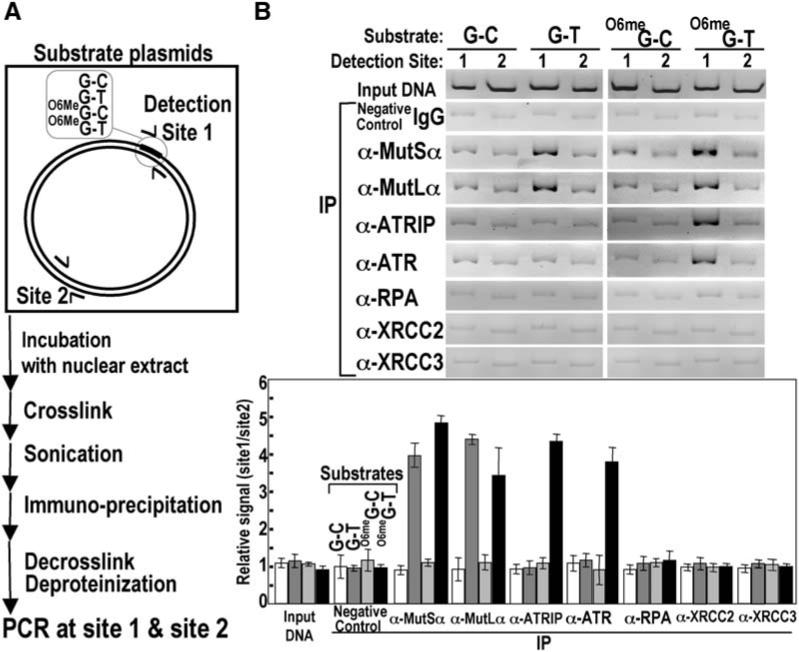
MutSα, MutLα, and ATR-ATRIP Localize to Regions of O6-meG/T Mispairs (A) Scheme for CrIP assay. See the Experimental Procedures for details. The preferred association of MutSα, MutLα, and ATR-ATRIP with various DNA mispairs was determined by the relative recovery of sites 1 and 2 after PCR. (B) MutSα, MutLα, and ATR-ATRIP associate preferentially with an O6-meG/T mispair. Relative recoveries of MutSα, MutLα, ATR/ATRIP, RPA, XRCC2, and XRCC3 at sites 1 and 2 were determined by the CrIP assay for G/C, G/T, O6-meG/C, and O6-meG/T substrates using HeLaS3 nuclear extracts and indicated antisera. Error bars represent the SDs of three independent experiments.
In vitro assays for mismatch excision repair require substrates containing a defined nick 5′ or 3′ to the mismatch. The covalently closed circular substrates used in CrIP assays do not support MMR as evidenced by the failure of PstI to cleave the G/T, O6-meG/T, and O6-meG/C substrates after incubation with HeLa nuclear extract (the PstI site resides at the mismatch position, Figure S6). As expected, the G/C control substrate was readily cleaved by PstI after incubation with HeLa extract.
Consistent with the results of in vitro DNA binding by MutSα described above, mismatched site 1 was preferentially recovered in the case of G/T and O6-meG/T mismatches by using antisera directed against either MutSα or MutLα. Preferential localization was not observed for either G/C or O6-meG/C substrates (Figure 4B). Strikingly, ATR and ATRIP were preferentially localized only to the O6-meG/T mismatch. These results indicate that although both G/T and O6-meG/T mismatches are preferred substrates in vitro for MutSα and MutLα they are differentiated in this assay with respect to recruitment of ATR-ATRIP. RPA exhibited nonspecific DNA binding in all four of the closed circular substrates (Figure 4B). Although preferential localization of RPA to damaged sites is not observed, it is possible that a subset of “activated” RPA molecules is preferentially localized to O6-meG adducts. Like RPA, XRCC2 and XRCC3 also failed to preferentially associate with O6-meG/T mismatches. These two proteins participate in homologous recombination. Whether other components of the recombination pathway are recruited requires further study.
Recruitment of ATR to O6-meG/T Requires MMR Proteins
MMR proteins are necessary for the localization of ATR-ATRIP to regions containing an O6-meG/T mispair. Nuclear extracts from MSH2-deficient LoVo and MLH1-deficient HCT116 cells failed to yield enhanced recovery of site 1 containing O6-meG/T mismatches with both anti-ATR and anti-ATRIP antisera (Figure 5). The preferential localization of ATR-ATRIP was restored upon the addition of 40 nM purified recombinant murine MutSα or MutLα (Figure S2) to nuclear extracts (Figure 5). These concentrations of MMR protein are very close to physiological concentrations as measured by quantitative Western blotting (Figure S5)(Genschel and Modrich, 2003). The ability of murine MMR proteins to function in place of human MMR proteins is consistent with extensive amino acid identity shared between murine and human MMR proteins and previous work demonstrating the restoration of MMR activity upon the addition of recombinant human MutLα protein to nuclear extracts from mouse embryo fibroblasts deficient in MLH1 or PMS2 (Tomer et al., 2002). The involvement of MMR proteins in the recruitment of ATR-ATRIP to sites of alkylation is further substantiated by the observation that MNNG treatment of HeLaS3 cells during S phase results in enhanced coimmunoprecipitation of MutSα with anti-ATRIP antibody (Figure S4).
Figure 5.
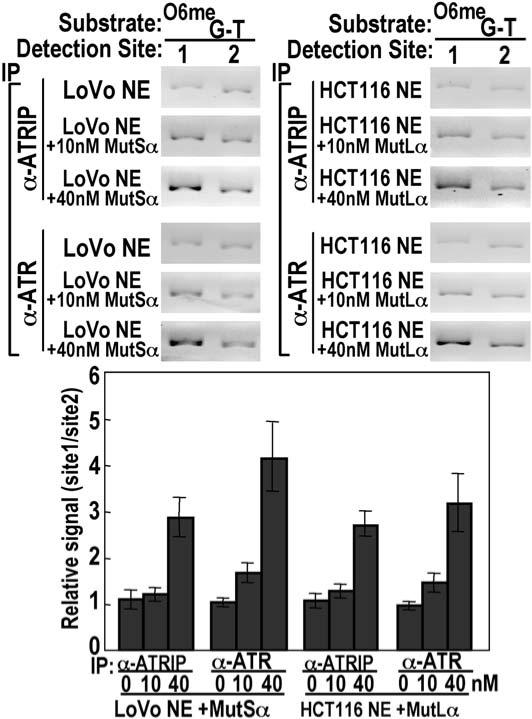
Association of ATR-ATRIP with O6-meG/T Requires Both MutSα and MutLα Recovery of ATR and ATRIP at O6-meG/T-containing DNA was determined in the CrIP assay by using LoVo or HCT116 nuclear extracts either alone or supplemented with purified, recombinant murine MutSα or MutLα as indicated. Error bars represent the SDs of three independent experiments.
ATR Kinase Activation by O6-meG/T Lesions in the Presence of MMR Proteins
Does formation of an ATR-ATRIP complex on O6-meG/T mismatches result in activation of ATR kinase and phosphorylation of a downstream target? To address this issue, we performed an in vitro kinase assay with HeLaS3 nuclear extract and the covalently closed circular DNAs containing a single mismatch used in CrIP experiments described above. Kinase activity was monitored by phosphorylation of recombinant His6-Chk1 at S317. We observed phosphorylated Chk1 only in the case of O6-meG/T-modified DNA; O6-meG/C-, G/T-, and G/C-containing substrates yielded only background levels of phosphorylated Chk1 (Figure 6A). The same results were obtained with a 100 bp duplex containing a single mispair or G/C base pair (Figure 6A). The phosphorylation of Chk1 is most likely attributable to ATR and not to other kinases such as DNA-PK because phosphorylation is specific for the O6-meG/T-containing substrate and is not triggered by the mere presence of double-strand ends in the DNA duplexes. We conclude that both recruitment of ATR to DNA and activation of ATR kinase activity occur only in the presence of O6-meG/T mispairs.
Figure 6.
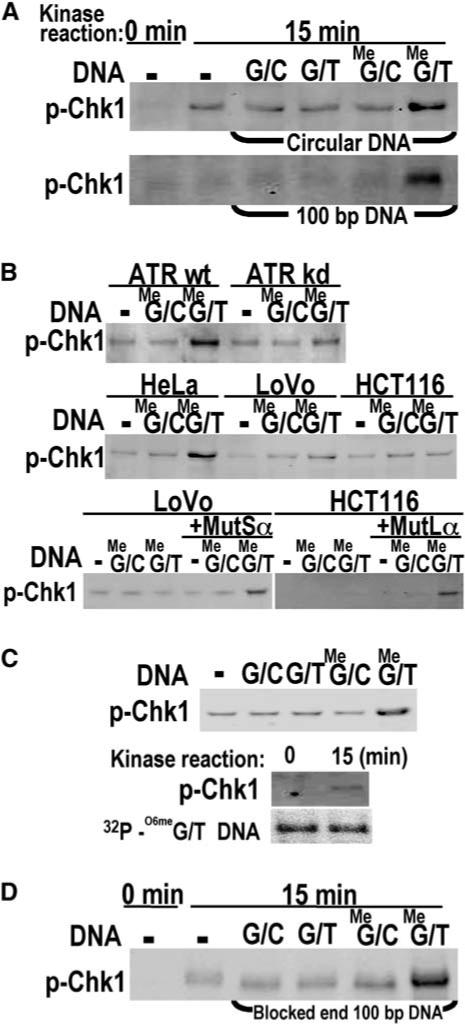
ATR Kinase Activation by O6-meG/T in the Presence of MutSα and MutLα (A) His6-Chk1 phosphorylation dependent on O6-meG/T. Kinase assays with 20 nM recombinant Chk1 and HeLaS3 nuclear extract were carried out with closed circular DNAs or 100 bp duplexes containing G/C, G/T, O6-meG/C, or O6-meG/T base pairs followed by Western blotting with anti-phosphoChk1 antibody. (B) O6-meG/T-dependent phosphorylation of Chk1 requires ATR, MutSα, and MutLα. Kinase assays were determined as in (A) in the absence or presence of 100 bp O6-meG/C or O6-meG/T DNA by using nuclear extracts from U2OS conditionally overexpressing wild-type (wt) or kinase-deficient (kd) ATR, or HeLaS3, LoVo, or HCT116 cells. Where indicated, LoVo or HCT116 nuclear extracts were supplemented with recombinant murine MutSα or MutLα. (C) Recovery of duplex DNA after ATR kinase assay. Top, Chk1 phosphorylation was assayed by using biotinylated 100 bp duplexes containing G/C, G/T, O6-meG/C, or O6-meG/T incubated with HeLaS3 nuclear extracts followed by a pull-down with streptavidin beads. Bottom, biotinylated 100 bp duplexes containing an O6-meG/T mispair and 32P 5′ labeled on both strands were used in a kinase assay after pull-down as described above. 32P 5′ labeled duplex DNA from kinase assays (15 min) or control samples (0 min) was also analyzed by denaturing PAGE. (D) O6-meG/T-dependent His6-Chk1 phosphorylation with blocked end DNA. Kinase assays contained streptavidin-dual-blocked-end-100 bp duplexes containing G/C, G/T, O6-meG/C, or O6-meG/T base pairs.
To confirm the involvement of ATR and MMR proteins in the O6-meG/T-dependent phosphorylation of Chk1, we repeated the kinase assay with nuclear extracts derived from U2OS cells conditionally overexpressing wild-type ATR or a dominant-negative kinase-deficient variant, ATR-kd, and with MSH2-deficient LoVo and MLH1-deficient HCT116 cells (Figure 6B). Phosphorylation at S317 of His6-Chk1 was observed in cells overex-pressing ATR-wt but substantially diminished in the ATR-kd nuclear extract (17% of wt level). Neither LoVo nor HCT116 nuclear extracts supported phosphorylation of Chk1. However, kinase activity was substantially restored upon the addition of 40 nM recombinant MutSα or MutLα, respectively (41% and 49% of HeLa levels, respectively, Figure 6B). These results together with the results of immunoprecipitation of protein-DNA complexes shown in Figure 4 suggest that the recruitment of ATR-ATRIP together with MutSα and MutLα at sites of O6-meG/T mispairs activates the ATR kinase resulting in phosphorylation of the downstream target Chk1.
Interestingly, the localization of ATR-ATRIP to O6-meG/T mispairs and ATR kinase activation both mediated by MMR proteins was observed with covalently closed circular DNAs that are not optimal substrates for in vitro mismatch repair assays in which excision is initiated from preexisting, defined nicks in the DNA substrate (Holmes et al., 1990; Thomas et al., 1991). To test the requirement for mismatch-provoked excision in the phosphorylation of Chk1 during the kinase reaction, we repeated kinase assays using a biotinylated, 100 bp O6-meG/T duplex that could be 32P 5′ end-labeled on both strands. As a control, the kinase assay was repeated with biotinylated, 100 bp duplexes containing G/T, O6-meG/C, O6-meG/T, or control G/C base pairs (Figure 6C, top). The biotinylated substrates yielded the same pattern of O6-meG/T–specific Chk1 phosphor-ylation observed previously. Kinase assays were then carried out after a streptavidin-biotin pull-down of nucleoprotein complexes containing the O6-meG/T duplex that was 32P 5′ end-labeled on both strands. After incubation of DNA bound nuclear proteins for 0 min or 15 min at 30°C under kinase assay conditions, the reaction was subjected to Western blotting for phosphorylated Chk1 or quenched by the addition of formamide and analyzed by denaturing urea PAGE. No discernible loss of the radiolabeled DNA was detected after 15 min incubation, and the DNA appeared to be full length. Kinase reactions were repeated with the 100 bp duplexes in which both ends were blocked with biotin-streptavidin to discourage exonucleolytic digestion (Figure 6D). Again, phosphorylation of Chk1 was enhanced ∼4-fold for O6-meG/T compared to the other three substrates. The absence of extensive DNA processing during the kinase reaction is consistent with the failure of RPA to preferentially localize to O6-meG/T in the immunoprecipitation experiments described above.
Discussion
The results of this study support a role for MMR proteins as direct sensors of DNA alkylation damage in which recognition of O6-meG/T lesions by MMR proteins results in recruitment of ATR-ATRIP and ATR kinase activation. This conclusion is supported by the following data. (1) Checkpoint signaling in response to cytotoxic O6-meG adducts requires MMR proteins, is largely constrained to cells in S phase, and requires DNA replication during which time O6-meG directs the misincorporation of T leading to O6-meG/T mismatches. (2) MutSα binds preferentially in vitro to O6-meG/T mismatches as compared to O6-meG/C base pairs. (3) ATR-ATRIP preferentially localizes in vitro to sites of O6-meG adducts, specifically O6-meG/T mismatches, but only when MMR proteins are present. Recruitment of ATR to O6-meG/T mismatches occurs without preferential binding of RPA to O6-meG/T mismatches and occurs in the absence of detectable MMR. Although G/T mismatches are preferred substrates for postreplication repair by the MMR system and are bound with high affinity by MutSα, G/T mismatches do not trigger preferential association of ATR-ATRIP. (4) Paralleling the specific recruitment of ATR to O6-meG/T mismatches, ATR kinase activation leading to phosphorylation of Chk1 in vitro is observed only in the presence of O6-meG/T mismatches and requires MMR proteins MutSα and MutLα. Extensive exonucleolytic processing of the DNA does not appear to be a requirement for kinase activation in the in vitro system.
Cell cycle arrest in response to MNNG is dependent on the relative levels of detoxifying MGMT activity in different cell types and concentrations of alkylating agent. Arrest in the second cell cycle postdamage is observed in response to low doses of MNNG in a variety of cells, including MMR-proficient 293T cells, and has led to a model for damage signaling in which an intermediate that accumulates as a result of aberrant MMR-provoked processing of O6-meG mispairs subsequently triggers replication stress in S phase of the next cell cycle (Stojic et al., 2004a). In this scenario, the role of MMR proteins in damage signaling is essential, albeit indirect. However, even in cases where low concentrations of MNNG induce delayed arrest in the second cell cycle posttreatment, ATR- and MMR-dependent phosphorylation of Chk1 is readily detected much earlier, within 12 hr postdamage (Stojic et al., 2004b). This rapid MMR-dependent ATR checkpoint signaling that precedes cell cycle arrest could readily be explained by a direct signaling mechanism involving MMR proteins. In addition, not all cells exhibit delayed arrest at low drug doses. An early block in cell cycle progression at a low dose of 0.4 μM MNNG is observed for WTK1 cells (di Pietro et al., 2003). Taken together, these findings suggest that there may exist more than one damage signaling pathway triggered in response to alkylation damage just as multiple ATM- and ATR-dependent pathways are activated after exposure to ionizing radiation.
Perhaps the strongest evidence to date supporting distinct pathways for MMR and damage signaling is separation of function alleles in murine Msh2 and Msh6 . These mutations disable mismatch repair but leave intact the apoptotic response to DNA damaging agents, including MNNG (Yang et al., 2004; Lin et al., 2004). The Msh2G674A missense mutation maps to the Walker “type A” motif in the ATP binding site of MSH2, whereas the Msh6T1217D mutation maps in a region adjacent to the ATP binding site of MSH6. Biochemical studies of mutant MutSα proteins containing these amino acid changes reveal that they bind to mismatches with normal affinity (Das Gupta and Kolodner, 2000; Lin et al., 2004; Yang et al., 2004). However, unlike wild-type MutSα, the mutant proteins are slow to dissociate from mismatched DNA in the presence of ATP. Thus, they are likely to be defective in the normal ATP-provoked mismatch signaling of downstream excision repair steps. Nevertheless, they support checkpoint signaling in response to DNA damage. These studies support the notion that MMR proteins can trigger a DNA damage response independently of MMR, which is supported by our findings.
How might MutSα and MutLα be involved in the in vitro localization of ATR and ATRIP to O6-meG/T mismatches? Obviously by virtue of its affinity for O6-meG/T mismatches, MutSα plays the pivotal role in initiating a DNA damage response that is specific for O6-meG/T mismatches. A constitutive physical interaction between ATR and human Msh2 has been observed in nuclear extracts of HeLa cells (Wang and Qin, 2003). Thus, it is possible that this interaction mediates the recruitment of ATR. The role of MutLα is more enigmatic. We know that, in both bacterial and eukaryotic cells, MutS and MutL proteins interact and modulate interactions on a DNA mismatch thereby signaling downstream excision steps of MMR. In addition, MutL and its homologs have been postulated to function as molecular chaperones recruiting proteins essential for downstream events in repair. In the case of damage signaling, a similar function for MutLα may exist. Finally, we cannot rule out the possibility that other as yet unidentified components are required for the recruitment and activation of ATR.
There is precedent for the notion that mismatch recognition by MSH2-MSH6 can trigger a pathway distinct from canonical mismatch excision repair. MMR plays an important role in inhibiting recombination between divergent sequences or homeologous recombination (reviewed in Schofield and Hsieh [2003]). In budding yeast, a DSB between two repeated sequences is subject to repair involving single-strand annealing that gives rise to a heteroduplex intermediate. Mismatches in this heteroduplex are subject to correction via normal mismatch excision repair involving MSH2-MSH6 and MLH1-PMS1. However, if the density of mismatches reaches a critical threshold, MSH2-MSH6 suppresses homeologous recombination and gross chromosomal rearrangements. Sgs1, a DNA helicase, appears to have a redundant role with MMR in this suppression (Myung et al., 2001). Genetic studies focusing on the fate of the heteroduplex DNA suggest a scenario in which the recognition of mismatches by MSH2-MSH6 is followed by unwinding of the DNA strand by Sgs1 helicase as opposed to nucleolytic degradation (Sugawara et al., 2004). Interactions between Msh2p and Msh6p and Sgs1p have been observed in budding yeast as have interactions between human Msh6 and Mlh1 and BLM, an Sgs1p homolog (Bachrati and Hickson, 2003; Gavin et al., 2006). Although the precise molecular mechanism of homeologous recombination suppression by MMR proteins remains under study, these findings support the idea that MutSα can recognize mismatches and initiate a pathway(s) distinct from mismatch excision repair.
The formation of a signaling complex at O6-meG/T mispairs containing as core components MutSα, MutLα, and ATR-ATRIP leads to activation of ATR kinase and phosphorylation of Chk1 in vitro. In the nucleus, checkpoint signaling would be damage specific by virtue of MutSα's interaction with O6-meG/T and would be triggered during S phase when O6-meG/T mismatches arise after DNA synthesis. A direct signaling mechanism could be responsible for the checkpoint signaling observed in the first cell cycle after alkylation damage. We posit that the formation of persistent signaling complexes on methylated DNA could disrupt DNA synthesis in the ensuing cell cycle, causing delayed cell cycle arrest. The development of an in vitro system will aid future studies of damage signaling mediated by MMR proteins and facilitate the identification of the full complement of proteins required to elicit the damage response.
Experimental Procedures
Cell Culture, Synchronization, and DNA Modification
HeLaS3 (American Type Culture Collection), LoVo (ATCC), and HCT116 (ATCC) cells were maintained at 37°C in DMEM (Mediatech) with 10% fetal bovine serum (Invitrogen), 50 U penicillin, and 50 μg/ml streptomycin (Invitrogen) in a 5% CO2 humidified atmosphere. U2OS cell lines conditionally overexpressing ATR-wt (GW33) or kinase-deficient (GK41) protein (Paul Nghiem, Massachusetts General Hospital, Boston, MA) were maintained in DMEM supplemented with 10% FCS, 200 μg/mL G418 (Sigma), and 200 μg/mL Hygromycin B (Calbiochem). Inductions of ATR-wt and ATR-kd were accomplished by supplementing the growth medium with 1 μg/mL doxycycline (Sigma) for 48 hr, as described (Nghiem et al., 2001). For cell synchronization, a double thymidine block was performed essentially as described (Bostock et al., 1971). Cells were grown for 18 hr in complete medium with 1 μM thymidine (Sigma), an additional 15 hr without thymidine, then an additional 16 hr with 1 μM thymi-dine. Harvested cells were analyzed by propidium iodide-flow cyto-metric analysis (Beckman Coulter, Inc., Fullerton, CA) after fixation with 70% ethanol, incubation in PBS containing RNase A (100 μg/ml, Sigma) for 1 hr at 37°C, staining with propidium iodide (20 μg/ml, Sigma), and incubation on ice in the dark for 30 min. Cells were incubated with 2 or 10 μM MNNG (Sigma) for 1 hr in serum-free medium, unless otherwise indicated, harvested, or washed with PBS and grown in complete medium. DNA synthesis was inhibited by treatment with 5 μM aphidicolin (Sigma).
Antibodies and Immunoprecipitation
Anti-MutSα (MSH2-His6-MSH6) and anti-MutLα (MLH1-His6-PMS2) polyclonal antibodies were generated by immunizing rabbits (Strategic Biosolutions) with purified recombinant mouse MutSα or MutLα protein. Serum was purified by passage over a Hi-Trap Protein G column (Amersham). Western blotting and immunoprecipitation were performed with anti-MutSα, anti-MutLα, anti-ATRIP (A300−095A Bethyl Laboratories), anti-ATR (A300−138A Bethyl Laboratories and ab2905 Abcam), anti-SMC1 (A300−055A Bethyl Laboratories), anti-SMC1-pS966 (A300−050A Bethyl Laboratories), anti-Chk1 (A300−298A Bethyl Laboratories), anti-Chk1-pS317 (A300−163A Bethyl Laboratories), anti-RPA (NA13 Calbiochem and ab2175 Abcam), anti-XRCC2 (ab2367 Abcam and sc5895 and sc5896 Santa Cruz Biotechnology), anti-XRCC3 (ab2367 and ab6494 Abcam), and negative control rabbit IgG (Sigma).
Recombinant MutSα, MutLα, and Chk1
Sequences corresponding to full-length mouse Msh2, Msh6, Mlh1, and Pms2 ORFs were amplified by PCR from a mouse embryonic cDNA library (Stratagene). The human Chk1 ORF was amplified by PCR from the cDNA (NM_001274 Open Biosystem). Sequences were cloned into pFastBac vectors (Invitrogen) and recombinant proteins were purified from lysates of Sf9 or HighFive cells (see Supplemental Data).
DNA Binding Assays
MutSα was incubated with 0.01 nM 5′ 32P labeled DNA in binding buffer (25 μM HEPES [pH 7.5], 100 μM KCl, 2 μM MgCl2, 1 μM dithiothreitol, and 5% glycerol) in 20 μl total volume. After 30 min at 25°C, 2 μl of 20% Ficoll was added, and samples were loaded onto preelectrophoresed 5% native polyacrylamide gels at 4°C in 0.5 × TBE with 5% glycerol. After electrophoresis at 4°C, gels were dried and quantitated on a Fuji BAS-2500 PhosphorImager. Sequences of oligonucleotides used for duplex DNA substrates were as follows (G denotes the guanine in G/C or G/T; X denotes the O6-meG in O6-meG/C or O6-meG/T): G/C or G/T, 5′-GCTTAGGATCATCGAGGATCGAGCTCGGTGCAATTCAGCGG-3′; and O6-meG/C or O6-meG/T, 5′-GCTTAGGATCATCGAGGATCXAGCTCGGTGCAATTCAGCGG-3′ and the corresponding complementary sequences.
Nuclear Extract Preparation and CrIP Assay
Nuclear extracts were prepared from nuclei of HeLaS3, LoVo (MSH2 deficient), or HCT116 (MLH1 deficient) as described (Dignam et al., 1983), flash frozen in liquid N2, and stored at −80°C. Covalently closed circular DNA substrates for CrIP assays containing a single G/T, O6-meG/T, or O6-meG/C mismatch or G/C control in nearly identical sequence contexts were constructed in a trimolecular annealing/ligation reaction (Lu et al., 1983). Mismatches reside at a unique PstI restriction endonuclease site for monitoring the presence of mismatches. pBC-KS+ plasmid DNA (Stratagene) was digested with XbaI and HindIII, and the larger fragment was purified on agarose gels. This double-strand fragment was incubated with the plus strand of pBC-KS+, and one of four 5′-phosphorylated, PAGE-purified oligodeoxynucleotides spanning the XbaI and HindIII sites of the minus strand of pBC-KS+ in a 1:3:6 molar ratio. After alkaline denaturation and high-salt neutralization, the DNAs were annealed at 65°C for 2 hr followed by incubation at 37°C and slow cooling. Nicked circular DNA, Form II, was purified on agarose gels followed by ligation with T4 DNA ligase (New England Biolabs) in the presence of 3.1 μM ethidium bromide. Closed circular, supercoiled dsDNA substrates were purified on 1% agarose gels in 1 × TAE by using GFX columns (GE Health), and DNA concentrations were determined spectrophotometrically. Retention of G/T, O6-meG/C, and O6-meG/T mismatches was confirmed by protection from PstI cleavage. The sequences of the four deoxyoligonucleotides were as follows (G denotes the guanine mispaired with thymine, X denotes the O6-meG paired with cytosine, and X denotes the O6-meG mispaired with thymine): G/C, 5′-CTAGAACTAGTGGATCCCCCGGGCTGCAGGAATTCGATATCA-3′; G/T, 5′-CTAGAACTAGTGGATCCCCCGGGCTGCGGGAATTCGATATCA-3′; O6-meG/C, 5′-CTA GAACTAGTGGATCCCCCGGGCTXCAGGAATTCGATATCA-3′; O6-meG/T, 5′-CTAGAACTAGTGGATCCCCCGGGCTGCXGGAATTCGA TATCA-3′. Nuclear extract (final protein concentration 2 μg/μl) was incubated with 0.3 ng/μl DNA substrate and 2 μg/μl sonicated salmon sperm DNA (Eppendorf) in a total volume of 65 μl in PBS for 1 hr at 4°C. After crosslinking in 1% formaldehyde for 15 min at 37°C, the solutions were brought to 200 μl with dilution buffer (final concentration 1.1% Triton X-100, 1.2 μM EDTA, 16.7 μM Tris-HCl [pH 8.1], and 167 μM NaCl) and sonicated. The solutions were diluted 10-fold with dilution buffer in the presence of BSA (final 15 μg/ml) and nonspecifically adsorbed with protein G plus/protein A agarose (Calbiochem). Immunoprecipitations were performed with the resulting supernatant with anti-MutSα, anti-MutLα, anti-ATR, anti-ATRIP, or anti-RPA antibodies or control IgG with protein G plus/protein A agarose. After washing once in low salt buffer (1% Triton X-100, 2 μM EDTA, 20 μM Tris-HCl [pH 8.1], and 150 μM NaCl), once in high salt buffer (1% Triton X-100, 2 μM EDTA, 20 μM Tris-Hcl [pH 8.1], and 500 μM NaCl), once in LiCl buffer (250 μM LiCl, 1% NP-40, 1 μM EDTA, and 10 μM Tris-HCl [pH 8.1]), and twice in TE, the precipitates were eluted twice with 50 μl elution buffer (10 μM DTT, 1% SDS, and 0.1 M NaHCO3) at room temperature for 20 min. Crosslinks were reversed by adding 4 μl 5M NaCl to 100 μl eluate and incubating for 16 hr at 65°C. For deproteinization, 2 μl 0.5M EDTA (pH 8.0), 4 μl 1M Tris-HCl (pH 6.5), and 0.6 μl of 6 μg/ml Proteinase K were added, followed by incubation for 1 hr at 45°C. After phenol/chloroform extraction, DNAs were precipitated with ethanol and redissolved in 20 μl H2O. Recovery was detected after 26 cycles of PCR, 94°C (30 s), 58°C (30 s), and 72°C (30 s) at site 1 containing O6-meG or G/T mismatched site (positions 627−722) and site 2 (internal control corresponding to positions 1969−059) by using LA-Taq polymerase (Takara). Amplified products were separated on 5% native polyacrylamide gels in 0.5 × TBE and stained with ethidium bromide. Quantitation of relative recoveries of site 1 versus site 2 was carried out by using a ChemiImager 5500 imaging system and AlphaEase FC software (Alpha Innotech). Each experiment was performed at least three times from which the standard deviation was determined. The PCR primer sequences were as follows: site 1 forward, 5′-GTAATACGACTCACTATAGGG-3′; site 1 reverse, 5′-GCTTGATATCGAATTCCTGCA-3′; site 2 forward, 5′-CAAACTGGCCTCAGGCATTT-3′; and site 2 reverse, 5′-GCCGCTTATGTCTATTGCTGGT-3′.
Kinase Assay
Kinase assays were performed in 25 μM HEPES (pH 7.5), 150 μM potassium chloride, 3 μM magnesium chloride, 10% glycerol, 0.5 μM ATP, and 1 μM dithiothreitol (DTT) for 15 min at 30°C in a volume of 15 μl. Nuclear extracts (final protein concentration 2 μg/μl) were incubated with 20 nM recombinant His6-Chk1 and 10 nM closed circular supercoiled DNA used in CrIP assays, 20 nM 100 bp linear DNA, or 20 nM 100 bp linear duplexes blocked at both 5′ ends with streptavidin-biotin (Sigma). Sequences of 100 bp linear DNAs were as follows (X denotes the guanine or O6-meG paired with cyto-sine for G/C or O6-meG/C substrates or mispaired with thymine for G/T or O6-meG/T substrates): 5′-CCACCGCGGTGGCGGCCGCTCTAGAACTAGTGGATCCCCCGGGCTGCAGXAATTCGATATCAAGCTTATCGATACCGTCGACCTCGAGGGGGGGCCCGGT-3′.
DNA pull-down assays were performed with biotin-labeled, 100 bp G/C, G/T, O6-meG/C, or O6-meG/T duplexes. After incubation with HeLaS3 nuclear extract in kinase buffer at 4°C for 1 hr, sonicated salmon sperm DNA (Eppendorf) at a final concentration of 2 μg/μl was added followed by further incubation at 4°C for 1 hr. Twenty microliters Streptavidin agarose (Novagen) was added followed by incubation at 4°C for 1 hr. The agarose beads were washed five times with 1 μl HNE buffer. The kinase activity of the pull-down complex was determined as described above.
DNA excision during kinase reactions was assessed with 5′ 32P labeled, biotinylated 100 bp O6-meG/T mismatch DNA using a biotinstreptavidin pull-down complex. After 15 min incubation at 30°C, the reactions were terminated by the addition of an equal volume of 50% formamide containing 50 μM EDTA. Samples were heated at 92°C for 3 min and analyzed on 5% denaturing polyacrylamide gels. After electrophoresis, gels were dried and quantitated on a Fuji BAS-2500 PhosphorImager.
Acknowledgments
We are grateful to Paul Nghiem, Massachusetts General Hospital, for providing ATR-wt and ATR-kd U2OS cells. We acknowledge Dan Camerini-Otero, Martin Gellert, Guo-Min Li, Tanya Paull, and anonymous reviewers for their helpful comments and suggestions. We also thank Rocky Cipriano for help in initial studies and George Poy for oligonucleotide syntheses. This work was funded by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health.
Supplementary Material
Supplemental Data
Supplemental Data include Supplemental Experimental Procedures, Supplemental References, and six figures and can be found with this article online at http://www.molecule.org/cgi/content/full/22/4/501/DC1/.
References
- Abraham RT. PI 3-kinase related kinases: ‘big’ players in stress-induced signaling pathways. DNA Repair (Amst.) 2004;3:883–887. doi: 10.1016/j.dnarep.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Adamson AW, Beardsley DI, Kim WJ, Gao Y, Baskaran R, Brown KD. Methylator-induced, mismatch repair-dependent G2 arrest is activated through Chk1 and Chk2. Mol. Biol. Cell. 2005;16:1513–1526. doi: 10.1091/mbc.E04-02-0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachrati CZ, Hickson ID. RecQ helicases: suppressors of tumorigenesis and premature aging. Biochem. J. 2003;374:577–606. doi: 10.1042/BJ20030491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball HL, Myers JS, Cortez D. ATRIP binding to replication protein A-single-stranded DNA promotes ATR-ATRIP localization but is dispensable for Chk1 phosphorylation. Mol. Biol. Cell. 2005;16:2372–2381. doi: 10.1091/mbc.E04-11-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berardini M, Mazurek A, Fishel R. The effect of O6-methylguanine DNA adducts on the adenosine nucleotide switch functions of hMSH2-hMSH6 and hMSH2-hMSH3. J. Biol. Chem. 2000;275:27851–27857. doi: 10.1074/jbc.M003589200. [DOI] [PubMed] [Google Scholar]
- Bomgarden RD, Yean D, Yee MC, Cimprich KA. A novel protein activity mediates DNA binding of an ATR-ATRIP complex. J. Biol. Chem. 2004;279:13346–13353. doi: 10.1074/jbc.M311098200. [DOI] [PubMed] [Google Scholar]
- Bostock CJ, Prescott DM, Kirkpatrick JB. An evaluation of the double thymidine block for synchronizing mammalian cells at the G1-S border. Exp. Cell Res. 1971;68:163–168. doi: 10.1016/0014-4827(71)90599-4. [DOI] [PubMed] [Google Scholar]
- Brown KD, Rathi A, Kamath R, Beardsley DI, Zhan Q, Mannino JL, Baskaran R. The mismatch repair system is required for S-phase checkpoint activation. Nat. Genet. 2003;33:80–84. doi: 10.1038/ng1052. [DOI] [PubMed] [Google Scholar]
- Cejka P, Stojic L, Mojas N, Russell AM, Heinimann K, Cannavo E, di Pietro M, Marra G, Jiricny J. Methylation-induced G(2)/M arrest requires a full complement of the mismatch repair protein hMLH1. EMBO J. 2003;22:2245–2254. doi: 10.1093/emboj/cdg216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cejka P, Mojas N, Gillet L, Schar P, Jiricny J. Homologous recombination rescues mismatch-repair-dependent cytotoxicity of S(N)1-type methylating agents in S. cerevisiae. Curr. Biol. 2005;15:1395–1400. doi: 10.1016/j.cub.2005.07.032. [DOI] [PubMed] [Google Scholar]
- Claij N, te Riele H. Msh2 deficiency does not contribute to cisplatin resistance in mouse embryonic stem cells. Oncogene. 2004;23:260–266. doi: 10.1038/sj.onc.1207015. [DOI] [PubMed] [Google Scholar]
- Cortez D, Guntuku S, Qin J, Elledge SJ. ATR and ATRIP: partners in checkpoint signaling. Science. 2001;294:1713–1716. doi: 10.1126/science.1065521. [DOI] [PubMed] [Google Scholar]
- Das Gupta R, Kolodner RD. Novel dominant mutations in Saccharomyces cerevisiae MSH6. Nat. Genet. 2000;24:53–56. doi: 10.1038/71684. [DOI] [PubMed] [Google Scholar]
- di Pietro M, Marra G, Cejka P, Stojic L, Menigatti M, Cattaruzza MS, Jiricny J. Mismatch repair-dependent transcriptome changes in human cells treated with the methylating agent N-methyl-n'-nitro-N-nitrosoguanidine. Cancer Res. 2003;63:8158–8166. [PubMed] [Google Scholar]
- Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson GE, Shi Y, Tibbetts RS. DNA replication defects, spontaneous DNA damage, and ATM-dependent checkpoint activation in replication protein A-deficient cells. J. Biol. Chem. 2004;279:34010–34014. doi: 10.1074/jbc.C400242200. [DOI] [PubMed] [Google Scholar]
- Dosanjh MK, Galeros G, Goodman MF, Singer B. Kinetics of extension of O6-methylguanine paired with cytosine or thymine in defined oligonucleotide sequences. Biochemistry. 1991;30:11595–11599. doi: 10.1021/bi00113a015. [DOI] [PubMed] [Google Scholar]
- Duckett DR, Drummond JT, Murchie AIH, Reardon JT, Sancar A, Lilley DM, Modrich P. Human MutSalpha recognizes damaged DNA base pairs containing O6-methylguanine, O4-methylthymine, or the cisplatin-d(GpG) adduct. Proc. Natl. Acad. Sci. USA. 1996;93:6443–6447. doi: 10.1073/pnas.93.13.6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duckett DR, Bronstein SM, Taya Y, Modrich P. hMutSα- and hMutLα-dependent phosphorylation of p53 in response to DNA methylator damage. Proc. Natl. Acad. Sci. USA. 1999;96:12384–12388. doi: 10.1073/pnas.96.22.12384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishel R. Signaling mismatch repair in cancer. Nat. Med. 1999;5:1239–1241. doi: 10.1038/15191. [DOI] [PubMed] [Google Scholar]
- Gavin AC, Aloy P, Grandi P, Krause R, Boesche M, Marzioch M, Rau C, Jensen LJ, Bastuck S, Dumpelfeld B, et al. Proteome survey reveals modularity of the yeast cell machinery. Nature. 2006;440:613–636. doi: 10.1038/nature04532. [DOI] [PubMed] [Google Scholar]
- Genschel J, Modrich P. Mechanism of 5’-directed excision in human mismatch repair. Mol. Cell. 2003;12:1077–1086. doi: 10.1016/s1097-2765(03)00428-3. [DOI] [PubMed] [Google Scholar]
- Holmes JJ, Clark S, Modrich P. Strand-specific mismatch correction in nuclear extracts of human and Drosophila melanogaster cell lines. Proc. Natl. Acad. Sci. USA. 1990;87:5837–5841. doi: 10.1073/pnas.87.15.5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaina B. Mechanisms and consequences of methylating agent-induced SCEs and chromosomal aberrations: a long road traveled and still a far way to go. Cytogenet. Genome Res. 2004;104:77–86. doi: 10.1159/000077469. [DOI] [PubMed] [Google Scholar]
- Karran P. Mechanisms of tolerance to DNA damaging therapeutic drugs. Carcinogenesis. 2001;22:1931–1937. doi: 10.1093/carcin/22.12.1931. [DOI] [PubMed] [Google Scholar]
- Kat A, Thilly WG, Fang WH, Longley MJ, Li GM, Mod-rich P. An alkylation-tolerant, mutator human cell line is deficient in strand-specific mismatch repair. Proc. Natl. Acad. Sci. USA. 1993;90:6424–6428. doi: 10.1073/pnas.90.14.6424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SM, Kumagai A, Lee J, Dunphy WG. Phosphorylation of Chk1 by ATM- and Rad3-related (ATR) in Xenopus egg extracts requires binding of ATRIP to ATR but not the stable DNA-binding or coiled-coil domains of ATRIP. J. Biol. Chem. 2005;280:38355–38364. doi: 10.1074/jbc.M508673200. [DOI] [PubMed] [Google Scholar]
- Kumagai A, Lee J, Yoo HY, Dunphy WG. TopBP1 activates the ATR-ATRIP complex. Cell. 2006;124:943–955. doi: 10.1016/j.cell.2005.12.041. [DOI] [PubMed] [Google Scholar]
- Kunkel TA, Erie DA. DNA mismatch repair. Annu. Rev. Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- Lettieri T, Marra G, Aquilina G, Bignami M, Crompton NE, Palombo F, Jiricny J. Effect of hMSH6 cDNA expression on the phenotype of mismatch repair-deficient colon cancer cell line HCT15. Carcinogenesis. 1999;20:373–382. doi: 10.1093/carcin/20.3.373. [DOI] [PubMed] [Google Scholar]
- Li GM. The role of mismatch repair in DNA damage-induced apoptosis. Oncol. Res. 1999;11:393–400. [PubMed] [Google Scholar]
- Lin DP, Wang Y, Scherer SJ, Clark AB, Yang K, Avdievich E, Jin B, Werling U, Parris T, Kurihara N, et al. An Msh2 point mutation uncouples DNA mismatch repair and apoptosis. Cancer Res. 2004;64:517–522. doi: 10.1158/0008-5472.can-03-2957. [DOI] [PubMed] [Google Scholar]
- Lu AL, Clark S, Modrich P. Methyl-directed repair of DNA base-pair mismatches in vitro. Proc. Natl. Acad. Sci. USA. 1983;80:4639–4643. doi: 10.1073/pnas.80.15.4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myung K, Datta A, Chen C, Kolodner RD. SGS1, the Saccharomyces cerevisiae homologue of BLM and WRN, suppresses genome instability and homeologous recombination. Nat. Genet. 2001;27:113–116. doi: 10.1038/83673. [DOI] [PubMed] [Google Scholar]
- Nghiem P, Park PK, Kim Y, Vaziri C, Schreiber SL. ATR inhibition selectively sensitizes G1 checkpoint-deficient cells to lethal premature chromatin condensation. Proc. Natl. Acad. Sci. USA. 2001;98:9092–9097. doi: 10.1073/pnas.161281798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield MJ, Hsieh P. DNA mismatch repair: molecular mechanisms and biological function. Annu. Rev. Microbiol. 2003;57:579–608. doi: 10.1146/annurev.micro.57.030502.090847. [DOI] [PubMed] [Google Scholar]
- Shechter D, Costanzo V, Gautier J. Regulation of DNA replication by ATR: signaling in response to DNA intermediates. DNA Repair (Amst.) 2004;3:901–908. doi: 10.1016/j.dnarep.2004.03.020. [DOI] [PubMed] [Google Scholar]
- Stojic L, Brun R, Jiricny J. Mismatch repair and DNA damage signalling. DNA Repair (Amst.) 2004a;3:1091–1101. doi: 10.1016/j.dnarep.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Stojic L, Mojas N, Cejka P, Di Pietro M, Ferrari S, Marra G, Jiricny J. Mismatch repair-dependent G2 checkpoint induced by low doses of SN1 type methylating agents requires the ATR kinase. Genes Dev. 2004b;18:1331–1344. doi: 10.1101/gad.294404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara N, Goldfarb T, Studamire B, Alani E, Haber JE. Heteroduplex rejection during single-strand annealing requires Sgs1 helicase and mismatch repair proteins Msh2 and Msh6 but not Pms1. Proc. Natl. Acad. Sci. USA. 2004;101:9315–9320. doi: 10.1073/pnas.0305749101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DC, Roberts JD, Kunkel TA. Heteroduplex repair in extracts of human HeLa cells. J. Biol. Chem. 1991;266:3744–3751. [PubMed] [Google Scholar]
- Tomer G, Buermeyer AB, Nguyen MM, Liskay RM. Contribution of human mlh1 and pms2 ATPase activities to DNA mismatch repair. J. Biol. Chem. 2002;277:21801–21809. doi: 10.1074/jbc.M111342200. [DOI] [PubMed] [Google Scholar]
- Unsal-Kacmaz K, Sancar A. Quaternary structure of ATR and effects of ATRIP and replication protein A on its DNA binding and kinase activities. Mol. Cell. Biol. 2004;24:1292–1300. doi: 10.1128/MCB.24.3.1292-1300.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Qin J. MSH2 and ATR form a signaling module and regulate two branches of the damage response to DNA methylation. Proc. Natl. Acad. Sci. USA. 2003;100:15387–15392. doi: 10.1073/pnas.2536810100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Scherer SJ, Shell SS, Yang K, Kim M, Lipkin M, Kucherlapati R, Kolodner RD, Edelmann W. Dominant effects of an Msh6 missense mutation on DNA repair and cancer susceptibility. Cancer Cell. 2004;6:139–150. doi: 10.1016/j.ccr.2004.06.024. [DOI] [PubMed] [Google Scholar]
- You Z, Kong L, Newport J. The role of single-stranded DNA and polymerase alpha in establishing the ATR, Hus1 DNA replication checkpoint. J. Biol. Chem. 2002;277:27088–27093. doi: 10.1074/jbc.M204120200. [DOI] [PubMed] [Google Scholar]
- Zhukovskaya N, Branch P, Aquilina G, Karran P. DNA replication arrest and tolerance to DNA methylation damage. Carcinogenesis. 1994;15:2189–2194. doi: 10.1093/carcin/15.10.2189. [DOI] [PubMed] [Google Scholar]
- Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data
Supplemental Data include Supplemental Experimental Procedures, Supplemental References, and six figures and can be found with this article online at http://www.molecule.org/cgi/content/full/22/4/501/DC1/.