

Abstract
Mitogen- activated protein kinases (MAPKs) are key signaling molecules that respond to mitogenic stimulation or environmental stress, resulting in the expression of target proteins. c-Jun N-terminal kinase (JNK) and p38 MAPKs are activated by inflammatory cytokines or environmental stress. Specific p38 MAPK inhibitors, such as SB202190 or SB203580, are widely used to dissect p38 MAPK-related signal transduction mechanisms. While Using SB202190 to inhibit p38 MAPK-related signaling, we observed that SB202190 treatment could activate JNK. Further experiments showed that treatingment of cells with SB202190 could phosphorylate JNK and activating transcription factor 2 (ATF-2), and increased AP-1 DNA binding. Using multiple cell lines and primary endothelial cells, we demonstrated that specific p38 MAPK inhibitors SB202190 or SB203580 induces the activation of the JNK pathway. Further, using with RNA interference and kinase-inactive expression of intermediates of the JNK pathway, we demonstrated SB202190- or SB203580-induced JNK activation is dependsent on the MLK-3-MKK4/MKK7 -dependent signal transduction pathway. Finally, we demonstrate that treatment of cells with SB202190 or SB203580 induces the phosphorylation and activation of MLK3.
Introduction
The Mitogen-activated protein kinases (MAPKs) are key signaling molecules that transduce a plethora of cellular signals to the effector molecules [1, 2]. They are also key targets of therapeutic interventions for inflammatory diseases. There are three major forms of MAPK modules, such as including extracellular signal-regulating kinases (ERKs), ); the stress-activated protein kinase (SAPK), also known as or c-Jun N-terminal kinase (JNK), ); and the p38 MAPK. The ERKs are activated by mitogens, whereas the JNKs and p38 MAPKs are activated by environmental stress, ultraviolet light, osmotic shock, and inflammatory cytokines [3].
A large body of evidence indicates a crucial role for p38 MAPK in inflammation [3, 4]. Several groups have reported that specific, selective p38a/b MAPK inhibitors block the production of interleukin 1 (IL-1), tumor necrosis factor α, TNFa and IL-6 in vitro and in vivo. In addition, the p38 MAPK pathway is involved in the induction of several other inflammatory molecules, such as cyclooxygenase-2 and inducible nitric oxide synthase. Therefore, p38 MAPK inhibitors are promising candidates for the treatment of inflammatory diseases.
An initial series of pyridinyl imidazole anti-inflammatory agents served as tools to elucidate the role of p38 in inflammation [4, 5]. The bicyclic imidazole SKF-86002 was first reported to inhibit lipopolysaccharide-stimulated cytokine production [1]. Subsequently, SB203580 [4-(4-fluorophenyl)-2-(4-methylsulfinyl phenyl)-5-(4-pyridyl) 1H-imidazole] and other 2,4,5-triaryl imidazoles were prepared to be used as tools to search for molecular targets involved in cytokine regulation. SB202190 [4-(4-Fluorophenyl)-2-(4-hydroxyphenyl)-5-(4-pyridyl)1H-imidazole] and SB203580 are widely used as p38 MAPK inhibitors [1].
JNK (or SAPK) is activated by the cellular stress response and is distinctly different from p38 MAPK. JNK has three isoforms (JNK1, JNK2, and JNK3), of which JNK1 and JNK2 are ubiquitously expressed. The JNK signaling cascade functions by activating an initiating kinase, such as MAPK kinase kinase 1 (MEKK1), which in turn phosphorylates a MAPK kinase, such as MKK4/JNKK1 or MKK7/JNKK2, which then activates JNK by phosphorylation on Thr 183 and Tyr 185 residues [2, 6]. Activated JNK phosphorylates the transcription factors c-Jun, ATF-2, Elk-1, p53, and c-Myc [7], as well as non-transcription factors, such as members of the Bcl2 family (Bcl2, BclxL, Bim, and BAD) [7]. In addition to MEKK1, mixed lineage kinase-3 (MLK3), a MAP2K, has also been shown to activate JNK or p38 MAPK via activation of MKK7 or MKK6.
MLK3 is a widely expressed mammalian serine/threonine kinase that functions as a mitogen activated protein kinase kinase kinase (MAPKKK) and can activate multiple MAPK pathways [8]. MLK3-induced JNK activation, through dual phosphorylation of MKK4 and/or MKK7 is implicated in apoptosis in neuronal cells [9, 10]. Targeted gene disruption of mlk3 decreases tumor necrosis factor a (TNFa)-induced JNK activation [11]. In addition to its catalytic domain MLK3 contains several other regions important for its regulation, including N-terminal Src homology 3 domain, and a centrally located sipper followed by a Cdc42/Rac-interactive binding motif [12]. Activated forms of small GTPases such as Cdc42 and Rac can bind to MLK3 and increase MLK3 catalytic activity and potentiate its signaling to JNK [13, 14]. Recent studies have shown that Cdc42 promotes phosphorylation of Thr277 and ser281 within the activation domain of MLK3, leading to increased MLK3 activity [14].
Because there are many common inducers of JNK and p38 MAPK, inhibitors specific for p38 MAPK, such as SB202190 or SB203580, or inhibitors specific for JNK, such as SP600125, have been used to inhibit respective MAPKs [15, 16]. These inhibitors are widely used to dissect the signaling mechanisms involved in MAPK activation. While evaluating the inhibition of p38 MAPK activation by SB202190 and SB203580, we observed that these inhibitors inhibited p38 MAPK, but activated JNK. Further probing of this unexpected finding demonstrated that SB202190 could activate AP-1 and initiate ATF-2 phosphorylation, some of the downstream consequences of JNK activation.
Materials and Methods
Chemicals and reagents
The AP-1 consensus oligonucleotide (5′ CGC TTG ATG AGT CAG CCG GAA 3′) was obtained from Promega Corp. (Madison, WI). SB202190 and SB203580 were purchased from Sigma Chemical Co. (St. Louis, MO). SP600125 was purchased from Tocaris, UK. All other reagents were of ultrapure grade or ACS grade. Buffers were made in water purified with the Milli-Q system (Millipore Corp., Billerica, MA).
Cell culture and transfection and RNA interference
A549 human lung alveolar epithelial cells and MCF-7 human breast adenocarcinoma cell line were obtained from ATCC, and were cultured in Kaigan's modified F-12K and Dolbeco’s modified medium (DMEM, Mediatech Co., Herndon, VA), respectively. Human microvascular endothelial cells (HMVEC) were obtained from Clonetics (San Diego, CA) and propagated in endothelial medium (Clonetics). MLK-3 and p38 MAPK siRNA were purchased from Dharmacon RNA Technologies (Lafayette, CO). For transfection, A549 cells were seeded in 6-well plates to obtain 30% confluence at the time of transfection. Xtreme siRNA transfection reagent (Roche Applied Science, Indianapolis, IN) was used to transfect siRNA to a final concentration of 100 nM. Inhibition of gene expression by siRNA was determined after 48 hours by Western analysis. Cells were harvested, and the nuclear extract or total cell lysate was assayed for AP-1 DNA binding or Western blotting, respectively. HEK293T cells were cultured in complete DMEM. We obtained pcDNA4TO-MLK3 from Dr Means (Duke University Medical Center [8], and cloned the MLK3 cDNA into phCMV2 expression vector (GTS Technologies, CA). phCMV2-HA-MLK3 was transfected into HEK293T cells using genejammer transfection reagent (Contech, CA) using manufacturer’s instructions. After 48 hours, cells were either untreated or treated with 5 or 10 µM SB202190 or SB203580 for 4 hours. Following treatment cell lysates were prepared using lysis buffer (50 mM Tris-HCl at pH 7.5, 5 mM EDTA, 150 mM NaCl, 1% Triton X-100, 50 mM NaF, 10 mM sodium pyrophosphate, 25 mM β- glycerophosphate, 1 mM PMSF, 30 µL/mL aprotinin [Sigma Chemical Company, St. Louis, MO], and 1 mM Na3VO4). 500 µg of total protein was immunoprecipitated with anti-HA-agarose conjugate. Phospho-MLK3 (Thr277/Ser281) was detected in western blotting using phosphospecific antibodies from Cell Signaling Technologies (Beverly, MA). PcDNA3-Flag-MKK7 was obtained form Dr. Roger Davis (University of Massachusetts, Worcester, MA). The expression vector was transfected into HEK293T cells using Genejammer as stated earlier. After 48 hours, cell lysates was prepared and Flag-MKK7 was immunoprecipitated using anti-Flag-agarose conjugate (Sigma Chemical Co). The Flag-MKK7 was used as a substrate for MLK3 kinase assay.
Nuclear extract preparation and EMSA
Nuclear extracts were prepared as described earlier [17]. For the EMSA, the consensus AP-1 binding sequence 5′ CGC TTG ATG AGT CAG CCG GAA 3′ was obtained from Promega Corporation (Madison, WI). The EMSA was performed as described earlier [18].
Western blotting
Protein lysates were prepared using radio immunoprecipitation assay (RIPA) buffer containing 5% sodium deoxycholate, 1% SDS, and 1% Igepal in PBS with protease inhibitors; protein concentration was determined using Biorad protein assay reagent (Biorad). Equal amounts of protein were resolved on 10% SDS-polyacrylamide gel electrophoresis, and transferred onto nitrocellulose membrane (Hybond-ECL, Amersham Pharmacia Biotech). The blot was treated with appropriate dilutions of primary antibody and visualized using either Lumiglo (Cell Signaling Technology, Beverly, MA) or ECL plus system (Amersham Pharmacia Biotech, Piscataway, New Jersey), with appropriate HRP-conjugated secondary antibody.
JNK kinase assay
Activation of JNK was performed by a non-radioactive assay kit as per the manufacturer's protocol (New England Biolabs, Beverly, MA). Briefly, SAPK/JNK was precipitated from the cell lysate by c-Jun fusion protein bound to glutathione sepharose beads. c-Jun contains a high affinity-binding site for SAPK/JNK, N-terminal to the two phosphorylation sites, Ser63 and Ser 73. After selectively pulling down JNK using c-jun fusion protein beads, the beads were extensively washed and the kinase reaction was carried out in the presence of cold ATP in a final volume of 25µl. The reaction was stopped with 25 µl of 2X SDS sample buffer and loaded onto a 10% polyacrylamide gel. Protein was transferred to nitrocellulose by electroblotting, and c-jun phosphorylation was selectively measured using phospho-c-Jun antibody. This antibody specifically measures JNK-induced phosphorylation of c-Jun at Ser63, a site important for the c-Jun-dependent transcriptional activity (20).
MLK3 Kinase Assay
HA-MLK3 was immunoprecipitated from the cell lysates of HEK293T that were untreated or treated with 5 or 10 µM SB202190 or SB203580. Immunoprecipitated HA-MLK3 (10 to 15µl/reaction) was incubated with Flag-MKK7 in the kinase reaction buffer [25 mM Tris (pH 7.5), 5 mM β-glycerophosphate, 2 mM DTT, 0.1 mM Sodium orthovanadate (Na3VO4), 10 mM MgCl2, 1 mM ATP]. The reaction mixture was incubated for 30 min at 30°C. The reaction was stopped by adding 3X SDS sample buffer (30 µl) to the reaction. Samples (10 to 15 µL) was loaded onto SDS-polyacrylamide gel, and subsequently transferred to nitrocellulose membrane. Phospho-MKK7-ser271/Thr275 was detected by phospho-specific antibodies that was obtained from Cell Signaling Technologies (Beverly, MA). The blot was stripped and probed for unphosphorylated MKK7 or β-actin. In some cases a new western analysis was performed using similar amounts of Flag-MKK7 to confirm that the stripping did not unevenly removed proteins from the membrane.
Results
P38 MAPK inhibitors SB202190 and SB203580 induces phosphorylation of JNK in a dose-and time-dependent manner
While evaluating the effect of p38 MAPK and JNK on gene expression, we unexpectedly observed activation of JNK in SB202190-treated cells. Therefore, we further analyzed the effect of specific p38 MAPK inhibitors, SB202190 or SB203580, on JNK phosphorylation. A dose–response study in A549 cells demonstrated that JNK is indeed phosphorylated due to treatment of cells with SB202190 (Fig. 1A and 1B). The response was dose-dependent up to about 6 µM. A similar response was also observed in response to SB203580 treatment (Fig. 1C). A time-course analysis revealed that JNK activation occurs as early as 10 minutes in response to SB202190 (Fig. 2A) or SB203580 (Fig. 2C), and remains until about 4 hours in response to SB202190 (Fig. 2B) or 6 hours with SB203580 (Fig. 2D); further incubation with SB202190 beyond 4 hours did not increase JNK phosphorylation (Fig. 2B). However, JNK remain activated in cells treated with SB203580 beyond 6 hours (Fig. 2D). There was no increase in the levels of p38 MAPK protein in response to either SB202190 or SB203580. In addition, there was no phosphorylation of p38 MAPK in response to SB202190 or SB203580 (data not shown). Additionally, the levels of unphosphorylated JNK remained unchanged in response to the treatment of cells with these inhibitors.
Figure 1. Phosphorylation of JNK by specific p38 MAPK inhibitors SB202190 and SB203580: Dose-Response.


A549 cells were treated with indicated concentration of SB202190 or SB203580 for indicated time periods, and total cell lysate was prepared as mentioned in the Methods section. Western analysis of phospho-JNK and unphosphorylated JNK was performed as described using specific antibodies for phospho-JNK and JNK. (A) Dose–response of JNK phosphorylation in response to SB202190 in A549 cells. Cells were treated with 1, 3, or 6 µM of SB202190 in triplicate for 2 hours. Following incubation, cell lysates were probed for pJNK or JNK. (B) Densitometry ratios of pJNK to JNK from Figure 1A. (C) Dose–response of JNK phosphorylation due to treatment of cells with SB203580. A549 cells were treated with 0.5 to 7 µM of SB203580 for 2 hours, and cell lysates were probed for pJNK or JNK.
Figure 2. Time course of JNK phosphorylation due to treatment of A549 cells with p38 MAPK inhibitors SB202190 or SB203580.



A549 cells were treated with DMSO control or with SB202190 or SB203580 for indicated time periods, and total cell lysate was prepared as mentioned in the Methods section. Western analysis of phospho-JNK and unphosphorylated JNK was performed as described using antibodies specific for phospho-JNK and JNK. (A) A549 cells were treated with DMSO control or 5 µM of SB202190 and incubated for 10, 15, 20, or 25 minutes. Phospho-JNK was detected as mentioned in the Methods section. Lower Panel: p38 MAPK was detected using anti-p38 MAPK antibodies by Western analysis. (B) A549 cells were treated with DMSO control or with 10 µM of SB202190 for 0.5, 1, 2, 3, 4, or 6 hours, and pJNK and unphosphorylated JNK were detected as described. (C) A549 cells were treated with DMSO control or 5µM of SB203580 and incubated for 10, 15, 20, or 25 minutes, followed by detection of pJNK (Top panel), JNK (middle panel), and p38 MAPK (lower panel), as mentioned in the Methods section. (D) A549 cells were treated with DMSO control or SB203580 (5µM) for 1, 2, 3, 4, or 6 hours, followed by detection of pJNK (Top panel), JNK (middle panel), and p38 MAPK (lower panel), as described.
To further evaluate whether data obtained with A549 adenocarcinoma cells could be reproduced in a primary cell culture, we treated HMVEC cells with 10 µM SB202190 and examined the time-course of JNK activation by detecting JNK phosphorylation. As demonstrated in Fig. 3A, strong phosphorylation of JNK was observed after 30 minutes and JNK remain phosphorylated for 3–4 hours. We also evaluated the effect of SB202190 on JNK activation in MCF-7 cells to evaluate whether data obtained with A549 cells also apply to another cancer cell line. As demonstrated in Figure 3B, JNK was maximally phosphorylated after 1 hour of treatment with SB202190. This activation of JNK could be abrogated by SP600126, a specific JNK inhibitor. The level of unphosphorylated JNK in these experiments remained unaffected due to treatment with SB202190, indicating that the increased phosphorylation of JNK was not due to an increase in JNK protein, but was due to activation of JNK. Similar results were obtained with SB203580, another p38MAPK inhibitor very similar in structure to that of SB202190 (data not shown). These data demonstrate that the specific p38 MAPK inhibitors SB202190 or SB203580 can indeed activate JNK.
Figure 3.


(A) SB202190 induces phosphorylation of JNK in human microvascular endothelial cells (HMVEC) HMVEC cells were treated with DMSO control or with 10 µM of SB202190 for 0.5, 1, 1.5, 2, 3, 4, or 6 hours, followed by detection of pJNK or JNK as described in the Methods section. (B) SB202190 induces phosphorylation of JNK in MCF-7 cells. MCF-7 cells were treated with SB202190 (5µM) for 0.5, 1, 2, 4, or 6 hours, and pJNK or JNK was detected as mentioned in the Methods section (lanes 1–6). In lane 7, MCF-7 cells were pretreated with 5 µM of SP600125 for 2 hours, followed by treatment with 5 µM of SB202190, and incubated for another 2 hours, followed by PJNK and JNK detection. In lane 8, cells were treated with anisomycin (1 µg/ml) for 2 hours as a positive control.
SB202190 induces phosphorylation of ATF-2 transcription factor
ATF-2 is a ubiquitously expressed member of the basic region-leucine zipper transcription factor family that regulates the expression of genes in response to various stress signals, and is a target for JNK and p38 MAPK pathways [19–21]. Cellular stress activates ATF-2 by phosphorylation on Thr69 and Thr71 residues. ATF-2 can form heterodimeric complexes with AP-1 DNA binding proteins such as c-Jun, resulting in the expression of various genes in response to cellular stress signals [19–21]. If JNK is phosphorylated and activated due to treatment of cells with SB202190, then it should also result in the phosphorylation of ATF-2. To examine this possibility, we treated A549 cells with indicated concentrations of SB202190 and evaluated phosphorylation of Thr71 using phospho-specific antibodies (Cell Signaling, MA). As demonstrated in Figure 4A (upper panel), ATF-2 was dose-dependently phosphorylated by treatment of cells with SB202190 in the nuclear extract of cells. However, the level of unphosphorylated ATF-2 (lower panel) remained unchanged. To evaluate whether this increase in ATF-2 phosphorylation was due to JNK activation, we treated cells with specific JNK inhibitor SP600125 in combination with SB202190 and evaluated ATF-2 phosphorylation. As demonstrated in Figure 4A, treatment of cells with SB202190 and SP600125 in combination did not phosphorylate ATF-2. This experiment demonstrated that SB202190 specifically induces activation of JNK, which phosphorylates ATF-2. We also demonstrated that a positive control anisomycin that activates both JNK and p38 MAPK also induces ATF-2 phosphorylation.
Figure 4.

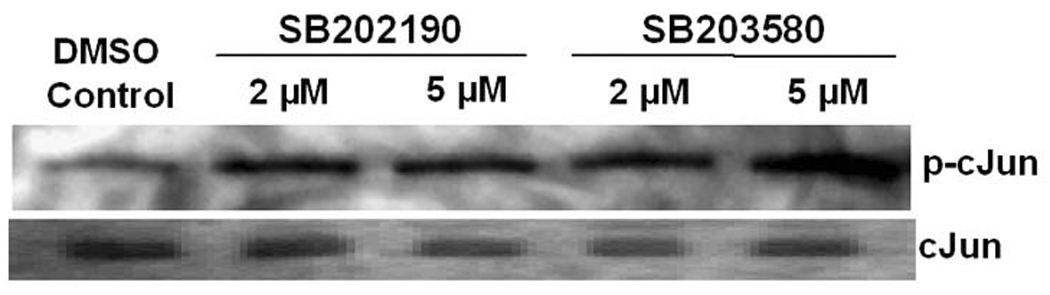
A. ATF-2 is phosphorylated by the p38 MAPK-specific inhibitor SB202190 A549 cells were treated with indicated concentrations of SB202190 or SP600125 for 16 hours. Anisomycin (1 µg/mL) was used as a positive control for ATF-2 phosphorylation. Cells were harvested after 16 hours of treatment; nuclear extract was prepared as mentioned in the Methods section. Nuclear extract (10 µg) was analyzed by Western analysis for phospho Thr71 using specific antibodies. Lower panel: Nuclear extracts were analyzed for unphosphorylated ATF-2 by Western analysis. Lane 1, untreated DMSO control cells; lanes 2–6, increasing concentrations of SB202190; lane 7, cells treated with only SP600125 (20 µM); lane 8, cells treated with 20 µM SP600125 and 1 µM SB202190; lane 9, cells treated with anisomycin (1 µM). (B) Phosphorylation of cJun by SB202190 or SB203580. A549 cells were treated with either 2 or 5 µM each of SB202190 or SB203580 for 1 hour. Cell lysates were prepared and JNK kinase assay was performed as described in the Methods section; lower panel, unphosphorylated cJun.
Increased JNK kinase activity in response to SB202190 or SB203580
Phosphorylation of c-Jun is a major effect of JNK activation. JNK-dependent phosphorylation of c-Jun activates AP-1 transcription factor, and has been implicated in several cellular stress responses. Therefore, we sought to determine whether treatment of cells with SB202190 or SB203580 could also activate JNK kinase activity. We used c-Jun as a substrate for determining JNK kinase activity in response to SB202190 or SB203580. As demonstrated in Figure 4B, treatment of cells with either SB202190 or SB203580 phosphorylated c-Jun, which suggests that JNK is activated due to treatment of cells with SB202190 or SB203580.
SB202190 induces activation of AP-1 DNA binding in a dose-dependent manner
Activator protein-1 (AP-1) is a sequence-specific transcription factor composed of either homo-or heterodimers between members within the c-Jun (c-Jun, JunB, and JunD) and c-Fos (c-Fos, FosB, Fra1, and Fra2) families [22]. Activated JNK phosphorylates c-Jun, which can form an AP-1 complex by homodimerization or heterodimerization. Therefore, if p38 MAPK inhibitor SB202190 induces activation of JNK, then we would expect AP-1 DNA binding due to phosphorylation and homo- or heteromerization of c-Jun proteins. In response to SB202190, A549 cells showed increased AP-1 DNA binding treatment that followed a dose–response pattern, as demonstrated in Figure 5A. The increase in AP-1 DNA binding due to SB202190 treatment was abrogated by simultaneous treatment of cells with the specific JNK inhibitor SP600125 (Fig. 5A, lane). In addition, we also used MCF-7 cells to evaluate whether the data obtained with A549 cells could be reproduced in another cell type. As demonstrated in Figure 5B, MCF-7 cells also demonstrated increased AP-1 DNA binding in response to SB202190 treatment. These experiments show that SB202190 can induce the activation of AP-1 DNA binding due to JNK activation.
Figure 5. Specific p38 MAPK inhibitor SB202190 induces AP-1 DNA binding.


Cells were treated with indicated concentrations of SB202190 for 16 hours, followed by nuclear extract preparation as mentioned in the Methods section. The AP-1 DNA binding assay was performed on nuclear extracts using an AP-1 consensus probe as described. (A) A549 cells: lane 1, DMSO controls; lanes 2–6, increasing concentrations of AB202190; lane 7, cells treated with only SP600125 (20 µM); lane 8, cells treated with SP600125 and 1 µM SB202190; lane 9, free probe only. (B) MCF-7 cells: lane 1, DMSO control; lanes 2–6, increasing doses of SB202190.
Decreased levels of p38 MAPK do not induce JNK activation due to SB202190 or SB203580 treatment
Because both JNK and p38 MAPK are activated simultaneously by many stimuli, it is possible that inhibition of p38 MAPK by SB202190 or SB203580 induces JNK as a compensatory mechanism in response to stress. Therefore, we sought to determine the effect of p38 downregulation by RNA interference on SB202190- or SB203580-dependent JNK activation. As demonstrated in Figure 6A (lower panel), treatment of A549 cells with siRNA effectively downregulated the level of p38 MAPK. However, treatment of these cells alone or with either SB202190 or SB203580 did not increase the level of JNK phosphorylation (Fig. 6A, upper panel). These data suggest that decreased p38 level or activation is not responsible for increased JNK activation due to treatment of cells with SB202190 or SB203580.
Figure 6.



(A) p38 MAPK down-regulation by RNA interference does not decrease JNK phosphorylation induced by SB202190 or SB203580 A549 cells were transfected with either non-targeting (NT) siRNA or p38MAPK siRNA, as described in the Methods section. After 48 hours, these cells were treated with either SB202190 or SB203580 (5 µM) for 2 hours. pJNK (upper panel) and p38 MAPK (lower panel) were detected as described. (B) Over-expression of MKK4 or MKK7 increased JNK phosphorylation in response to SB202190 or SB203580. A549 cells were transfected with pcDNA3 vector or with pcDNA3-MKK4 or pcDNA3-MKK7 over-expression vectors, as described. After 48 hours, these cells were treated with either SB202190 or SB203580 (2 µM each) for 2 hours. pJNK (upper panel) and JNK (lower panel) were detected, as described. (C) Expression of kinase-dead (Kd) MKK4 or MKK7 in a dominant-negative manner decreases JNK phosphorylation in response to SB202190 or SB203580. A549 cells were transfected with pcDNA3 vector or with pcDNA3-MKK4(Kd) or pcDNA3-MKK7(Kd) over-expression vectors, as described. After 48 hours, these cells were treated with either SB202190 or SB203580 (2 µM each) for 2 hours. pJNK (upper panel) and JNK (lower panel) were detected, as described.
MKK4/MKK7-dependent activation of JNK due to treatment of cells with SB202190 or SB203580
To determine the activation mechanism of JNK due to treatment of cells with SB202190 or SB203580, we first analyzed the effect of either overexpression of wildtype MKK4/MKK7 or kinase-inactive MKK4/MKK7 expression on SB202190- or SB203580-induced JNK activation. As demonstrated in Figure 6B, transfection of cells with wildtype MKK4 or MKK7 followed by treatment of cells with SB202190 or SB203580, increased the level of JNK phosphorylation compared to vector-only transfected cells. In addition, transfection of wildtype MKK4 or MKK7 alone could phosphorylate JNK, as expected. However, the level of unphosphorylated JNK remained unchanged.
In converse experiments, we over-expressed kinase-inactive MKK4 or MKK7 and treated these cells with SB202190 or SB203580. As demonstrated in Figure 6C, cells transfected with vector control and treated with SB202190 or SB203580 showed increased JNK phosphorylation. However, cells either transfected with MKK4 (Kd) or MKK7 (Kd) demonstrated lower levels of JNK phosphorylation in response to SB202190 or SB203580 treatment (Fig. 6C). These studies suggest that SB202190 or SB203580 induces JNK in a MKK4/MKK7-dependent manner.
SB202190 or SB203580 induces activation of JNK in a MLK-3 -dependent manner
Kinases MEKK1, apoptosis signal regulating kinase 1(ASK1), or MLK-3 could each activate MKK4/MKK7, resulting in activation of JNK [4, 13, 23]. Because SB202190- or SB203580-induced JNK activation was dependent on MKK4/MKK7 activation, we analyzed for the possible upstream activator of MKK4/MKK7 in response to SB202190 or SB203580. First, we overexpressed the level of wild-type MEKK1 using an over-expression construct and then treated these cells with SB202190 or SB203580. As demonstrated in Figure 7A, SB202190- or SB203580-induced JNK activation did not result in over-expression of wild-type MEKK1. In converse experiments, we also used a kinase-inactive MEKK1 construct to over-express kinase-inactive MEKK1 in a dominant-negative manner. As shown in Figure 7A, there was no effect on the level of JNK phosphorylation induced by treatment of cells with either SB202190 or SB203580. These data indicate that MEKK1 does not play a role in the activation of MKK4/MKK7 induced in response to SB202190 or SB203580.
Figure 7. MLK-3-, but not MEKK1-mediated phosphorylation of JNK by SB202190 or SB203580.


(A) A549 cells were transfected with either wildtype MEKK1 or kinase-inactive MEKK1 (dnMEKK1) expression vectors using the transfection protocol as described in the Methods section. After 48 hours, these cells were treated with either SB202190 or SB203580 (2 µM each) for 2 hours. Cell lysates were prepared and phosphoJNK was detected, as described. (B) MLK-3 is required for the phosphorylation of JNK due to treatment of cells with SB202190 or SB203580. A549 cells were either transfected with NT siRNA or with MLK-3 siRNA as described in the Methods section. After 48 hours, these cells were treated with either SB202190 or with SB203580 (1µM) for 2 hours. Cell lysates were prepared and phospho-JNK (top panel), unphosphorylated JNK (second panel from top), MLK-3 (third panel from top), and β-actin (fourth panel from top) were detected using respective antibodies, as described in the Methods section.
Finally, we examined the role of MLK-3 in JNK activation using siRNA approach. Cells were transfected with MLK-3 siRNA as described in the Methods section, followed by treatment of these cells with SB202190 or SB203580. As shown in Figure 7B (third panel from the top), MLK-3 siRNA transfection downregulated the level of MKL-3, but transfection of non-targeting (NT) siRNA did not downregulate MLK-3. Treatment of cells with SB202190 or SB203580 resulted in higher expression of phosphorylated JNK in NT siRNA-transfected cells, but lower levels of phosphorylated JNK in MLK-3 siRNA-treated cells. These data suggest that SB202190- or SB203580-induced MKK4/MKK7–JNK activation could involve an MLK-3-dependent pathway.
Downregulation of MLK-3 by RNA interference decreases the expression of phospho-cJun, AP-1 and phospho-ATF2 in response to SB202190 or SB203580
If SB202190 or SB203580 induces MLK-3-dependent JNK activation then inhibition of MLK-3 protein expression by RNA interference should demonstrate decreased JNK-dependent effects such as phosphorylation of cJun, phosphorylation of ATF-2 or activation of AP-1 DNA binding. Therefore, we determined the expression of phospho-cJun, phospho-ATF2 or AP-1 DNA binding in MLK-3 siRNA transfected cells treated with SB202190 or SB203580. As demonstrated in Fig 8A, downregulation of MLK-3 protein almost abolished the expression of phospho-cJun in response to SB202190 or SB203580. However, there was no significant change in the levels of unphosphorylated cJun in MLK-3 siRNA treated cells compared to non-targeting siRNA treated cells. Similarly, the DNA binding of AP-1 was also decreased in cells transfected with MLK-3 siRNA and treated with SB202190 or SB203580 (Fig 8B upper panel). Additionally, the phosphorylation of ATF2 also followed a similar pattern suggesting that MLK-3 mediates cJun phosphorylation, ATF2 phosphorylation and AP-1 DNA binding in response to SB202190 or SB203580.
Figure 8. MLK-3 downregulation by RNA interference decreases the phosphorylation of cJun, ATF-2 and AP-1 DNA binding activated by SB202190 or SB203580.


(A) A549 cells were either transfected with NT siRNA or with MLK-3 siRNA as described in the Methods section. After 48 hours, these cells were treated with either SB202190 or with SB203580 (1µM) for 2 hours. Cell lysates were prepared and western analysis of phospho-cJun (upper panel) or unphosphorylated cJun (lower panel) was performed as described in the methods section. (B) A549 cells were either transfected with NT siRNA or with MLK-3 siRNA as described in the Methods section. After 48 hours, these cells were treated with either SB202190 or with SB203580 (1µM) for 16 to 18 hours. Nuclear extract was prepared as described in the methods. AP-1 EMSA was performed as described in the methods (upper panel). Nuclear extracts was also probed for the presence of phospho-ATF2 (middle panel) or non-phosphoATF2 (lower panel) by western analysis.
Phosphorylation and activation of MLK3 by SB202190 or SB203580
MLK3 is known to be activated by phosphorylation on Thr277/ser281 [13]. Therefore, we sought to determine whether MLK3 phosphorylation is induced by SB202190 or SB203580. As shown in Fig 9A & B, HEK293T cells treated with SB202190 or SB203580 show enhanced phosphorylation of transiently overexpressed MLK3 on ser277/ser281. This data shows that MLK3 is phosphorylated in intact cells by treatment of SB202190 or SB203580. Further, we sought to determine whether activated MLK3 immunoprecipitated from SB202190 or SB203580 treated cells could phosphorylated its substrate MKK7 in a kinase assay. When MLK3 and MKK7 are included in a kinase assay, MLK3 immunoprecipitated from SB202190 or SB203580 treated cells phosphorylated MKK7, but MLK3 immunoprecipitated from untreated cells did not phosphorylate MKK7. These results indicate that MLK3 is activated by treatment of cells with SB202190 or SB203580. However, the inhibitors could either directly stimulate the kinase activity of MLK3 or could activate upstream G-Protein coupled receptors such as Cdc42 or Rac that in turn can activate MLK3. Thus, the inhibitors could directly induce phosphorylation of MLK3 or they could indirectly activate cdc42/Rac resulting in phosphorylation and activation of MLK3. Therefore, we performed MLK3 activation using purified MLK3 and using MKK7 as substrate as described in the methods. Flag-MKK7 was not phosphorylated in presence of HAMLK3 and SB202190 or SB203580 in the kinase reaction, therefore, we could not detect phospho-MKK7 in the kinase reaction (data not shown). These data demonstrate that MLK3 is not phosphorylated in vitro by either SB202190 or SB203580. Therefore, our data demonstrate that cellular factors are essential for activation of MLK3 in response to SB202190 or SB203580.
Figure 9. MLK3 is phosphorylated on ser277/281 in response to SB202190 or SB203580 treatment.
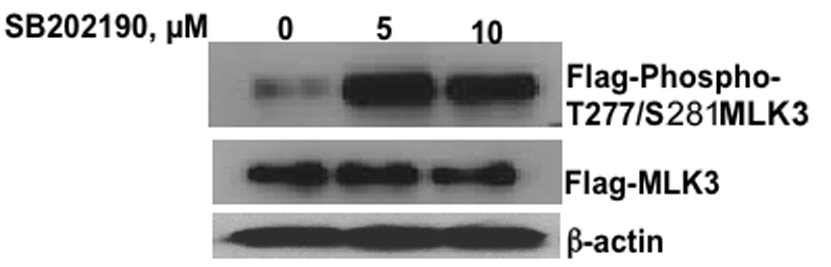
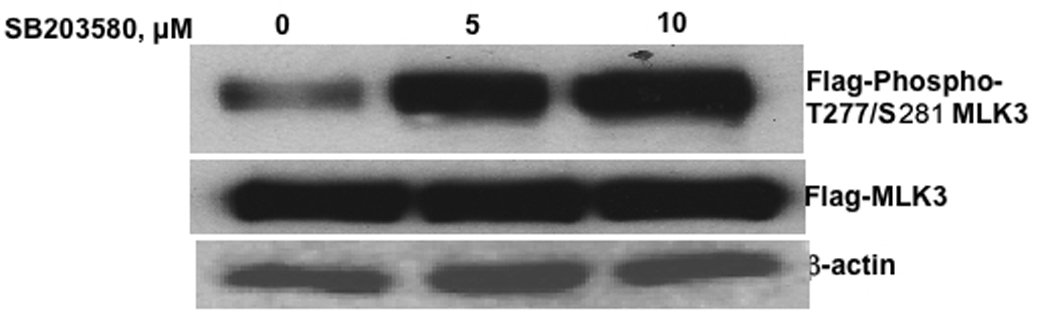
(A) Effect of SB202190 on MLK3 phosphorylation. HEK 293T cells were transfected with phCMV2-HA-MLK3 as described in the methods. After 48 hours cells were treated with SB202190 for 4 hours. Cellular lysates were probed with anti-phospho-MLK3 antibody as described in the methods. Lane 1, DMSO treated control; lane 2, 5 µM SB202190; Lane3, 10 µM SB202190; Bottom Panel, Flag-MLK3; Lower Panel, β-actin. (B) Effect of SB203580 on MLK3 phosphorylation. HEK 293T cells were transfected with pCMV2-HA-MLK3 as described in the methods. After 48 hours cells were treated with SB203580 for 4 hours. Cellular lysates were probed with anti-phospho-MLK3 antibody as described in the methods. Lane 1, DMSO treated control; lane 2, 5 µM SB203580; Lane3, 10 µM SB203580; Bottom Panel, Flag-MLK3; Lower Panel, β-actin.
Discussion
The discovery of selective and potent inhibitors of p38 MAPK has aided our understanding of the role of p38 MAPK in signal transduction and regulation of inflammatory cellular responses [1]. It is also widely anticipated that p38 MAPK inhibitors will have efficacy in arthritic and inflammatory diseases, and some compounds have reached phase I and phase II clinical trials [1]. Our study provides compelling evidence that a specific p38 MAPK inhibitor, SB202190, activates JNK, phosphorylates ATF-2, and increases AP-1 DNA binding. The data obtained with multiple cell lines and primary endothelial cells suggest that SB202190 and SB203580 activate JNK. Because SB202190 and related p38 MAPK inhibitors are widely used to dissect the JNK/p38MAPK signaling pathways, and conclusions are made based on the inhibition of p38 MAPK by SB202190, our study provides an important piece of information in the examination of whether the effects observed are due to the use of SB202190 or SB203580, or are consequences of p38 MAPK inhibition or JNK activation.
The phosphorylation of JNK induced by SB202190 or SB203580 could be observed as early as 10 minutes and remained for 4–6 hours in A549 cells, and for more than 6 hours in endothelial cells. Additionally, these data demonstrate that the duration of activation could be cell-type specific. An optimal concentration of 3 µM of SB202190 or SB203580 was sufficient to induce strong JNK phosphorylation (Fig. 1A and 1C). These data suggest that treating cells with a specific p38 MAPK inhibitor can induce JNK activation. Activation of JNK by SB202190 or SB203580 was not limited to A549 cells, but was also observed in primary endothelial cells and in MCF-7 breast cancer cells. Both JNK and p38 MAPK have been shown to phosphorylate ATF-2 on Thr71 [19, 20]. When cells were treated with SB202190, we observed a dose-dependent increase in ATF-2 phosphorylation that could be abrogated by treating cells with JNK inhibitor SP600125 and p38 MAPK inhibitor SB202190. These data demonstrate that the phosphorylation of ATF-2 by SB202190 is specifically mediated by activated JNK. In addition, SB202190 or SB203580 can also induce JNK kinase activity as determined by c-Jun phosphorylation. Although 1 µM of SB202190 seems to be optimal in inducing JNK, a nanomolar range of SB202190 can strongly induce AP-1 DNA binding activity (Fig. 5A and B). These data indicate that physiological effects of SB202190 treatment can take place at much lower levels of SB202190.
Because JNK and p38 MAPK are induced simultaneously by many stress signals, and one of the upstream activators of JNK and p38 MAPK is MKK4, we reasoned that inhibition of p38 MAPK could induce JNK activation due to a compensatory mechanism. Therefore, we downregulated the expression of endogenous p38 MAPK by RNA interference and evaluated its effect on JNK activation; additionally, we evaluated JNK activation in response to SB202190 or SB203580 in these p38 MAPK-down-regulated cells. As demonstrated in Figure 6A, downregulation of the p38 MAPK level did not induce JNK activation. In addition, there was no effect of p38 downregulation on SB202190- or SB203580-induced JNK activation. Therefore, inhibition of p38 MAPK activation does not induce JNK activation. Thus, SB202190- or SB203580-induced JNK activation is a specific property of these inhibitors.
Because JNK is activated by its upstream activator MKK4/MKK7, we sought to determine the role of MKK4/MKK7 on JNK activation by treating the cells with SB202190 or SB203580. As demonstrated in Figures 6B and 6C, SB202190- or SB203580-induced JNK activation was dependent on MKK4/MKK7 activation. MKK4/MKK7 was activated by the activation of MEKK1, ASK1, or MLK-3. Therefore, we determined the role of each of these upstream kinases on the activation of JNK by SB202190 or SB203580. Our data indicate that MLK-3, but not MEKK1 or ASK1, mediates SB202190- or SB203580-induced JNK activation. A recent report has also indicated that treatment of cells with SB203580 enhances phosphorylation of MLK3[24]. Additionally, our study is further strengthened by the fact that the levels of phosphocJun, phospho-ATF2 or AP-1 DNA binding was decreased or abrogated in MLK-3 downregulated cells treated with SB202190 or SB203580. These studies show that MLK-3 mediates the downstream events of JNK activation in response to SB202190 or SB203580.
Activated form of small GTPases, Cdc42 and Rac can bind to MLK3 and has been shown to promote phosphorylation of MLK3 on serine 277/281 residues [10, 13]. This phosphorylation increases the catalytic activity of MLK3, and potentiate its signaling to JNK [10, 13]. Therefore, we evaluated the phosphorylation of MLK3 in response to SB202190 or SB203580. Our data demonstrate that MLK3 is phosphorylated in response to SB202190 or SB203580. Further, we also observed that MLK3 immunoprecipited from SB202190 or SB203580 treated cells could phosphorylate MKK7, used as a substrate in the kinase reaction, demonstrating that MLK3 is activated by SB202190 or SB203580. However, we did not observe phosphorylation of MKK7 in a kinase assay using HA-MLK3 from unstimulated cells with SB202190 or SB203580. These studies suggest that SB202190 or SB203580 activates MLK3 in intact cells, but not using purified MLK3. Because Cdc42/Rac activates MLK3 SB202190 or SB203580 may directly activate Cdc2/Rac that in turn activates MLK3. These studies demonstrate a novel property of p38 MAPK inhibitors SB202190 or SB203580 in the induction of JNK activation that was not recognized before.
Figure 10. Activation of MLK3 in response to SB202190 or SB203580.
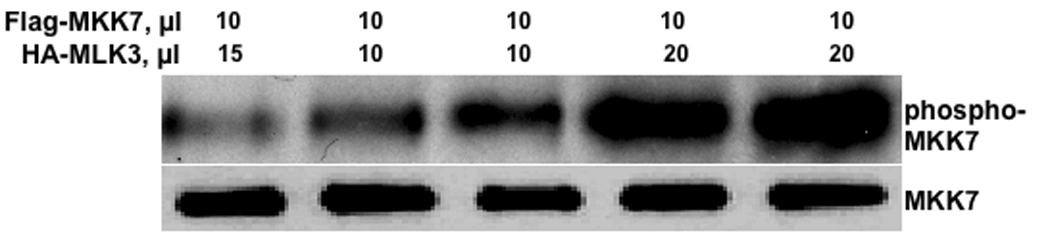
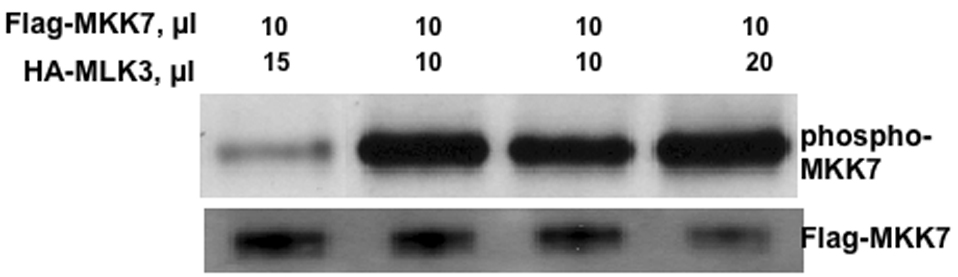
(A) Effect of SB202190 on MLK3 activity in intact cells. HEK 293T cells were transfected with pCMV2- HA-MLK3. After 48 hours cells were treated with SB202190 for 4 hours. Following incubation, MLK3 was immunoprecipitated using Anti-HA antibody as described in the methods. 10 or 20 microliters of immunoprecipitate was used in the kinase assay using Flag-MKK7 as a substrate as described in the methods. Lane 1, 15 µl of HA-MKK3 from control DMSO treated cells and 10 µl of Flag MKK7; lane 2, 10 µl of HA-MLK3 from 5µM SB202190 treated cell and 10 µl of MKK7; lane 3, 10 µl of HA-MLK3 from 10 µM SB202190 treated cells and 10 µl of Flag-MKK7; lane 4, 20 µl of HA-MLK3 from 5µM SB202190 treated cell and 10 µl of MKK7; lane 5, 20 µl of HA-MLK3 from 10 µM SB202190 treated cells and 10 µl of Flag-MKK7; Lower Panel, unphosphorylated MKK7. (B) Effect of SB203580 on MLK3 activity in intact cells. HEK 293T cells were transfected with phCMV2-HA-MLK3. After 48 hours cells were treated with SB203580 for 4 hours. Following incubation, MLK3 was immunoprecipitated using Anti-HA antibody as described in the methods. 10 or 20 microliters (µl) of immunoprecipitate was used in the kinase assay using Flag-MKK7 as a substrate as described in the methods. Lane 1, 15 µl of HA-MKK3 from control DMSO treated cells and 10 ul of Flag MKK7; lane 2, 10 ul of HA-MLK3 from 5µM SB203580 treated cell and 10 µl of MKK7; lane 3, 10 µl of HA-MLK3 from 10 µM SB203580 treated cells and 10 µl of Flag-MKK7; lane 4, 20 µl of HA-MLK3 from 5µM SB203580 treated cell and 10 µl of MKK7; Lower Panel, un-phosphorylated MKK7.
Acknowledgement
The authors gratefully acknowledge Dr. Tom Maniatis, Harvard University, and Dr. Roger Davies, University of Massachusetts, Worcester, MA for the generous gifts of over-expression plasmids used in this study. KCD was supported by NIH grant 7R01 HL071558. We also thank the Office of Grants and Scientific Publications at the University of Arkansas for Medical Sciences for editorial assistance during the preparation of this manuscript.
Abbreviations
- AP
activator Protein
- ASK
apoptosis signal regulating kinase
- ATF
activating transcription factor
- EMSA
electrophoretic mobility shift assay
- ERKs
extracellular signal-regulating kinases
- HMVEC
human Microvascular microvascular endothelial cells
- IL
interleukin
- JNK
c-Jun N-terminal kinase
- MAPKs
Mitogen- activated protein kinases
- MEKK
MAPK kinase kinase
- MLK
mixed lineage kinase
- RIPA
radio immunoprecipitation assay
- SAPK
stress-activated protein kinase
- SB90
SB202190
- SB80
SB203580
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kumar S, Boehm J, Lee JC. Nat Rev Drug Discov. 2003;2(9):717–726. doi: 10.1038/nrd1177. [DOI] [PubMed] [Google Scholar]
- 2.Karin M, Gallagher E. IUBMB Life. 2005;57(4–5):283–295. doi: 10.1080/15216540500097111. [DOI] [PubMed] [Google Scholar]
- 3.Davis RJ. Cell. 2000;103(2):239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 4.Kaminska B. Biochim Biophys Acta. 2005;1754(1–2):253–262. doi: 10.1016/j.bbapap.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 5.Karin M. Proc Am Thorac Soc. 2005;2(4):386–390. doi: 10.1513/pats.200504-034SR. discussion 394-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davis RJ. Biochem Soc Symp. 1999;64:1–12. doi: 10.1515/9781400865048.1. [DOI] [PubMed] [Google Scholar]
- 7.Liu J, Lin A. Cell Res. 2005;15(1):36–42. doi: 10.1038/sj.cr.7290262. [DOI] [PubMed] [Google Scholar]
- 8.Swenson KI, Winkler KE, Means AR. Mol Biol Cell. 2003;14(1):156–172. doi: 10.1091/mbc.E02-02-0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rana A, Gallo K, Godowski P, Hirai S, Ohno S, Zon L, Kyriakis JM, Avruch J. J Biol Chem. 1996;271(32):19025–19028. doi: 10.1074/jbc.271.32.19025. [DOI] [PubMed] [Google Scholar]
- 10.Teramoto H, Coso OA, Miyata H, Igishi T, Miki T, Gutkind JS. J Biol Chem. 1996;271(44):27225–27228. doi: 10.1074/jbc.271.44.27225. [DOI] [PubMed] [Google Scholar]
- 11.Brancho D, Ventura JJ, Jaeschke A, Doran B, Flavell RA, Davis RJ. Mol Cell Biol. 2005;25(9):3670–3681. doi: 10.1128/MCB.25.9.3670-3681.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang H, Gallo KA. J Biol Chem. 2001;276(49):45598–45603. doi: 10.1074/jbc.M107176200. [DOI] [PubMed] [Google Scholar]
- 13.Tibbles LA, Ing YL, Kiefer F, Chan J, Iscove N, Woodgett JR, Lassam NJ. Embo J. 1996;15(24):7026–7035. [PMC free article] [PubMed] [Google Scholar]
- 14.Burbelo PD, Drechsel D, Hall A. J Biol Chem. 1995;270(49):29071–29074. doi: 10.1074/jbc.270.49.29071. [DOI] [PubMed] [Google Scholar]
- 15.Zhang YL, Dong C. Cell Mol Immunol. 2005;2(1):20–27. [PubMed] [Google Scholar]
- 16.Wang W, Shi L, Xie Y, Ma C, Li W, Su X, Huang S, Chen R, Zhu Z, Mao Z, Han Y, Li M. Neurosci Res. 2004;48(2):195–202. doi: 10.1016/j.neures.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 17.Das KC. J Biol Chem. 2001;276(7):4662–4670. doi: 10.1074/jbc.M006206200. [DOI] [PubMed] [Google Scholar]
- 18.Das KC, Lewis-Molock Y, White CW. Am J Physiol. 1995;269(5 Pt 1):L588–L602. doi: 10.1152/ajplung.1995.269.5.L588. [DOI] [PubMed] [Google Scholar]
- 19.Gupta S, Campbell D, Derijard B, Davis RJ. Science. 1995;267(5196):389–393. doi: 10.1126/science.7824938. [DOI] [PubMed] [Google Scholar]
- 20.Livingstone C, Patel G, Jones N. Embo J. 1995;14(8):1785–1797. doi: 10.1002/j.1460-2075.1995.tb07167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Dam H, Wilhelm D, Herr I, Steffen A, Herrlich P, Angel P. Embo J. 1995;14(8):1798–1811. doi: 10.1002/j.1460-2075.1995.tb07168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karin M. J Biol Chem. 1995;270(28):16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- 23.Takeda K, Matsuzawa A, Nishitoh H, Ichijo H. Cell Struct Funct. 2003;28(1):23–29. doi: 10.1247/csf.28.23. [DOI] [PubMed] [Google Scholar]
- 24.Schachter KA, Du Y, Lin A, Gallo KA. J Biol Chem. 2006;281(28):19134–19144. doi: 10.1074/jbc.M603324200. [DOI] [PubMed] [Google Scholar]