

SUMMARY
Antibiotics are typically more effective against replicating rather than nonreplicating bacteria. However, a major need in global health is to eradicate persistent or nonreplicating subpopulations of bacteria such as Mycobacterium tuberculosis (Mtb). Hence, identifying chemical inhibitors that selectively kill bacteria that are not replicating is of practical importance. To address this, we screened for inhibitors of dihydrolipoamide acyltransferase (DlaT), an enzyme required by Mtb to cause tuberculosis in guinea pigs and used by the bacterium to resist nitric oxide-derived reactive nitrogen intermediates, a stress encountered in the host. Chemical screening for inhibitors of Mtb DlaT identified select rhodanines as compounds that almost exclusively kill nonreplicating mycobacteria in synergy with products of host immunity, such as nitric oxide and hypoxia, and are effective on bacteria within macrophages, a cellular reservoir for latent Mtb. Compounds that kill nonreplicating pathogens in cooperation with host immunity could complement the conventional chemotherapy of infectious disease.
INTRODUCTION
Most antibiotics target biosynthetic processes that bacteria need to increase their biomass (Walsh, 2003). It is not surprising that such antibiotics are more effective against replicating than nonreplicating bacteria (Hobby and Lenert, 1957; Levin and Rozen, 2006). However, a major need in global health is to eradicate persistent or nonreplicating subpopulations of bacteria such as Mtb (McCune et al., 1966; Munoz-Elias et al., 2005; Rogerson et al., 2006). Worldwide, an estimated 1 person in 3 is infected with Mtb; in about 9 of every 10 infected, Mtb persist in a largely nonreplicating (“latent”) state throughout the lifetime of the host. If the immune response flags, Mtb can resume replication and give rise to tuberculosis, a contagious disease that kills more people than any other bacterial infection. Nonreplicating Mtb are also problematic in clinically active tuberculosis (Boshoff and Barry, 2005). The acidic phagosomal environment and “nitroxidative” (Lancaster, 2004) chemistry generated by the macrophages in which Mtb resides and the deprivation of oxygen and nutrients that may result from the accumulation of inflammatory cells at infected sites can each keep Mtb from replicating (Voskuil et al., 2003). Nonreplicating Mtb display nonheritable antibiotic resistance, also called phenotypic tolerance, a phenomenon that pertains to most members of a bacterial population starved for nutrients, as well as to a small, nonreplicating fraction of a population undergoing logarithmic expansion (Levin and Rozen, 2006). Durable cure of tuberculosis requires eradication of both replicating and nonreplicating Mtb (McCune et al., 1966). During treatment of tuberculosis, nonreplicators termed “persisters” may be responsible for relapse rates that only fall below 5% when chemotherapy is extended for many months. Such prolonged treatment is difficult to sustain, and its interruption fosters the emergence of mutants with heritable drug resistance.
That most antibiotics act preferentially against replicating bacteria may be a consequence of the way these compounds have been sought: by screening against pathogens in vitro under conditions that sustain rapid growth and assaying for inhibition of that growth (Nathan, 2004). In contrast, here we screened for inhibitors of an enzyme that Mtb uses to resist a stress it encounters in the host, nitric oxide-derived reactive nitrogen intermediates (RNI) (Nathan and Ehrt, 2004). In vitro, RNI at low levels or short exposures prevent Mtb from replicating, while higher concentrations or longer exposures kill it (Nathan and Ehrt, 2004).
One mycobacterial defense against RNI is a peroxynitrite reductase and peroxidase (Bryk et al., 2000). Its four components are a peroxiredoxin, alkylhydroperoxide reductase subunit C (AhpC) (Bryk et al., 2000); a thioredoxin-related oxidoreductase, AhpC-neighboring protein D (AhpD) (Bryk et al., 2002); DlaT (formerly SucB) (Bryk et al., 2002; Tian et al., 2005b); and lipoamide dehydrogenase (Lpd) (Bryk et al., 2002). DlaT and Lpd are named for their primary roles as the E2 and E3 components of Mtb's pyruvate dehydrogenase (PDH) (Tian et al., 2005b). By supplying acetyl coenzyme A (CoA), PDH joins glycolysis to the citric acid cycle, which generates high-energy phosphate bonds, reducing equivalents for biosynthesis and precursors of amino acids and heme. Acetyl CoA also serves as a building block for the lipids in Mtb's cell wall and a substrate for the glyoxylate shunt, a pathway required for Mtb to persist in macrophages and in mice (Munoz-Elias and McKinney, 2005). Thus, DlaT may contribute to Mtb's antinitroxidative defense by helping to detoxify peroxynitrite (Bryk et al., 2002) and by sustaining metabolic pathways required for Mtb to survive when exposed to RNI (Rhee et al., 2005).
Herein, we demonstrated DlaT's contribution to virulence in experimental tuberculosis, identified a DlaT inhibitor, synthesized and characterized analogs with improved properties, and discovered a compound that can be preferentially bactericidal against a pathogen that is not replicating.
RESULTS
Lack of Pathogenicity of DlaT-Deficient Mtb
Consistent with DlaT's postulated roles, ΔdlaT (DlaT-deficient) Mtb grew poorly in vitro, were hypersusceptible to RNI, died in mouse macrophages, and left mice with minimal histologic evidence of tuberculosis by 22 weeks after infection (Shi and Ehrt, 2006). Because the guinea pig may model human tuberculosis better than the mouse (McMurray, 2001), we studied the course of infection in guinea pigs after inhalation of wild-type Mtb (H37Rv), the ΔdlaT mutant, and the latter strain complemented with the wild-type allele. The two dlaT-expressing strains each caused severe tuberculosis. In contrast, the ΔdlaT mutant produced low bacillary loads (Figure 1A) and did not lead to detectable disease (Figure 1B). All guinea pigs infected with the ΔdlaT mutant had inhaled Mtb because they developed positive tuberculin skin tests (mean induration, 9 mm). To test the effect of comparable initial bacterial loads, we then infected guinea pigs with 100-fold more colony-forming units (CFU) of the ΔdlaT mutant and 3-fold more of the complemented strain than of the wild-type. The ensuing 4 weeks led to a 3.1 log10 increase in CFU of wild-type Mtb in the lungs and a 3.1 log10 increase in CFU of the complemented mutant, but only a mean 0.11 log10 increase for the ΔdlaT mutant (Figure 1C). Results were similar in the spleen. Thus, DlaT appears to be essential for pathogenesis by Mtb in guinea pigs.
Figure 1. ΔdlaT Is Severely Attenuated in Guinea Pigs.
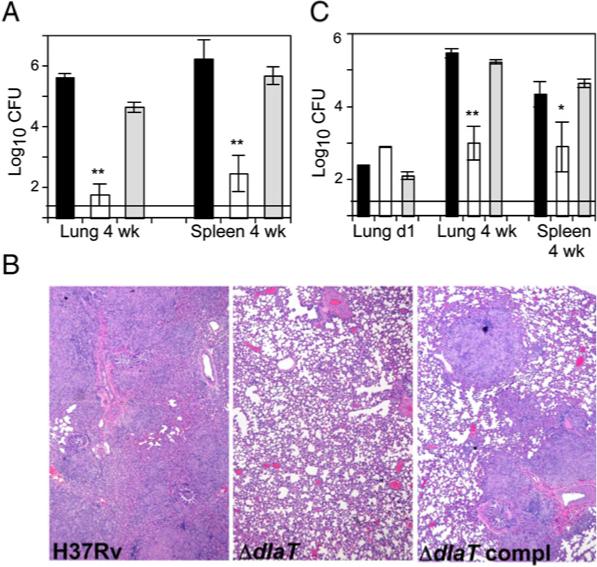
(A) Organ CFU in guinea pigs infected with equal inocula of Mtb H37Rv (black bars), ΔdlaT (white bars), and ΔdlaT complemented with the wild-type allele (gray bars), sufficient to allow recovery one day later of 135−358 CFU in lungs of animals infected with either of the two strains encoding DlaT. In guinea pigs infected with the ΔdlaT strain, no CFU were detected in 4 of 5 animals' lungs on day 1, or in 4 of 5 additional animals' lungs or spleens at week 4. Horizontal line denotes limit of detection (1.35 log10).
(B) Photomicrographs of lungs at 4 weeks postinfection.
(C) Organ CFU in guinea pigs infected with the same inoculum of wild-type Mtb as in (A) but a 100-fold higher dose of ΔdlaT Mtb and a 3-fold higher dose of the ΔdlaT complemented strain.
In (A) and (C), the value of the limit of detection was assigned to samples with undetectable CFU. **p < 0.001 and *p < 0.01 by Student-Newman-Keuls test. In (A) and (C), results are mean ± SD (n = 5) in single experiments, each of which was repeated at least two times.
Chemical Screen for DlaT Inhibitors
Because ΔdlaT Mtb grows in bacteriologic medium (Shi and Ehrt, 2006) and Mtb's DlaT is 29% and 39% identical to the E2s of human PDH and α-ketoglutarate dehydrogenase, respectively, DlaT would normally be dismissed as a drug target. Nonetheless, the inability of ΔdlaT Mtb to cause disease in guinea pigs prompted us to try to develop Mtb-selective DlaT inhibitors. We devised a colorimetric assay for DlaT (see Figure S1 available online) in which Lpd initiates sequential electron transfer from NADH to DlaT to AhpD to the colorless acceptor dithionitrobenzene (DTNB), generating yellow TNB. From a combinatorial library of 15,000 small molecules (Chemical Diversity, Inc.) with drug-like characteristics (Lipinski and Hopkins, 2004), we identified 34 (0.23%) as affording >50% inhibition at 10 μM or >75% inhibition at 25 μM. Of these, 13 inhibited DlaT alone. The most potent were rhodanines (2-thioxo-thiazolidi-4-ones) linked through a chain to a carboxylic acid and joined by a methylene bridge to a furan substituted with a halogenated aromatic. We selected those that inhibited not only DTNB reductase (Lpd+ DlaT+AhpD) but also Mtb PDH (Lpd+DlaT+AceE). However, the compounds in the screening collection also inhibited porcine PDH and bound bovine serum albumin (BSA), as judged by a loss of inhibitory activity in the presence of 100 μg/ml BSA (data not shown). BSA that had been reduced and alkylated no longer interfered with these rhodanines (data not shown); thus, their binding to BSA appeared to be sulfhydryl-dependent.
Structure-Activity Relationship and Mechanism of Inhibition
To develop more selective inhibitors, we synthesized over 1000 additional rhodanines and analyzed their structure-activity relationship (SAR) (Figure 2A; Table S1). We assessed potency by determining the concentrations affording 50% inhibition (IC50s) of DTNB reductase and Mtb PDH. We evaluated nonreactivity with sulfhydryls by the relative lack of increase in IC50s for DTNB reductase in the presence of 100 μg/ml BSA and lack of inhibition of bovine thioredoxin reductase, an enzyme dependent on active site cysteine and seleno-cysteine residues (Zhong et al., 2000). Finally, we evaluated species selectivity by determining IC50s for porcine PDH. Chemical features favoring efficacy and selectivity are summarized in Figure 2 and Table S1.
Figure 2. Structures of Inhibitors.

(A) Structure-activity relationship. Regioisomer (Z) orientation is based on X-ray crystallography of D155931. Substituents are listed in order from most to least potent (activity profile) or most to least selective for Mtb versus mammalian PDH (selectivity profile). Activity profile: R1 = 3-COOH > > 3-(CH2)2COOH > 4-SO2NHCOCH3 > 4-OH > 4-SO2NH2 > 3-SO2NH2 > 3-OH > 3-COCH3 > 4-COOH; R2 = H > 4-OH > 4-Cl > 6-Cl > 6-F > 6-CH3 > 6-OCH3; R4 = H > CH3 > > OH; Het = 2,5-furyl > > 2,5-thiazolyl > 2,6-pyridinyl > 2,5-oxazolyl > 2,4-isoxazolyl; R3 = 4-alkyl [Alkyl:CH3,ethyl,isopropyl, tert-butyl] > > 3-isopropyl > 2, 3 or 4-Cl > 2-OCH3 > 2-methyl or ethyl > 4-Br. Selectivity profile: R1 = 3-COOH; R2 = H; R4 = H; R3: site of substituent = 2 > > 3 > 4, substituents = Cl ∼F>CH3 > > OCH3 > OH > NH2; Het = 2,5-furyl.
(B) Structure of D155931 (3-((Z)-5-((5-(2-chlorophenyl)furan-2-yl)methylene)-4-oxo-2-thioxothiazolidin-3-yl)benzoic acid).
(C) Structure of D157070 (3-hydroxypropyl 3-((Z)-5-((5-(2-chlorophenyl)furan-2-yl)methylene)-4-oxo-2-thioxothiazolidin-3-yl)benzoate).
We selected compound D155931 for further study (Figure 2B; Figure S1). Its IC50 for DTNB peroxidase (21 μM) changed little (24 μM) in the presence of 100 μg/ml BSA. It inhibited Mtb PDH (IC50,45 mM) more potently than porcine PDH or thioredoxin reductase (IC50s, >75 mM, near the limit of solubility in the reaction buffer). For reasons explained later, we also studied its propanolic ester, D157070 (Figure 2C), a less potent inhibitor of DTNB peroxidase (IC50,47 μM).
Kinetic analysis demonstrated that D155931 inhibited DlaT competitively with AhpD (Figure 3A). Inhibition by D155931 required that DlaT be incubated with Lpd and NADH (Figure 3B), conditions supporting DlaT's reduction. Inhibition was time dependent (Figure 3B) and irreversible upon dialysis (Figure S2). These results suggested that D155931 bound tightly to reduced DlaT in such a way as to interfere with DlaT's interaction with AhpD, while also blocking DlaT's ability to participate in the PDH reaction.
Figure 3. D155931 Is an Irreversible Inhibitor of DlaT Competitive with AhpD.

(A) D155931 is a competitive inhibitor of DlaT against AhpD. DTNB assay was conducted with variable amounts of AhpD (10, 20, 30, 40, 60, 100, and 150 nM) without (black circles) or with 7 μM (white squares), 15 μM (black triangles), and 45 μM (white triangles) D155931. Experiments were performed without preincubation and were started immediately upon addition of inhibitors; initial rates were taken from the first 5 min. Ki values of 15−18 μM were calculated from the replots of slopes versus inhibitor concentration.
(B) D155931 is time- and substrate-dependent inhibitor of DlaT. A mixture of Lpd, DlaT, and AhpD was preincubated with D155931 at 10 μM (diamonds), 20 μM (triangles), or 40 μM (squares) or with the vehicle control (DMSO; circles), in each case with (white symbols) or without (black symbols) 250 mM NADH. Aliquots were withdrawn over time and tested for remaining activity by the DTNB assay.
Selective Killing of Mycobacteria Whose Replication Was Halted by Acid, Nitric Oxide, Hypoxia, or Tissue Culture Medium
Next, we selected physiologic conditions that inhibited mycobacterial replication without reducing recoverable CFU. The following conditions are thought to exist in the Mtb-containing phagosome in immunocompetent hosts: a pH of 4.5 (MacMicking et al., 2003); NO diffusing from the cytosol and/or locally generated after acidification of its auto-oxidation product, nitrite (Nathan and Ehrt, 2004); lack of leucine and lysine (Hondalus et al., 2000; Sambandamurthy et al., 2002); minimal carbohydrate (Munoz-Elias and McKinney, 2005); decreased oxygen (Voskuil et al., 2003); and limiting iron (Ferreras et al., 2005). These inferences are supported by gene expression profiles of Mtb recovered from macrophages in vitro (Schnappinger et al., 2003) and from lungs (Schnappinger et al., 2003; Shi et al., 2003, 2005; Timm et al., 2003), compared to Mtb cultured in media altered in the specified manner, along with the inability of amino acid auxotrophs of Mtb to survive in macrophages (Hondalus et al., 2000; Sambandamurthy et al., 2002). However, exposure to RNI alone elicits many of the same adaptive transcriptional responses in Mtb seen with depletion of amino acids, carbohydrates, iron, or oxygen (Schnappinger et al., 2003). The ability of NO to kill Mtb depends on concentration and time, as illustrated with the NO-donating compound 2, 2-(hydroxynitrosohydrazino)-bis-ethanamine (DETANO) (Figure S3). Thus, a minimal set of conditions that mimics the phagosomal milieu is the combination of weak acid and RNI. For this purpose, we used Middlebrook 7H9 medium at pH 5.5 (approximately midway between the pH of Mtb-containing phagosomes in nonactivated and fully activated macrophages) (MacMicking et al., 2003) and the generation of small fluxes of NO from 0.5 mM nitrite. In 7H9 broth at pH 5.5, Bacille Calmette-Guérin (BCG), an attenuated variant of M. bovis, stopped replicating. Mtb continued to divide slowly unless the pH was lowered to 4.5. With the addition of 0.5 mM nitrite to 7H9 medium at pH 5.5, Mtb also ceased replicating. Under the latter conditions, BCG and Mtb neither increased nor decreased in CFU over 4 days, while they increased >20-fold in 7H9 broth at pH 6.6 without nitrite (data not shown).
At pH 5.5, D155931 (10 μM) reduced the CFU of nonreplicating BCG by 2 to 3 log10 without nitrite and by 4 to 5 log10 with nitrite in a time- and concentration-dependent manner (Figures 4A and 4B). Mtb resisted killing by nitrite (0.5 mM) alone and by D155931 alone, but the combination led to a 2 log10 reduction in CFU (Figure 4C). D155931 had no effect on BCG or Mtb under replicating conditions at pH 6.6 (data not shown).
Figure 4. D155931 Kills Nonreplicating Mycobacteria.
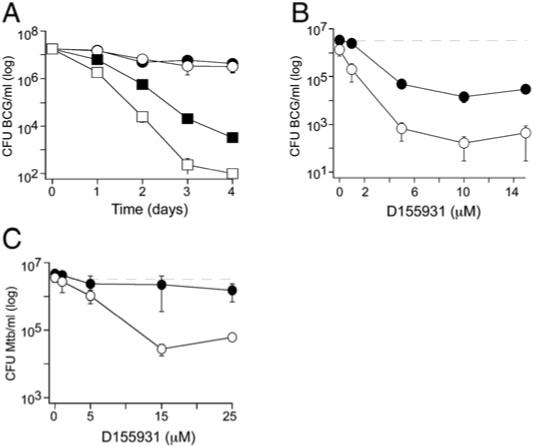
(A) Killing of BCG by D155931 was time dependent and synergistic with nitrite. BCG was treated at pH 5.5 (which prevents its replication) with 10 μM D155931 (squares) or DMSO (circles) without (black symbols) or with (white symbols) 0.5 mM nitrite for 4 days. Nitrite alone at this concentration caused no killing.
(B) Killing of BCG by D155931 was concentration-dependent. BCG was treated with D155931 at pH 5.5 without (black symbols) or with 0.5 mM nitrite (white symbols) for 4 days. Dashed line denotes inoculum.
(C) D155931 only killed nonreplicating Mtb synergistically with nitrite. Mtb H37Rv was treated for 4 days with D155931 at pH 5.5 (which does not prevent its replication) without (black symbols) or with 0.5 mM nitrite (white symbols). Dashed line denotes inoculum. Results are means ± SD of triplicates in single experiments, each of which was repeated at least two times.
However, D155931 failed to kill intracellular BCG within primary mouse macrophages (data not shown). After comparing several esters, we selected D157070 for study in the expectation that masking the anionic character and increasing the hydro-phobicity of the compound might improve its entry into the phagosome, and that mycobacterial esterases might then cleave it to release D155931. To test whether D157070 may serve as a pro-drug for D155931, we quantified D157070 and D155931 by LC/MS/MS after incubation with BCG. D157070 (25 mM; 4.5 mmoles in 180 ml) incubated with 2 3 108 CFU/ml of BCG turned the bacteria yellow (Figure 5A), and we only recovered 94% of D157070 after 24 hr and 83% after 48 hr. Setting the recovered portion to 100%, the compound was distributed as follows: 30% in the medium, 2%−3% in washes of the bacteria, 0.1% in the soluble portion of the bacterial lysate, and 67% in the sedimentable cell walls and membranes. We were not able to detect any D155931 in BCG incubated with D157070. However, when we incubated BCG with D155931 itself, the bacteria turned yellow, indicating the presence of the rhodanine chromophore, yet recovery of D155931 was negligible (2% to 3%). Thus, D155931 appears to associate irreversibly with bacterial constituents. If D157070 did give rise to D155931 inside BCG, we would not have detected it. Although these experiments neither demonstrated nor excluded de-esterification of D157070, they revealed a marked proclivity of D157070 to accumulate in mycobacteria, particularly in the cell wall. Considering only the D157070 that we could not recover (6% at 24 hr; 17% at 48 hr), the concentration attained in BCG cell water would be estimated at 2 to 3 mM. We used D157070 in the remaining studies.
Figure 5. D157070 Kills Nonreplicating Mycobacteria.
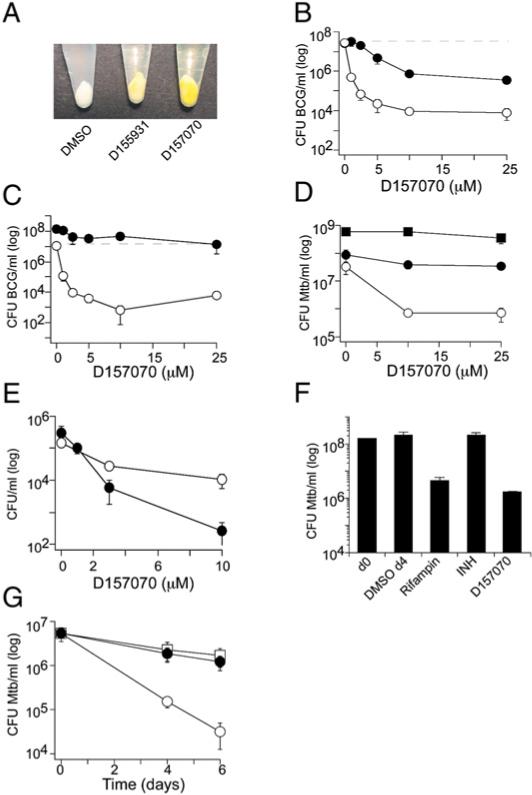
(A) The yellow compounds D155931 (25 μM) and D157070 (25 μM) partitioned from the medium into BCG over 24 hr at pH 6.6, as evidenced by the color of the bacteria collected by centrifugation as compared to BCG exposed to vehicle alone.
(B) Killing of BCG by D157070 was concentration dependent and synergistic with nitrite. BCG was treated at pH 5.5 (which prevents its replication) without (black symbols) or with 0.5 mM nitrite (white symbols) for 4 days. Nitrite alone at this concentration caused no killing. Dashed line denotes inoculum.
(C) D157070 killed BCG at pH 6.6 in the presence of 50 μM DETANO (white symbols), which generates sublethal NO, but not DETA (black symbols). Dashed line denotes inoculum. DETANO or DETA were added at 0, 24, 48, and 72 hr.
(D) D157070 killed Mtb synergistically with nitrite. Mtb was treated with D157070 at pH 5.5 (black circles), pH 5.5 + 0.5 mM nitrite (white circles), or at pH 6.6 (black squares) for 4 days. Inocula were 6.6 × 107 (pH 5.5) and 5.9 × 107 (pH 6.6) CFU/ml.
(E) D157070 killed mycobacteria in Dulbecc's modified Eagle's medium with 10% FBS (“complete medium”), which did not support mycobacterial replication. BCG (black symbols) or Mtb (white symbols) were inoculated at 8 × 105 (BCG) or 1.5 × 105 (Mtb) CFU/ml into complete medium and treated with D157070 for 4 days.
(F) D157070 killed hypoxic Mtb. Mtb was treated with rifampin (1 μg/ml), isoniazid (INH) (0.4 μg/ml), D157070 (12.5 μg/ml), or DMSO for 4 days after reaching stage 2 nonreplicative persistence (Wayne and Hayes, 1996). Results are means ± SD of triplicates in single experiments, each of which was repeated at least two times.
(G) D157070, but not isoniazid, killed hypoxic Mtb with butyrate instead of carbohydrate as a carbon source. Mtb was treated with D157070 (12.5 μg/ml) (white circles), isoniazid (0.1 μg/ml) (black circles), or DMSO (white squares) for 4 or 6 days in environmental chambers at 1% O2, pH 5.5 with 0.3 mM NO2. Results are mean ± SD of triplicates in single experiments, each of which was repeated at least two times.
In 7H9 medium at pH 5.5 (conditions in which BCG does not replicate), D157070 reduced BCG CFU by 1.5 log10 in the absence and by 2.5 to 3 log10 in the presence of 0.5 mM nitrite (Figure 5B). D157070 also killed BCG at nearly neutral pH when replication was inhibited by sublethal concentrations of DETANO (Figure 5C). Like D155931, D157070 had no effect on Mtb at pH 5.5 (conditions in which Mtb replicates) but reduced the number of viable Mtb by 1.5 log10 in the presence of sublethal nitrite (conditions in which Mtb does not replicate) (Figure 5D).
Under growth-sustaining conditions in bacteriologic medium 7H9 at pH 6.6, D157070 had no effect on the viability of BCG (data not shown) or Mtb (Figure 5D). In contrast, D157070 killed BCG and Mtb in Dulbecco's modified Eagle's medium with 10% fetal bovine serum at pH 7.4 (Figure 5E), a medium that mimics human extracellular fluid but does not support the replication of Mtb.
To further test the dependence of bactericidal activity on the microbe's nonreplicative state, we added D157070 to Mtb in Dubos broth at pH 6.6 in the Wayne model of hypoxia (Wayne and Hayes, 1996). Under conditions of stage 2 nonreplicative persistence, D157070 (25 μM; 12.5 μg/ml) reduced the number of viable Mtb by 2 log10 within 4 days of exposure (Figure 5F). As a positive control, we included rifampin (1.2 μM; 1 μg/ml), the only standard chemotherapeutic effective against hypoxic Mtb in vitro (Zhang, 2005). Isoniazid (2.9 μM; 0.4 μg/ml), which is effective only against replicating Mtb, served as a negative control (Figure 5F).
Finally, we subjected Mtb to a model of nonreplicating persistence that we have developed in our lab. It comprises four conditions introduced simultaneously, each of which can prevent Mtb from replicating and each of which is thought to mimic the environment encountered by some populations of Mtb in an infected host: moderate hypoxia (1% O2), replacement of carbohydrate with fatty acid as a carbon source (modified Sauton's medium with 0.05% butyrate), mild acidity (pH 5.5), and low-level nitrite (0.3 mM). In combination, these conditions caused 5 × 106 Mtb Erdman to decline in viability about 0.4 log10 over 6 days. Exposure to isoniazid (0.1 μmg/ml) during that period had no discernible effect. In contrast, D157070 (25 μM; 12.5 μg/ml) reduced the number of viable Mtb by nearly another 2 log10 (Figure 5G).
Evidence that D157070 Kills Mycobacteria in a DlaT-Dependent Manner
In the presence of sublethal nitrite at pH 5.5, Mtb lacking DlaT was almost completely spared by 10 μM D157070 (Figure 6A), while wild-type Mtb (Figure 5D) and the mutant strain complemented with the wild-type allele of dlaT (Figure 6B) were both susceptible. Thus, an Mtb strain with a genetic lack of DlaT was resistant to killing by D157070, and sensitivity was restored when DlaT was expressed.
Figure 6. Killing of Mtb by D157070 Is DlaT-Dependent.
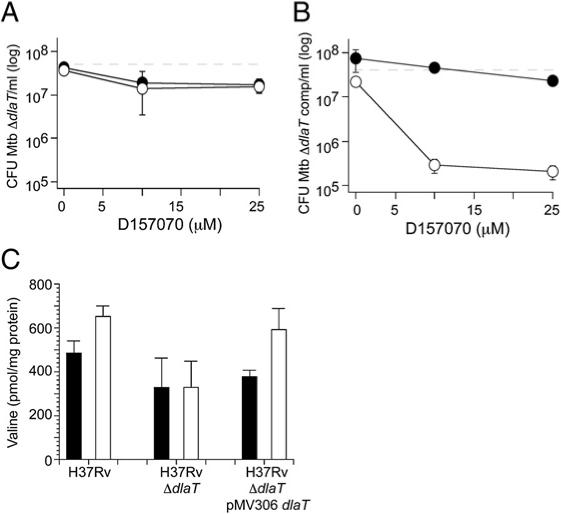
ΔdlaT Mtb (A) or the complemented mutant (ΔdlaT pMV306dlaT) (B) were treated for 4 days with D157070 at pH 5.5 without (black symbols) or with 0.5 mM nitrite (white symbols). Dashed lines denote inocula (6.6 × 107 CFU/ml for ΔdlaT and 5.7 × 107 CFU/ml for ΔdlaT pMV306dlaT). Results in (A) and (B) are mean ± SD of triplicates in single experiments, each of which was repeated at least two times.
(C) Intracellular levels of valine increased in wild-type and ΔdlaT pMV306dlaT Mtb, but not in ΔdlaT, upon treatment with D157070 (10 mM; white bars) as compared to DMSO-treated controls (black bars). Results are mean ± SD from three independent experiments. The following differences in levels of va-line were statistically significant (p < 0.05) by Student's t test: D157070-treated wild-type Mtb versus untreated wild-type; and D157070-treated complemented strain versus untreated complemented strain.
We considered three possible explanations for the fact that susceptibility to D157070 was DlaT-dependent. First, DlaT could be activating the compound to its bactericidal form. However, upon treatment of mycobacteria with D157070, we could quantitatively recover the parent compound, but no metabolites arising from it. Second, after reacting with D157070, DlaT might acquire the ability to inhibit another enzyme, such as its binding partner, Lpd. We could find no evidence for this (data not shown), but cannot exclude such an effect on an enzyme we did not test. Third, mycobacteria that have adapted to the presence of DlaT may be killed when DlaT is acutely inhibited, while those that can only grow by adapting to the absence of DlaT may not be vulnerable to a DlaT inhibitor.
To explore the possibility that DlaT is inhibited in vivo by D157070, we tested whether we could detect accumulation of pyruvate in Mtb grown on glucose and glycerol after treatment with D157070. We reasoned that inhibition of DlaT would result in PDH inhibition, followed by accumulation of pyruvate and intermediary metabolites upstream of PDH. Given the lability of many of the glycolytic and tricarboxylic acid (TCA) cycle metabolites (Tian et al., 2005a), we were unable to detect pyruvate but instead quantified free amino acids, many of which arise from the TCA cycle. We could quantify 13 of the 20 amino acids in Mtb ly-sates. When we added D157070 to wild-type Mtb or the complemented mutant (ΔdlaT pMV306dlaT) grown on glucose and glycerol in the presence of sublethal nitrite at pH 5.5, the only consistently observed change was elevation of valine, a metabolite of pyruvate (Figure 6C). In ΔdlaT Mtb, the valine level did not change upon exposure to D157070 (Figure 6C), though the baseline level of alanine, another major metabolite of pyruvate, was higher than in wild-type or complemented strains (data not shown). These results support the inference that DlaT is inhibited by D157070 in intact Mtb. These results also underscore that genetic absence of DlaT affected Mtb's intermediary metabolism differently than acute treatment with a DlaT inhibitor.
Impact of D157070 on Mycobacteria within Macrophages
When applied to cultures of infected bone marrow-derived mouse macrophages (BMM) for 2 days, D157070 (5 μM) reduced the number of viable Mtb by ∼2.5 log10 CFU (Figure 7A), thus overcoming the ineffectiveness of the parent compound, D155931, on mycobacteria residing in macrophages. D157070 also killed BCG in mouse BMM in a dose- (data not shown) and time-dependent manner (Figure 7B). Although D157070 (5 μM) achieved 2 log10 reduction in CFU by day 1, BCG appeared to regain some of its replicative potential when recovered from macrophages on day 4. This suggested that upon exposure to D157070, BMM may upregulate multidrug resistance (MDR) export pumps, so that intracellular concentrations of D157070 in macrophages initially rose and then declined. Treatment of BCG-infected BMM with a combination of D157070 and verapamil, an MDR pump inhibitor, led to >2 log10 killing of BCG by day 1, and this was sustained for 4 days, the duration of the experiment (Figure 7B). Verapamil alone had no effect on BCG in culture or in BMM. Enhanced intracellular accumulation of D157070 in the presence of verapamil was confirmed microscopically by the coloration of the cells (data not shown). D157070 was nontoxic to the BMM during the treatment, as judged by morphology and trypan blue exclusion (data not shown). Similarly, there was little or no morphologic or metabolic evidence of toxicity of D157070 to primary human lung and skin fibroblasts and Vero monkey kidney cells at up to 30 μM over 48 hr (Figure 7C).
Figure 7. D157070 Kills BCG and Mtb in Mouse Bone Marrow Macro phages but Spares Mammalian Cells.

(A) BMM infected with Mtb were treated with D157070 at 5 μM (black bar) or with DMSO (white bar) beginning after a 4 hr period of infection (day 0) with Mtb (3.8 × 104 CFU/ml). Medium and compound were replaced on day 1. On day 2, the monolayers were lysed, and CFU were determined.
(B) BMM infected with BCG were treated with D157070 at 5 μM (white circles) or with DMSO (black circles) or with a combination of D157070 (5 μM) and verapamil (50 μM) (black squares) beginning after a 4 hr period of infection (day 0) with BCG (1 × 104 CFU/ml). Medium and compounds were replaced on day 2.
(C) Primary human fibroblasts from lung (triangles) or skin (squares) and monkey kidney Vero cells (circles) were treated with D157070 at the indicated concentrations for 48 hr and assessed for viability by MTS assay.
Results are mean ± SD of triplicates in single experiments, each of which was repeated at least two times. *p < 0.05 by unpaired t test.
DISCUSSION
The inability of ΔdlaT Mtb to cause tuberculosis in the guinea pig validates DlaT as a target. We could not have predicted this degree of attenuation from the phenotype of ΔdlaT Mtb in bacteriologic growth medium or mice (Shi and Ehrt, 2006). It will be important to learn more about the intermediary metabolism of Mtb in mice, guinea pigs, and humans and the levels of nitroxidative stress that Mtb faces in these hosts.
We have identified the rhodanine D155931 as an irreversible, substrate-dependent, time-dependent inhibitor of Mtb's DlaT and DlaT-dependent peroxynitrite reductase-peroxidase and PDH. Its propanolic ester, D157070, partitioned from aqueous media into mycobacteria and killed Mtb within macrophages. D155931 and D157070 killed mycobacteria only when the bacteria were precluded from replicating (Table 1). Inhibition of replication could be afforded by sublethal levels of acid, RNI, or hypoxia; by combinations of these with deprivation of carbohydrate and provision of fatty acid as a carbon source; or by incubation in a culture medium optimized for mammalian cells. These findings suggest new approaches to the search for chemotherapeutics useful in persistent infections. For example, these inhibitors could not have been identified had we screened for mycobactericidal activity in a bacteriologic growth medium, as anti-infectives are customarily sought. In contrast, we presumably could have identified them by screening for compounds that kill mycobacteria in a medium resembling human extracellular fluid.
Table 1.
Summary of Rhodanine Effects on Mycobacteria
| D155931 (μM)a |
D157070 (μM)a |
|||
|---|---|---|---|---|
| Condition | BCG | Mtb | BCG | Mtb |
| 7H9, pH 6.6 | NE | NE | NE | NE |
| 7H9, pH 6.6 + DETANO | - | - | 1 | - |
| 7H9, pH 5.5 | 10 | NE | 15 | NE |
| 7H9, pH 5.5 + NO2− | 3 | 15 | 2 | 10 |
| DMEM-10%FBS, pH 7.4 | - | - | 4 | >10 |
| Hypoxia, NRP-2 | - | - | - | 25 |
| Mouse BMMs | NE | NE | 5 | 5 |
-, not tested; NE, no effect.
Concentration at which 2 log10 killing is observed.
Anti-infectives may kill a nonreplicating pathogen in at least two ways: by inhibiting metabolic processes that the pathogen needs to sustain viability or by inhibiting pathways of detoxification or repair that the pathogen uses to protect itself from sterilization by the host chemistry that prevents it from replicating. Inhibitors of Mtb's DlaT may act by both mechanisms. To our knowledge, the synergy between the compounds studied here and host antimicrobial factors such as RNI and acid has only one precedent among clinically approved anti-infectives, in that pyrazinamide kills Mtb preferentially at low pH (Zhang, 2005). Pyrazinamide sterilizing activity is also remarkable in that it increases as Mtb enters dormancy in very late stationary phase, as modeled by Hu, Coates, and Mitchison (Hu et al., 2006). Only one standard anti-mycobacterial agent, rifampin, is effective under hypoxic conditions in vitro, although some experimental compounds share this property (Stover et al., 2000).
Our studies raise the surprising possibility that PDH may be less critical to the survival of replicating than nonreplicating mycobacteria. Perhaps alternative metabolic pathways are more readily available to mycobacteria under growth conditions than under conditions stringent enough to preclude replication. However, such inferences are complicated by partial discrepancies between the phenotypes of ΔdlaT Mtb and wild-type Mtb treated with D157070. Both ΔdlaT Mtb and wild-type Mtb treated with D157070 survived at pH 5.5 in the absence of RNI. At pH 5.5, 3 mM nitrite killed the ΔdlaT mutant more extensively than wild-type Mtb (Shi and Ehrt, 2006). D157070-treated wild-type Mtb succumbed to 0.5 mM nitrite, which spared untreated Mtb. Thus, knockout of the gene and treatment with an inhibitor of the enzyme both increased Mtb's sensitivity to nitrite, but not to the same extent. D157070 had very little mycobactericidal effect on Mtb that lacked DlaT. Likewise, the level of valine in ΔdlaT Mtb did not rise in response to D157070 as it did in wild-type Mtb and the complemented mutant. Yet, the basal level of valine differed markedly in ΔdlaT Mtb from that in the D157070-treated wild-type and complemented strains.
At least two explanations for these discrepancies are likely. First, the ΔdlaT mutant may have adapted to prolonged DlaT deficiency by upregulating compensatory pathways of metabolism and anti-RNI defense. Second, it is common that chemical inhibitors have more than one target in living organisms. Various rhodanines with different substituents than those studied here were reported to inhibit a wide range of targets, including protein tyro-sine phosphatases (Cutshall et al., 2005), Klebsiella UDP-galactopyranose mutase (Soltero-Higgin et al., 2004), viral (Powers et al., 2006) and bacterial (Gualtieri et al., 2006) RNA polymerase, and Plasmodium enoyl-acyl carrier protein reductase (Kumar et al., 2007). D157070 is likely to have (an) additional target(s) in Mtb besides DlaT, in particular because it accumulates in myco-bacteria to very high concentrations. Efforts to identify additional targets are underway. Irrespective of the specific antimyco-bacterial targets of D157070, it is clear that the ability of D157070 to kill nonreplicating mycobacteria is DlaT-dependent.
Some authorities have argued that “the dependence of front-line anti-TB drugs on actively replicating cells for activity is probably the greatest limitation of current therapy” (Warner and Mizrahi, 2006). According to this view, more agents that kill nonreplicating Mtb are needed to shorten the time it takes to treat active tuberculosis (Duncan and Barry, 2004). Treatment of latent Mtb infection is a critical need in its own right, because latent Mtb infection sustains the pandemic: nearly 9 million latently infected people develop active tuberculosis per year, each of whom infects an estimated 10−15 other people per year before being isolated, responding to treatment, or dying (World Health Organization, 2006).
Several clinically proven (rifampin) (Zhang, 2005) and experimental agents (Stover et al., 2000) kill nonreplicating Mtb. However, they kill replicating Mtb better. This is a useful property. Nonetheless, it has reinforced the presumption that anti-infective compounds must necessarily be more effective against replicating than nonreplicating bacterial pathogens. The most important result of this work is to show that the opposite is possible, lending strong support to the argument of Coates and Hu (Coates and Hu, 2007) that screens should be undertaken for agents that kill nonreplicating pathogens. Given suitable pharmaceutical properties, such agents could be used for prophylaxis in individuals with latent Mtb infection, and could be combined with conventional agents to shorten the treatment of clinically active tuberculosis.
EXPERIMENTAL PROCEDURES
Recombinant AhpD, DlaT, and Lpd from Mtb were expressed in E. coli and purified as reported (Argyrou and Blanchard, 2001; Bryk et al., 2002). A combinatorial library of 15,000 small molecules with drug-like characteristics covering 125 different templates from Chemical Diversity, Inc. was used for screen. Compounds in DMSO or the vehicle alone as a control were robotically dispensed to Falcon Microtest 384-well plates containing a mixture of the 3 enzymes (Lpd, 200 nM; DlaT, 350 nM; AhpD, 36 nM). After a 30 min preincubation with inhibitors, an equal volume of the reaction mix (NADH, 200 μM; DTNB, 150 μM;EDTA,2mM;potassiumphosphate,100mM;pH7.0)wasadded.Optical density was recorded at 405 nm at 0 and 30 min. We selected compounds producing 50% (7, 000 compounds tested at 10 μM) or 75% (8,000 compounds tested at 25 μM) inhibition, confirmed their activity after resupply and resynthesis, and identified each as inhibiting either AhpD, Lpd, or DlaT by separate as-says. Lpd inhibitors were identified by inhibition of lipoamide-dependent NADH consumption. DlaT inhibitors were identified in Lpd/DlaT-dependent DTNB assay. Kinetic analyses were carried out with the DTNB peroxidase assay. Bovine thioredoxin reductase was purified as described (Zhong et al., 2000) and assayed with 100 μM NADPH and 75 μM DTNB. PDH activity was measured in 50 μM KPi, pH 7.0, 1 μM NAD+, 200 μM thiamine pyrophosphate, 1 mM MgCl2, 1 mM pyruvate and 170 μM CoA with a mixture of 100 nM Lpd, 175 nM DlaT and 100 nM AceE (for Mtb's PDH), or with porcine PDH (Sigma) after removing BSA on Sephadex G100. NADH formation was monitored at 340 nm for 5 min.
Mtb H37Rv (ATCC 25618) and M. bovis BCG (ATCC 35734) were cultivated in Middlebrook 7H9 broth, pH 6.6 with 0.2% glycerol, 0.05% Tween 80 and 10% ADN (0.5% BSA, 2% dextrose, 0.85% NaCl). Where indicated, we also tested Mtb Erdman. CFU were enumerated on Middlebrook 7H11 agar plates with 10% Middlebrook OADC enrichment. Midlog cultures (A580 0.8−1.0) were prepared as single cell suspensions in 7H9 at pH 5.5, diluted to 0.1 (A580). Mycobacteria were incubated under indicated test conditions in 96-well plates in 200 ml and plated for CFU after serial dilution in 7H9, pH 6.6. Lysates were prepared as described (Bryk et al., 2002), and protein was determined by the Bradford method. Hypoxic conditions were achieved as specified by Wayne and Hayes (Wayne and Hayes, 1996). Decoloration of methylene blue in control tubes by 12−14 days indicated stage 2 nonreplicative persistence. On day 18, control tubes were cultured and experimental tubes received DMSO, D157070, isoniazid, or rifampin by injection through seals, which were then reinforced with wax. Experimental tubes were plated 4 days after injection of compounds. Hypoxic conditions (1% O2) in experiments with butyrate as a carbon source were achieved in environmental chambers from BD Biosciences. For these experiments, Mtb Erdman was treated with D157070 (25 μM) or isoniazid (0.1 μg/ml) for 4 and 6 days in modified Sauton's medium (0.1% NH4Cl, 0.005% ferric ammonium citrate) with 10% AN (0.5% BSA, 0.85% NaCl), 0.02% tyloxapol, 0.05% butyrate at pH 5.5 in the presence of 0.3 mM NaNO2.
Intracellular levels of amino acids (AA) were determined on 15 ml cultures of Mtb inoculated at A580 0.1 in 7H9 at pH 5.5 with 0.5 mM NaNO2 with or without 10 μM D157070. After 24 hr, metabolism was interrupted by rapid cooling. Bacteria were pelleted at 4°C and washed twice with ice-cold PBS, 0.05% Tween 80. No AA were detectable in supernatants or washes. Lysates were prepared by mechanical disruption and normalized according to protein concentration by Bradford assay. AA were extracted as described (Khan et al., 1991), and levels were determined by o-phthaldialdehyde derivatization and reverse phase high-performance liquid chromatography with fluorescent detection (Henderson et al., 2000).
BMMs differentiated for 7 days as described (Shi and Ehrt, 2006) were removed from the original cultures and replated at 1 × 105 cell/well in 48-well plates in 0.5 ml of DMEM with 4.5 g/l glucose, 0.584 g/l L-glutamine, 1 mM pyruvate, 10% FBS, and 10% L-cell conditioned medium (“complete medium”). One day later, ∼105 mycobacteria were added. Four hours later, monolayers were washed twice in PBS. Complete medium (0.5 ml) with inhibitors or DMSO as a control was added and replaced daily (for Mtb-infected cells) or on days 2 and 4 (for BCG-infected cells). On indicated days, monolayers were lysed in PBS with 0.5% Triton X-100 for CFU determination. Macrophage viability was monitored by photomicroscopy, trypan blue exclusion, and/or staining with ethidium homodimer and calcein AM (Invitrogen). Primary, diploid human lung (ATCC CCL-171) and skin (ATCC CRL-2522) fibroblasts and monkey kidney Vero 76 cells (ATCC CRL-1587) were incubated with test compounds for 24 hr in complete medium, and viability was assessed by photomicroscopy and MTS tetrazolium reduction assay (Invitrogen).
Guinea pig studies and the synthesis, structural confirmation, purity, and assay of D155931 and D157070 are described in the Supplemental Data available with this article online.
SUPPLEMENTAL DATA
The Supplemental Data include Supplemental Experimental Procedures, three supplemental figures, and one supplemental table and can be found with this article online at http://www.cellhostandmicrobe.com/cgi/content/full/3/3/137/DC1/.
ACKNOWLEDGMENTS
This work was supported by NIH (UC1-A1062559 and the TARGET consortium, N01-AI30036) and the Milstein Program in Chemical Biology of Infectious Disease. The Department of Microbiology and Immunology is supported by the William Randolph Hearst Foundation. We thank T.M. Kapoor (Rockefeller University) for invaluable collaboration; W. Bishai (Johns Hopkins University) and D. McMurray (Texas A&M University) for access to the TARGET program; T. Andresson, S. Hrafnsdóttir and I. Gylfadóttir (deCODE Genetics, Inc., Reykjavik, Iceland) for high-throughput screening results not included here; M. Patel (State University of New York at Buffalo) for a kind gift of PDH; A. Zubkina, J. Roberts (Weill Medical College) and E. Onua, M. Keyvan, and J. Zhang (de-CODE Chemistry) for technical assistance; M. Fuortes for photomicroscopy; and A. Ding and G. Vogt (Weill Medical College) for critical comments. R.B., J.S., M.G., and C.N. are listed as coinventors on a patent application for compounds described in this report under a policy by which any resulting product would be distributed on a not-for-profit basis in low and middle income countries. None of the authors has any other financial interest bearing on this work.
Supplementary Material
REFERENCES
- Argyrou A, Blanchard JS. Mycobacterium tuberculosis lipoamide dehydrogenase is encoded by Rv0462 and not by the lpdA or lpdB genes. Biochemistry. 2001;40:11353–11363. doi: 10.1021/bi010575o. [DOI] [PubMed] [Google Scholar]
- Boshoff HI, Barry CE,, III Tuberculosis - metabolism and respiration in the absence of growth. Nat. Rev. Microbiol. 2005;3:70–80. doi: 10.1038/nrmicro1065. [DOI] [PubMed] [Google Scholar]
- Bryk R, Griffin P, Nathan C. Peroxynitrite reductase activity of bacterial peroxiredoxins. Nature. 2000;407:211–215. doi: 10.1038/35025109. [DOI] [PubMed] [Google Scholar]
- Bryk R, Lima CD, Erdjument-Bromage H, Tempst P, Nathan C. Metabolic enzymes of mycobacteria linked to antioxidant defense by a thioredoxin-like protein. Science. 2002;295:1073–1077. doi: 10.1126/science.1067798. [DOI] [PubMed] [Google Scholar]
- Coates AR, Hu Y. Novel approaches to developing new antibiotics for bacterial infection. Br. J. Pharmacol. 2007;152:1147–1154. doi: 10.1038/sj.bjp.0707432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutshall NS, O'Day C, Prezhdo M. Rhodanine derivatives as inhibitors of JSP-1. Bioorg. Med. Chem. Lett. 2005;15:3374–3379. doi: 10.1016/j.bmcl.2005.05.034. [DOI] [PubMed] [Google Scholar]
- Duncan K, Barry CE,, III Prospects for new antitubercular drugs. Curr. Opin. Microbiol. 2004;7:460–465. doi: 10.1016/j.mib.2004.08.011. [DOI] [PubMed] [Google Scholar]
- Ferreras JA, Ryu JS, Di Lello F, Tan DS, Quadri LE. Small-molecule inhibition of siderophore biosynthesis in Mycobacterium tuberculosis and Yersinia pestis. Nat. Chem. Biol. 2005;1:29–32. doi: 10.1038/nchembio706. [DOI] [PubMed] [Google Scholar]
- Gualtieri M, Bastide L, Villain-Guillot P, Michaux-Charachon S, Latouche J, Leonetti JP. In vitro activity of a new antibacterial rhodanine derivative against Staphylococcus epidermidis biofilms. J. Antimicrob. Chemother. 2006;58:778–783. doi: 10.1093/jac/dkl314. [DOI] [PubMed] [Google Scholar]
- Henderson J, Ricker R, Bidlingmeyer B, Woodward C. Application bulletin 5980–1193E. Agilent Technologies; Palo Alto, CA: 2000. Rapid, accurate, sensitive and reproducible HPLC analysis of amino acids. [Google Scholar]
- Hobby GL, Lenert TF. The in vitro action of antituberculous agents against multiplying and non-multiplying microbial cells. Am. Rev. Tuberc. 1957;76:1031–1048. doi: 10.1164/artpd.1957.76.6.1031. [DOI] [PubMed] [Google Scholar]
- Hondalus MK, Bardarov S, Russell R, Chan J, Jacobs WR,, Jr., Bloom BR. Attenuation of and protection induced by a leucine auxotroph of Mycobacterium tuberculosis. Infect. Immun. 2000;68:2888–2898. doi: 10.1128/iai.68.5.2888-2898.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Coates AR, Mitchison DA. Sterilizing action of pyrazinamide in models of dormant and rifampicin-tolerant Mycobacterium tuberculosis. Int. J. Tuberc. Lung Dis. 2006;10:317–322. [PubMed] [Google Scholar]
- Khan K, Blaak E, Elia M. Quantifying intermediary metabolites in whole blood after a simple deproteinization step with sulfosalicylic acid. Clin. Chem. 1991;37:728–733. [PubMed] [Google Scholar]
- Kumar G, Parasuraman P, Sharma SK, Banerjee T, Karmodiya K, Surolia N, Surolia A. Discovery of a rhodanine class of compounds as inhibitors of Plasmodium falciparum enoyl-acyl carrier protein reductase. J. Med. Chem. 2007;50:2665–2675. doi: 10.1021/jm061257w. [DOI] [PubMed] [Google Scholar]
- Lancaster JJ. Nitroxidative stres: The dominant process of reactive nitrogen species chemistry under biological conditions. Free Radic. Biol. Med. 2004;37:S98. [Google Scholar]
- Levin BR, Rozen DE. Non-inherited antibiotic resistance. Nat. Rev. Microbiol. 2006;4:556–562. doi: 10.1038/nrmicro1445. [DOI] [PubMed] [Google Scholar]
- Lipinski C, Hopkins A. Navigating chemical space for biology and medicine. Nature. 2004;432:855–861. doi: 10.1038/nature03193. [DOI] [PubMed] [Google Scholar]
- MacMicking JD, Taylor GA, McKinney JD. Immune control of tuberculosis by IFN-gamma-inducible LRG-47. Science. 2003;302:654–659. doi: 10.1126/science.1088063. [DOI] [PubMed] [Google Scholar]
- McCune RM, Feldmann FM, Lambert HP, McDermott W. Microbial persistence. I. The capacity of tubercle bacilli to survive sterilization in mouse tissues. J. Exp. Med. 1966;123:445–468. doi: 10.1084/jem.123.3.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurray DN. Disease model: Pulmonary tuberculosis. Trends Mol. Med. 2001;7:135–137. doi: 10.1016/s1471-4914(00)01901-8. [DOI] [PubMed] [Google Scholar]
- Munoz-Elias EJ, McKinney JD. Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence. Nat. Med. 2005;11:638–644. doi: 10.1038/nm1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Elias EJ, Timm J, Botha T, Chan WT, Gomez JE, McKinney JD. Replication dynamics of Mycobacterium tuberculosis in chronically infected mice. Infect. Immun. 2005;73:546–551. doi: 10.1128/IAI.73.1.546-551.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan C. Antibiotics at the crossroads. Nature. 2004;431:899–902. doi: 10.1038/431899a. [DOI] [PubMed] [Google Scholar]
- Nathan C, Ehrt S. Nitric oxide in tuberculosis. In: Rom WN, Garay SM, editors. Tuberculosis. Lippincott Williams & Wilkins; Philadelphia: 2004. pp. 215–235. [Google Scholar]
- Powers JP, Piper DE, Li Y, Mayorga V, Anzola J, Chen JM, Jaen JC, Lee G, Liu J, Peterson MG, et al. SAR and mode of action of novel non-nucleoside inhibitors of hepatitis C NS5b RNA polymerase. J. Med. Chem. 2006;49:1034–1046. doi: 10.1021/jm050859x. [DOI] [PubMed] [Google Scholar]
- Rhee KY, Erdjument-Bromage H, Tempst P, Nathan CF. S-nitroso proteome of Mycobacterium tuberculosis: Enzymes of intermediary metabolism and antioxidant defense. Proc. Natl. Acad. Sci. USA. 2005;102:467–472. doi: 10.1073/pnas.0406133102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogerson BJ, Jung YJ, LaCourse R, Ryan L, Enright N, North RJ. Expression levels of Mycobacterium tuberculosis antigen-encoding genes versus production levels of antigen-specific T cells during stationary level lung infection in mice. Immunology. 2006;118:195–201. doi: 10.1111/j.1365-2567.2006.02355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambandamurthy VK, Wang X, Chen B, Russell RG, Derrick S, Collins FM, Morris SL, Jacobs WR,, Jr. A pantothenate auxotroph of Mycobacterium tuberculosis is highly attenuated and protects mice against tuberculosis. Nat. Med. 2002;8:1171–1174. doi: 10.1038/nm765. [DOI] [PubMed] [Google Scholar]
- Schnappinger D, Ehrt S, Voskuil MI, Liu Y, Mangan JA, Monahan IM, Dolganov G, Efron B, Butcher PD, Nathan C, Schoolnik GK. Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: Insights into the phagosomal environment. J. Exp. Med. 2003;198:693–704. doi: 10.1084/jem.20030846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Jung YJ, Tyagi S, Gennaro ML, North RJ. Expression of Th1-mediated immunity in mouse lungs induces a Mycobacterium tuberculosis transcription pattern characteristic of nonreplicating persistence. Proc. Natl. Acad. Sci. USA. 2003;100:241–246. doi: 10.1073/pnas.0136863100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Sohaskey CD, Kana BD, Dawes S, North RJ, Mizrahi V, Gennaro ML. Changes in energy metabolism of Mycobacterium tuberculosis in mouse lung and under in vitro conditions affecting aerobic respiration. Proc. Natl. Acad. Sci. USA. 2005;102:15629–15634. doi: 10.1073/pnas.0507850102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi S, Ehrt S. Dihydrolipoamide acyltransferase is critical for Mycobacterium tuberculosis pathogenesis. Infect. Immun. 2006;74:56–63. doi: 10.1128/IAI.74.1.56-63.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltero-Higgin M, Carlson EE, Phillips JH, Kiessling LL. Identification of inhibitors for UDP-galactopyranose mutase. J. Am. Chem. Soc. 2004;126:10532–10533. doi: 10.1021/ja048017v. [DOI] [PubMed] [Google Scholar]
- Stover CK, Warrener P, VanDevanter DR, Sherman DR, Arain TM, Langhorne MH, Anderson SW, Towell JA, Yuan Y, McMurray DN, et al. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature. 2000;405:962–966. doi: 10.1038/35016103. [DOI] [PubMed] [Google Scholar]
- Tian J, Bryk R, Itoh M, Suematsu M, Nathan C. Variant tricarboxylic acid cycle in Mycobacterium tuberculosis: Identification of alpha-ketoglutarate decarboxylase. Proc. Natl. Acad. Sci. USA. 2005a;102:10670–10675. doi: 10.1073/pnas.0501605102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian J, Bryk R, Shi S, Erdjument-Bromage H, Tempst P, Nathan C. Mycobacterium tuberculosis appears to lack alpha-ketoglutarate dehydrogenase and encodes pyruvate dehydrogenase in widely separated genes. Mol. Microbiol. 2005b;57:859–868. doi: 10.1111/j.1365-2958.2005.04741.x. [DOI] [PubMed] [Google Scholar]
- Timm J, Post FA, Bekker LG, Walther GB, Wainwright HC, Manganelli R, Chan WT, Tsenova L, Gold B, Smith I, et al. Differential expression of iron-, carbon-, and oxygen-responsive mycobacterial genes in the lungs of chronically infected mice and tuberculosis patients. Proc. Natl. Acad. Sci. USA. 2003;100:14321–14326. doi: 10.1073/pnas.2436197100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voskuil MI, Schnappinger D, Visconti KC, Harrell MI, Dolganov GM, Sherman DR, Schoolnik GK. Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis dormancy program. J. Exp. Med. 2003;198:705–713. doi: 10.1084/jem.20030205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh C. Where will new antibiotics come from? Nat. Rev. Microbiol. 2003;1:65–70. doi: 10.1038/nrmicro727. [DOI] [PubMed] [Google Scholar]
- Warner DF, Mizrahi V. Tuberculosis chemotherapy: The influence of bacillary stress and damage response pathways on drug efficacy. Clin. Microbiol. Rev. 2006;19:558–570. doi: 10.1128/CMR.00060-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayne LG, Hayes LG. An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect. Immun. 1996;64:2062–2069. doi: 10.1128/iai.64.6.2062-2069.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization Tuberculosis: Infection and Transmission. 2006 ( http://www.who.int/mediacentre/factsheets/fs104/en/)
- Zhang Y. The magic bullets and tuberculosis drug targets. Annu. Rev. Pharmacol. Toxicol. 2005;45:529–564. doi: 10.1146/annurev.pharmtox.45.120403.100120. [DOI] [PubMed] [Google Scholar]
- Zhong L, Arner ES, Holmgren A. Structure and mechanism of mammalian thioredoxin reductase: The active site is a redox-active selenolthiol/selenenylsulfide formed from the conserved cysteine-selenocysteine sequence. Proc. Natl. Acad. Sci. USA. 2000;97:5854–5859. doi: 10.1073/pnas.100114897. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.