

Abstract
A short synthesis of (±)-brazilin is reported. This synthesis uses several interesting and underutilized transformations including a regioselective dirhodium-catalyzed aryl C–H insertion, a regioselective IBX phenol → o-quinone oxidation, a tautomerization of an o-quinone to a p-quinone methide, and an intramolecular aryl cyclization with a p-quinone methide.
The structurally related natural products (+)-brazilin (1) and (−)-haematoxylin (2)1 and their reduced counterparts (+)-brazilane (3) and haematoxylane (4) have been known for some time.2 Brazilide A (5) has been proposed as the newest constituent of this natural product family.3 Recent efforts culminated in the first enantioselective synthesis of a member of this family, a tris-methylated derivative of 1.4 Compounds 1–5 (Figure 1) are all found in the alcoholic extracts of trees collectively referred to as Brazil wood located in the equator regions.5 These ethanolic tinctures were formally used in medicines, but have since found application in the production of dyes and inks. The origins of their pink color resides in the ability of these catechol products to oxidize to their o-quinone and tautomeric p-quinone methide counterparts. Beyond this redox chemistry, our interest in devising a general synthesis for this family emerged after reading a report on the ability of brazilin (1) to serve as a micromolar telomerase inhibitor6 and to nick DNA.7 These properties are fairly common among anticancer agents. Therefore, we devised a strategy that would enable an asymmetric preparation of 1 by desymmetrization. Herein, we report the successful conclusion of the initial phase of our plan, a racemic synthesis of brazilin (1) using o-iodoxybenzoic acid (IBX) for o-quinone formation followed by tautomerization and a desymmetrizing cyclization.
Figure 1.
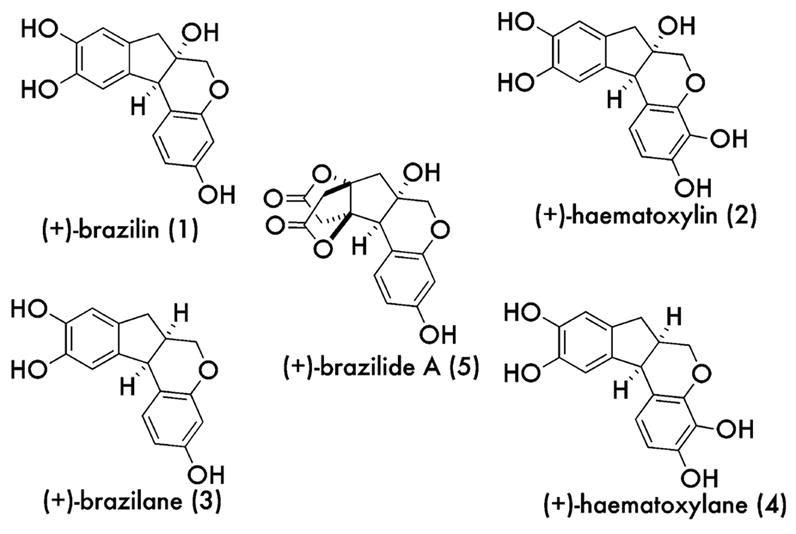
Brazilin family of natural products.
Our retrosynthetic plan for brazilin (1) is depicted in Scheme 1. The brazilin skeleton 6 would emerge from the symmetric o-quinone 7 by a tautomerization to the corresponding p-quinone methide and an intramolecular aryl cyclization. The o-quinone 7 would form upon the oxidation of the phenol 8 using a method developed by our group.8 The phenol 8 should appear upon addition of the metal species 10 to the indanone 9, a ring system that seemed accessible from 3-hydroxy phenyl acetic acid 11 by homologation and aryl C-H insertion.
Scheme 1.
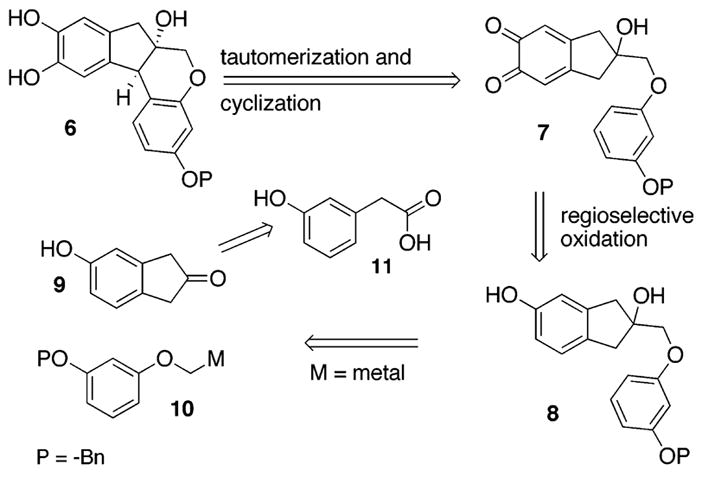
Retrosynthesis of Brazilin (1)
Some time ago, we revealed the first convenient method for regioselective formation of an o-quinone such as 7 from a phenol such as 8 and established the first step of this mechanism to be an intramolecular electrophilic aromatic substitution.8 Unlike the Fremy’s salt procedure, our IBX procedure proves successful in oxidizing phenols to o-quinones even in the absence of a para substituent. Our disclosure of this transformation was followed by several publications from others. These included the development of a benzoic acid stabilized and supposedly nonexplosive IBX reagent,9 as well as the use of Dess-Martin periodane.10 In addition, our method found application in several syntheses.11 We expect that it should enable improved synthetic access to a variety of biologically active catechols, such as those that inhibit protein tyrosine phosphatase 1B,12 decrease anxiety,13 and serve as a 5-HT2C receptor agonists.14 Given our considerable experience using this reagent for o-quinone formation, we were surprised by a recent article reporting low yields and poor selectivity when IBX was applied to the 2,3-dimethylphenol 12 and a tetralin system similar to 14 shown in Scheme 2.15 Because of our intended strategy for the synthesis of 1, we paused to consider the cause of these potentially problematic findings. We began by evaluating literature examples of other electrophillic aromatic substitution reactions. As expected, trends in regioselective substitution have been known for some time. Among the most comprehensive and enlightening investigations are studies examining the regiochemistry for bromination16 and amino-methylation.17 An abbreviated excerpt from these studies is shown in Scheme 2 for 2,3-dimethyl phenol 12, 2,3-dihydro-1H-inden-5-ol 13, and 5,6,7,8-tetrahydro-naphthalen-2-ol 14.
Scheme 2.
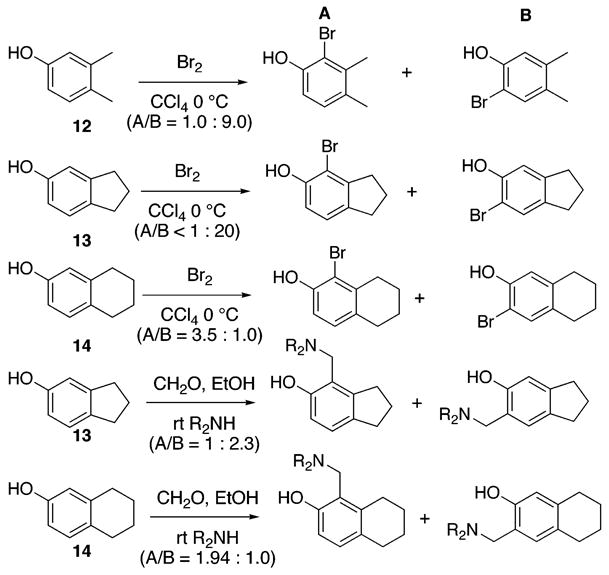
Some Literature Examples of Regioselective Electrophillic Aromatic Substitutions
Our results for IBX oxidation of the three skeletons 12–14 are summarized in Scheme 3. Reactions were conducted in deuterated dimethylformamide (0.1 M, 0 °C) and monitored with 1H NMR using a methylene chloride standard. Oxidation of the phenol 12 results in a 1.0:2.3 ratio of 15:16 in a 63% overall yield after 90 min. The indene skeleton 13 favors the regioisomer 18 over 17 with a 4.4:1.0 ratio and 70% overall yield after 75 min. Of some interest is the oxidation of the tetralin 14, which favors the regioisomer 19 over 20 in a 3.5:1.0 ratio with a combined yield of 59% after 90 min. However, this reversal of selectivity was expected given the examples of electrophilic aromatic substitution shown in Scheme 2. It should be noted that when reactions were left unattended for lengthier periods of time without in situ reduction to the corresponding catechol, the o-quinone products began to decompose at different rates. As the yield decreases, the regiomeric ratio can appear to improve or worsen.
Scheme 3.
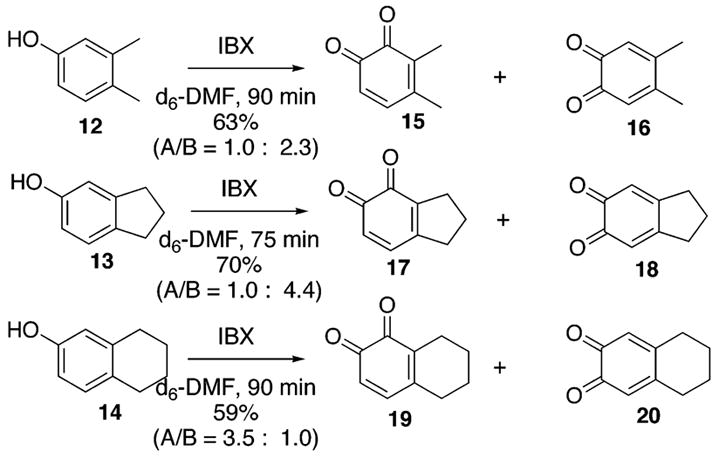
Regioselective Oxidations of 12–14 with IBX
With our worries concerning the oxidation of phenol 8 laid to rest, we began our synthesis of brazilin 1 in earnest. Sequential treatment of the 2-(3-hydroxyphenyl)acetic acid 11 (0.07 M in CH2Cl2/THF (10:1)) with oxalyl chloride (1.05 equiv), dimethylformamide (0.03 equiv), and diazomethane (2.2 equiv in ether) smoothly affords the phenolic diazoketone 21 in 68% yield. However, acid-promoted cyclization of the diazoketone 21 proved unsatifactory; trifluoroacetic acid affords the 2,2,2-trifluoroacetate adduct 22 (90% yield) from 21 and a small amount of the 4-hydroxy-1H-inden-2(3H)-one 9.18 On the other hand, addition of boron trifluoride etherate led to unidentified products and a 1:1 mixture of compounds 23 and 9 in a 40% overall yield. We were surprised to find that addition of rhodium acetate (0.02 equiv) to the diazoketone 21 (0.045 M in CH2Cl2) exclusively furnishes the more congested 4-hydroxy-1H-inden-2(3H)-one 23 in a 53% yield.19 Formation of product 23 suggests to us that the site of cyclization is governed by the unprotected phenol residue. This suspicion was confirmed by subjecting the 3-(methoxymethoxy)phenyl derivative 24, prepared from 21, to rhodium acetate for cyclization. The ensuing regioselective cyclization affords the 5-(meth-oxymethoxy)-1H-inden-2(3H)-one 25 in 74% yield. The desired indanone skeleton 9 was also prepared by treatment of the diazoketone 26, derived from 4-hydroxyphenylacetic acid in 71% yield, with rhodium acetate. However, the resulting 28% yield implies that the cyclization suffers without the appropriately positioned and protected oxygen substituent.
Next, we examined nucleophiles in 1,2-addition reactions with the ketones 9 and 25. The acidity of the methylene residues in the inden-2(3H)-ones 9 and 25 was an anticipated problem. Alkylation of the commerically available 3-(ben-zyloxy)phenol with (bromomethyl)-tributyl-stannane affords the stannane 27.20 After considerable experimentation, we found that adducts 8 (Scheme 4) and 28 are accessible in yields of 26% and 61%, respectively, by the addition of solid 9 (0.42 equiv) or solid 25 (0.83 equiv) directly to a solution of the lithium species (0.6 M in THF, precooled to −100 °C) derived from addition of n-butyllithium to the stannane 27. Although the former sequence precludes protection and deprotection, we chose the latter. Compound 28 undergoes quantitative deprotection to phenol 8 (MeOH, concentrated HCl). Oxidation of the phenol 8 (0.03 M in DMF, room temperature) with IBX (1.05 equiv) affords the o-quinone 7 in 67% yield as determined by 1H NMR. Inspection of the crude 1H NMR spectrum reveals an 84% yield of a 4:1 ratio of 7 and what we tentativelly assign as the regiomeric o-quinone. However, over 1–2 h the undesired material succumbs to selective destruction most likely through polymerization. After 2 h, in situ reduction of the crude reaction mixture with sodium hydrosulfite (3 equiv) affords the benzylated catechol 29 in 58% isolated yield without contamination by the corresponding regiomeric catechol. After chromotography, the o-quinone 7 can be reformulated by oxidation (0.03 M in THF) with bis-trifluoroacetate-phenyliodine (PIFA, 1.00 equiv)21 or by oxidation (0.03 M in DMSO) with silver oxide (5 equiv). Without further purification, the o-quinone 7 (0.03 M in THF) is readily trapped through a 1,6-conjugate addition with phenyl thiol (1.05 equiv) to produce the isolable catechol (±)-31 in 91% yield from 29, thereby confirming the identity of the transitory intermediate.
Scheme 4.
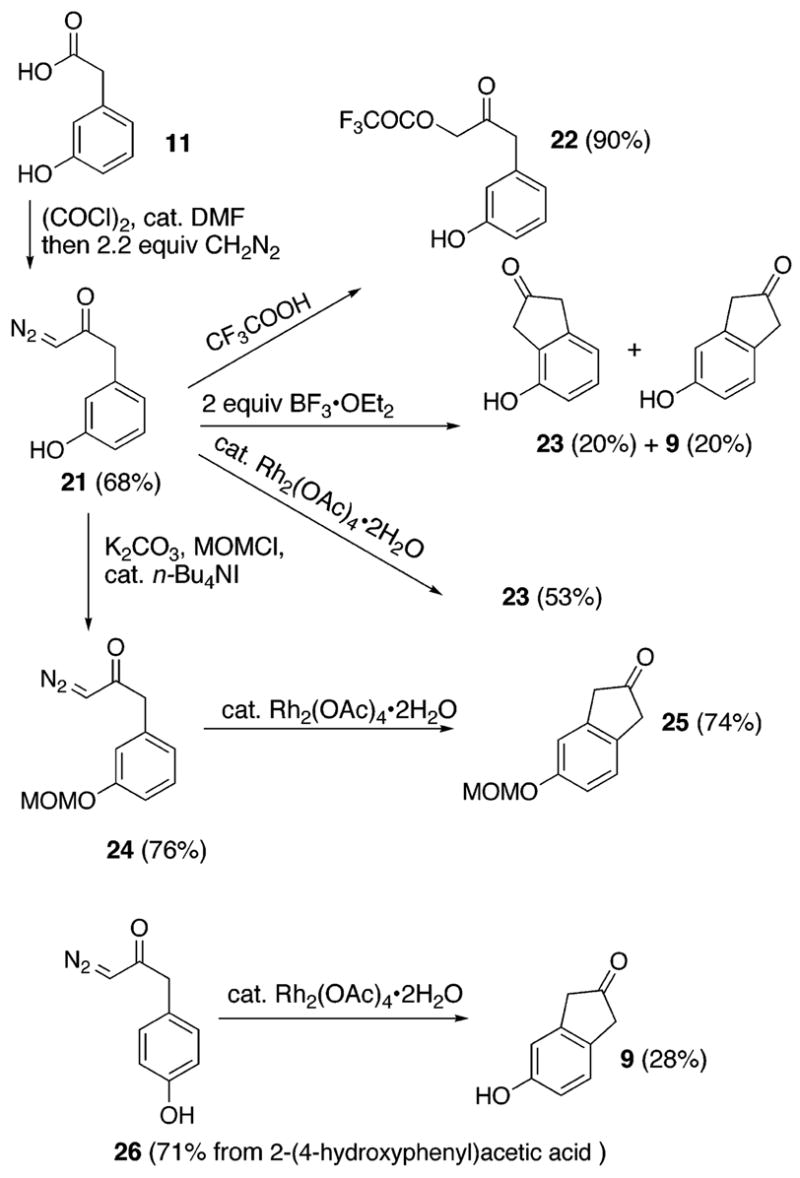
Synthesis of Indenones and Unusual Proximity Effects of Phenols; Comparison of H+- and Rh2+-Promoted Cyclizations
The o-quinone 7 (0.03 M in THF) suffers tautomerization upon the addition of Hünig’s base (1.0 equiv) to afford the thermodynamically more stable p-quinone methide (±)-30.22 The existence of (±)-30 was substantiated by 1H NMR and by isolation of its thioether adduct (±)-32, which emerges as a single diastereomer in 81% from 29. In the absence of a thiol or other nucleophile, the p-quinone methide (±)-30 undergoes cyclization to produce the tetracycle (±)-6 in 41% overall yield from 29 (70% BORSM 29). Lithium iodide appears to catalyze the cyclization processes. Subsequent hydrogenolysis of the benzyl ether 6 (0.01 M in EtOAc), 1 atm H2, Pd/C) affords synthetic (±)-brazilin (1), identical in every respect with the known natural product. We were surprised to find that (±)-brazilin (1) also forms in an 8% yield when the catechol 29 is subjected to hydrogenolysis (0.01 M in EtOAc/EtOH (1:1), 1 atm H2, Pd/C). This transformation most likely proceeds through similar quinone intermediates. As yet, we are unable to improve the yield of brazilin (1) in this one-pot oxidation-tautomerization-cyclization-hydrogenolysis process.
In conclusion, we have disclosed a unique route to a useful class of compounds. During this endeavor, we have employed several unusual and underutilized methods for constructing catechols, o-quinones. and p-quinone methides. While most syntheses of brazilin (1) have stopped at the corresponding tris-methylated derivative, ours does not and so it appears well positioned to address the entire family of natural products in their unprotected form. With access to sizable amounts of these compounds and their analogues, it should be possible to determine the process by which this family inhibits telomerase activity, as well as the mechanism by which compound 1 nicks the telomeric region of DNA. These insights should enable the development of a second series of more potent compounds. Last, it should be noted that during the tautomerization process, the achiral compound 7 becomes the chiral compound (±)-30. This transfomation seems irreversible, which suggests a process for the enantioselective synthesis of brazilin (1) and its derivatives through desymmetrization. Our findings in these studies and their application toward (±)-brazilide (5) will be reported in due course.
Scheme 5.
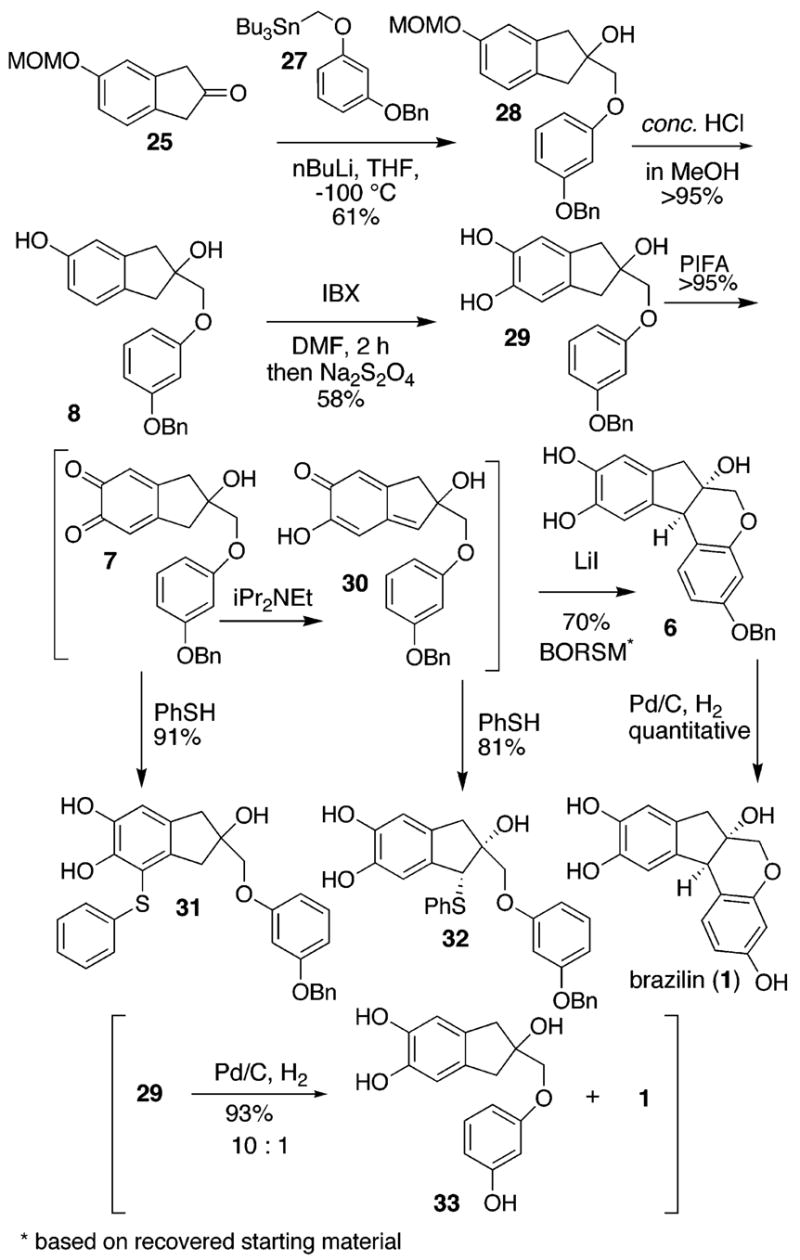
o-Quinone Formation, Reduction, Tautomerization, 1,4-Conjugate Addition, and Cyclization
Acknowledgments
Research Grants from the National Science Foundation (Career-0135031), National Institutes of Health (GM-64831), and the University of California Cancer Research Coordinating Committee are greatly appreciated. Y.H. is thankful for a dissertation fellowship granted by the University of California Tobacco-Related Disease Research Program.
Footnotes
Supporting Information Available: Experimental procedures for the five-step sequence [p-hydroxyphenylacetic acid → 26 → 9 → 8 → 29 → 1] (0.24% yield) and more efficient nine-step sequence [11 → 21 → 24 → 25 → 28 → 8 → 29 → 7 → 6 → 1] (8.5% yield) and key spectroscopic data for select compounds (1, 6–9, 21–29, 31–33) and o-quinones. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Robinson R. Bull Soc Chim Fr. 1958:125–134. [Google Scholar]
- 2.Xu J, Yadan JC. Tetrahedron Lett. 1996;37:2421–2424. [Google Scholar]
- 3.Yang BO, Ke CQ, He ZS, Yang YP, Ye Y. Tetrohedron Lett. 2002;43:1731–1733. [Google Scholar]
- 4.Davis FA, Chen B-C. J Org Chem. 1993;58:1751–1753. [Google Scholar]
- 5.Livingstone R. Nat Prod Rep. 1987:25–33. [Google Scholar]
- 6.Tolman RL, Chin AC. Telomerase inhibitors and methods of their use. 0,193,864. WO Patent. 2001 Dec 13;
- 7.Mar W, Lee H-T, Je K-H, Choi H-Y, Seo E-K. Arch Pharm Res. 2003;26:147–150. doi: 10.1007/BF02976661. [DOI] [PubMed] [Google Scholar]
- 8.Magdziak D, Rodriguez AA, Van De Water RW, Pettus TRR. Org Lett. 2002;4:285–288. doi: 10.1021/ol017068j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Quideau S, Pouysegu L, Deffieux D, Ozanne A, Gagnepain J, Fabre I, Oxoby M. ARKIVOC. 2003:106–119. [Google Scholar]
- 10.Luo HB, Xie YY. Chin Chem Lett. 2003;14:555–556. [Google Scholar]
- 11.(a) Tisdale EJ, Vong BG, Li HM, Kim SH, Chowdhury C, Theodorakis EA. Tetrahedron. 2003;59:6873–6887. [Google Scholar]; (b) Yuan Y, Men HB, Lee CB. J Am Chem Soc. 2004;126:14720–14721. doi: 10.1021/ja0447154. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhu JL, Germain AR, Porco JA. Angew Chem, Int Ed. 2004;43:1239–1243. doi: 10.1002/anie.200353037. [DOI] [PubMed] [Google Scholar]
- 12.Ahn JH, Cho SY, Ha JD, Kang SK, Jung SH, Kim H-M, Kim SS, Kim KR, Cheon HG, Yang S-D, Choi J-K. Bull Korean Chem Soc. 2003;24:1505–1508. [Google Scholar]
- 13.Kikumoto R, Tober A, Fukami H, Egawa M. J Med Chem. 1983;26:246–250. doi: 10.1021/jm00356a024. [DOI] [PubMed] [Google Scholar]
- 14.Adams DR, Duncton MAJ. Synth Commun. 2001;31:2029–2036. [Google Scholar]
- 15.Pezzella A, Lista L, Napolitano A, d’Ischia M. Tetrahedron Lett. 2005;46:3541–3544. [Google Scholar]
- 16.Nilsson JLG, Selander H, Sievertsson H, Skanberg I, Svensson K-G. Acta Chem Scand. 1971;25:94–100. [Google Scholar]
- 17.Lange J, Hoogeveen S, Veerman W, Wals H. Heterocycles. 2000;53:197–204. [Google Scholar]
- 18.Rosnati V, Di Vonna ML, Pusino A, Saba A. Tetrahedron Lett. 1988;29:4193–4196. [Google Scholar]
- 19.Watanabe N, Ogawa T, Ohtaka Y, Ikegami Y, Hashimoto S. Synlett. 1996:85–86. [Google Scholar]
- 20.Wipf P, Kim Y, Fritch PC. J Org Chem. 1993;58:7195–7203. [Google Scholar]
- 21.When compound 29 is exposed to an excess of PIFA or prolonged oxidation with excess Ag2O, the catechol ring appears to rupture. This chemistry may prove useful for the synthesis of brazilide 5.
- 22.Land EJ, Ramsden CA, Riley PA, Yoganathan G. Tetrahedron. 2003;59:9547–9554. [Google Scholar]