

Abstract
Redox modification of thiol/disulfide interchange in proteins by selenium could lead to protein unfolding. When this occurs in the endoplasmic reticulum (ER), a process known as unfolded protein response (UPR) is orchestrated for survival through activation of PERK–eIF2α(PERK: double-stranded RNA-activated protein kinase-like ER kinase; eIF2α: eucaryotic initiation factor 2α), ATFα(ATFα: activating transcription factor 6) and inositol requiring 1 (IRE1)-x-box-binding protein 1 (XBP1) signalings. All three UPR transducer pathways were upregulated very rapidly when PC-3 cells were exposed to selenium. These changes were accompanied by increased expression of UPR target genes, including immunoglobulin heavy chain-binding protein/glucose-regulated protein, 78 kDa and CCAAT/enhancer binding protein-homologous protein/growth arrest- and DNA damage- inducible gene (CHOP/GADD153). Induction of BiP/GRP78, an ER-resident chaperone, is part of the damage control mechanism, while CHOP/GADD153 is a transcription factor associated with growth arrest and apoptosis in the event of prolonged ER stress. Knocking down BiP/GRP78 induction by small interference RNA produced a differential response of the three transducers to selenium, suggesting that the signaling intensity of each transducer could be fine-tuned depending on BiP/GRP78 availability. In the presence of selenium, CHOP/GADD153 expression was raised even higher by BiP/GRP78 knockdown. Under this condition, the selenium effect on wild-type p53-activated fragment p21 (p21WAF), cyclin-dependent kinase (CDK)1 and CDK2 was also magnified in a manner consistent with enhanced cell growth arrest. Additional experiments with CHOP/GADD153 siRNA knockdown strongly suggested that CHOP/GADD153 may play a positive role in upregulating the expression of p21WAF in a p53-independent manner (PC-3 cells are p53 null). Collectively, the above findings support the idea that UPR could be an important mechanism in mediating the anticancer activity of selenium.
Keywords: selenium, unfolded protein response, BiP/GRP78 knockdown, ER stress signalings
Introduction
The anticancer activity of selenium is well documented (Ip et al., 2002; El Bayoumy and Sinha, 2004). Selenomethionine is the reagent of choice in a number of human intervention trials with prostate cancer. As proposed originally by Ip and Ganther (1990) and Ip et al. (1991), the metabolism of selenomethionine to methylselenol (CH3SeH) is essential for anticancer activity. For reasons that have been elaborated in detail previously, neither selenomethionine nor methylselenol is suitable for use, as in cell culture experiments, to investigate the mechanism of action of selenium (Ip, 1998). A stable metabolite called methylseleninic acid (CH3SeO2H, abbreviated to MSA) was developed specifically for in vitro studies (Ip et al., 2000). Once taken up by cells, MSA is reduced rapidly to CH3SeH through nonenzymatic reactions. Exposure of human prostate cancer cells to physiological concentrations of MSA results in cell cycle arrest and apoptosis (Dong et al., 2003; Zu and Ip, 2003).
Owing to its strong nucleophilicity, methylselenolate (the anion of methylselenol) readily reacts with protein sulfhydryl groups to cause thiol/disulfide interchange. By using a thiol-proteomics approach coupled to matrix-assisted laser desorption ionization-time-of-flight (MALDI-TOF) and electrospray ionization (ESI)-tandem mass spectrometry, we recently showed that MSA caused global thiol/disulfide redox modification of numerous proteins that are distributed in various subcellular compartments, including cytosol, mitochondria, nucleus, and endoplasmic reticulum (ER) (Park et al., 2005). These changes are expected to lead to protein unfolding or misfolding, especially for the newly synthesized proteins.
The ER is an intracellular organelle where newly synthesized proteins undergo post-translational modifications to form their proper tertiary structure. This process is tightly supervised by a host of ER-resident molecules, which are charged with performing specific tasks, as exemplified by immunoglobulin heavy chain-binding protein/glucose-regulated protein, 78 kDa (BiP/GRP78) for maintaining proteins in a folding-competent state (Kuznetsov et al., 1994), protein disulfide isomerase for catalysing the protein folding reaction (Jessop et al., 2004; Tu and Weissman, 2004), as well as calnexin and calreticulin for quality control monitoring (Ellgaard and Helenius, 2003). Normal functions of the ER could be impaired under various stressful conditions, including suppression of protein glycosylation, disruption of calcium homeostasis, or alterations in redox status. These ER stress signals cause accumulation of misfolded/unfolded proteins in the ER lumen, and in turn initiate a series of transducer pathways as a selfprotective mechanism. This so-called unfolded protein response (UPR) is characterized by an immediate stoppage of new protein synthesis and growth arrest, followed by adaptive survival, or apoptosis if stress is prolonged (Kadowaki et al., 2004; Ma and Hendershot, 2004; Shen et al., 2004).
The UPR is mediated primarily by one protein chaperone, BiP/GRP78, and three transmembrane ER stress transducers: double-stranded RNA-activated protein kinase-like ER kinase (PERK), inositol requiring 1 (IRE1), and activating transcription factor 6 (ATF6). In the unstressed ER, BiP/GRP78 binds to the ER luminal domains of the transducers and keeps them inactive in sequestration. Upon sensing the accumulation of misfolded/unfolded proteins, BiP/GRP78 dissociates from its clients and translocates to the ER lumen to help protein folding (Bertolotti et al., 2000; Liu et al., 2000, 2002, 2003). Once released from sequestration, PERK is activated by oligomerization and autophosphorylation. It then phosphorylates eucaryotic initiation factor 2α (eIF2α), thereby shutting off general protein translation (Harding et al., 1999). Similar to PERK, IRE1 is fully activated after dimerization and autophosphorylation. The site-specific endoribonuclease (RNase) activity of IRE1 mediates the removal of a 26-nucleotide intron from x-box-binding protein 1 (XBP1) mRNA (Tirasophon et al., 1998, 2000; Yoshida et al., 2001; Calfon et al., 2002; Lee et al., 2002). The spliced form of XBP1 subsequently binds to the ER stress response element (ERSE) and upregulates the transcription of UPR target genes, such as BiP/GRP78 and endoplasmic reticulum degradation-enhancing alpha-mannosidase-like protein (EDEM). The latter is meant to accelerate the degradation of misfolded proteins (Yoshida et al., 1998, 2003). After dissociation from BiP/GRP78, ATF6 translocates to the Golgi, where the active form of ATF6, ATF6 p50, is generated by proteolysis (Haze et al., 1999; Chen et al., 2002; Shen et al., 2002; Okada et al., 2003). As a transcription factor, ATF6 p50 binds to ERSE, resulting in the induction of BiP/GRP78, XBP1, CCAAT/enhancer binding protein-homologous protein/growth arrest- and DNA damage-inducible gene (CHOP/GADD153), and protein kinase inhibitor p58 (p58IPK) (Yoshida et al., 1998, 2000; van Huizen et al., 2003). In summary, a circuitry of signaling molecules are functioning cooperatively to alleviate the accumulation of unfolded/misfolded proteins in the ER lumen. When the survival response fails to adapt under prolonged ER stress, cells will eventually undergo apoptosis, although the mechanism behind this decision has not been elucidated.
The objectives of the present study were (i) to examine systemically the activation of signature UPR transducers and target genes by MSA in PC-3 human prostate cancer cells, (ii) to investigate the sensitivity of various UPR transducers to knockdown of BiP/GRP78 induction by MSA, and (iii) to study how BiP/GRP78 availability might modulate the growth arrest effect of MSA. These experiments were designed to test the hypothesis that UPR is an important mechanism in mediating the anticancer action of selenium.
Results
Induction of signature UPR transducer pathways and target genes by MSA
To assess the effect of MSA on UPR, all three transducer pathways were examined systematically in PC-3 cells with or without MSA treatment. As shown in Figure 1a, signal transduction of the PERK–eIF2α pathway and activation of ATF6 were evaluated by Western blot analysis. At the 6 h time point, MSA treatment increased the levels of phospho-PERK and cleaved ATF6, that is, ATF6 p50. The active forms of both PERK and ATF6 remained elevated at 12 h, but returned to their basal level or below by 24 h. Phospho-eIF2α changed in a direction similar to that of phospho-PERK, although the former still remained above its basal level at the 24 h time point. The total protein level of eIF2α was not affected by MSA. Phosphorylation of eIF2α by PERK inhibits its activity as an initiation factor in protein translation. Since total eIF2α did not change while phospho-eIF2α (inactive) increased with MSA treatment, the inference is that there would be less unphosphorylated eIF2α (active), thus leading to suppression of protein translation.
Figure 1.
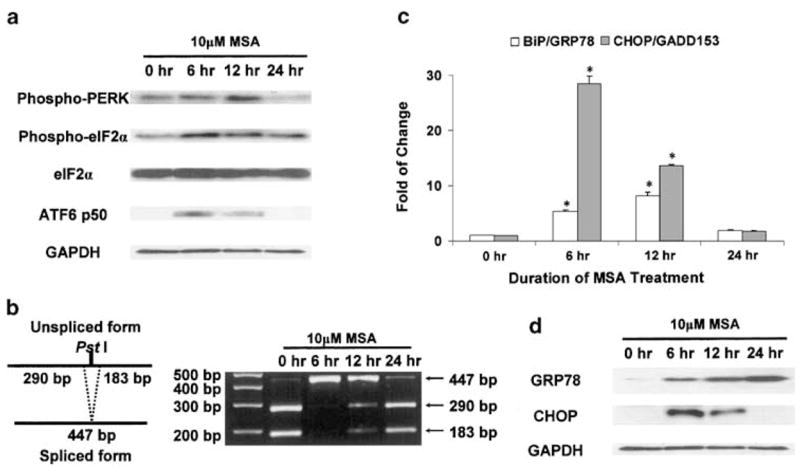
Induction of signature UPR transducer pathways and target genes by MSA. The Western blot and DNA electrophoresis data are representative of the results from three independent experiments. The real-time RT–PCR results are presented as mean±standard error (n = 3). *statistically different (P < 0.05) compared to the untreated control. (a) MSA-induced signal transduction of the PERK–eIF2α pathway and activation of ATF6 by Western blot analysis. Phosphorylation of PERK and eIF2α was determined by phosphospecific antibodies. (b) Analysis of XBP1 splicing as a surrogate marker for activation of the IRE1 pathway. The principle of this analysis (Calfon et al., 2002) is presented in a schematic illustration (left panel). XBP1 cDNA fragments were amplified by RT–PCR using XBP1-specific primers. After digestion with the PstI restriction enzyme at 37°C for 3 h, the cDNA fragments were separated on a 2% agarose gel (right panel). (c) MSA induction of UPR target genes, BiP/GRP78 and CHOP/GADD153, as determined by real-time RT–PCR. The relative quantitation of gene expression (fold of change) was calculated as described in Materials and methods. (d) MSA induction of BiP/GRP78 and CHOP/GADD153 as determined by Western blot analysis.
The antibody to phospho-IRE1 is not available commercially. The effect of MSA on IRE1 activation was assessed by using IRE1-mediated XBP1 splicing as a surrogate marker (Calfon et al., 2002). A schematic illustration of the principle of this analysis is presented in the left panel of Figure 1b. The PCR fragments of XBP1 cDNA contain a 473-bp unspliced form and a 447-bp spliced form. The unspliced form encompasses a PstI restriction site, which is lost in the spliced form. After digestion of the PCR fragments with PstI, the unspliced XBP1 cDNA is detected as two digestion products of 290 and 183 bp, whereas the spliced form remains as a 447-bp fragment due to its resistance to PstI. The results of MSA-induced XBP1 splicing are shown in the right panel of Figure 1b. The unspliced XBP1 fragment was the dominant form in the control. With MSA treatment, almost all XBP1 cDNAs became spliced at the 6 h time point. The appearance of spliced XBP1 suggested that the IRE1 pathway was activated. The level of spliced XBP1 decreased gradually as the unspliced form recovered steadily with longer treatment.
In order to assess the transcriptional control of target genes by activated ATF6 and XBP1, the expression of BiP/GRP78 and CHOP/GADD153 was examined at the mRNA level by quantitative real-time reverse transcription–polymerase chain reaction (RT–PCR) (Figure 1c), and at the protein level by Western blot analysis (Figure 1d). Similar to the activation of UPR transducers, the mRNA level of BiP/GRP78 and CHOP/GADD153 increased markedly following a short exposure to MSA, and returned to the basal level by 24 h. The change in CHOP/GADD153 protein level was consistent with the change in mRNA. However, the accumulation of BiP/GRP78 protein continued to rise with the duration of treatment, despite the drop in mRNA level at 24 h. This discrepancy between mRNA and protein levels may reflect the long half-life of BiP/GRP78 protein (Satoh et al., 1993).
Differential sensitivity of UPR transducers to knockdown of BiP/GRP78 induction by MSA
As a master negative regulator of UPR, BiP/GRP78 binds to PERK, ATF6, and IRE1 to keep them inactive. BiP/GRP78 also exists in a free form to facilitate protein folding in the ER lumen. UPR signal transduction is regulated through a delicate balance between free and bound BiP/GRP78. During ER stress, BiP/GRP78 is induced in order to increase the folding capacity of the ER and to compensate for the depletion of free BiP/GRP78. In an attempt to investigate the role of BiP/GRP78 induction by MSA, small interference RNA (siRNA) technique was used to knock down the increased expression of BiP/GRP78. A concentration of 75nM siRNA was used since it was the highest nontoxic dose based on preliminary titration experiments. As shown in Figure 2a, transient transfection with siRNA against BiP/GRP78 was able to tone down significantly the robust induction of this gene by MSA at both the mRNA (upper panel) and protein levels (lower panel). The comparison was made against control cells transfected with scramble siRNA and similarly treated with MSA. The UPR transducer pathways and downstream signalings were then examined in this BiP/GRP78 knockdown model.
Figure 2.
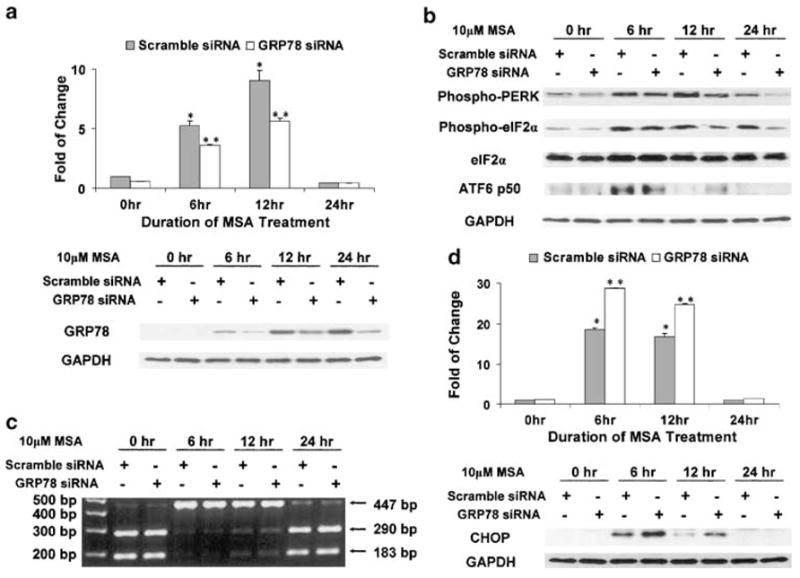
Differential sensitivity of UPR transducers to knockdown of BiP/GRP78 induction by MSA. The Western blot and DNA electrophoresis data are representative of the results from three independent experiments. The real-time RT–PCR results are presented as mean±standard error (n = 3). *statistically different (P < 0.05) compared to the untreated control. ** BiP/GRP78 knockdown results are statistically different (P < 0.05) compared to the scramble control. (a) SiRNA knockdown of BiP/GRP78 induction by MSA. Upper panel: real-time RT–PCR; lower panel: Western blot analysis. (b) Effect of BiP/GRP78 knockdown on MSA-induced signal transduction of the PERK–eIF2α pathway and activation of ATF6. (c) Effect of BiP/GRP78 knockdown on MSA-induced XBP1 splicing. XBP1 cDNA fragments were amplified by RT–PCR using XBP1-specific primers. After digestion with the PstI restriction enzyme at 37°C for 3 h, the cDNA fragments were separated on a 2% agarose gel. (d) Enhanced MSA induction of CHOP/GADD153 by BiP/GRP78 knockdown. Upper panel: real-time RT–PCR; lower panel: Western blot analysis.
As shown in Figure 2b, the level of phospho-PERK in both the control and BiP/GRP78 knockdown samples was increased by MSA treatment. However, at the later time points (12 and 24 h), the magnitude of the increase was dampened in the BiP/GRP78 knockdown samples. Likewise, the modulation of eIF2α followed a similar pattern of a lesser amount of phospho-eIF2α in the GRP78 siRNA-transfected cells. In contrast, the induction of ATF6 p50 was maintained for a longer period of time by reduced expression of BiP/GRP78.
Analysis of IRE1-mediated XBP1 splicing in the GRP78 siRNA-transfected cells is shown in Figure 2c. After 6 h of MSA treatment, XBP1 mRNA was present primarily in the spliced form. As observed previously, the unspliced form recovered gradually with time. However, there was no significant difference in XBP1 splicing between the control and BiP/GRP78 knockdown samples.
The expression of CHOP/GADD153 in cells transfected with scramble siRNA or BiP/GRP78 siRNA was examined at the mRNA level by quantitative real-time RT–PCR (Figure 2d, upper panel) and at the protein level by Western blot analysis (Figure 2d, lower panel). At the 6 h time point, the mRNA level was elevated by MSA treatment, and this increase was sustained at 12 h. More importantly, the effect was significantly magnified in the BiP/GRP78 knockdown samples. The boost in MSA induction of CHOP/GADD153 by BiP/GRP78 knockdown could be accounted for in part by the prolonged activation of ATF6, as shown in Figure 2b.
Enhanced MSA effect on growth arrest by BiP/GRP78 knockdown
Next, we used two different end points to examine the biological consequence of BiP/GRP78 knockdown in cells treated with MSA. Cell proliferation was measured by the 5-bromo-2′-deoxyuridine (BrdU) incorporation assay (Figure 3a). After exposure to 10 μM MSA for 12 h, DNA synthesis was suppressed by ~38% in cells transfected with scramble siRNA. The inhibition was significantly greater (~52%, P < 0.05) in cells with reduced BiP/GRP78 expression.
Figure 3.
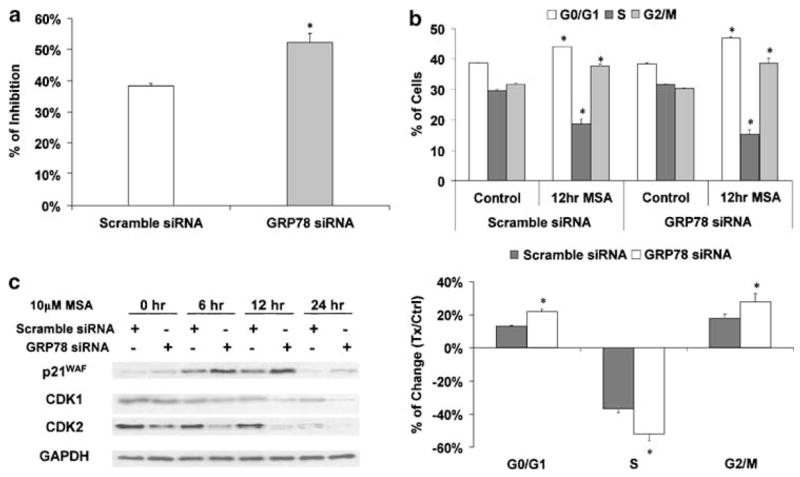
Enhanced MSA effect on growth arrest by BiP/GRP78 knockdown. The Western blot data are representative of the results from three independent experiments. (a) Enhanced MSA effect on DNA synthesis suppression by BiP/GRP78 knockdown. Percentage of inhibition was calculated based on the percentages of BrdU-positive cells in MSA-treated samples and untreated control. The results are presented as mean±standard error (n = 3). *BiP/GRP78 knockdown results are statistically different (P < 0.05) compared to the scramble control. (b) Enhanced MSA effect on cell cycle arrest by BiP/GRP78 knockdown. The upper panel shows the cell cycle distribution data in cells transfected with scramble or BiP/GRP78 siRNA, with or without 10 μM MSA treatment. The results are presented as mean±standard error (n = 3). *MSA-treated results are statistically different (P < 0.05) compared to the untreated control. The lower panel shows the difference between the scramble control and BiP/GRP78 knockdown samples. *BiP/GRP78 knockdown results are statistically different (P < 0.05) compared to the scramble control. (c) Enhanced MSA effect on expression of cell cycle regulatory molecules by BiP/GRP78 knockdown. The protein levels of p21WAF, CDK1, and CDK2 were examined by Western blot analysis.
The results of the cell cycle distribution analysis are shown in Figure 3b. Without MSA treatment, no difference was observed in cell cycle distribution between transfection with scramble siRNA or BiP/GRP78 siRNA. After cells were treated with 10 μM MSA for 12 h, the percentage of S-phase cells was significantly decreased, while the percentages of both G0/G1- and G2/M-phase cells were increased (Figure 3b, upper panel). The comparative changes in cell cycle distribution profile between the scramble siRNA and BiP/GRP78 siRNA-transfection groups are highlighted in the lower panel of Figure 3b. The effect of MSA on cell cycle arrest was notably enhanced by BiP/GRP78 knockdown.
Enhanced MSA effect on expression of cell cycle regulatory molecules by BiP/GRP78 knockdown
In order to gain further insight into the mechanism of UPR-induced cell cycle arrest, we examined the expression of several key cell cycle regulatory molecules by Western blot analysis. These molecules were selected based on our previous MSA data in PC-3 cells. As shown in Figure 3c, MSA treatment downregulated cyclin-dependent kinase (CDK)1 and CDK2, but upregulated their inhibitor wild-type p53-activated fragment p21 (p21WAF). The effect of MSA on the expression of these molecules was significantly enhanced by knocking down BiP/GRP78, that is, more pronounced decreases with CDK1 and CDK2, and greater increase with p21WAF.
Reduced MSA induction of p21WAF expression by CHOP/GADD153 knockdown
As shown in Figures 2d and 3c, the expression of CHOP/GADD153 and p21WAF was induced by MSA in a parallel manner as a function of time. The protein levels of these two molecules were further elevated by knocking down BiP/GRP78. In order to investigate the role of CHOP/GADD153 in upregulating the expression of p21WAF, we used the siRNA technique to knockdown the induction of CHOP/GADD153. A concentration of 100 nM siRNA was used since it was the highest nontoxic dose based on preliminary titration experiments. As shown in Figure 4a, the induction of CHOP/GADD153 by MSA was knocked down by more than 50% at both the mRNA (upper panel) and protein levels (lower panel) over a course of 12 h. The expression of p21WAF was then examined in this CHOP/GADD153 knockdown model (Figure 4b). At the 6 h time point, the induction of p21WAF mRNA (upper panel) and protein (lower panel) by MSA was reduced markedly in cells transfected with siRNA against CHOP/GADD153. At the 12 h time point, the difference in p21WAF mRNA levels between the CHOP/GADD153 knockdown and scramble control cells became marginal, while the difference in p21WAF protein levels still remained significant. Based on the above data, we conclude that CHOP/GADD153 may play a positive role in upregulating the expression of p21WAF in a p53-independent manner (PC-3 cells are p53 null).
Figure 4.
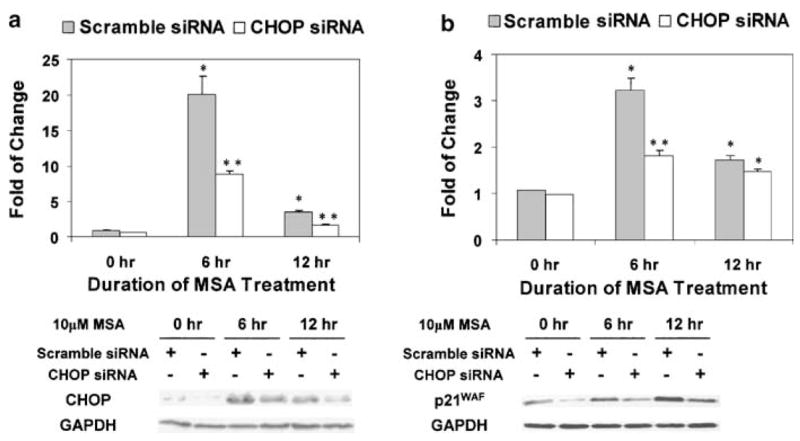
Reduced MSA induction of p21WAF by CHOP/GADD153 knockdown. The Western blot data are representative of the results from three independent experiments. The real-time RT–PCR results are presented as mean±standard error (n = 3). *statistically different (P < 0.05) compared to the untreated control. **CHOP/GADD153 knockdown results are statistically different (P < 0.05) compared to the scramble control. (a) SiRNA knockdown of CHOP/GADD153 induction by MSA. Upper panel: real-time RT–PCR; lower panel: Western blot analysis. (b) Reduced MSA induction of p21WAF by CHOP/GADD153 knockdown. Upper panel: real-time RT–PCR; lower panel: Western blot analysis.
Discussion
In this study, we systematically investigated the induction of UPR by MSA in an effort to link ER stress to the anticancer action of selenium. All three UPR transducer pathways, PERK–eIF2α, ATF6, and IRE1–XBP1, are activated very rapidly, leading to increased expression of UPR target genes. Activation of PERK–eIF2α signaling is meant to block general protein synthesis in order to reduce the burden of more unfolded proteins. Knocking down BiP/GRP78 by siRNA elicits a differential response of the three stress transducer pathways; the net outcome is an enhanced selenium effect on cell growth arrest. The above findings suggest that UPR may be an important done in mediating the anticancer activities of selenium.
As a negative regulator of UPR, BiP/GRP78 sequesters PERK, ATF6, and IRE1 on the ER membrane by binding to their luminal domains. There is also free BiP/GRP78 in the ER lumen, cycling between a monomeric and an oligomeric state (Freiden et al., 1992; Blond-Elguindi et al., 1993). Only the monomeric BiP/GRP78 associates with newly synthesized proteins to facilitate their folding. The oligomeric BiP/GRP78 represents a storage pool, from which monomeric BiP/GRP78 is recruited in the presence of unfolded protein accumulation. Depleting this reservoir during ER stress causes BiP/GRP78 to dissociate from PERK, ATF6, and IRE1, and leads to downstream UPR signaling. As a target gene, BiP/GRP78 is then upregulated to replenish the oligomeric pool.
Since the increased expression of BiP/GRP78 is designed to help cells cope with ER stress, our strategy was to suppress the induction of BiP/GRP78 by RNA interference in order to intensify the stress signal in the ER. Interestingly, the three transducer pathways respond differently to BiP/GRP78 knockdown, suggesting that there might be distinctive mechanisms regulating their activation. PERK and IRE1 are both type I transmembrane protein kinases. Their ER luminal domains are homologous and interchangeable (Bertolotti et al., 2000). However, the oligomerization and BiP/GRP78 binding domains of IRE1 overlap partially (Liu et al., 2003), whereas those of PERK are distinctive (Ma et al., 2002). When excessive unfolded proteins are present in the ER, BiP/GRP78 is titrated competitively from the luminal domains of PERK and IRE1 to effect their oligomerization and activation (Schroder and Kaufman, 2005). On the other hand, ATF6 is a type II transmembrane protein. There are two independent and redundant Golgi localization sequences (GLSs), GLS1 and GLS2, in the ER luminal domain of ATF6 (Shen et al., 2002). BiP/GRP78 only binds to GLS1 and keeps ATF6 in an inactive state. After the release of BiP/GRP78 from GLS1, GLS2 becomes dominant and regulates the translocation of ATF6 to the Golgi, where ATF6 is activated through proteolysis. Thus, the three signaling arms of the UPR may have distinctive sensitivities to fluctuations of the free BiP/GRP78 pool. Crosstalks also exist among the three transducer pathways. For example, p58IPK, a downstream target of ATF6, has been reported to inhibit the activity of PERK (Yan et al., 2002; van Huizen et al., 2003). Our results showed that a modest knockdown of BiP/GRP78 induction is sufficient to cause a prolonged activation of ATF6, which may account for the decrease in PERK activity (potentially mediated by p58IPK) at the later time points. During ER stress, the PERK-mediated translational block is short-lived, since the primary adaptive mechanism is to upregulate the transcription of UPR target genes. It is possible that differences in the binding affinity of BiP/GRP78 to each of the three transducers, as well as the interactions between downstream signaling pathways, are critical factors in fine-tuning UPR signalings.
CHOP/GADD153 is a member of the CCAAT/enhancer binding protein (C/EBP) family and is present normally at a very low expression level. It is induced rapidly when the functions of the ER are perturbed. The induction of CHOP/GADD153 is regulated primarily at the transcriptional level. All three transducer pathways of ER stress are involved in this process (Harding et al., 2000; Wang et al., 2000; Gotoh et al., 2002). The overexpression of CHOP/GADD153 has been shown to be involved in ER stress-induced cell cycle arrest and/or apoptosis (Barone et al., 1994; Friedman, 1996). However, very little information is available on the signaling events or the downstream targets of CHOP/GADD153. Based on our experience, CHOP/GADD153 is one of the most highly inducible genes in a number of cancer cell lines treated with MSA. Previously, we investigated the gene expression profile in MSA-treated PC-3 cells by oligonucleotide array analysis (Dong et al., 2003). Hundreds of genes are affected by MSA, among which is a large set of cell cycle regulatory genes. They are mostly modulated in a manner consistent with cell cycle arrest. In this study, we examined the expression of CHOP/GADD153, p21WAF, CDK1, and CDK2 in the context of UPR. Our data showed that in the presence of MSA, the mRNA and protein levels of CHOP/GADD153 are raised even higher by knocking down BiP/GRP78. The effect of MSA on p21WAF, CDK1, and CDK2 is also magnified by a muted induction of BiP/GRP78, suggesting that the modulation of these genes by MSA is associated with ER stress response. In particular, p21WAF is upregulated in a pattern that similar to that of CHOP/GADD153. The upregulation in p21WAF is severely dampened by CHOP/GADD153 knockdown. PC-3 cells are p53 null. Therefore, the increase of p21WAF gene expression in PC-3 cells during ER stress is likely to be mediated by CHOP/GADD153 in a p53-independent manner. Future studies will investigate the possible transcriptional control of p21WAF by CHOP/GADD153.
ER stress has been studied mostly in neuropathology, such as Parkinson’s disease and Alzheimer’s disease (Lehotsky et al., 2003). Scanty information is available regarding UPR in cancer research. Since the key selenium metabolite, methylselenol, is generated endogenously, the induction of UPR by selenium is likely to be a universal phenomenon and is not cell type specific. That is to say, proteins in normal cells are vulnerable to redox modification by selenium as well. However, different cells may have different abilities to manage and cope with stress. The selectivity of selenium as a primary chemopreventive agent has been well documented (Ip et al., 2002). A recent study reported that preadministration of selenium increases the therapeutic efficacy of irinotecan in the nude mice tumor xenograft model (Cao et al., 2004). More importantly, selenium is highly protective of normal cells, and is able to overcome the dose-limiting toxicity of the drug. The above finding implies that selenium may favor survival response in normal cells, but facilitates apoptotic response in cancer cells. Indeed, the protective role of pre-conditioned ER stress response against cytotoxicity has been reported in normal cells (Hung et al., 2003; Bednard et al., 2004). Many factors may impact on ER stress response to selenium. For example, the microenvironment may determine whether the outcome of UPR is survival or death. Hypoxia is a known inducer of ER stress (Koumenis et al., 2002; Tajiri et al., 2004). The hypoxic condition of a solid tumor could sensitize cancer cells to selenium. Our laboratory has preliminary data (unpublished) showing that hypoxia significantly enhances selenium induction of apoptosis. The genetic background of a particular cell may be another factor in tipping the balance toward either survival or death in responses to the same stress signal. The growth arrest induced by ER stress can be viewed not only as the primary effect of selenium in cancer prevention, but also as a priming mechanism in potentiating the therapeutic selectivity of therapeutic drugs.
Materials and methods
Cell culture and treatments
PC-3 human prostate cancer cells were obtained from ATCC (Manassas, VA, USA). These cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2mM glutamine, and were maintained in an atmosphere of 5% CO2 in a 37°C humidified incubator. Cells were exposed to 10 μM MSA for different periods of time, either at 48 h after seeding or 12 h after transfection.
Transient siRNA-transfection
The annealed siRNAs were synthesized by Ambion Inc. (Austin, TX, USA). The BiP/GRP78-specific siRNA (sense sequence, 5′-GGACAUCAAGUUCUUGCCGtt-3′; antisense sequence, 5′-CGGCAAGAACUUGAUGUCCtg-3′) was used to knock down the induction of BiP/GRP78 by MSA. The CHOP/GADD153-specific siRNA (sense sequence, 5′-CCAG GAAACGGAAACAGAGtt-3′; antisense sequence, 5′-CUC UGUUUCCGUUUCCUGGtt-3′) was used to knock down the induction of CHOP/GADD153 by MSA. The scramble nonsense siRNA (sense sequence, 5′-AGUACUGCUUAC GAUACGGtt-3′; antisense sequence, 5′-CCGUAUCGUAA GCAGUACUtt-3′) that has no homology to any known genes was used as control. In the flow cytometry experiments, carboxyfluorescein (FAM)-labeled siRNAs (synthesized by Ambion Inc.) were used in combination with unlabeled siRNAs at a ratio of 1:5 to target the transfected population.
PC-3 cells were placed in six-well cell culture plates at a density of 6 × 104 cells/cm2. At 48 h after seeding, the cells were transfected with GRP78 siRNA or scramble siRNA by using Oligofectamine Reagent (Invitrogen) according to the manufacturer’s protocol. Briefly, for each well in a six-well plate, 4 μl of oligofectamine was diluted in 11 μl of Opti-MEM I medium (Invitrogen). This mixture was carefully added to a solution containing 75 or 100 nmol of siRNA in 185 μl of Opti-MEM I medium. The solution was incubated for 30 min at room temperature, and then gently overlaid onto 80% confluent PC-3 cells in 800 μl of Opti-MEM I medium. After 6 h of transfection, cells were refed with regular growth medium for 12 h before exposure to different treatments.
Cell lysis and Western blot analysis
Whole cell lysate was prepared by using 1 × cell lysis buffer (Cell Signaling Technology, Beverly, MA, USA), and protein concentration was determined by using the BCA Protein Assay Kit (Pierce Biotechnology, Rockford, IL, USA). Whole cell lysates were then resolved over 8–15% SDS–PAGE and transferred to a PVDF membrane. The blot was blocked in blocking buffer (5% nonfat dry milk, 10mM Tris, pH 7.5, 10mM NaCl, and 0.1% Tween 20) at 37°C for 1 h, incubated with the primary antibody overnight at 4°C, followed by incubation with a horseradish peroxidase-conjugated secondary antibody (Bio-Rad, Hercules, CA, USA) at room temperature for 30 min. Individual proteins were visualized by an enhanced chemiluminescence kit obtained from Amersham Pharmacia Biotech (Piscataway, NJ, USA).
The antibodies (source) to the following proteins were used in this study: phospho-PERK (Thr980), eIF2α, phospho-eIF2α (Ser51), and cleaved PARP (Cell Signaling Technology, Beverly, MA, USA), BiP/GRP78, CHOP/GADD153, and p21WAF (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), CDK1 and CDK2 (BD Transduction Laboratories, San Jose, CA, USA), and GAPDH (Chemicon, Temecula, CA, USA). The rabbit antiserum raised against human ATF6 p50 (activated form) was purified as described previously (Haze et al., 1999).
XBP1 splicing analysis
Total RNA was isolated with the TRIzol reagent (Life Technologies Inc.). First-strand cDNA was synthesized from 100 ng of total RNA by SuperScript II reverse transcriptase (Invitrogen) following the manufacturer’s protocol. The XBP1-specific primers were synthesized by Integrated DNA Technologies Inc. (Coralville, IA, USA). PCR with the sense primer (5′-AAACAGAGTAGCAGCTCAGACTGC-3′) and the antisense primer (5′-TCCTTCTGGGTAGACCTCTGGG AG-3′) amplified a 473-bp cDNA product encompassing the IRE1-mediated splicing site. This fragment was further digested by PstI to reveal a restriction site that is lost after IRE1-mediated splicing of the XBP1 mRNA. The cDNA fragments were resolved on a 2% agarose gel containing ethidium bromide and then visualized with an AlphaImager 1220 Documentation and Analysis system (Alpha Innotech, San Leandro, CA, USA).
Quantitative real-time RT–PCR
First-strand cDNA was synthesized as described previously. The PCR primers and TaqMan probes for β-actin, BiP/GRP78, and CHOP/GADD153 were Assays-on-Demand products from Applied Biosystems (Foster City, CA, USA). An aliquot of 2 μl of first-strand cDNA was mixed with 25 μl of 2 × Taqman Universal PCR Master Mix (Applied Biosystems) and 2.5 μl of 20 × primers/probe mixture in a 50 μl final volume. Temperature cycling and real-time fluorescence measurements were performed using an ABI prism 7700 Sequence Detection System (Applied Biosystems). The PCR conditions were as follows: initial incubation at 50°C for 2 min, then denaturation at 95°C for 10 min, followed by 40 cycles of 95°C for 15s and 60°C for 1 min.
The relative quantitation of gene expression was performed by using the comparative CT (ΔΔCT) method. Briefly, the threshold cycle number (CT) was obtained as the first cycle at which a statistically significant increase in fluorescence signal was detected. Data normalization was carried out by subtracting the CT value of β-actin from that of the target gene. The ΔΔCT was calculated as the difference of the normalized CT values (ΔCT) of the treatment and the control samples: ΔΔCT = ΔCT treatment−ΔCT control. Finally, ΔΔCT was converted to fold of change by the following formula: fold of change = 2−ΔΔCT.
BrdU incorporation and cell cycle distribution analysis
After transfection with scramble siRNA or BiP/GRP78 siRNA (combined with FAM-labeled siRNAs at a ratio of 5:1), cells were exposed to regular growth medium with or without 10 μM of MSA for 12 h. The BrdU incorporation and cell cycle distribution analysis was then performed by using the BrdU Flow Kit from BD Pharmigen (San Diego, CA, USA) according to the manufacturer’s protocol. Briefly, cells were labeled with 10 μM BrdU during the last hour of treatment. They were then trypsinized, fixed, treated with DNase I, and stained with phycoerythrin (PE)-conjugated anti-BrdU antibody and 7-amino-actinomycin D (7AAD). Stained cells were then subjected to flow cytometric analysis, and the data were analysed with the WinList software (Variety Software House, Topsham, ME, USA).
Statistical analysis
The Student’s t-test was used to determine statistical differences between treatments and controls, and P < 0.05 was considered significant.
Acknowledgments
This work was supported by Grant CA 09796 from the National Cancer Institute and Grant 62-2198 from the Roswell Park Alliance Foundation, and was partially supported by core resources of the Roswell Park Cancer Institute Cancer Center Support Grant P30 CA 16056 from the National Cancer Institute.
Abbreviations
- ATF6
activating transcription factor 6
- BiP/GRP78
immunoglobulin heavy chain-binding protein/glucose-regulated protein, 78 kDa
- BrdU
5-bromo-2′-deoxyuridine
- CDK
cyclin- dependent kinase
- CHOP/GADD153
CCAAT/enhancer binding protein-homologous protein/growth arrest- and DNA damage-inducible gene
- EDEM
endoplasmic reticulum degradation- enhancing alpha-mannosidase-like protein
- eIF2α
eucaryotic initiation factor 2α
- ER
endoplasmic reticulum
- ERSE
ER stress response element
- ESI
electrospray ionization
- GLS
Golgi localization sequences
- IRE1
inositol requiring 1
- MSA
methylseleninic acid
- MALDI-TOF
matrix-assisted laser desorption ionization-time-of-flight
- p21WAF
wild-type p53-activated fragment p21
- p58IPK
protein kinase inhibitor p58
- PERK
double-stranded RNA-activated protein kinase-like ER kinase
- RNase
ribonuclease
- RT–PCR
reverse transcription–polymerase chain reaction
- siRNA
small interference RNA
- UPR
unfolded protein response
- XBP1
x-boxbinding protein 1
References
- Barone MV, Crozat A, Tabaee A, Philipson L, Ron D. Genes Dev. 1994;8:453–464. doi: 10.1101/gad.8.4.453. [DOI] [PubMed] [Google Scholar]
- Bednard K, MacDonald N, Collins J, Cribb A. Basic Clin Pharmacol Toxicol. 2004;94:124–131. doi: 10.1111/j.1742-7843.2004.pto940305.x. [DOI] [PubMed] [Google Scholar]
- Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Nat Cell Biol. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- Blond-Elguindi S, Fourie AM, Sambrook JF, Gething MJ. J Biol Chem. 1993;268:12730–12735. [PubMed] [Google Scholar]
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, et al. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- Cao S, Durrani FA, Rustum YM. Clin Cancer Res. 2004;10:2561–2569. doi: 10.1158/1078-0432.ccr-03-0268. [DOI] [PubMed] [Google Scholar]
- Chen X, Shen J, Prywes R. J Biol Chem. 2002;277:13045–13052. doi: 10.1074/jbc.M110636200. [DOI] [PubMed] [Google Scholar]
- Dong Y, Zhang H, Hawthorn L, Ganther HE, Ip C. Cancer Res. 2003;63:52–59. [PubMed] [Google Scholar]
- El Bayoumy K, Sinha R. Mutat Res. 2004;551:181–197. doi: 10.1016/j.mrfmmm.2004.02.023. [DOI] [PubMed] [Google Scholar]
- Ellgaard L, Helenius A. Nat Rev Mol Cell Biol. 2003;4:181–191. doi: 10.1038/nrm1052. [DOI] [PubMed] [Google Scholar]
- Freiden PJ, Gaut JR, Hendershot LM. EMBO J. 1992;11:63–70. doi: 10.1002/j.1460-2075.1992.tb05028.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman AD. Cancer Res. 1996;56:3250–3256. [PubMed] [Google Scholar]
- Gotoh T, Oyadomari S, Mori K, Mori M. J Biol Chem. 2002;277:12343–12350. doi: 10.1074/jbc.M107988200. [DOI] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, et al. Mol Cell. 2000;6:1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Ron D. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mol Biol Cell. 1999;10:3787–3799. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung CC, Ichimura T, Stevens JL, Bonventre JV. J Biol Chem. 2003;278:29317–29326. doi: 10.1074/jbc.M302368200. [DOI] [PubMed] [Google Scholar]
- Ip C. J Nutr. 1998;128:1845–1854. doi: 10.1093/jn/128.11.1845. [DOI] [PubMed] [Google Scholar]
- Ip C, Dong Y, Ganther HE. Cancer Metast Rev. 2002;21:281–289. doi: 10.1023/a:1021263027659. [DOI] [PubMed] [Google Scholar]
- Ip C, Ganther HE. Cancer Res. 1990;50:1206–1211. [PubMed] [Google Scholar]
- Ip C, Hayes C, Budnick RM, Ganther HE. Cancer Res. 1991;51:595–600. [PubMed] [Google Scholar]
- Ip C, Thompson HJ, Zhu Z, Ganther HE. Cancer Res. 2000;60:2882–2886. [PubMed] [Google Scholar]
- Jessop CE, Chakravarthi S, Watkins RH, Bulleid NJ. Biochem Soc Trans. 2004;32:655–658. doi: 10.1042/BST0320655. [DOI] [PubMed] [Google Scholar]
- Kadowaki H, Nishitoh H, Ichijo H. J Chem Neuroanat. 2004;28:93–100. doi: 10.1016/j.jchemneu.2004.05.004. [DOI] [PubMed] [Google Scholar]
- Koumenis C, Naczki C, Koritzinsky M, Rastani S, Diehl A, Sonenberg N, et al. Mol Biol Cell. 2002;22:7405–7416. doi: 10.1128/MCB.22.21.7405-7416.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsov G, Chen LB, Nigam SK. J Biol Chem. 1994;269:22990–22995. [PubMed] [Google Scholar]
- Lee K, Tirasophon W, Shen X, Michalak M, Prywes R, Okada T, et al. Genes Dev. 2002;16:452–466. doi: 10.1101/gad.964702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehotsky J, Kaplan P, Babusikova E, Strapkova A, Murin R. Physiol Res. 2003;52:269–274. [PubMed] [Google Scholar]
- Liu CY, Schroder M, Kaufman RJ. J Biol Chem. 2000;275:24881–24885. doi: 10.1074/jbc.M004454200. [DOI] [PubMed] [Google Scholar]
- Liu CY, Wong HN, Schauerte JA, Kaufman RJ. J Biol Chem. 2002;277:18346–18356. doi: 10.1074/jbc.M112454200. [DOI] [PubMed] [Google Scholar]
- Liu CY, Xu Z, Kaufman RJ. J Biol Chem. 2003;278:17680–17687. doi: 10.1074/jbc.M300418200. [DOI] [PubMed] [Google Scholar]
- Ma K, Vattem KM, Wek RC. J Biol Chem. 2002;277:18728–18735. doi: 10.1074/jbc.M200903200. [DOI] [PubMed] [Google Scholar]
- Ma Y, Hendershot LM. J Chem Neuroanat. 2004;28:51–65. doi: 10.1016/j.jchemneu.2003.08.007. [DOI] [PubMed] [Google Scholar]
- Okada T, Haze K, Nadanaka S, Yoshida H, Seidah NG, Hirano Y, et al. J Biol Chem. 2003;278:31024–31032. doi: 10.1074/jbc.M300923200. [DOI] [PubMed] [Google Scholar]
- Park EM, Choi KS, Park SY, Zu K, Wu Y, Zhang H, et al. Cancer Genom Proteom. 2005;2:25–36. [PubMed] [Google Scholar]
- Satoh M, Nakai A, Sokawa Y, Hirayoshi K, Nagata K. Exp Cell Res. 1993;205:76–83. doi: 10.1006/excr.1993.1060. [DOI] [PubMed] [Google Scholar]
- Schroder M, Kaufman RJ. Mutat Res. 2005;569:29–63. doi: 10.1016/j.mrfmmm.2004.06.056. [DOI] [PubMed] [Google Scholar]
- Shen J, Chen X, Hendershot L, Prywes R. Dev Cell. 2002;3:99–111. doi: 10.1016/s1534-5807(02)00203-4. [DOI] [PubMed] [Google Scholar]
- Shen X, Zhang K, Kaufman RJ. J Chem Neuroanat. 2004;28:79–92. doi: 10.1016/j.jchemneu.2004.02.006. [DOI] [PubMed] [Google Scholar]
- Tajiri S, Oyadomari S, Yano S, Morioka M, Gotoh T, Hamada JI, et al. Cell Death Differ. 2004;11:403–415. doi: 10.1038/sj.cdd.4401365. [DOI] [PubMed] [Google Scholar]
- Tirasophon W, Lee K, Callaghan B, Welihinda A, Kaufman RJ. Genes Dev. 2000;14:2725–2736. doi: 10.1101/gad.839400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirasophon W, Welihinda AA, Kaufman RJ. Genes Dev. 1998;12:1812–1824. doi: 10.1101/gad.12.12.1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu BP, Weissman JS. J Cell Biol. 2004;164:341–346. doi: 10.1083/jcb.200311055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Huizen R, Martindale JL, Gorospe M, Holbrook NJ. J Biol Chem. 2003;278:15558–15564. doi: 10.1074/jbc.M212074200. [DOI] [PubMed] [Google Scholar]
- Wang Y, Shen J, Arenzana N, Tirasophon W, Kaufman RJ, Prywes R. J Biol Chem. 2000;275:27013–27020. doi: 10.1074/jbc.M003322200. [DOI] [PubMed] [Google Scholar]
- Yan W, Frank CL, Korth MJ, Sopher BL, Novoa I, Ron D, et al. Proc Natl Acad Sci USA. 2002;99:15920–15925. doi: 10.1073/pnas.252341799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Haze K, Yanagi H, Yura T, Mori K. J Biol Chem. 1998;273:33741–33749. doi: 10.1074/jbc.273.50.33741. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Hosokawa N, Kaufman RJ, Nagata K, Mori K. Dev Cell. 2003;4:265–271. doi: 10.1016/s1534-5807(03)00022-4. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Okada T, Haze K, Yanagi H, Yura T, Negishi M, et al. Mol Cell Biol. 2000;20:6755–6767. doi: 10.1128/mcb.20.18.6755-6767.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu K, Ip C. Cancer Res. 2003;63:6988–6995. [PubMed] [Google Scholar]