

Abstract
Fibrodysplasia ossificans progressiva (FOP), a rare and disabling genetic condition of congenital skeletal malformations and progressive heterotopic ossification (HO), is the most catastrophic disorder of HO in humans. Episodic disease flare-ups are precipitated by soft tissue injury, and immobility is cumulative. Recently, a recurrent mutation in activin receptor IA/activin-like kinase 2 (ACVR1/ALK2), a bone morphogenetic protein (BMP) type I receptor, was reported in all sporadic and familial cases of classic FOP, making this one of the most highly specific disease-causing mutations in the human genome. The discovery of the FOP gene establishes a critical milestone in understanding FOP, reveals a highly conserved target for drug development in the TGF-β/BMP signalling pathway, and compels therapeutic approaches for the development of small molecule signal transduction inhibitors for ACVR1/ALK2. Present management involves early diagnosis, assiduous avoidance of iatrogenic harm, and symptomatic amelioration of painful flare-ups. Effective therapies for FOP, and possibly for other common conditions of HO, may potentially be based on future interventions that block ACVR1/ALK2 signalling.
Keywords: fibrodysplasia ossificans progressiva (FOP), heterotopic ossification, bone morphogenetic protein, BMP, ACVR1, ALK2
Fibrodysplasia ossificans progressiva (FOP), a rare and catastrophic genetic disorder of progressive heterotopic ossification (HO), is the most disabling condition of extraskeletal ossification known in humans. FOP causes immobility through progressive metamorphosis of skeletal muscle and soft connective tissue into a second skeleton of heterotopic bone. At the present time, there is no effective treatment.1–6
HISTORICAL DESCRIPTIONS OF FOP
Possible cases of FOP date back to antiquity. FOP, known by many names throughout history, was first described in detail more than 250 years ago by a London physician. In a letter to The Royal Society of Medicine, dated 14 April 1736 (published in 1740), John Freke of Saint Bartholomew’s Hospital, London wrote: ‘There came a boy of healthy look, and about 14 years of age, to ask of us at the hospital, what should be done to cure him of many large swellings on his back, which began about 3 years since, and have continued to grow as large on many parts as a penny loaf particularly on the left side. They arise from all the vertebrae of the neck and reach down to the os sacrum; they likewise arise from every rib of his body, and joining together in all parts of his back, as the ramifications of coral do, they make as it were, a fixed bony pair of bodice’.7
Nearly 200 years later in 1918, Jules Rosenstirn from Mount Zion Hospital in San Francisco, USA wrote: ‘One does not wonder that a disease, so baffling in its course from the first causes to its ultimate state, should invite the speculative as well as the patiently investigating observer to lift the obscuring veil and solve this embarrassing puzzle’.8
FOP was, until recently, one of medicine’s most elusive mysteries. To patients who suffer from FOP, it is a painful metamorphosis into progressive immobility and a lifelong obstacle to physical freedom. While definitive treatments and cures are not yet available, the goals of FOP research are well articulated: to establish the genetic, molecular and cellular basis of FOP; and to use that knowledge to establish effective prevention, treatment and eventually cure.
CLASSIC CLINICAL FEATURES OF FOP
Two clinical features define classic FOP: malformation of the great toes; and progressive HO in specific spatial patterns (Figure 1). Individuals with FOP appear normal at birth except for the characteristic malformations of the great toes which are present in all classically affected individuals.9 During the first decade of life, children with FOP develop painful and highly inflammatory soft tissue swellings (or flare-ups) that transform soft connective tissues, including aponeuroses, fascia, ligaments, tendons and skeletal muscles, into an armament-like encasement of bone.10,11 Ribbons, sheets and plates of heterotopic bone replace skeletal muscles and connective tissues through a process of endochondral ossification that leads to permanent immobility.12–15 Minor trauma such as intramuscular immunizations, mandibular blocks for dental work, muscle fatigue and blunt muscle trauma from bumps, bruises, falls or influenza-like illnesses can trigger painful new flare-ups of FOP leading to progressive HO.16–20 Surgical attempts to remove heterotopic bone commonly lead to episodes of explosive and painful new bone growth.1,3,5,6 HO in FOP progresses in characteristic anatomical and temporal patterns that mimic the patterns of normal embryonic skeletal formation. FOP involvement is typically seen first in the dorsal, axial, cranial and proximal regions of the body and later in the ventral, appendicular, caudal and distal regions.1,3,5,10,11 Several skeletal muscles including the diaphragm, tongue and extra-ocular muscles are enigmatically spared from FOP. Cardiac muscle and smooth muscle are not involved in the FOP process.1,3,5
Figure 1.
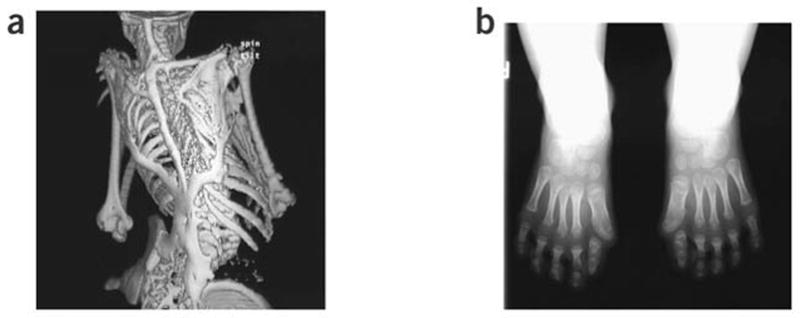
Characteristic clinical features of fibrodysplasia ossificans progressive (FOP). (a) Extensive heterotopic bone formation typical of FOP is seen by three-dimensional reconstructed computed tomography scan of the back of a 12-year-old child. (b) Anteroposterior radiograph of the feet of a 3-year-old child shows symmetrical great toe malformations. Source: Shore et al. Nature Genet 2006; 38: 525–527. Copyright held by the authors.
The clinical features of early lesional involvement in the axial regions are often different from those seen in the appendicular regions.21 Axial lesions may appear very rapidly, more rapidly than almost any neoplasm. In the axial regions, swelling is often mistaken for tumours, as large bulbous lesions may appear on the neck and back, whereas in the limbs, the swelling is often diffuse and may be mistaken for acute thrombophlebitis; a complication that can occur in patients with FOP due to generalized immobility and associated venous stasis.21 The qualitative differences in swelling in the axial versus the appendicular regions in patients with FOP may reflect regional differences in the anatomy of the subaponeurotic spaces, as well as differences in the anatomy of the fascial compartments.
Bone formation in FOP is episodic, but disability is cumulative. Most patients with FOP are confined to a wheelchair by the third decade of life, and require lifelong assistance in performing activities of daily living.1–6 Severe weight loss may result following ankylosis of the jaw, and pneumonia or right-sided heart failure may complicate rigid fixation of the chest wall.22 The severe disability of FOP results in low reproductive fitness, and fewer than 10 multigenerational families are known worldwide.23 The median age of survival is approximately 45 years, and death often results from complications of thoracic insufficiency syndrome (TIS).22
MISDIAGNOSIS OF FOP
FOP is commonly misdiagnosed, as clinicians often fail to associate the rapidly developing soft tissue swellings that appear on the head, neck and upper back with the malformed great toes.24 The correct diagnosis of FOP can be made clinically even before radiographic evidence of HO is seen if rapidly waxing and waning soft tissue lesions are associated with symmetrical malformations of the great toes. When such associations are not made, FOP is commonly misdiagnosed as aggressive juvenile fibromatosis (extra-abdominal desmoid tumours), lymphoedema or soft tissue sarcomas. Children often undergo unnecessary and harmful diagnostic biopsies that exacerbate progression of the condition.24 This can be particularly dangerous at any anatomical site, but especially so in the neck or back where asymmetric HO can lead to rapidly progressive spinal deformity and exacerbation of TIS.
CERVICAL SPINE ANOMALIES IN FOP
In addition to malformations of great toes and thumbs, early developmental anomalies are frequently observed in the cervical spine.25 Stiffness of the neck is an early finding in most patients and can precede the appearance of HO at that site. Characteristic anomalies of the cervical spine include large posterior elements, tall narrow vertebral bodies, and fusion of the facet joints between C2 and C7; findings that are strikingly similar to those seen in mice with homozygous deletions of the gene encoding noggin, a secreted bone morphogenetic protein antagonist.25
OTHER SKELETAL ANOMALIES IN FOP
Other skeletal anomalies often associated with FOP include short malformed thumbs, clinodactyly, short broad femoral neck and proximal medial tibial osteochondromas. The latter two findings are reminiscent of patients who have multiple hereditary exostoses, although the genes associated with multiple hereditary exostoses are not mutated in patients who have FOP. Nevertheless, these shared clinical findings may illuminate common pathway anomalies.1–3,5
THE TEMPOROMANDIBULAR JOINT IN FOP
Patients with FOP may have developmental anomalies of the temporomandibular joints (TMJs), although a comprehensive study of TMJ anatomy has not yet been undertaken in the FOP community. Spontaneous or post-traumatic extra-articular ankylosis of the TMJs is common, and leads to severe disability with resultant difficulties in eating and poor oral hygiene.1–3,5
SUBMANDIBULAR SWELLING IN FOP
Submandibular swelling can be a life-threatening complication, especially when associated with massive anterior neck swelling and difficulty in swallowing.17 Special measures to decrease swelling, including a course of glucocorticoids and respiratory support, may be warranted.17
HEARING IMPAIRMENT IN FOP
Hearing impairment is a common feature of FOP and occurs in approximately 50% of patients. The onset is usually in childhood or adolescence, and is generally slowly progressive. Hearing loss is usually conductive in nature and may be due to middle ear ossification; however, in some patients, the hearing impairment is neurological in nature.26
CARDIOPULMONARY FUNCTION IN FOP
Patients with FOP develop TIS that can lead to life-threatening complications. Features contributing to TIS in patients with FOP include: costovertebral malformations with orthotopic ankylosis of the costovertebral joints; ossification of intercostal muscles, paravertebral muscles and aponeuroses; and progressive spinal deformity including kyphoscoliosis or thoracic lordosis. Pneumonia and right-sided heart failure are the major life-threatening hazards that result from TIS in patients with FOP. Prophylactic measures to maximize pulmonary function, minimize respiratory compromise, and prevent influenza and pneumonia are helpful in decreasing the morbidity and mortality from TIS in patients with FOP.22,27,28
Assiduous attention should be directed towards the prevention and therapy of intercurrent chest infections. Such measures should include prophylactic pneumococcal pneumonia and influenza vaccinations (given subcutaneously), chest physiotherapy and prompt antibiotic treatment of early chest infection. Upper abdominal surgery should be avoided if possible, as it interferes with diaphragmatic respiration. Sleep studies to assess sleep apnoea may be helpful, and positive pressure assisted breathing devices such as bipap masks without the use of supplemental oxygen may also be helpful.22
Patients with FOP who have advanced TIS and who use unmonitored oxygen have a high risk of sudden death. Sudden correction of oxygen tension in the presence of chronic carbon dioxide retention suppresses respiratory drive. Patients who have FOP and severe TIS should not use supplemental oxygen in an unmonitored setting.22
Additional understanding of the complex chest wall dynamics in a true genetic model of FOP should greatly enhance understanding of the pathophysiology of these dreaded complications.
RADIOGRAPHIC FEATURES OF FOP
Joint malformations and soft tissue ossification are the characteristic radiographic features of FOP. Malformation of the great toes, thumbs, cervical spine and proximal femurs, along with the presence of proximal medial tibial osteochondromas, can make the diagnosis more certain.1–3,5
Radiographic and bone scan findings suggest normal modelling and remodelling of heterotopic bone.29,30 The incidence of fractures is not increased in patients with FOP, although fracture healing is characteristically accelerated in heterotopic bone.31 Bone scans are abnormal before HO can be detected by conventional radiographs. Computerized tomography and magnetic resonance imaging of early lesions have been described, but are superfluous.5,6 The definitive diagnosis of FOP can be made by simple clinical evaluation that associates progressively ossifying soft tissue lesions with malformations of the great toes.5,24 Clinical diagnosis of FOP can be confirmed by DNA diagnostic testing of the ACVR1 gene (see below).
LABORATORY FINDINGS IN FOP
Routine biochemical evaluations of bone mineral metabolism are usually normal, although serum alkaline phosphatase activity may be increased, especially during disease flare-ups.3,5,6 Urinary basic fibroblast growth factor levels may be elevated during disease flare-ups coinciding with the pre-osseous angiogenic phase of fibroproliferative lesions.32 Nephrolithiasis is more common in older patients with FOP, and may be due to increased immobilization and dehydration in the setting of generalized increased bone remodelling and mineral turnover.5
HISTOPATHOLOGY OF FOP LESIONS
The histological stages of FOP lesions have been well described.12–15 Early FOP lesions contain an intense perivascular B-cell and T-cell lymphocytic infiltrate. Subsequent migration of mononuclear inflammatory cells into affected muscle precedes widespread myonecrosis.13
Following a brief inflammatory stage, an intense fibroproliferative reaction associated with robust angiogenesis and neovascularity is noted.13,14 These early- to intermediate-stage lesions are microscopically indistinguishable from aggressive juvenile fibromatosis. As the lesion matures, fibroproliferative tissue undergoes an avascular condensation into cartilage followed by a revascularization stage and osteogenesis in a characteristic process of endochondral ossification. The resultant HO is normal, histologically mature lamellar bone with marrow elements.12–15
Mast cells have been identified at every histological stage, and are found in much greater abundance compared with normal skeletal muscle and non-lesional FOP muscle. In fact, during the intense fibroproliferative stage of the lesion, mast cells are found at a density much higher than in any other inflammatory myopathy.33
All stages of histological development are present in an active FOP lesion, indicating that different regions within the lesion mature at different rates. Although heterotopic bone formation in FOP is similar in some respects to bone formation in embryonic skeletal development and postnatal fracture healing, important differences are the lack of inflammation in embryonic skeletal induction and the relative absence of lymphocytic inflammatory cells in early fracture healing.15,34
EPIDEMIOLOGIC, GENETIC AND ENVIRONMENTAL FACTORS IN FOP
FOP is extremely rare with a worldwide prevalence of approximately one in two million. There appears to be no ethnic, racial, gender or geographic predisposition.3,6 Most cases arise as a result of a spontaneous new mutation. When observed, genetic transmission is autosomal dominant and can be inherited from either mothers or fathers.23,35
Both genetic and environmental factors affect the phenotype of FOP. A study of three pairs of monozygotic twins with FOP found that within each pair, congenital toe malformations were identical. However, postnatal HO varied greatly depending on life history and environmental exposure. This study indicated that genetic determinants strongly influence disease phenotype during prenatal development, and that environmental factors strongly influence postnatal progression of HO36
FOP AND THE BMP SIGNALLING PATHWAY
The classic and invariable FOP phenotype of great toe malformations and progressive heterotopic endochondral ossification suggested that the primary molecular pathology involves the bone morphogenetic protein (BMP) signalling pathway.37 A number of seminal discoveries provided evidence of profound dysregulation of the BMP signalling pathway in cells from patients who had FOP.38–47
DISCOVERY OF THE FOP GENE
In order to identify the chromosomal locus for the FOP gene, a conservative genome-wide linkage analysis was conducted using a subset of five families with the most stringent and unambiguous features of FOP. This approach identified linkage of FOP to 2q23–24.9 The gene encoding activin receptor IA (ACVR1) [also known as activin-like kinase 2 (ALK2)], a BMP type I receptor, was identified in the linkage interval. DNA sequencing of the ACVR1 gene determined that the same heterozygous mis-sense mutation in the glycine–serine (GS) activation domain (c.617G>A;R206H) occurs in all classically affected individuals examined.9 The discovery of the FOP gene was the culmination of a monumental 15-year search.
PROTEIN MODELLING OF THE FOP MUTATION
ACVR1/ALK2 is a BMP type I receptor, and protein structure homology modelling of the recurrent mutation predicts destabilization of the GS domain, consistent with an overactive BMP signalling pathway as the underlying cause of the ectopic chondrogenesis, osteogenesis and joint fusion seen in FOP. This mutation is consistent with a wealth of previous findings of an overactive BMP signalling pathway in FOP cells, and provides a rational basis for understanding both the postnatal HO and the congenital skeletal malformations that are ignominious signatures of this devastating disease.9
Hypothetical protein structure models are being developed to understand both inter-and intramolecular interactions of the mutant receptor. The GS domain of all TGF-β/BMP type I receptors is a critical site for binding and activation of pathway-specific Smad signalling proteins, and is a binding site of FKBP12, an inhibitory protein that prevents leaky activation of the type I receptor in the absence of ligand.48,49 FKBP12 also recruits a Smad7-Smurf1 ubiquitin ligase that functions normally to regulate the abundance of the receptor at the membrane.50 Both leaky activation of BMP signalling and accumulation of BMP type I receptors at the cell membrane are seen in FOP cells, suggesting possible aberrant association with FKBP12 in FOP. The most likely possibility is that FKBP12 interactions with the GS domain become altered, leading to promiscuous ACVR1/ALK2 activity (Figure 2). However, exactly how the R206H mutation in ACVR1/ALK2 specifically perturbs BMP signalling in FOP remains undetermined but could involve dysregulation of BMP receptor oligomerization, internalization, degradation and/or activation of downstream signalling. This is presentlythe subject of intense investigation.
Figure 2.

Hypothetical schema of bone morphogenetic protein (BMP) signalling in fibrodysplasia ossificans progressiva (FOP) cells. In control cells (A), in the absence of ligand, the Smad/Smurf-FKBP12 (SM-FKBP12) complex binds activin receptor IA (ACVR1; a BMP type I receptor) and prevents its promiscuous phosphorylation by the constitutively active type II BMP receptor (not shown). SM-FKBP12 also promotes ubiquitin-associated degradation of ACVR1 in the absence of ligand, thus maintaining low steady-state levels of ACVR1 at the cell membrane. Following ligand binding in control cells (B), SM-FKBP12 dissociates from ACVR1, thus allowing the constitutively active BMP type II receptor (not shown) to phosphorylate ACVR1, and promote Smad 1, 5 and8 phosphorylation and downstream BMP signalling. In FOP cells, SM-FKBP12 does not appear to bind appropriately to the mutant receptor [ACVR1 (R206H)]. Thus, inhibition of BMP signalling is impaired in the absence of ligand, and basal leakiness of BMP signalling occurs (C). Additionally, it is suspected that since the SM-FKBP12 complex cannot properly target the mutant ACVR1 (R206H) receptor for ubiquitin-associated degradation, ACVR1 may be expected to accumulate at the cell surface. Thus, in the presence of ligand (D), hyper-responsive BMP signalling may be predicted to occur. Arrows, signalling promoted; blunt-end lines, signalling inhibited; lock, SM-FKBP12 binding to ACVR1; dashed lines, SM-FKBP12 binding to ACVR1 impaired; open cups, extracellular ligand-binding domain of ACVR1; filled-in circles, BMP ligand; filled-in circles inside open cups, BMP ligand binding to ACVR1.
ACVR1/ALK2: A DRUGGABLE TARGET FOR THE SECOND SKELETON
The ultimate goal of FOP research is the development of treatments that will prevent, halt or even reverse progression of the condition. The prevention and treatment of HO in FOP, as for any of the more common forms of HO, will be based on at least one of four principles: disrupting the relevant inductive signalling pathways; suppressing the immunological and/or inflammatory triggers; altering the relevant osteoprogenitor cells in the target tissues; and/or modifying the tissue environment conducive to heterotopic osteogenesis.
The discovery of the FOP gene identifies ACVR1/ALK2 as a specific druggable target for FOP.51 The identification of the recurrent heterozygous mis-sense point mutation that causes FOP in all classically affected individuals provides a specific druggable target and a rational point of intervention in a critical signalling pathway. Plausible therapeutic approaches to inhibiting BMP signalling in FOP include inhibitory RNA technology, monoclonal antibodies directed against ACVR1/ALK2, and (most plausibly) orally available small molecule selective signal transduction inhibitors (STIs) of ACVR1/ALK2.51
Small molecule STIs have proven to be invaluable for investigating signal transduction pathways. Such molecules also have the potential for development into powerful therapeutic agents. The development of specific STIs for promiscuous ACVR1/ALK2 signalling in FOP have the potential to modify the natural history of the disease. Residues close to the ATP-binding site of ACVR1/ALK2 could be exploited to achieve selectivity, even among closely related receptor serine threonine kinases such as ALK3 (BMPRIA) and ALK6 (BMPRIB). Small soluble molecule inhibitors designed to specifically block ACVR1/ALK2 signalling intracellularly will need to be designed, screened and tested in cell and animal models of FOP. ACVR1/ALK2 STIs will need to have sufficient efficacy, tolerance to resistance, and acceptable safety profiles.51
Selective inhibitors have been developed for the ALKs that signal through Smads 2 and 3 [ALK4, 5 (TβRI) and 7]. At the present time, there are no known selective inhibitors of ACVR1/ALK2 or the other three BMP pathway type I receptors (ALK 1, 3 and 6) that signal through the BMP-pathway-specific Smads 1, 5 and 8. Such selective inhibitors are desperately needed.51
ANIMAL MODELS OF FOP
Animal models of FOP will be important for understanding the pathophysiology of FOP and for testing possible therapies.52 Laboratory-generated animal models with some features of FOP have provided the opportunity to better understand the biology of HO and to study the effectiveness and safety of currently available and emerging therapies. Development of a knock-in mouse model carrying the specific FOP-disease-causing mutation in ACVR1/ALK2 will be necessary to establish specificity of treatment in FOP. Such a genetically engineered knock-in mouse is presently being developed.
CURRENT MANAGEMENT OF FOP
The rarity, variable severity and episodic clinical course of FOP pose substantial uncertainties when evaluating experimental therapies.53 Accordingly, medical intervention is currently supportive. Surgical release of joint contractures is generally unsuccessful and risks new, trauma-induced HO. Osteotomy of heterotopic bone or surgical removal of heterotopic bone to mobilize joints is generally counterproductive because additional HO develops at the operative site. Rarely, a joint may be repositioned surgically to improve the patient’s overall functional status. Spinal bracing is ineffective and surgical intervention is associated with numerous complications.27
Guidelines for symptomatic management of disease flare-ups have been published, and highlight the anecdotal utility of glucocorticoids in managing new flare-ups affecting the function of major joints in the appendicular skeleton.53 Non-steroidal anti-inflammatory medications, cyclo-oxygenase-2 inhibitors, leukotriene inhibitors and mast cell stabilizers are useful anecdotally in managing chronic discomfort and ongoing flare-ups, but to date there is no proven efficacy with any therapy in altering the natural history of the disease.53 A recent report documented the failure of bone marrow transplantation to cure the condition, but suggested that chronic immunosuppression may have some utility, although its general use is not recommended.34
PROPHYLACTIC ISSUES IN FOP
Dental therapy must involve assiduous attention to prophylaxis of caries and must avoid intramuscular injection of local anesthetics, especially mandibular blocks and stretching of the jaw.54 All intramuscular injections must be avoided.16 Prevention of falls is crucial.19 Prophylaxis against influenza and pneumonia, as well as measures to prevent respiratory infection and cardiopulmonary complications of restrictive chest well disease, are vitally important.20
ANAESTHESIA IN PATIENTS WITH FOP
General anaesthesia is particularly dangerous in patients with FOP. Guidelines for general anaesthesia have been reported.54 Overstretching of the jaw for intubation may cause additional trauma to the TMJs, and lead to disease flare-ups. In older patients whose TMJs are ankylosed, oral access for intubation may not be possible. General anaesthesia in FOP patients should be accomplished through an awake fibre-optic nasal intubation under light sedation so that the patient can control secretions. This should be performed by well-trained anaesthesia teams who are familiar and experienced with this type of procedure.54
REHABILITATION ISSUES IN FOP
As heterotopic bone accumulates in FOP, range of motion is progressively lost leading to near-complete immobility. Present and future rehabilitation approaches should be focused on enhancing activities of daily living. Occupational therapy and vocational education consultations may be useful. Despite the widespread HO and progressive disability, most patients lead productive and fulfilling lives.55
THE INTERNATIONAL FOP ASSOCIATION
The International FOP Association (IFOPA) was founded in June 1988 to educate patients, doctors and the public about FOP; to support medical research into FOP; and to support patients with FOP and their families by providing a network of communication to help end the isolation that accompanies this rare and severely disabling condition. Additional information can be found on the IFOPA website (www.ifopa.org). In recent years, many regional FOP organizations have arisen worldwide to support patient-related activities.
RESEARCH AGENDA AND SUMMARY
While the mutation that causes classic FOP has been discovered, much work remains to elucidate the molecular mechanism by which this mutation leads to the complex disease phenotype of skeletal malformations and episodic progression of HO.
It will be essential to fully understand the role of the inflammatory pathways in triggering flare-ups of the disease, and to better understand the interaction of the immune system with the as-yet-unidentified connective tissue progenitor cells that are mobilized by disease flare-ups.56 Additionally, the molecular micro-environment in which HO develops needs to be more fully understood in the context of the disease-causing mutation that underlies the pathophysiology of the episodic flare-ups. The critical relationships between the mutant receptor, the environmental triggers, the responsive stem cells and the micro-environmental niches in which this renegade skeletal metamorphosis takes place will be vitally important to understand in order to design and develop the most effective treatment and prevention strategies. Accurate and clinically available premonitory markers of FOP flare-ups are desperately needed to assess potential therapies.
All of these important goals, and of course the ultimate goal of using this knowledge to develop better treatments and eventually a cure, will require the development of relevant cell and animal models.
A complete understanding of the genetic, molecular and cellular basis of HO in FOP will likely have broad therapeutic implications for patients with more common forms of HO, such as non-genetic forms of HO that may occur following total hip replacement, head injuries, spinal cord injuries, athletic injuries, blast injuries from war, and end-stage valvular heart disease.57,58
It may even be possible some day to harness the gene mutation that causes the renegade bone formation in FOP to create bone and new skeletal elements in a controlled way for patients who have osteoporosis, for those with severe bone loss from trauma or neoplasms, for those with fractures that fail to heal or spinal fusions that are slow to heal, or for those with congenital malformations of the spine and limbs. With the recent identification of the mutation responsible for FOP,9 we have reached a monumental milestone on our epic journey to understand FOP; knowledge which is needed to help the children with FOP and that has the potential to help many others. For the moment, the clinical management of FOP is focused in the prevention of flare-ups, the symptomatic management of disease symptoms and the optimization of function. The pathway to more effective management of FOP is through the research laboratory.
Practice points.
This very brief guide will summarize the current symptomatic management of FOP.
Activities: avoid soft tissue injuries, contact sports, overstretching of soft tissues and muscle fatigue. Avoid biopsies, surgical removal of heterotopic bone and all non-emergent surgical procedures
Anaesthesia: if general anaesthesia is required, perform awake intubation by nasotracheal fibre-optic technique
Falls: locked upper limbs may accentuate head trauma from falls. Epidural haematomas are common (surgical emergency). Use protective headgear in children who have upper limb involvement
Flare-up (back/chest): use non-steroidal anti-inflammatory medications with gastrointestinal precautions. Use analgesics and/or muscle relaxants, as needed
Flare-up (limbs/throat): prednisone – 2 mg/kg PO once daily for 4 days; begin within first 24 h of flare-up. Keep medication on-hand for emergencies. Use analgesias and/or muscle relaxants, as needed, with gastrointestinal precautions
Flare-ups (protection): most flare-ups result from over-use and soft tissue injuries. Prednisone 2 mg/kg PO once daily for 3 days to prevent flare-up after severe soft tissue injury. Do not use after minor bumps or bruises
Hearing: conductive hearing impairment is common. Perform periodic audiology evaluations. Hearing aids may improve conductive hearing loss
Immunizations: avoid all intramuscular immunizations. Subcutaneous immunizations are acceptable when FOP is quiescent. Avoid any immunizations during flare-ups
Influenza: administer influenza vaccines subcutaneously, but never during flare-ups. Avoid live attenuated flu vaccine; it may cause flu-like symptoms and exacerbate FOP. Household contacts of FOP patients should be immunized annually.
IVs: superficial IV access and venepuncture is acceptable. Traumatic IVs and arterial punctures may cause HO
Limb swelling: lymphoedema and transient neuropathy may occur with flare-ups of limbs. Elevate legs while sleeping and recumbent. Use support stockings. Take one baby aspirin daily with food. Rule-out deep vein phlebitis with Doppler ultrasound
Occupational therapy: perform periodic occupational therapy evaluations as activities of daily living change
Physiotherapy: avoid passive range of motion. Warm water hydrotherapy may be helpful
Pulmonary function: perform baseline pulmonary function tests and echocardiogram. Repeat periodically. Supplemental oxygen should not be used in an unmonitored setting
School: use school aides to protect and assist children. Request medical letter and preschool evaluation
Surgery: avoid surgery, except in emergencies
Teeth: avoid mandibular blocks, overstretching of the jaw and muscle fatigue
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Frederick S. Kaplan, Departments of Orthopedic Surgery & Medicine, The University of Pennsylvania School of Medicine, c/o Hospital of The University of Pennsylvania, Philadelphia, PA, USA.
Martine Le Merrer, U781 INSERM, Hopital Necker-Enfants Malades, Paris, France.
David L. Glaser, Department of Orthopedic Surgery, The University of Pennsylvania School of Medicine, c/o Hospital of the University of Pennsylvania, Philadelphia, PA, USA.
Robert J. Pignolo, Department of Medicine, The University of Pennsylvania School of Medicine, Philadelphia, PA, USA.
Robert Goldsby, Department of Pediatrics and Cardiovascular Research Institute, The University of California San Francisco, San Francisco, CA, USA.
Joseph A. Kitterman, Department of Pediatrics and Cardiovascular Research Institute, The University of California San Francisco, San Francisco, CA, USA.
Jay Groppe, Department of Biochemistry, University of Texas Health Science Center at San Antonio, San Antonio, TX, USA.
Eileen M. Shore, Departments of Orthopedic Surgery and Genetics, The University of Pennsylvania School of Medicine, Philadelphia, PA, USA.
References
- ❖1.Connor JM, Evans DA. Fibrodysplasia ossificans progressiva. The clinical features and natural history 34 patients. J Bone Joint Surg Br. 1982;64:76–83. doi: 10.1302/0301-620X.64B1.7068725. [DOI] [PubMed] [Google Scholar]
- 2.Smith R. Fibrodysplasia (myositis) ossificans progressiva: clinical lessons from a rare disease. Clin Orthop Rel Res. 1988;346:7–14. [PubMed] [Google Scholar]
- 3.Kaplan FS, Shore EM, Connor JM. Fibrodysplasia ossificans progressiva (FOP) In: Royce PM, Steinmann B, editors. Connective Tissue and Its Heritable Disorders: Molecular, Genetic, and Medical Aspects. 2. New York: Wiley-Liss, John Wiley & Sons, Inc.; 2002. pp. 827–840. [Google Scholar]
- 4.Kaplan FS, Glaser DL, Hebela N, Shore EM. Heterotopic ossification. J Am Acad Orthop Surg. 2004;12:116–125. doi: 10.5435/00124635-200403000-00007. [DOI] [PubMed] [Google Scholar]
- ❖5.Kaplan FS, Glaser DL, Shore EM, et al. The phenotype of fibrodysplasia ossificans progressiva. Clin Rev Bone Miner Metab. 2005;3:183–188. [Google Scholar]
- 6.Kaplan FS, Glaser DL, Shore EM. Fibrodysplasia (myositis) ossificans progressiva. In: Favus MJ, editor. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 6. Washington, DC: The American Society for Bone and Mineral Research; 2006. pp. 450–453. [Google Scholar]
- 7.Kaplan FS. Fibrodysplasia ossificans progressiva: an historical perspective. Clin Rev Bone Miner Metab. 2005;3:179–181. [Google Scholar]
- 8.Rosenstirn JA. A contribution to the study of myositis ossificans progressiva. Ann Surg. 1918;68:485–520. 591–637. doi: 10.1097/00000658-191811000-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ❖9.Shore EM, Xu M, Feldman GJ, et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nature Genet. 2006;38:525–527. doi: 10.1038/ng1783. [DOI] [PubMed] [Google Scholar]
- 10.Cohen RB, Hahn GV, Tabas J, et al. The natural history of heterotopic ossification in patients who have fibrodysplasia ossificans progressiva. J Bone Joint Surg Am. 1993;75:215–219. doi: 10.2106/00004623-199302000-00008. [DOI] [PubMed] [Google Scholar]
- 11.Rocke DM, Zasloff M, Peeper J, Cohen RB, Kaplan FS. Age and joint-specific risk of initial heterotopic ossification in patients who have fibrodysplasia ossificans progressiva. Clin Orthop Rel Res. 1994;301:243–248. [PubMed] [Google Scholar]
- 12.Kaplan FS, Tabas J, Gannon FH, Finkel G, Hahn GV, Zasloff MA. The histopathology of fibrodysplasia ossificans progressiva: an endochondral process. J Bone Joint Surg Am. 1993;75:220–230. doi: 10.2106/00004623-199302000-00009. [DOI] [PubMed] [Google Scholar]
- 13.Gannon FH, Valentine BA, Shore EM, Zasloff MA, Kaplan FS. Acute lymphocytic infiltration in an extremely early lesion of fibrodysplasia ossificans progressiva. Clin Orthop Rel Res. 1998;346:19–25. [PubMed] [Google Scholar]
- 14.Glaser DL, Economides AN, Wang L, et al. In vivo somatic cell gene transfer of an engineered noggin mutein prevents BMP4-induced heterotopic ossification. J Bone Joint Surg Am. 2003;85:2332–2342. doi: 10.2106/00004623-200312000-00010. [DOI] [PubMed] [Google Scholar]
- 15.Pignolo RJ, Suda RK, Kaplan FS. The fibrodysplasia ossificans progressiva lesion. Clin Rev Bone Miner Metab. 2005;3:195–200. [Google Scholar]
- 16.Lanchoney TF, Cohen RB, Rocke DM, Zasloff MA, Kaplan FS. Permanent heterotopic ossification at the injection site after diphtheria-tetanus-pertussis immunizations in children who have fibrodysplasia ossificans progressiva. J Pediatrics. 1995;126:762–764. doi: 10.1016/s0022-3476(95)70408-6. [DOI] [PubMed] [Google Scholar]
- 17.Janoff HB, Zasloff MA, Kaplan FS. Submandibular swelling in patients with fibrodysplasia ossificans progressiva. Otolaryngol Head Neck Surg. 1996;114:599–604. doi: 10.1016/S0194-59989670253-X. [DOI] [PubMed] [Google Scholar]
- 18.Luchetti W, Cohen RB, Hahn GV, et al. Severe restriction in jaw movement after routine injection of local anesthetic in patients who have fibrodysplasia ossificans progressiva. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;81:21–25. doi: 10.1016/s1079-2104(96)80141-7. [DOI] [PubMed] [Google Scholar]
- 19.Glaser DL, Rocke DM, Kaplan FS. Catastrophic falls in patients who have fibrodysplasia ossificans progressiva. Clin Orthop Rel Res. 1998;346:110–116. [PubMed] [Google Scholar]
- 20.Scarlett RF, Rocke DM, Kantanie S, Patel JB, Shore EM, Kaplan FS. Influenza-like viral illnesses and flare-ups of fibrodysplasia ossificans progressiva (FOP) Clin Orthop Rel Res. 2004;423:275–279. doi: 10.1097/01.blo.0000129557.38803.26. [DOI] [PubMed] [Google Scholar]
- 21.Moriatis JM, Gannon FH, Shore EM, Bilker W, Zasloff MA, Kaplan FS. Limb swelling in patients who have fibrodysplasia ossificans progressiva. Clin Orthop Rel Res. 1997;336:247–253. doi: 10.1097/00003086-199703000-00033. [DOI] [PubMed] [Google Scholar]
- 22.Kaplan FS, Glaser DL. Thoracic insufficiency syndrome in patients with fibrodysplasia ossificans progressiva. Clin Rev Bone Miner Metab. 2005;3:213–216. [Google Scholar]
- 23.Shore EM, Feldman GJ, Xu M, Kaplan FS. The genetics of fibrodysplasia ossificans progressiva. Clin Rev Bone Miner Metab. 2005;3:201–204. [Google Scholar]
- ❖24.Kitterman JA, Kantanie S, Rocke DM, Kaplan FS. Iatrogenic harm caused by diagnostic errors in fibrodysplasia ossificans progressiva. Pediatrics. 2005;116:654–661. doi: 10.1542/peds.2005-0469. [DOI] [PubMed] [Google Scholar]
- 25.Schaffer AA, Kaplan FS, Tracy MR, et al. Developmental anomalies of the cervical spine in patients with fibrodysplasia ossificans progressiva are distinctly different from those in patients with Klippel-Feil syndrome. Spine. 2005;30:1379–1385. doi: 10.1097/01.brs.0000166619.22832.2c. [DOI] [PubMed] [Google Scholar]
- 26.Levy CE, Lash AT, Janoff HB, Kaplan FS. Conductive hearing loss in individuals with fibrodysplasia ossificans progressiva. Am J Audiol. 1999;8:29–33. doi: 10.1044/1059-0889(1999/011). [DOI] [PubMed] [Google Scholar]
- 27.Shah PB, Zasloff MA, Drummond D, Kaplan FS. Spinal deformity in patients who have fibrodysplasia ossificans progressiva. J Bone Joint Surg Am. 1994;76:1442–1450. doi: 10.2106/00004623-199410000-00002. [DOI] [PubMed] [Google Scholar]
- 28.Kussmaul WG, Esmail AN, Sagar Y, Ross J, Gregory S, Kaplan FS. Pulmonary and cardiac function in advanced fibrodysplasia ossificans progressiva. Clin Orthop Rel Res. 1998;346:104–109. [PubMed] [Google Scholar]
- 29.Kaplan FS, Strear CM, Zasloff MA. Radiographic and scintigraphic features of modeling and remodeling in the heterotopic skeleton of patients who have fibrodysplasia ossificans progressiva. Clin Orthop Rel Res. 1994;304:238–247. [PubMed] [Google Scholar]
- 30.Mahboubi S, Glaser DL, Shore EM, Kaplan FS. Fibrodysplasia ossificans progressiva. Pediatr Radiol. 2001;31:307–314. doi: 10.1007/s002470100447. [DOI] [PubMed] [Google Scholar]
- 31.Einhorn TA, Kaplan FS. Traumatic fractures of heterotopic bone in patients who have fibrodysplasia ossificans progressiva. Clin Orthop Rel Res. 1994;308:173–177. [PubMed] [Google Scholar]
- 32.Kaplan FS, Sawyer J, Connors S, et al. Urinary basic fibroblast growth factor: a biochemical marker for preosseous fibroproliferative lesions in patients with FOP. Clin Orthop Rel Res. 1998;346:59–65. [PubMed] [Google Scholar]
- 33.Gannon FH, Glaser D, Caron R, Thompson LD, Shore EM, Kaplan FS. Mast cell involvement in fibrodysplasia ossificans progressiva. Hum Pathol. 2001;32:842–848. doi: 10.1053/hupa.2001.26464. [DOI] [PubMed] [Google Scholar]
- ❖34.Kaplan FS, Glaser DL, Shore EM, et al. Hematopoietic stem-cell contribution to ectopic skeletogenesis. J Bone Joint Surg Am. 2007;89:347–357. doi: 10.2106/JBJS.F.00472. [DOI] [PubMed] [Google Scholar]
- 35.Kaplan FS, McCluskey W, Hahn G, Tabas J, Muenke M, Zasloff MA. Genetic transmission of fibrodysplasia ossificans progressiva. J Bone Joint Surg Am. 1993;75:1214–1220. doi: 10.2106/00004623-199308000-00011. [DOI] [PubMed] [Google Scholar]
- 36.Hebela N, Shore EM, Kaplan FS. Three pairs of monozygotic twins with fibrodysplasia ossificans progressiva: the role of environment in the progression of heterotopic ossification. Clin Rev Bone Miner Metab. 2005;3:205–208. [Google Scholar]
- 37.Kaplan FS, Tabas JA, Zasloff MA. Fibrodysplasia ossificans progressiva: a clue from the fly? Calcif Tiss Int. 1990;47:117–125. doi: 10.1007/BF02555995. [DOI] [PubMed] [Google Scholar]
- ❖38.Shafritz AB, Shore EM, Gannon FH, et al. Over-expression of an osteogenic morphogen in fibrodysplasia ossificans progressiva. N Engl J Med. 1996;335:555–561. doi: 10.1056/NEJM199608223350804. [DOI] [PubMed] [Google Scholar]
- 39.Gannon FH, Kaplan FS, Olmsted E, Finkel G, Zasloff MA, Shore EM. Bone morphogenetic protein 2/4 in early fibromatous lesions of fibrodysplasia ossificans progressiva. Hum Pathol. 1997;28:339–343. doi: 10.1016/s0046-8177(97)90133-7. [DOI] [PubMed] [Google Scholar]
- 40.Olmsted EA, Kaplan FS, Shore EM. Bone morphogenetic protein 4 regulation in fibrodysplasia ossificans progressiva. Clin Orthop Rel Res. 2003;408:331–343. doi: 10.1097/00003086-200303000-00044. [DOI] [PubMed] [Google Scholar]
- ❖41.Ahn J, Serrano de la Peña L, Shore EM, Kaplan FS. Paresis of a bone morphogenetic protein-antagonist response in a genetic disorder of heterotopic skeletogenesis. J Bone Joint Surg Am. 2003;85:667–674. doi: 10.2106/00004623-200304000-00013. [DOI] [PubMed] [Google Scholar]
- 42.Hegyi L, Gannon FH, Glaser DL, Shore EM, Kaplan FS, Shanahan CM. Stromal cells of fibrodysplasia ossificans progressiva lesions express smooth muscle lineage markers and the osteogenic transcription factor Runx2/Cbfa-1: clues to a vascular origin of heterotopic ossification. J Pathol. 2003;201:141–148. doi: 10.1002/path.1413. [DOI] [PubMed] [Google Scholar]
- ❖43.Serrano de la Peña L, Billings PC, Fiori JL, Ahn J, Shore EM, Kaplan FS. Fibrodysplasia ossificans progressiva (FOP), a disorder of ectopic osteogenesis, misregulates cell surface expression and trafficking of BMPRIA. J Bone Miner Res. 2005;20:1168–1176. doi: 10.1359/JBMR.050305. [DOI] [PubMed] [Google Scholar]
- 44.Kaplan FS, Fiori JL, Ahn J, Billings PC, Shore EM. Dysregulation of BMP4 receptor trafficking and signaling in fibrodysplasia ossificans progressiva. Clin Rev Bone Miner Metab. 2005;3:217–223. [Google Scholar]
- ❖45.Fiori JL, Billings PC, Serrano de la Peña L, Kaplan FS, Shore EM. Dysregulation of the BMP-p38 MAPK signaling pathway in cells from patients with fibrodysplasia ossificans progressiva (FOP) J Bone Miner Res. 2006;21:902–909. doi: 10.1359/jbmr.060215. [DOI] [PubMed] [Google Scholar]
- 46.Kaplan FS, Fiori J, Serrano de la Peña L, Ahn J, Billings PC, Shore EM. Dysregulation of the BMP4 signaling pathway in fibrodysplasia ossificans progressiva. Ann NY Acad Sci. 2006;1068:54–65. doi: 10.1196/annals.1346.008. [DOI] [PubMed] [Google Scholar]
- 47.O’Connell MP, Billings PC, Fiori JL, et al. HSPG modulation of BMP signaling in fibrodysplasia ossificans progressiva cells. J Cell Biochem. 2007 doi: 10.1002/jcb.21370. [E pub ahead of print] [DOI] [PubMed] [Google Scholar]
- 48.Wang T, Li B-Y, Danielson PD, et al. The immunophilin FKBP12 functions as a common inhibitor of the TGF-β family type I receptors. Cell. 1996;86:435–444. doi: 10.1016/s0092-8674(00)80116-6. [DOI] [PubMed] [Google Scholar]
- 49.Chen Y-G, Liu F, Massagué J. Mechanism of TGF-β receptor inhibition by FKBP12. EMBO J. 1997;16:3866–3876. doi: 10.1093/emboj/16.13.3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yamaguchi T, Kurisaki A, Yamakawa N, Minakuchi K, Sugino H. FKBP12 functions as an adaptor of the Smad7-Smurf1 complex on activin type I receptor. J Mol Endocrinol. 2006;36:569–579. doi: 10.1677/jme.1.01966. [DOI] [PubMed] [Google Scholar]
- ❖51.Kaplan FS, Glaser DL, Pignolo RJ, Shore EM. A new era for fibrodysplasia ossificans progressiva: a druggable target for the second skeleton. Expert Opin Biol Ther. 2007;7:705–712. doi: 10.1517/14712598.7.5.705. [DOI] [PubMed] [Google Scholar]
- 52.Kaplan FS, Shore EM, Pignolo RJ, Glaser DL. Animal models of fibrodysplasia ossificans progressiva. Clin Rev Bone Miner Metab. 2005;3:229–234. [Google Scholar]
- 53.Glaser DL, Kaplan FS. Treatment considerations for the management of fibrodysplasia ossificans progressiva. Clin Rev Bone Miner Metab. 2205;3:243–250. [Google Scholar]
- 54.Nussbaum BL, Grunwald Z, Kaplan FS. Oral and dental healthcare and anesthesia for persons with fibrodysplasia ossificans progressiva. Clin Rev Bone Miner Metab. 2005;3:239–242. [Google Scholar]
- 55.Levy CE, Berner TF, Bendixen R. Rehabilitation for individuals with fibrodysplasia ossificans progressiva. Clin Rev Bone Miner Metab. 2005;3:251–256. [Google Scholar]
- 56.Kaplan FS, Shore EM, Gupta R, et al. Immunological features of fibrodysplasia ossificans progressiva and the dysregulated BMP4 pathway. Clin Rev Bone Miner Metab. 2005;3:189–193. [Google Scholar]
- 57.Mohler ER, 3rd, Gannon F, Reynolds C, Zimmerman R, Keane MG, Kaplan FS. Bone formation and inflammation in cardiac valves. Circulation. 2001;20:1522–1528. doi: 10.1161/01.cir.103.11.1522. [DOI] [PubMed] [Google Scholar]
- 58.Kaplan FS. The key to the closet is the key to the kingdom: a common lesson of rare diseases. Orph Dis Update. 2006;24:1–9. [Google Scholar]