

Abstract
Previous studies show that acute choline deficiency (CD) triggers apoptosis in cultured rat hepatocytes (CWSV-1 cells). We demonstrate that prolonged EGF stimulation (10 ng/mL x 48 hrs) restores cell proliferation, as assessed by BrdU labeling, and protects cells from CD-induced apoptosis, as assessed by TUNEL labeling and cleavage of poly(ADP-ribose) polymerase. However, EGF rescue was not accompanied by restoration of depleted intracellular concentrations of choline, glycerphosphocholine, phosphocholine, or phosphatidylcholine. In contrast, we show that EGF stimulation blocks apoptosis by restoring mitochondrial membrane potential (ΔΨm), as determined using the potential-sensitive dye chloromethyl-X-rosamine, and by preventing the release and nuclear localization of cytochrome c. We investigated whether EGF rescue involves EGF receptor phosphorylation and activation of the down-stream cell survival factor Akt. Compared to cells in control medium (CT, 70 μmol choline x 48hrs), cells in CD medium (5 μmol choline) were less sensitive to EGF-induced (0–300 ng/mL x 5 min) receptor tyrosine phosphorylation. Compared to cells in CT medium, cells in CD medium treated with EGF (10 ng/mL x 5 min) exhibited higher levels of phosphatidylinositol 3-kinase (PI3K)-dependent phosphorylation of AktSer473. Inactivation of PI3K was sufficient to block EGF-stimulated activation of Akt, restoration of mitochondrial ΔΨm, and prevention of cytochrome c release. These studies indicate that stimulation with EGF activates a cell survival response against CD-apoptosis by restoring mitochondrial membrane potential and preventing cytochrome c release and nuclear translocation which are mediated by activation of Akt in hepatocytes.
Keywords: Apoptosis, Choline, Epidermal growth factor, Hepatocytes, Mitochondria, Phosphatidylcholine, Phosphorylcholine
Introduction
Previous studies have emphasized the role for choline, an essential nutrient [1], in normal cell function, including synthesis of membrane phosphatidylcholine (PtdCho), methyl group-donation, and DNA synthesis [2–4]. An intriguing feature of choline metabolism is that acute choline deficiency (CD) results in dysregulation of liver cell proliferation and decreased cell survival [5–9], whereas prolonged CD results in acquisition of resistance to apoptosis and in malignant transformation [10, 11]. Thus, although CD is associated with diverse pathophysiological events in liver, the precise role for choline metabolites in the intracellular signaling pathways mediating these responses is not clear.
Membrane receptors and membrane derived signaling intermediates have been implicated in the response to CD. In rats fed a choline deficient diet, liver cells accumulated 1,2-sn-diacylglycerol, and exhibited prolonged activation of protein kinase C (PKC) [12]. Activation of PKC downstream of CD is important for an array of cellular processes, including modulation of cytoskeletal organization and cell motility [13], as well as for transduction of growth factor-mediated inhibitory (e.g., transforming growth factor beta) [14] and stimulatory cell signaling (e.g., epidermal growth factor, EGF) [15]. In animals made choline deficient, liver cells expressed fewer membrane receptors for epidermal growth factor (EGF) [16]. Although decreased cell survival in cells exposed to acute CD is accompanied by the loss of mitochondrial membrane potential (ΔΨm) [9], the interrelationship between mitochondrial function and EGF in CD-apoptosis is not well understood. In this study, we have determined that CD is a regulator of the EGF signaling pathway in CWSV-1 rat hepatocytes and that stimulation with EGF receptor activates a cell survival response in these cells that is mediated by activation of Akt and results in restoration of mitochondrial functions independent of restoration of choline metabolites.
Materials and Methods
Cell Culture
CWSV-1 cells originated from normal male F344 rat hepatocytes, and are routinely grown in serum-free medium [17] and express liver-specific proteins. Transfection of cells with SV40 large T-antigen inactivates p53 function, and CWSV-1 cells do not respond to γ-radiation, a p53-dependent apoptotic trigger [18]. CWSV-1 cells were maintained in RPCD defined medium: RPMI 1640 medium (Atlanta Biologicals, Norcross, GA) containing 11.5 μmol bovine serum albumin (Sigma Chemical, St. Louis, MO), 2 mmol L-glutamine (Sigma), 20 μmol oleate (Sigma), 7.2 μmol linoleate (Sigma), 67 μmol ethanolamine (Sigma), 1.27 μmol transferrin (Sigma), 10.4 nmol insulin (Sigma), 1 μmol dexamethasone (Sigma), 10 nmol glucagon (Sigma), 15 mmol HEPES, pH 7.4 (Sigma), trace metals (Sigma), 100 μg/mL penicillin (Gibco-BRL, Grand Island, NY), 100 μg/mL streptomycin and choline chloride as indicated.
Assessment of Apoptosis
For morphological assessment of apoptosis, cells were seeded at 2.2 x 104 cells/cm2 in 6-well multi-well plates in 70 μM choline-supplemented RPCD medium for 4 days before rinsing with phosphate-buffered saline (PBS) and switching them to fresh, control (CT; 70 μmol choline), or choline-deficient (CD; 5 μmol choline) RPCD medium for an additional 2 days as described in figures. To investigate the role of EGF receptor function in apoptosis, cells were maintained for 2 days in CT or CD medium with or without EGF (human recombinant, >97% purity by SDS-PAGE; Sigma Chemical Co., St. Louis, MO). Cells with classical morphological features of apoptosis were detected in hematoxylin stained plates as described previously [5]. The presence of apoptosis was confirmed by Tdt-mediated dUTP nick end labeling (TUNEL) assay [5, 11], and by cleavage of poly(ADP-ribose)polymerase (PARP) (6).
Assessment of Cell Proliferation
Cells grown in LabTek® chamber slides on day 2 of treatment with experimental media were incubated with 100 μmol 5-bromo-2′-deoxyuridine (BrdU) (Sigma) as described previously [19] and then fixed in ice-cold 70 percent ethanol. Sites of BrdU incorporation were visualized using an indirect streptavidin-peroxidase immunocytochemical method and a commercially available kit (Zymed, South San Francisco, CA), followed by counterstaining with Gill hematoxylin. The BrdU labeling index was determined by counting at least 200 cells in 3 replicates per experiment.
Choline Metabolites
Cell samples (5–6 x 106 cells per sample) were harvested and suspended in PBS following treatment as described above. DNA content was measured as a basis for normalization using a previously described fluorometric method [20]. For choline and related metabolites, an aliquot of cell suspension was extracted [21] and [2H-methyl]-labeled internal standard of each metabolite was added to permit correction for recovery. After separating the extracts into organic and aqueous phases, both hydrophilic choline metabolites (betaine, choline, glycerophosphocholine, and phosphocholine), and hydrophobic metabolites (phosphatidylcholine and sphingomyelin) were quantified using liquid chromatography-electrospray ionization-isotope dilution mass spectrometry [22].
Immunoblotting
CWSV-1 cells in the log phase of growth were treated with agonists for the indicated times, washed x2 in phosphate-buffered saline containing 1 mmol vanadate (PBS-V), then placed immediately in lysis buffer (1% NP40, 20 mmol Tris-HCl (pH 8.0), 150 mmol NaCl, 1 mmol EDTA, 0.1% NaN3, 2% SDS, 0.1 mmol Na3VO4, 1 mmol PMSF, 10 μg/mL aprotinin, 1 μmol pepstatin, 16.4 μg/mL leupeptin, and 10% glycerol). Lysates were heated at 95°C and protein concentrations determined, with BSA used to generate a standard curve, prior to adding 100 mmol DTT tracking dye. Samples were heated again for 5 min at 95°C, and the proteins separated using 8–10% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and electrotransferred onto nitrocellulose membranes (Schleicher & Schuell, Keene, NH). The membranes were incubated in blocking buffer containing 1% BSA for blotting with anti-phosphotyrosine antibodies. Membranes were subsequently incubated in RC20 antibody (detects EGF receptor tyrosine; Transduction Laboratories, Lexington, KY). Immunoblots were developed using a horseradish peroxidase conjugated secondary antibody and an ECL method (Amersham Pharmacia, Piscataway, NJ).
Mitochondrial function
We showed that CD disrupts mitochondrial transmembrane potential (ΔΨm), measured with the potential-sensitive dye JC-1, in CWSV-1 cells prior to the appearance of apoptosis [9]. To determine whether EGF-rescue of CD-apoptosis could involve restoration of mitochondrial function, we measured ΔΨm using the potential-sensitive dye chloromethyl-X-Rosamine (CMX-Ros; 200 nmol; Molecular Probes, Eugene, OR) [23]. CMX-Ros, unlike JC-1, has the advantage that changes in ΔΨm can be stabilized by formalin fixation for measurement at later time intervals. CMX-Ros (200 nmol) was added to the cell cultures for a 30 min incubation period at 37°C. The incubation was stopped by washes with PBS followed by fixation with 4% formaldehyde in PBS. 4′,6-Diamidino-2-phenylindole (DAPI), 0.1 μg/mL, for 20 min was used to counterstain nuclear DNA. Cells were mounted using GelMount® and a No. 1 thickness coverglass. Fluorescence images were obtained with a Nikon FX microscope using 595/615-nm filters. The levels of CMX-Ros were determined using an unbiased image analysis method and Scion Image as described previously [24].
Release of cytochrome c
CWSV-1 cells were grown on glass chamber slides, switched for an additional 48 hr to control (70 μmol choline) or choline deficient medium (5 μM choline) with or without EGF (10 ng/mL). The cells were then fixed in 4% formaldehyde in PBS, washed three times with PBS containing 0.1% Tween 20 (PBS-T), and blocked for 1 hr with 5% normal goat serum. After incubating with rabbit anticytochrome c (clone H-104, Santa Cruz Biotechnology, Santa Cruz, CA) at a dilution of 1:500 in 5% blocking serum in PBS-T for 1 hr at room temperature, the cells were washed three times with PBS-T, and then incubated with goat antirabbit Alexa Fluor® 546 conjugate secondary antibody at a dilution of 1:1000 in 5% blocking serum in PBS-T for 1 hr. The cells were washed three times with PBS, and coverslips were mounted onto slides with aqueous mounting medium. Fluorescence was visualized using a Zeiss LSM 5 Pascal confocal microscope equipped with a band-pass filter (excitation 535 nm, emission 570 nm) and fluorescent images were captured using Scion Image as described previously [24]. For immunoblotting analysis of cytochrome c release, total cell lysates were prepared as described below, loaded onto SDS-polyacrylamide gels and Western blotted for cytochrome c release using rabbit anticytochrome c (Santa Cruz Biotechnology) and an enhanced chemiluminescence method (Amersham Pharmacia Biotechnology, Piscataway, NJ).
EGF receptor autophosphorylation
CWSV-1 cells maintained for 48hr in CT or CD medium were treated with EGF (0–300 ng/mL x 5 min), cell lysates were prepared, and the levels of tyrosine phosphorylation of EGF (EGF pTyr) were assayed by immunoblotting with anti-phosphotyrosine antibody (RC-20, Transduction Lab.) and quantified by densitometry. The band intensity of the heat shock protein 90 kDa product (hsp 90) was measured as a basis for normalization of the corresponding 170 kDa EGF receptor tyrosine band intensities in CT and CD samples.
PI3 kinase and Akt
Cells in CT or CD medium were treated with EGF (10 ng/mL x 48 hr) in the presence or absence of LY294002 (2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one; Sigma), a specific inhibitor of phosphatidylinositol 3-kinase (PI3K). Proteins in whole cell lysates were subjected to SDS-PAGE and transferred to nitrocellulose membranes as described above. The blots were probed with the following antibodies: antiphospho-Akt (Ser473) and total Akt, antiphospho-FKHR (Ser256) and total FKHR, anti-AFX, and antiphospho-BAD (Ser136) (all from Cell Signaling Technology, Beverly, MA). Immunoblots were developed as described above.
Statistical analysis
Statistical differences among group means for apoptosis were assessed using Student’s t test. Analysis of variance and appropriate multiple comparisons procedures were used to determine statistical significance among group means for choline and its metabolites, mitochondrial transmembrane potential (ΔΨm), and for cell proliferation (BrdU labeling) (JMP Version 2, SAS Institute, Cary, NC).
Results
EGF and Choline Deficiency Apoptosis
Previous evidence demonstrated that feeding an extremely methyl-deficient diet (i.e., choline, methionine, folic acid and vitamin B12 deficient) to rats decreased EGF receptor mRNA expression and increased cell death in their livers, although the mode of cell death was not determined [25]. This evidence, coupled with later studies showing that feeding a diet deficient in choline alone is sufficient to induce apoptosis in rat liver hepatocytes [26], led us to investigate the role of EGF in CD apoptosis in CWSV-1 rat hepatocytes. Cells cultured in low choline-containing medium (5 μmol choline) exhibited increased levels of the 89 kDa PARP-cleaved product (Figure 1), and a nearly 8-fold increased rate of apoptosis (apoptotic morphology, TUNEL (+)) compared to cells in control medium (CT, 70 μmol choline) (Figure 1). In contrast, the rate of CD-apoptosis and the level of PARP cleavage product were dramatically reduced when cells were grown in CD medium containing EGF (10 ng/mL x 48 hrs).
Fig. 1.
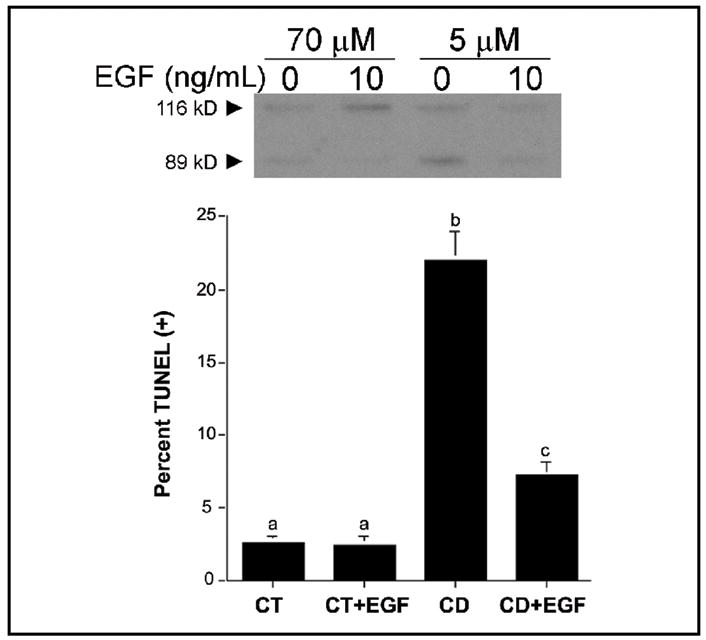
Epidermal growth factor rescues p53-defective CWSV-1 cells from apoptosis induced by acute withdrawal of choline. CWSV-1 hepatocytes in the log phase of growth were switched for 2 days to RPMI medium containing 70 μmol choline (CT) or 5 μmol choline (CD) with or without epidermal growth factor (EGF) as described in the Materials and Methods. The occurrence of apoptosis was determined by (A) PARP cleavage and (B) a TUNEL assay for apoptosis-associated DNA fragmentation. Mean ± SE, n = 6/point; bars identified with different letters are significantly different from one another (p < 0.01, t-test).
EGF and Mitochondrial Function
CWSV-1 cells switched to CD medium for 2 days exhibited a significant reduction in the incorporation of CMX-Ros, consistent with disruption in ΔΨm. Loss of this transmembrane potential was prevented in cells that had been treated for 2 days with EGF (10 ng/ml) in the presence of CD (Figure 2). To further investigate the role of mitochondrial function in CD-apoptosis, we localized cytochrome c in CWSV-1 cells in control and CD cells that had been treated for 2 days with EGF (10 ng/mL) (Figure 3A–D). For cells in control medium, cytochrome c staining was localized in a punctate perinuclear pattern (Figure 3A), consistent with mitochondrial localization of cytochrome c, and was unaffected by the addition of EGF (Figure 3B). In contrast, CD cells exhibited a diffuse pattern of cytochrome c staining throughout the cytoplasm and nucleus (Figure 3C), consistent with mitochondrial release of cytochrome c that was translocated to the nucleus. Stimulation of CD cells with EGF restored cytochrome c to a punctate and perinuclear staining pattern (Figure 3D), consistent with prevention of cytochrome c release from mitochondria. Western blotting for the presence of cytochrome c in total cell lysates was performed. Cytochrome c was detected in similar amounts independent of the amount of choline or EGF in the medium (Figure 3E).
Fig. 2.
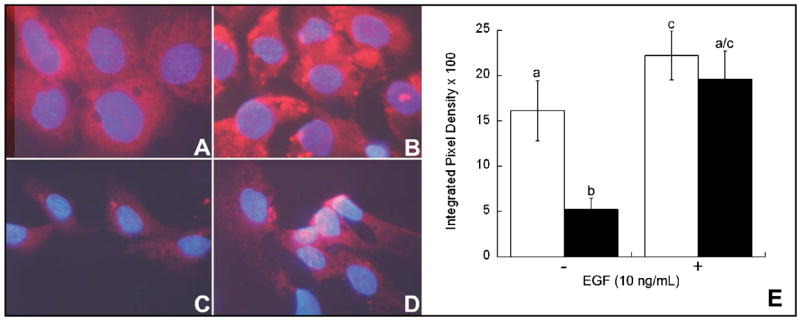
Effects of choline availability and EGF on mitochondrial function. Mitochondrial transmembrane potential (ΔΨm) was studied as described in the Materials and Methods with the potential-sensitive probe CMX-Ros (red fluorescence) and the nuclear counterstain DAPI (blue fluorescence). Compared to CWSV-1 cells maintained in control medium (70 μmol choline) (A) cells maintained in choline deficient medium (5 μmol choline) exhibited decreased incorporation of CMX-Ros consistent with disruption of ΔΨm (C). Treatment with EGF (10ng/mL) for 48 hr restored MPT in CWSV-1 cells maintained in control (B) and choline deficient medium (D). The relative levels of CMX-Ros (E) were measured using an objective image analysis method as described in the Materials and Methods. In the absence of EGF, cells maintained in control medium (white bars) exhibited a 3-fold increase in the amount of CMX-Ros incorporated compared to cells in choline deficient medium (black bars). Treatment with EGF (10 ng/mL x 48hr) restored the level of incorporation of CMX-Ros in choline deficient cells to control levels. Consistent with restoration of ΔΨm. Mean ± SE, n = 4/treatment group; columns identified with different letters differ significantly from one another (p < 0.01, t-test).
Fig. 3.
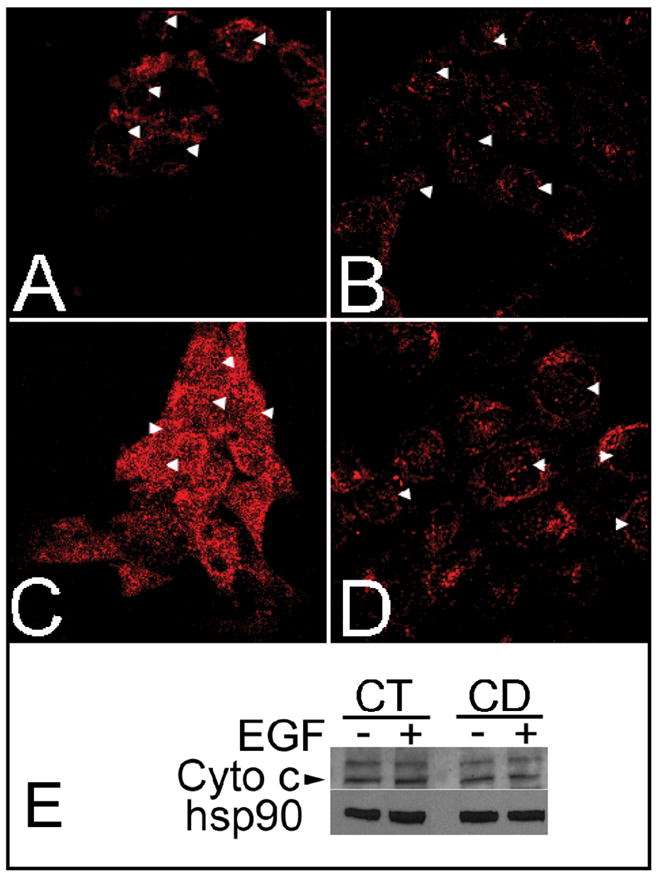
Effects of choline availability and epidermal growth factor on cytochrome c release. CWSV-1 cells were grown and treated as described in Materials and Methods. Cells were fixed in formaldehyde and immunocytochemically stained for cytochrome c (red). The cells were visualized on a Nikon FX microscope and images captured by a digital camera. (A, B) Localization of cytochrome c staining in CT cells (70 μmol choline) grown in the absence (A) or presence (B) of EGF (10 ng/mL x 48 hrs). (C, D) Localization of cytochrome c staining in CD cells grown in the absence (C) or presence (D) of EGF. In the absence of EGF, CD cells exhibited a diffuse and cytoplasmic and nuclear pattern of cytochrome c staining, unlike the punctate perinuclear staining of cytochrome c in CT cells. Treatment with EGF (10 ng/mL x 48 hr) restored the punctate perinuclear pattern of cytochrome c staining in CD cells maintained in choline deficient medium (D) consistent with the pattern of staining observed in CT cells (B). (E) Cytochrome c levels in total cell lysates were unaffected by choline availability or EGF. Arrows: location of cytoplasm overlying nucleus.
EGF and Choline Metabolite
We measured the levels of choline metabolites in control and CD cells that had been treated for 2 days with EGF (10 ng/mL). Compared to controls, cells maintained for 2 days in CD medium in the absence of EGF had approximately 67%, 90%, and 14% less choline, phosphocholine (PCho), and PtdCho, respectively, which was not restored by the addition of EGF to the medium (Table 1).
Table 1.
Effects of choline and epidermal growth factor (EGF) on choline metabolites in CWSV-1 hepatocytes. CWSV-1 cells were grown in control (CT, 70 μmol choline) or choline deficient (CD, 5 μmol choline) medium for 2 days with or without EGF (10 ng/mL), and biochemical parameters measured as described in the Materials and Methods. Data are expressed as mean ± SE, n = 4/treatment group; for each metabolite, data identified with different letters differ significantly (p < 0.01, ANOVA/Tukey-Kramer test). 1nmol/mg DNA. GPCho = glycerophosphocholine; PCho = phosphocholine; PtdCho = phosphatidylcholine; SM = sphingomyelin.
| CT | CD | |||
|---|---|---|---|---|
| Metabolite1 | (−) EOF | (+) EGF | (−) EGF | (+) EGF |
| Choline | 27.9 ± 2.0a | 35.5 ± 8.1a | 9.0 ± 0.9b | 10.7 ± 1.6b |
| GPCho | 129.2 ± 12.5a | 133.4 ± 3.1a | 106.8 ± 1.7b | 92.4 ± 5.6b |
| PCho | 619.5 ± 49.6a | 534.8 ± 31.5b | 61.4 ± 2.6c | 53.4 ±2.1c |
| PtdCho | 1680.2 ± 118.9a | 1581.6 ± 54.9a,b | 1443.8 ± 38.2b,c | 1339.5± 53.8c |
| SM | 210.8 ± 107.la | 250.9 ± 23.8a | 258.9 ± 25.9a | 242.0 ± 17.8a |
EGF and Inhibition of Cell Proliferation by CD
One of the key choline metabolites involved in the regulation of cell proliferation is PCho. It is a mitogenic factor for some types of cells [27]. Since EGF did not restore PCho levels in CD cells (Table 1), we examined whether DNA synthetic activity was modulated during CD and whether the depleted levels of PCho reduced the ability of EGF to stimulate DNA synthetic activity in CD cells, as determined by the incorporation of BrdU. As expected, CD treatment alone for 48 hr (Figure 4) reduced DNA synthetic activity. Restoration of DNA synthetic activity was observed when CD CWSV-1 cells were treated with EGF, even though the level of PCho remained at 10% of control levels (Table 1).
Fig. 4.
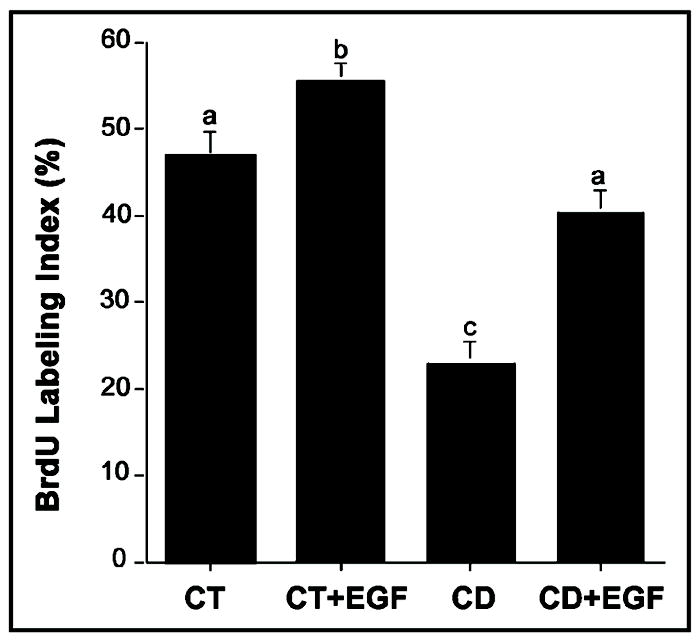
Epidermal growth factor restores cell proliferation in CD-CWSV-1 cells. Cells in the log phase of growth were switched for an additional 2 days to control (70 μmol choline) or CD medium (5 μmol choline) with or without EGF, and the percentage of cells incorporating BrdU (BrdU labeling index) was determined as described in the Materials and Methods. Mean ± SE; columns identified with different letters differ significantly from one another (p < 0.01, ANOVA/Sheffe’s test).
CD Alters EGF Receptor Phosphorylation
Ligand binding with EGFR results in autophosphorylation of intracellular EGFR tyrosine residues which regulate multiple biological processes. Since EGF stimulated cell proliferation and inhibited apoptosis in CD-cells, we examined the effects of EGF on EGFR tyrosine phosphorylation. Compared to CWSV-1 cells in control medium (70 μmol choline), cells in CD medium exhibited decreased sensitivity to EGF-induced receptor tyrosine phosphorylation when band intensities were normalized to hsp90 kDa protein loading controls (Figure 5). Thus, treatment with EGF increased EGF pTyr by 1.8-fold in CWSV-1 cells maintained in CT medium compared to 1.1-fold in cells in grown in CD medium. This is consistent with EGF receptor-mediated activation of downstream signal transduction important for proliferation and survival in CD cells.
Fig. 5.
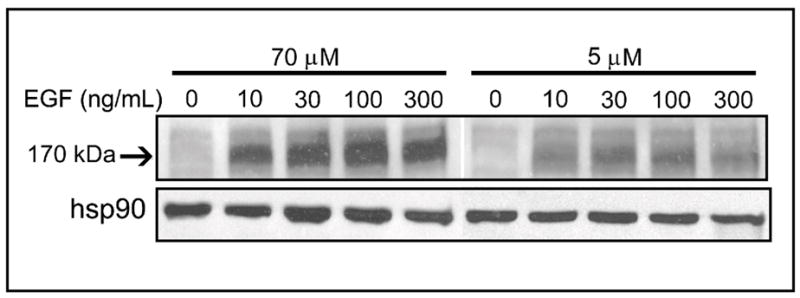
Effects of choline availability on EGF receptor tyrosine phosphorylation. CWSV-1 cells were treated with EGF (0–300 ng/mL x 5 min) and the levels of tyrosine phosphorylation of EGF (EGF pTyr) was assayed immunoblotting with anti-phosphotyrosine antibody (RC-20) and quantified by densitometry, as described in the Materials and Methods. Hsp90 was used to normalize band intensities for protein loading. The arrow indicates the EGF pTyr band at ~ 170 kDa.
Akt activation, forkhead transcription factors, and EGF protection
Activation of the Akt pathway by EGF is regulated by catalytically active PI3K, resulting in phosphorylation of Akt on Ser473. The expression of AktSer473 was increased by EGF (10ng/mL x 5 min) in both CT and CD cells (Figure 6), and was undetectable in cells treated with EGF in the presence of LY294002 (Figure 6). PI3K signaling and AktSer473 are involved in phosphorylation and inactivation of Forkhead transcription factors, thereby regulating cell cycle entry and apoptosis in some types of cells. However, CD and CT cells maintained in the absence or presence of EGF showed no detectable differences in the levels of activated or inactivated Forkheads AFX or FKHR (i.e., AFXSer193, FKHRSer256) (Figure 7), or in the levels of phosphorylated (inactivated) BadSer136 (data not shown). Importantly, inactivation of PI3K using LY294002 blocked EGF restoration of mitochondrial cytochrome c (Figure 3) and ΔΨm (data not shown). Taken together, these results indicated that stimulation with EGF activates a cell survival response that is mediated, in part, by PI3K-dependent inhibition of CD-induced early pro-apoptotic mitochondrial alterations (Figure 8).
Fig. 6.
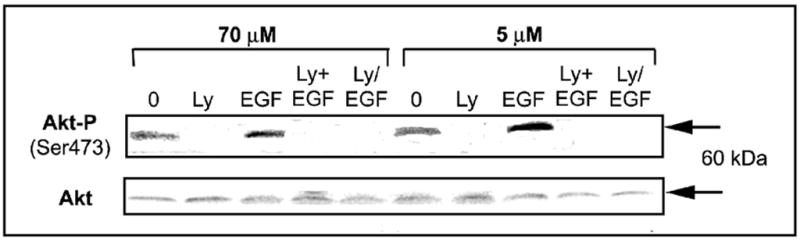
Effects of choline availability and activation of PI3 kinase on the expression of Akt. CWSV-1 cells were treated with EGF (10 ng/mL x 5 min) in the absence or presence of LY294002 and the levels of phosphorylated Akt and total Akt were assayed by immunoblotting with anti-Akt473 and total Akt antibodies, as described in the Materials and Methods. Treatment with EGF increased AktSer473 in CD medium (5 μM choline) cells more than in cells maintained in control medium (70 μM choline). Treatment with the LY294002 (50 μM) either simultaneous with (Ly+EGF) or for 1 hr before the addition of EGF (Ly/EGF) suppressed the expression of Akt, consistent with PI3 kinase-dependent activation of AktSer473 in response to EGF.
Fig. 7.
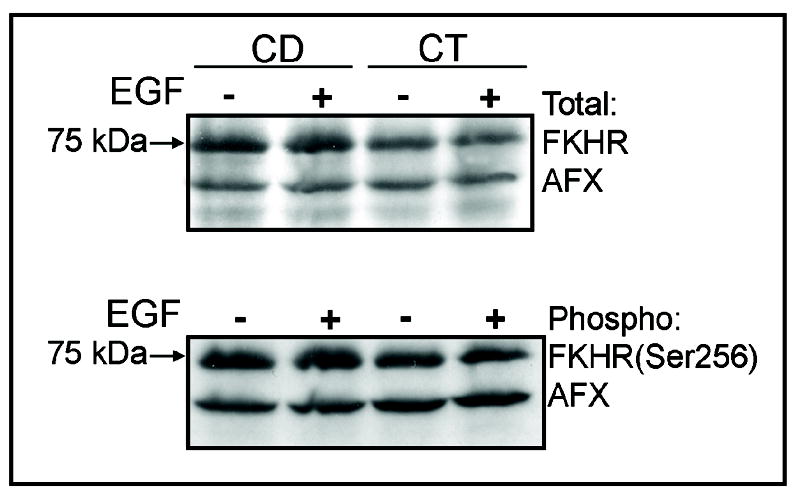
Effects of choline availability and EGF on the expression of Forkhead-related transcription factors. CWSV-1 cells were treated with EGF (10 ng/mL x 5 min) and the levels of phosphorylated and total FKHR and AFX were assayed by immunoblotting with antibodies that recognize FKHRSer256, total FKHR, AFXSer193, and total AFX., as described in the Materials and Methods. Cells in control (CT) and choline deficient medium (CD) exhibited similar levels of FKHR and AFX and treatment with EGF had no effect on the expression of phosphorylated (inactivated) or total Forkhead-related proteins.
Fig. 8.
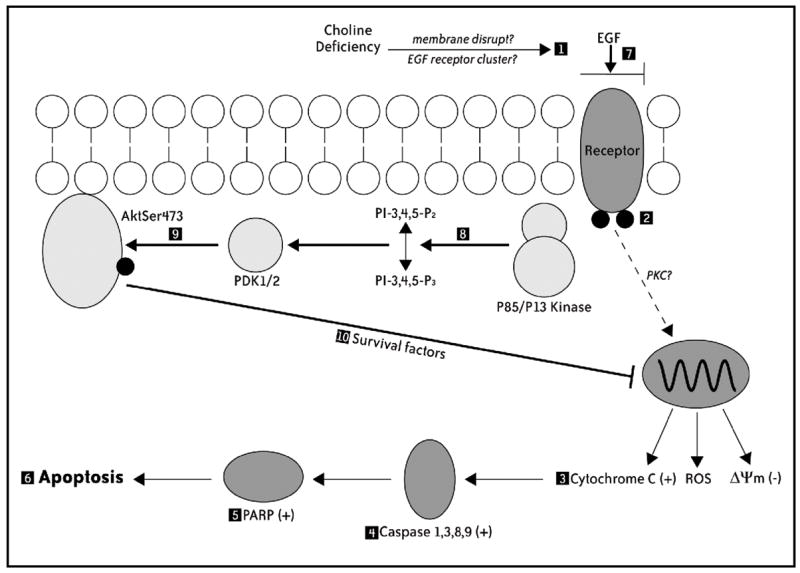
Model for EGF-induced rescue against CD apoptosis in rat hepatocytes. Cells switched acutely to CD medium (1) exhibit decreased sensitivity to EGF-induced receptor tyrosine phosphorylation (2), and (3) proapoptotic mitochondrial alterations (i.e., loss of ΔΨm, cytochrome c release, increased generation of ROS) that result in (4) activation of a caspases cascade, (5) cleavage of PARP, and (6) the completion of apoptosis. In the presence of added EGF (7), CD cells exhibit (8) PI3 kinase-dependent activation of AktSer473 (9), and (10) restoration of early mitochondrial pro-apoptotic alterations (i.e., ΔΨm, cytochrome c), independent of the expression of Forkhead-related proteins (i.e., FKHR, AFX) and phosphorylated Bad.
Discussion
Previous studies showed that, when CWSV-1 cells are deprived of extracellular choline, they become depleted of intracellular choline, PCho, and PtdCho, and undergo mitotic growth inhibition and apoptosis [5, 7, 11]. PtdCho, a phospholipid generated from the CDP-choline pathway, and from the phosphatidylethanolamine N-methyltransferase (PEMT) pathway, plays a significant role in membrane function [28] and is a source of important intracellular signaling intermediates (e.g., diacylglycerol) [12] and bioactive lipids (e.g., arachidonic acid, platelet activating factor) [29]. In addition to affecting intracellular signaling, earlier studies suggested choline metabolism could influence transmembrane signal transduction. In fact, feeding rats an extremely methyl-deficient diet decreased the expression of EGF receptor mRNA [16], suggesting a link between choline availability and EGF receptor-mediated signaling. The present study shows that CD cells compared to controls are less sensitive to EGF-stimulation of EGF receptor tyrosine phosphorylation. EGF receptor tyrosine phosphorylation is needed for ligand-stimulation of downstream signaling pathways that regulate cell proliferation and survival [30]. We demonstrate that stimulation with EGF activates a PI3 kinase-dependent cell survival response that results in prevention of apoptosis and restoration of DNA synthetic activity in CD cells.
Studies show that cell proliferation and survival are coordinated with metabolism of choline. Thus, in lymphocytes, the rate of synthesis of PCho was higher in proliferating cells than in growth arrested cells [31] or in cells undergoing apoptosis [32], consistent with the effects of CD on CWSV-1 cells. Competitive interference with diacylglycerol for CTP:cholinephosphate cytidylyltransferase (CT), the final enzyme in the biosynthesis of PtdCho, or mutational down regulation of the CT enzyme, impairs mitotic cell division and induces apoptosis [33, 34]. Importantly, exogenous PtdCho, lysophosphatidylcholine, or PCho, but not expression of PEMT or exogenous methyl-group donors, were able to restore DNA synthetic activity and to rescue primary neuronal cells, fibroblasts, or CWSV-1 hepatocytes from apoptosis [7, 33, 35, 36].
Links have been identified between EGF and choline metabolism in some types of cells. In lung and HeLa cells, EGF increased the activity of choline kinase [37] and CT [38], thereby increasing the rate of synthesis of PtdCho. In HeLa cells, increased expression of CT under the control of a tetracycline-responsive promoter prevented apoptosis induced by ET-18-OCH3 [27], a known trigger of apoptosis in CWSV-1 cells [9]. In T51B rat liver cells, EGF stimulation of DNA synthesis was preceded by a slow increase in the activity of phospholipase D (PLD) and the level of free choline [39]. EGF receptor tyrosine kinase activity is required for the activation of PLD [40]. In the present studies, we show that continuous treatment of cells with EGF at the time they are switched to CD medium rescues them from CD-apoptosis and restores DNA synthetic activity without restoring PCho or PtdCho levels. This suggests the involvement of one or more alternative pathways for effecting cell survival despite inadequate levels of choline.
Previously we demonstrated that acute CD increased apoptosis and inhibited mitotic cell division in CWSV-1 cells, as well as in whole rat liver [26, 41]. Recent studies showed that non-hepatocyte epithelial cells deprived of EGF [42] or prevented from activating the EGF receptor tyrosine kinase underwent cell cycle arrest that was reversed by the addition of EGF [43]. Consistent with these studies, EGF stimulated DNA synthetic activity in hepatocytes maintained in adequate levels of choline[44]. Here we show that stimulation of hepatocytes with EGF at the time that they are switched to CD medium, increases EGF receptor tyrosine phosphorylation, resulting in PI3 kinase-dependent activation of Akt and restoration of DNA synthetic activity. These findings are consistent with previous studies showing that activation of the PI3 kinase pathway, and not the MAPK pathway, is necessary and sufficient to explain the induction of DNA synthesis by EGF in primary hepatocytes [45].
In addition to inhibition of DNA synthesis, CD in the absence of EGF resulted in loss of mitochondrial ΔΨm, consistent with earlier studies showing that TGFβ1, a mediator of CD-apoptosis, caused dissipation of ΔΨm [46] and increased generation of reactive oxygen species from both mitochondrial and microsomal compartments [47]. We now show that, in addition to dissipation of ΔΨm, CD causes mitochondrial release of cytochrome c that is known to control interactions between early pro-apoptotic signaling molecules (e.g., Apaf-1, caspase-9) that are responsible for activation of a cascade of caspases [48]. Previously we have shown that CD causes activation of a caspase cascade early during apoptosis [8]. However, the mechanisms causing the late-stage nuclear alterations in CD-apoptosis are not well understood. Interestingly, when HeLa cells were treated with UV, cytochrome c release and accumulation in the nucleus correlated temporally with release of acetylated histone H2A into the cytosol and chromatin condensation in cells undergoing apoptosis [49]. Our data are consistent with this report and suggest that nuclear localization of cytochrome c may play a role in the end-stage nuclear changes associated with CD-apoptosis. Importantly, we demonstrate that stimulation with EGF prevents early (e.g., ΔΨm, cytochrome c release and nuclear translocation) and late manifestations of CD-apoptosis (i.e., PARP cleavage, TUNEL).
We have evidence that EGF stimulation of a cell survival response in CD cells involves PI3 kinase-dependent activation of Akt and restoration of mitochondrial function. Indeed, the restoration of mitochondrial ΔΨm and prevention of cytochrome c release and nuclear translocation observed in EGF-treated CD cells was abolished by LY294002, a known inhibitor or PI3 kinase [50]. Following activation, PI3 kinase phosphorylates Akt at Ser473, leading to the phosphorylation of Akt substrates that could play a role in EGF stimulation of a cell survival response in CD cells. Studies in quiescent non-hepatocyte type cells have shown that PI3 kinase/Akt-dependent phosphorylation and inactivation of Forkhead transcription factors (e.g., FKHR, AFX) contributes to increased incorporation of BrdU that is abolished by LY294002 [51]. Accordingly, we investigated whether changes in the activation of Forkhead transcription factors could underlie the EGF cell survival response in CD cells. However, we found no effect of EGF on the levels of activated or total FKHR and AFX in CT or CD cells. In addition, while Akt473 is capable of phosphorylating Bad at Ser136, which is responsible for preventing cytochrome c release and the activation of early apoptosis signaling molecules [52], phosphorylated BadSer136 was not detected in CT or CD cells either in the absence or presence of EGF stimulation. Nonetheless, LY294002 abolished EGF restoration of mitochondrial functions. Thus further studies are required to better understand the role of PI3 kinase/Akt in EGF rescue of CD-apoptosis.
In summary, we found that CD decreases the sensitivity of EGF receptors to EGF-induced receptor tyrosine phosphorylation and induces a form of apoptosis in rat hepatocytes that is mediated by dissipation of mitochondrial ΔΨm and release and nuclear translocation of cytochrome c. CD-apoptosis was prevented by stimulation with EGF, and this action is mediated by PI3 kinase/Akt-dependent restoration of ΔΨm and prevention of mitochondrial release and nuclear translocation of cytochrome c. Our studies provide novel insights on the mechanism of CD-apoptosis, and suggest that restoration of mitochondrial functions plays a more important role than restoration of choline metabolites in EGF-stimulation of a cell survival response.
Acknowledgments
This work was funded by a grant from the National Institutes of Health (DK55865, AG09525) awarded to SHZ, and by a grant from the NIH to the UNC Clinical Nutrition Research Unit (DK56350, ES10126). Additional support for this work was provided by grants from American Institute for Cancer Research (99B074) and the Association for International Cancer Research awarded to CDA. We thank Dr. H. Shelton Earp for advice and generous help with receptor tyrosine phosphorylation assays.
References
- 1.Food and Nutrition Board, Institute of Medicine. Dietary References Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin and Choline. National Academy Press; Washington, D.C: 1998. [PubMed] [Google Scholar]
- 2.Blusztajn JK. Choline: a vital amine. Science. 1998;281:794–795. doi: 10.1126/science.281.5378.794. [DOI] [PubMed] [Google Scholar]
- 3.Jackowski S. Coordination of membrane phospholipid synthesis with the cell cycle. J Biol Chem. 1994;269:3858–3867. [PubMed] [Google Scholar]
- 4.Zeisel SH. Choline: essential for brain development and function. Adv Pediatr. 1997;44:263–295. [PubMed] [Google Scholar]
- 5.Albright CD, Liu R, Bethea TC, da Costa K-A, Salganik RI, Zeisel SH. Choline deficiency induces apoptosis in CWSV-1 hepatocytes in culture. FASEB J. 1996;10:510–516. doi: 10.1096/fasebj.10.4.8647350. [DOI] [PubMed] [Google Scholar]
- 6.Albright CD, Borgman C, Craciunescu CN. Activation of a caspase-dependent oxidative damage response mediates TGFβ1 apoptosis in rat hepatocytes. Exp Mol Pathol. 2002;74:256–261. doi: 10.1016/s0014-4800(03)00002-9. [DOI] [PubMed] [Google Scholar]
- 7.Shin OH, Mar M-H, Albright CD, Citarella MT, da Costa K-A, Zeisel SH. Methyl-group donors cannot prevent apoptotic death of rat hepatocytes induced by choline deficiency. J Cell Biochem. 1997;64:196–208. doi: 10.1002/(sici)1097-4644(199702)64:2<196::aid-jcb3>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 8.Yen CL, Mar M-H, Zeisel SH. Choline deficiency-induced apoptosis in PC12 cells is associated with diminished membrane phosphatidylchoine and sphingomyelin, accumulation of ceramide and diacylglycerol, and activation of a caspase. FASEB J. 1999;13:135–142. [PubMed] [Google Scholar]
- 9.Vrablic AS, Albright CD, Craciunescu CN, Salganik RI, Zeisel SH. Altered mitochondrial function and over-generation of reactive oxygen species precede the induction of apoptosis by 1-O-octadecyl-2-methyl-rac-glycero-3-phosphocholine in p53-defective hepatocytes. FASEB J. 2001;15:1739–1744. doi: 10.1096/fj.00-0300com. [DOI] [PubMed] [Google Scholar]
- 10.Chandar N, Lombardi B. Liver cell proliferation and incidence of hepatocellular carcinomas in rats fed consecutively a choline-devoid and a choline-supplemented diet. Carcinogenesis. 1988;9:259–263. doi: 10.1093/carcin/9.2.259. [DOI] [PubMed] [Google Scholar]
- 11.Zeisel SH, Albright CD, Shin OH, Mar MH, Salganik RI, da Costa KA. Choline deficiency selects for resistance to p53-independent apoptosis and causes tumorigenic transformation of rat hepatocytes. Carcinogenesis. 1997;18:731–738. doi: 10.1093/carcin/18.4.731. [DOI] [PubMed] [Google Scholar]
- 12.da Costa KA, Cochary EF, Blusztajn JK, Garner SC, Zeisel SH. Accumulation of 1,2-sn-diradylglycerol with increased membrane-associated protein kinase C may be the mechanism for spontaneous hepatocarcinogenesis in choline deficient rats. J Biol Chem. 1993;268:2100–2105. [PubMed] [Google Scholar]
- 13.Carter CA, Albright CD, Kaufman DG. Differential effects of dioctanoylglycerol on fibronectin localization in normal, partially transformed, and malignant human endometrial stromal cells. Exp Cell Res. 1992;201:262–272. doi: 10.1016/0014-4827(92)90273-b. [DOI] [PubMed] [Google Scholar]
- 14.Jones CDRT, Hudson EA, Fontana JA, Trump BF, Resau JH. Transformed human bronchial epithelial cells (BEAS-Albright CD, Grimley PM, Jones RT, Fontana JA, Keenan KP, Resau JH: Cell-to-cell communication: A differential response to TGFβ in normal and transformed (BEAS-2B) human bronchial epithelial cells. Carcinogenesis. 1991;12:1993–1999. doi: 10.1093/carcin/12.11.1993. [DOI] [PubMed] [Google Scholar]
- 15.Pai R, Tarnawski A. Signal transduction cascades triggered by EGF receptor activation: relevance to gastric injury repair and ulcer healing. Diges Dis Sci. 1998;43:14S–22S. [PubMed] [Google Scholar]
- 16.Gupta C, Hattori A, Betschart JM, Virji MA, Shinozuka H. Modulation of epidermal growth factor receptors in rat hepatocytes by two liver tumor-promoting regimens, a choline-deficient and a phenobarbital diet. Cancer Res. 1988;48:1162–1165. [PubMed] [Google Scholar]
- 17.Woodworth CD, Secott T, Isom HC. Transformation of rat hepatocytes by transfection with simian virus 40 DNA to yield proliferating differentiated cells. Cancer Res. 1986;46:4018–4026. [PubMed] [Google Scholar]
- 18.Albright CD, Salganik RI, Kaufmann WK, Vrablic AS, Zeisel SH. A p53-dependent G1 checkpoint function is not required for induction of apoptosis by acute choline deficiency in immortalized rat hepatocytes in culture. J Nutr Biochem. 1998;9:476–481. [Google Scholar]
- 19.Albright 2B) alter the growth and morphology of normal human bronchial epithelial cells in vitro. Cell Biol Toxicol. 1990;6:379–388. doi: 10.1007/BF00120804. [DOI] [PubMed] [Google Scholar]
- 20.Labarca C, Paigen K. A simple, rapid and sensitive DNA assay procedure. Anal Biochem. 1980;102:344–352. doi: 10.1016/0003-2697(80)90165-7. [DOI] [PubMed] [Google Scholar]
- 21.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 22.Koc H, Mar M-H, Ranasinghe A, Swenberg JA, Zeisel SH. Quantitation of Choline and Its Metabolites in Tissues and Foods by Chromatography/Electrospray Ionization-Isotope Dilution Mass Spectrometry. Anal Chem. 2002;74:4734–4740. doi: 10.1021/ac025624x. [DOI] [PubMed] [Google Scholar]
- 23.Métivier D, Dallaporta B, Zamzami N, Larochette N, Susin SA, Marzo I, Kroemer G. Cytofluorometric detection of mitochondrial alterations in early CD95/Fas/APO-1-triggered apoptosis of Jurkat T Lymphoma cells. Comparison of seven mitochondrion-specific fluorochromes. Immunol Lett. 1998;61:157–163. doi: 10.1016/s0165-2478(98)00013-3. [DOI] [PubMed] [Google Scholar]
- 24.Albright CD, Friedrich CB, Brown EC, Mar M-H, Zeisel SH. Maternal dietary choline availability alters mitosis, apoptosis and the localization of TOAD-64 protein in the developing fetal rat septum. Devel Brain Res. 1999;115:123–129. doi: 10.1016/s0165-3806(99)00057-7. [DOI] [PubMed] [Google Scholar]
- 25.Shinozuka H, Masuhara M, Kubo Y, Katyal SL. Differential expression of hepatocyte growth factor, transforming growth factor-alpha and transforming growth factor-beta 1 messenger RNAs in two experimental models of liver cell proliferation. J Nutr Biochem. 1993;4:610–617. [PubMed] [Google Scholar]
- 26.Albright CD, Zeisel SH. Choline deficiency causes increased localization of TGFβ1 signaling proteins and apoptosis in rat liver. Pathobiology. 1997;65:264–270. doi: 10.1159/000164137. [DOI] [PubMed] [Google Scholar]
- 27.Baburina I, Jackowski S. Apoptosis triggered by 1-O-octadecyl-2-O-methyl-rac-glycero-3-phosphocholine is prevented by increased expression of CTP:phosphocholine cytidylyltransferase. J Biol Chem. 1998;273:2169–2173. doi: 10.1074/jbc.273.4.2169. [DOI] [PubMed] [Google Scholar]
- 28.Araki W, Wurtman RJ. Control of membrane phosphatidylcholine biosynthesis by diacylglycerol levels in neuronal cells undergoing neurite outgrowth. Proc Natl Acad Sci USA. 1997;94:11946–11950. doi: 10.1073/pnas.94.22.11946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kent C, Carman GM. Interactions among pathways for phosphatidylcholine metabolism, CTP synthesis and secretion through the Golgi apparatus. Trends Biochem Sci. 1999;24:146–150. doi: 10.1016/s0968-0004(99)01365-1. [DOI] [PubMed] [Google Scholar]
- 30.Justman QA, Clinton GM. Herstatin, an autoinhibitor of the human epidermal growth factor receptor 2 tyrosine kinase, modulates epidermal growth factor signaling pathways resulting in growth arrest. J Biol Chem. 2002;277:20618–20624. doi: 10.1074/jbc.M111359200. [DOI] [PubMed] [Google Scholar]
- 31.Flores I, Jones DR, Merida I. Changes in the balance between mitogenic and antimitogenic lipid second messengers during proliferation, cell arrest, and apoptosis in T-lymphocytes. FASEB J. 2000;14:1873–1875. doi: 10.1096/fj.99-1066fje. [DOI] [PubMed] [Google Scholar]
- 32.Al-Saffir NM, Titley JC, Robertson D, Clarke PA, Jackson LE, Leach MO, Ronen SM. Apoptosis is associated with triacylglycerol accumulation in Jurkat T-cells. Br J Cancer. 2002;86:963–970. doi: 10.1038/sj.bjc.6600188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tercé F, Brun H, Vance DE. Requirement of phosphatidylcholine for normal progression through the cell cycle in C3H/10T1/2 fibroblasts. J Lipid Res. 1994;35:2130–2142. [PubMed] [Google Scholar]
- 34.Wright MM, Henneberry AL, Lagace TA, Ridgway ND, McMaster CR. Uncoupling farnesol-induced apoptosis from its inhibition of phosphatidylcholine synthesis. J Biol Chem. 2001;276:25254–25261. doi: 10.1074/jbc.M011552200. [DOI] [PubMed] [Google Scholar]
- 35.Yen CL, Mar M-H, Meeker RB, Fernandez A, Zeisel SH. Choline deficiency induces apoptosis in primary cultures of fetal neurons. FASEB J. 2001;15:1704–1710. doi: 10.1096/fj.00-0800com. [DOI] [PubMed] [Google Scholar]
- 36.Ramos B, Salido GM, Campo ML, Claro E. Inhibition of phosphatidylcholine synthesis precedes apoptosis induced by C2-ceramide: protection by exogenous phosphatidylcholine. NeuroReport. 2000;11:3103–3108. doi: 10.1097/00001756-200009280-00013. [DOI] [PubMed] [Google Scholar]
- 37.Uchida T. Stimulation of phospholipid synthesis in HeLa cells by epidermal growth factor and insulin: activation of choline kinase and glycerophosphate acyltransferase. Biochim Biophys Acta. 1996;1304:89–104. doi: 10.1016/s0005-2760(96)00109-9. [DOI] [PubMed] [Google Scholar]
- 38.Mallampalli RK, Salome RG, Hunninghake GW. Epidermal growth factor is a positive in vivo regulator of CTP:cholinephosphate cytidylyltransferase. Exp Lung Res. 1994;20:1–11. doi: 10.3109/01902149409064369. [DOI] [PubMed] [Google Scholar]
- 39.Dean NM, Boynton AL. EGF-induced increase indiacylglycerol, choline release, and DNA synthesis is extracellular calcium dependent. J Cell Physiol. 1995;164:449–458. doi: 10.1002/jcp.1041640302. [DOI] [PubMed] [Google Scholar]
- 40.Slaaby R, Jensen T, Hansen HS, Frohman MA, Seedorf K. PLD2 complexes with the EGF receptor and undergoes tyrosine phosphorylation at a single site upon agonist stimulation. J Biol Chem. 1998;273:33722–33727. doi: 10.1074/jbc.273.50.33722. [DOI] [PubMed] [Google Scholar]
- 41.Chou JL, Fan Z, DeBlasio T, Koff A, Rosen N, Mendelsohn J. Constitutive overexpression of cyclin D1 in human breast epithelial cells does not prevent G1 arrest induced by deprivation of epidermal growth factor. Breast Cancer Res Treat. 1999;55:267–283. doi: 10.1023/a:1006217413089. [DOI] [PubMed] [Google Scholar]
- 42.Wu S, Rubin M, Fan Z, DeBlasio T, Soos T, Koff A, Mendelsohn J. Involvement of p27Kip1 in G1 arrest mediated by an anti-epidermal growth factor receptor monoclonal antibody. Oncogene. 1996;12:1397–1403. [PubMed] [Google Scholar]
- 43.Ye D, Mendelsohn J, Fan Z. Androgen and epidermal growth factor down-regulate cyclin-dependent kinase inhibitor p27Kip1 and costimulate proliferation of MDA Pca 2a and MDA Pca 2b prostate cancer cells. Clin Cancer Res. 1999;5:2171–2177. [PubMed] [Google Scholar]
- 44.Loyer P, Ilvin G, Cariou S, Glaise D, Corlu A, Guguen-Guillouzo C. Progression through G1 and S phases of adult rat hepatocytes. Prog Cell Cycle Res. 1996;2:37–47. doi: 10.1007/978-1-4615-5873-6_4. [DOI] [PubMed] [Google Scholar]
- 45.Band CJ, Mounier C, Posner BI. Epidermal growth factor and insulin-induced deoxyribonucleic acid synthesis in primary rat hepatocytes is phosphatidylinositol 3-kinase dependent and dissociated from protooncogene induction. Endocrinology. 1999;140:5626–5634. doi: 10.1210/endo.140.12.7188. [DOI] [PubMed] [Google Scholar]
- 46.Albright CD, Kuo J, Jeong S. cAMP enhances Cx43 gap junction formation and function and reverses choline deficiency-apoptosis. Exp Molec Pathol. 2001;71:34–39. doi: 10.1006/exmp.2001.2375. [DOI] [PubMed] [Google Scholar]
- 47.Albright CD, Salganik RI, Craciunescu CN, Mar M-H, Zeisel SH. Mitochondrial and microsomal derived reactive oxygen species mediate apoptosis induced by transforming growth factor-β1 in immortalized rat hepatocytes. J Cell Biochem. 2003;89:254–261. doi: 10.1002/jcb.10498. [DOI] [PubMed] [Google Scholar]
- 48.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:4050413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- 49.Nur-E-Kamal A, Gross SR, Pan Z, Balklava Z, Ma J, Liu JF. Nuclear translocation of cytochrome c during apoptosis. J Biol Chem. 2004;279:24911–24914. doi: 10.1074/jbc.C400051200. [DOI] [PubMed] [Google Scholar]
- 50.Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidlyinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) J Biol Chem. 1994;269:5241–5248. [PubMed] [Google Scholar]
- 51.Kops GJPL, Medema RH, Glassford J, Essers MAG, Kijkers PF, Coffer PJ, Lam EW-F, Burgering BMT. Control of cell cycle exit and entry by protein kinase B-regulated Forkhead transcription factors. Molec Cell Biol. 2002;22:2025–2036. doi: 10.1128/MCB.22.7.2025-2036.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou H, Li X-M, Meinkoth J, Pittman RN. Akt regulates cell survival and apoptosis at a postmitochondrial level. J Cell Biol. 2000;151:483–494. doi: 10.1083/jcb.151.3.483. [DOI] [PMC free article] [PubMed] [Google Scholar]