

Abstract
The surface protein Shp of Streptococcus pyogenes rapidly transfers its hemin to HtsA, the lipoprotein component of the HtsABC transporter, in a concerted two-step process with one kinetic phase. The structural basis and molecular mechanism of this hemin transfer has been explored by mutagenesis and truncation of Shp. The heme-binding domain of Shp is in the amino-terminal region and is functionally active by itself, although inclusion of the C-terminal domain speeds up the process ~10-fold. Single alanine replacements of the axial methionine 66 and 153 ligands (ShpM66A and ShpM153A) cause formation of pentacoordinate hemin-Met complexes. The association equilibrium constants for hemin binding to wild-type, M66A, and M153A Shp are 5,300, 22,000, and 38 μM−1, respectively, showing that the Met153-Fe bond is critical for high affinity binding and that Met66 destabilizes hemin binding to facilitate its rapid transfer. ShpM66A and ShpM153A rapidly bind to hemin-free HtsA (apoHtsA), forming stable transfer intermediates. These intermediates appear to be Shp-hemin-HtsA complexes with one axial ligand from each protein and decay to the products with rate constants of 3 to 0.4 s−1. Thus, the M66A and M153A replacements alter the kinetic mechanism and unexpectedly slow down hemin transfer by stabilizing the intermediates. These results, in combination with the structure of the Shp heme-binding domain, allow us to propose a “plug-in” mechanism in which side chains from apoHtsA are inserted into the axial positions of hemin in Shp to extract it from the surface protein and pull it into the transporter active site.
Numerous acquisition machineries have been identified in bacterial pathogens for heme as a preferred iron source from mammals. Specific ATP-binding cassette (ABC) transporters, which transport heme across the cytoplasmic membrane, are common components of the uptake machineries in both Gram-positive and Gram-negative pathogens (1–3). However, the transfer events and proteins involved prior to the action of the ABC transporters are different due to the distinct extracellular structures between these two types of bacteria. Gram-negative bacteria utilize an outer-membrane receptor protein to acquire heme from host hemoproteins directly or through a hemophore and bring the captured heme to the periplasmic space for the ABC transporter in a TonB-dependent process (4–6). Gram-positive bacteria produce cell surface proteins to relay heme from host proteins to the ABC transporter (7–9).
Heme1 [Fe(II)-protoporphyrin IX complex] or hemin [Fe(III)-protoporphyrin IX complex] exchange from one protein to another has been demonstrated biochemically in only a few bacterial systems, including transfers from hemoglobin to Serratia marcescens hemophore HasA (10), the cell surface protein Shp to HtsA, the lipoprotein component of the HtsABC transporter, in Streptococcus pyogenes and Streptococcus equi (11, 12), HasA to its outer membrane receptor HasR (10), and hemoglobin to Shigella dysenteriae outer-membrane receptor ShuA (5). A detailed kinetic mechanism has only been proposed for the S. pyogenes Shp/HtsA system (13). This process occurs in a single kinetic phase with transfer rate constants that are ~100,000 times greater than that for simple hemin dissociation from Shp. The structural basis for this rapid and concerted heme transfer is unknown.
In some hemoproteins, iron is hexacoordinate, with four ligands from protoporphyrin IX and two axial ligands from the side chains of His, Lys, Tyr, Met, and/or Cys. Combinations of the strong ligands, His, Lys, Met, and Cys, usually result in the low spin ferrous and ferric states with an intense Soret absorption peak and two Qov or αβ bands in the visible wavelength region (14, 15). The axial ligands of heme iron in HasA (16), HasR (6), ShuA (5), and Porphyromonas gingivalis heme receptor HmuR (17) are critical for hemin transfer and acquisition. However, it is unclear whether these axial ligands contribute to just binding affinity or have additional catalytic roles in heme and hemin transfer. Thus, detailed examination of the roles of the axial ligands in hemin binding and transfer should provide insight into the molecular mechanisms of these processes.
We have recently determined the crystal structure of the heme-binding domain of Shp (18), which reveals two methionine thiol ether S atoms (Met66 and Met153) as the axial ligands of the iron atom. In order to gain insight into the structural mechanism of rapid hemin transfer from Shp to HtsA, we examined these processes for Shp mutants containing only the N-terminal heme-binding domain or full length Shp in which the Met axial ligands were replaced with alanine (Ala) or histidine (His). Both the heme-binding domain and C-terminal region contribute to rapid heme transfer. Met153, but not Met66, appears to be critical for the high affinity of Shp for hemin, whereas both Met66 and Met153 are critical for rapid hemin transfer. The replacements of either Met66 or Met153 with Ala result in detection of an intermediate in hemin transfer to hemin-free HtsA (apoHtsA) indicating multiple first order reaction steps. Taken together, these data suggest a mechanism in which the two axial Met ligands in wild-type Shp are simultaneously displaced by groups from apoHtsA after the two proteins have formed a transient binary complex.
EXPERIMENTAL PROCEDURES
Protein Preparation
The preparation of the heme-binding domain of Shp containing amino acids 30 to 180 (designated Shp180) has been described elsewhere (18). Ala and His replacement mutants of Met66 and Met153 of Shp (designated ShpM66A, ShpM153A, ShpM66H, and ShpM153H) were generated by site-directed mutagenesis using the Stratagene QuickChange site-directed mutagenesis kit. The mutant proteins were expressed in Escherichia coli (DE3) containing the appropriate plasmid. The majority of the mutant proteins were expressed in inclusion bodies. Except for ShpM153H, hemin-binding form (holo-form) of these mutant proteins were purified from inclusion precipitates. All solutions were buffered with 20 mM Tris-HCl, pH 8.0 (Tris-HCl), unless otherwise specified. The cell pellet from 6 l culture was resuspended in 50 ml Tris-HCl, sonicated for 15 min, and centrifuged to obtain the pellet. The inclusion bodies were dissolved in 50 ml of 8 M urea. Each denatured protein was refolded by diluting with 40-fold Tris-HCl in the presence of excess hemin. The sample was loaded onto a DEAE column (2.5 × 10 cm), and the column was washed with 100 ml Tris-HCl and eluted with a 100-ml linear gradient of 0–0.25 M NaCl. Each protein was dialyzed against 3 l of 10 mM acetate buffer, pH 5.5, and loaded onto a SP Sepharose column (1.5 × 6 cm). The column was eluted with a 100-ml linear gradient of 0 to 0.25 M NaCl in the acetate buffer. The fractions containing >95% mutant protein were pooled, dialyzed against 3 l Tris-HCl, concentrated using Centricon Plus 20 filtration devices. HoloShpM153H was prepared by incubating purified apoShpM153H with excess hemin, loading the mixture onto a Sephadex G-25 column (1 × 20 cm), and eluting with Tris-HCl.
ApoShp and mutant proteins were prepared from inclusion bodies as described above except that hemin was absent in the refolding step. ApoHtsA was prepared, as described previously (13).
EPR Measurement
EPR spectra of wild-type and mutant Shp were measured with a Bruker EMX spectrometer using the following conditions: frequency, 9.6 GHz; power, 3 mW; modulation amplitude, 10 G; the modulation frequency, 100 kHz; and temperature, 10 K. The high-spin signal at g = 6 was quantified by double integration between 800 and 1700 G and comparison with the signal of a high-spin sperm whale metmyoglobin at pH 7. Quantification of the low spin signals was based on comparison of the area of the g = 3 absorption-like signal with the analogous low-spin signal of metmyoglobin at pH 9.5.
Rates of Hemin Dissociation from Shp Mutants
The rates of hemin dissociation from ShpM66A, ShpM153A, and ShpM153H were measured using H64Y/V68F apomyoglobin as a hemin scavenger as described previously (19). Each Shp mutant protein (3 μM) was incubated with 58 μM apomyoglobin in 1 ml of 20 mM Tris-HCl, and the changes in absorbance at 410 nm for ShpM153A and at both 405 and 410 nm for ShpM66A were monitored. The ΔA410 and Δ(A410–A405) time courses were fit to a single exponential equation to obtain the rate constants for hemin dissociation from ShpM153A and ShpM66A, respectively.
Kinetic Analyses
A stopped-flow spectrophotometer equipped with a photodiode array detector (SX20; Applied Photophysics) was used to measure the rates of hemin transfer from Shp mutants to apoHtsA and the binding of hemin to apoShp mutants. In these measurements, 2.4 μM holoShp or 2 μM hemin in one syringe was mixed with apoprotein at ≥ 5x [apoHtsA] or [apoShp], respectively, in another syringe. Spectra were recorded with time in each reaction. Changes in absorbance at appropriate wavelengths were fitted to a single or double exponential expression, yielding pseudo-first-order rate constants for each reaction step for further analysis as described in Results.
RESULTS
The Heme-Binding Domain of Shp
Shp and its homologue in S. equi share 75% and 25% identify in amino acid sequence in the regions over amino acids 28–173 and 174–291, respectively (12), suggesting that the heme-binding domain of Shp is located in the amino-terminal region. To test this idea, Shp180, consisting of amino acids 30 to 180, was prepared. The spectra of oxidized and reduced Shp180 are almost identical to those reported for full length Shp (13). Like Shp, reduced Shp180 is stable in air. These data indicate that the heme-binding domain of Shp is located in the region of amino acids 30–180. This result led us to try Shp180 in successful crystallization and structure determination studies (18).
Hemin and Heme Transfer from Shp180 to apoHtsA
When reduced Shp180 reacts with excess apoHtsA, the absorbance at 424 nm (A424) apidly increases on milliseconds time scales due to heme transfer from Shp to apoHtsA and slowly decreases on seconds time scales due to autoxidation of the heme bound to HtsA, as is also seen for heme transfer from full length Shp to apoHtsA (13). The time course for hemin transfer from oxidized Shp180 to apoHtsA measured at 414 nm also resembles that for transfer from full length Shp. The basis for these optical changes is the difference in the maximum wavelengths of the Soret band of reduced and oxidized Shp180 and HtsA, which are 428, 419, 524, and 412 nm, respectively. Both heme and hemin transfers are pseudo-first order processes at excess [apoHtsA], and the observed rate constants depend hyperbolically on [apoHtsA] as in the full-length Shp-apoHtsA reactions (13). Thus, the mechanism established for the full length Shp-apoHtsA reactions (13) applies to the reactions of apoHtsA with Shp180. In both cases, the observed time courses can be analyzed by Scheme I which proposes rapid formation of a Shp180-apoHtsA complex (Kd = 90 and 107 μM for the oxidized and reduced Shp180-apoHtsA complexes, respectively, Table I) and a first order decay involving intra-complex heme or hemin transfer (ktransfer values, Table I).
Scheme 1.

TABLE I.
Kinetic parameters for hemin or heme transfer from Shp proteins to apoHtsA
| Kinetic parameter
|
||||||
|---|---|---|---|---|---|---|
| Heme or hemin Donor | k2/k1 or Kd (μM) | ktransfer (s−1) | kt1 (s−1) | kt2 (s−1) | cktransfer/Kd or kt1/Kd (μM−1s−1) | Reference |
| a Ox. Shp | 48 ± 7 | 43 ± 3 | 0.8 | 13 | ||
| a Re. Shp | 120 ± 18 | 28 ± 6 | 0.3 | 13 | ||
| a Ox. Shp30–180 | 90 ± 5 | 2.9 ± 0.9 | 0.03 | This work | ||
| a Re. Shp30–180 | 107 ± 12 | 4.4 ± 0.4 | 0.04 | This work | ||
| a Ox. ShpM153H | 6.6 ± 0.5 | 3.5 ± 0.4 | 0.5 | This work | ||
| b Ox. ShpM66A | 11.4 ± 0.3 | 8.7 ± 0.3 | 0.38 ± 0.08 | 0.76 | This work | |
| b Ox. ShpM153A | 170 ± 8 | 120 ± 30 | 2.5 ± 0.2 | 0.7 | This work | |
The values were determined according to Scheme I as described in reference 13. Note that no hemin transfer could be detected in the ShpM66H–apoHtsA reaction, indicating either complete inhibition of formation of the Shp-HtsA complex or that affinity of apoShpM66H is ≥ 10 to 100 fold larger than that of wild-type apoHtsA. Ox. and Re. stand for Oxidized and Reduced, respectively.
The values were determined according to Scheme II. The values of Kd and kt1 were calculated from fits of the dependence of the observed rates of the formation of the transfer intermediate to Equation 2, and the values of kt2 were obtained directly from the double-exponential fits of the primary stopped-flow data.
Apparent bimolecular rate constants at low [apoHtsA] for heme transfer from Shp and Shp180 and for the intermediate formation in the ShpM66A- and ShpM153A-apoHtsA reactions.
Truncation to residues 30–180 does slow the rate of intra-complex hemin transfer from 43 to 2.9 s−1 and that for heme transfer from 26 to 4.4 s−1 compared to full length Shp. Thus, although the heme-binding domain retains the ability to directly and efficiently transfer heme and hemin to apoHtsA, the C-terminal domain does play a significant role by enhancing 6 to 15-fold the intra-complex transfer rate.
Ala and His Replacement Mutants of Shp Met66 and Met153
To assess the contributions of the Shp axial ligands to hemin binding and transfer, Met66 and Met153 were replaced by either a non-coordinating Ala or the strong ligand His. The Soret absorption peak shifts from 420 nm in wild-type oxidized Shp to 406 nm and 402 nm, respectively, in ShpM66A and ShpM153A (Fig. 1). In addition, there was a marked alternation in the visible absorbance spectrum (470–700 nm) with a new peak at around 600 nm indicative of a high spin Fe(III)-protoporphyrin IX complex. The UV-visible spectra of ShpM66A and ShpM153A are similar to those of H64V and H64L human metmyoglobin mutants (20). The crystal structures of the analogous sperm whale metmyoglobin show pentacoordinate hemin complexes with no coordinated water (21). Thus, the spectral features of the Shp mutants suggest that loss of one of the Met axial ligands leads to formation of a pentacoordinate hemin-Met complex. This interpretation is supported by the spectra of the reduced Shp Ala mutants. The well-resolved and intense α and β bands of reduced wild-type Shp, which is typical of hexacoordinate heme complexes with two strong axial ligands, is replaced with a single broad band for the Ala mutants (Fig. 1), which is similar to that of pentacoordinate deoxyhemoglobin. This result indicates strongly that the reduced Shp mutants also form pentacoordinate heme complexes.
FIGURE 1.
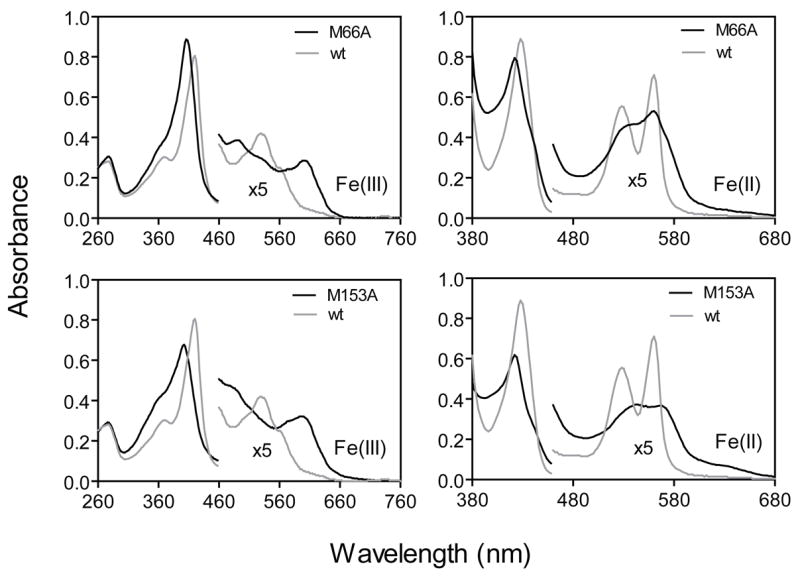
Optical absorption spectra of oxidized and reduced wild-type Shp, ShpM66A, and ShpM153A each at 7.7 μM.
Our interpretation of the coordinate state of ShpM66A and ShpM153A is further supported by the EPR spectra shown in Fig. 2A. The mutants both show EPR spectra with 100% high spin signals in the g=6 region. In both cases multiple derivative signals occur, which are similar to those reported by Ikeda-Saito et al. (20) for pentacoordinate H64V and H64L metmyoglobin. The latter authors suggested an increase in rhombic symmetry in the apolar metmyoglobin mutants due to increased anionic character of the proximal imidazole and/or mixing of S=3/2 and 5/2 spin states due to the pentacoordinate character of the hemin iron atom. The latter explanation probably applies to the spectra of the Shp mutants, and in addition, some partial coordination with solvent water could occur and account for the very sharp feature at g=5.81 (Fig. 2A). In contrast, wild-type Shp exhibits an EPR spectrum with dominant low spin signals (>86%) due to strong coordination by the sulfur atoms of Met66 and Met153 (18).
FIGURE 2. EPR spectra of oxidized Shp proteins.
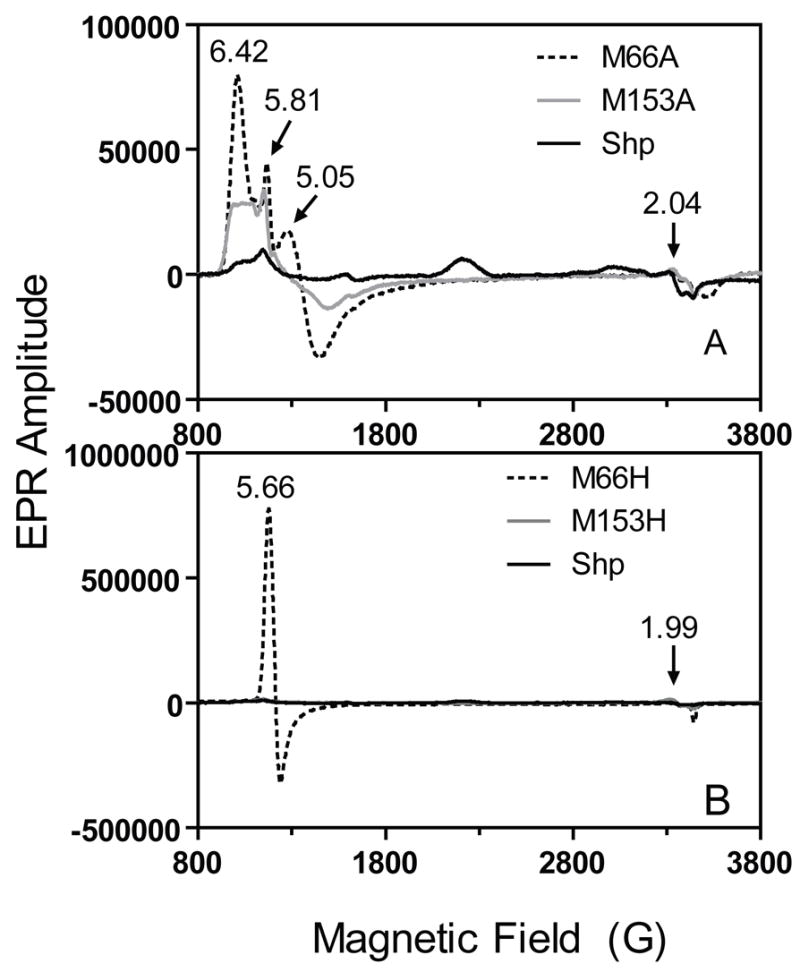
Proteins (each at 100 μM): (A), ShpM66A, ShpM153A, and Shp; (B), ShpM66H, ShpM153H, and Shp. The numbers in both panels are the g values of the indicated EPR peaks.
ShpM153H appears to exhibit normal wild-type-like hemichrome and hemochrome spectra (Fig. 3A and 3B), indicating axial His and Met coordination in both oxidation states. This conclusion is supported by the EPR spectrum of oxidized ShpM153H which shows ~60% low spin character (Fig. 2B, solid line). Although reduced ShpM66H displays a wild-type-like spectrum indicative of His and Met coordination (Fig. 3B), oxidized ShpM66H shows an optical spectrum (Fig. 3A) similar to that of high-spin aquometmyoglobin (19, 20), suggesting hexacoordinate Met-Fe-OH2 coordination. This coordination is supported by the EPR spectrum of oxidized ShpM66H, which shows a single g=5.66 signal indicative of a purely high spin aquohemin form (Fig. 2B). Thus, replacement of Met66 by His results in a coordination of the hemin iron to a solvent water molecule, instead His66, in the oxidized mutant, further supporting the idea that Met66 is only weakly coordinated to the heme iron atom.
FIGURE 3. Optical absorption spectra of ShpM66H and ShpM153H.

Absorption spectra of 7.7 μM ShpM66H, ShpM153H, and wild-type (wt) Shp in the oxidized (A) and reduced (B) states are shown.
Hemin Binding to and Dissociation from the Met66 and Met153 Mutants
To examine the individual contributions of the Met ligands to the overall affinity of Shp for hemin, association and dissociation rate constants for hemin binding to ShpM66A, ShpM153A, ShpM66H, and ShpM153H were measured and compared to the corresponding parameters for wild-type Shp (Table II). Association equilibrium constants for hemin binding to all five proteins were calculated from these parameters as described previously (13).
TABLE II.
Rate and Equilibrium Constants for Hemin Binding to and Dissociation from Shp Proteins
| Protein
|
|||||
|---|---|---|---|---|---|
| Kinetic parameter | Shp | ShpM66A | ShpM153A | ShpM66H | ShpM153H |
| k2/k1 or Kd (hemin binding)a (μM) | 22 ± 2 | 22 ± 2 | 62 ± 7 | 19 ± 5 | 58 ± 7 |
| kcoordinationa (s−1) | 35 ± 4 | 145 ± 47 | 490 ± 24 | 36 ± 8 | 48 ± 6 |
| K′hemin≈ kcoordination/Kd, apparent bimolecular rate constant at low [Protein] (μM−1s−1) | 1.6 | 6.6 | 7.9 | 1.9 | 0.82 |
| k-heminb (s−1) | 0.0003 | 0.0003 | 0.21 | ≤0.00001c | 0.005 |
| Khemin ≈k′hemin/k-hemin (μM−1) | 5,300 | 22,000 | 38 | ≥200,000c | 11,600 |
The hemin binding reaction at 25°C in 20 mM Tris-HCl at pH8.0 appears to occur by a two-step process involving an initial hemin binding step followed by first order iron coordination. In this case, values for k2/k1 and kcoordination were obtained from fits of the dependence of the observed rates of transfer on [apoprotein] to a hyperbolic one site-binding model.
The hemin dissociation rate constants from oxidized Shp were determined by the H64Y/V68F apomyoglobin assay (19).
These parameters were estimated as k-hemin less than that for H64Y/V68F metmyoglobin and Khemin greater than 10 times the affinity of H64Y/V68F apomyoglobin for hemin because no loss of hemin was observed, even when the concentration of the hemin scavenger was > 30 times that of ShpM66H
When hemin was mixed with each apoShp mutant, the spectrum of the reaction shifted from that of free hemin to those of the holoShp mutants within 1 second (Fig. 4A and 4B). The time course for each reaction could be described by a single exponential function, and the observed pseudo-first order rate constants were hyperbolically dependent on [apoprotein] (Fig. 4C). The apparent bimolecular rate constant for hemin association was estimated from the slope at low apoprotein concentration, which mathematically is the limiting rate constant divided by the apparent Kd for the initial hemin-apoprotein complex (13). Hemin dissociation from the mutant Shp proteins was measured using H64Y/V68F apomyoglobin, and again, the observed time courses could be described by a single exponential expression with the rate of hemin dissociation from ShpM153A being much larger than that for ShpM66A (Fig. 4D).
FIGURE 4. Hemin association to and dissociation from ShpM66A and ShpM153A.

(A and B) Spectral shifts in the reactions of 1.0 μM hemin with 6.0 μM apoShpM66A and apoShpM153A. The arrows in these panels indicate the direction of the spectral shifts with time. (C) The observed pseudo first order rate constants for hemin binding to apoShpM66A and apoShpM153A as a function of [apoprotein]. The curves are theoretical lines obtained by fitting the results to a hyperbolic one site-binding model. (D) Time courses for hemin dissociation from 3 μM ShpM66A and ShpM153A using 58 μM H64Y/V68F apomyoglobin as a hemin scavenger.
The apparent equilibrium association constants, Khemin, for ShpM66A and ShpM153A were 22,000 and 38 μM−1, respectively, which are 3-fold higher and 138-fold lower than Khemin for wild-type Shp. Surprisingly, both Ala mutants had much greater hemin association rate constants than wild-type Shp, and ShpM153A had a 700-fold higher dissociation rate constant than either wild-type Shp or ShpM66A. Although ShpM153H had a hemin dissociation rate constant similar to that for wild-type Shp, we could detect no hemin dissociation from ShpM66H at excess H64Y/V68F apomyoglobin, indicating that Khemin for ShpM66H is at least 10 times greater than that for the hemin scavenger, i.e. ≥ 200,000 μM−1, and that the dissociation rate constants is ≤ 0.00001s−1 (19).
Kinetics of Hemin Transfer from Shp Mutants to ApoHtsA
The loss and replacement of the Shp axial bonds were expected to have profound effects on the mechanism of its heme transfer to HtsA. Elucidation of these effects should provide insights into the molecular mechanism of the heme transfer. Thus, we performed the spectral and kinetic analyses of the Shp mutant-apoHtsA reactions. When ShpM153A or ShpM66A was mixed with excess apoHtsA, the A600 peak of the Shp mutants decreased rapidly with a concurrent red shift of the Soret peak. These initial changes were followed by a slower additional red shift of the Soret band and an increase in the A530 peak, resulting in the spectrum of the holoHtsA product (Fig. 5A and 5B). The difference between the absorbance at 418 nm and 406 nm (ΔA406–418 = A406 − A418) in these two reactions fits to a two-exponential equation, with easily resolved phases (Fig. 6A). This result is in contrast to the wild-type Shp-apoHtsA reaction, for which the ΔA425–406 time course fits well to a single exponential expression (insert in Fig. 6A). Thus, replacement of either Met66 or Met153 with Ala causes the internal heme transfer in the Shp:apoHtsA complex to become a two-step process.
FIGURE 5. Spectral shifts demonstrating the existence of an intermediate in hemin transfer from ShpM66A or ShpM153A to apoHtsA.
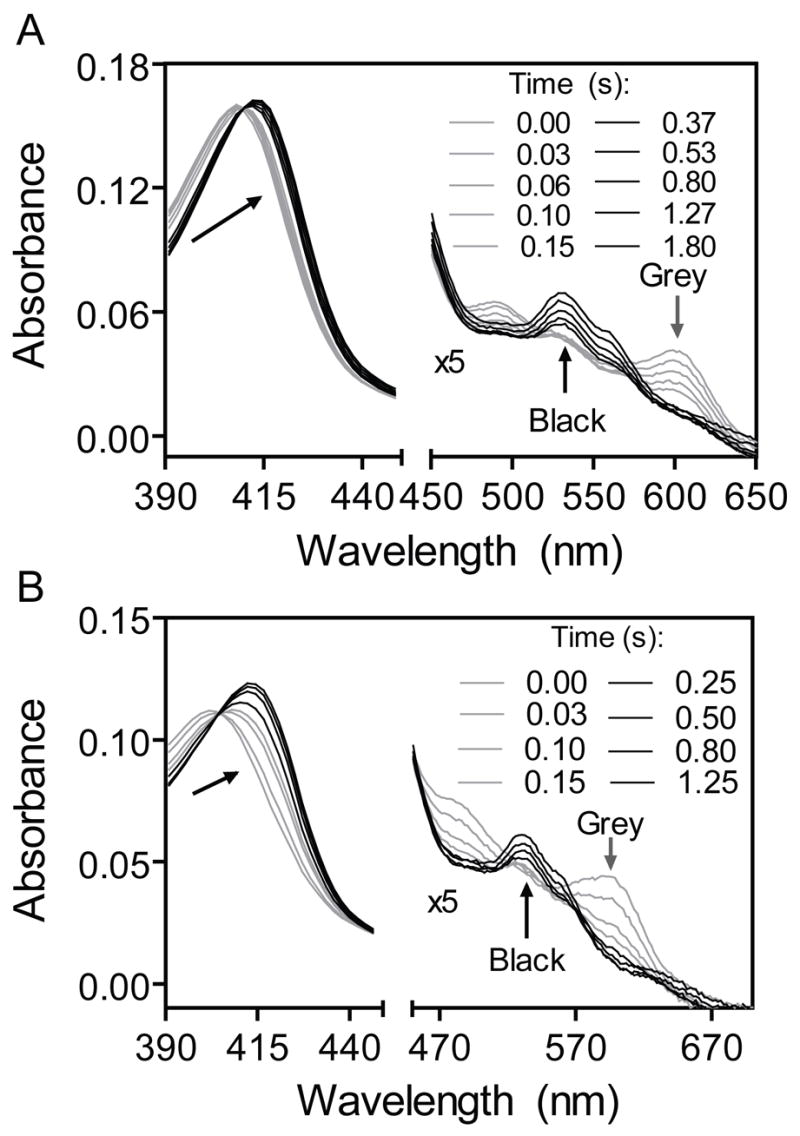
Absorption spectra of 1.3 μM ShpM66A (A) or ShpM153A (B) are presented as a function of time for their reactions with12 μM apoHtsA. The arrows indicate the directions of the spectral shifts.
FIGURE 6. Kinetic analysis of hemin transfer from ShpM66A and ShpM153A to apoHtsA.

(A) Absorption time courses at the indicated wavelengths for the reactions of 1.3 μM ShpM66A, ShpM153A, or wt Shp (insert) with 40 μM apoHtsA. Dotted black curves are the observed data, and the grey solid curves are theoretical lines obtained by fitting the data for the mutant and wt reactions to two-exponential and single exponential equations, respectively. (B) The observed rate constants kt1obs and kt2obs plotted as a function of [apoHtsA] in the ShpM66A-apoHtsA reaction. The rate constants at different [apoHtsA] were obtained from double-exponential fitting as in panel A. (C) The observed rate constants kt1obs and kt2obs as functions of [apoHtsA] in the ShpM153A-apoHtsA reaction.
Fitting the ΔA406–418 time courses for the Shp mutant-apoHtsA reactions to a two-exponential equation results in two observed rate constants designated kt1obs and kt2obs for the rates of the fast and slow spectral changes, respectively. The values of kt1obs depend on [apoHtsA] hyperbolically and almost linearly for the ShpM66A/apoHtsA and ShpM153A/apoHtsA reactions, respectively. In contrast, kt2obs does not change with [apoHtsA] (Fig. 6B and 6C) and represents a simple first order process. These results can be interpreted in terms of the minimal model shown in Scheme II. In this model, ShpM66A or ShpM153A (ShpA) first forms a complex with apoHtsA, and hemin transfer begins with the formation of an intermediate, which we propose to be hexacoordinate ShpA-hemin-HtsA with one ligand from each protein. This true ternary complex then converts into apoShpA and holoHtsA in a simple first order process.
Scheme 2.

k1 and k2 are the rate constants for bimolecular formation and unimolecular dissociation of the initial Shp mutant-apoHtsA complex, respectively, and kt1 and kt2 are the first order rate constants for the formation of the intermediate and the products, respectively. When the initial [apoHtsA] is ≥ 5[ShpA], the time course for ΔA406–418 can be represented by Equation 1,
| (Eq. 1) |
where t is time, and kt1obs is given by Equation 2.
| (Eq. 2) |
Kd equals k2/k1, the dissociation constant of the ShpA-apoHtsA complex. According to this model, kt2obs is directly equal to the rate constant kt2 for the final transfer step to form holoHtsA. Kd, kt1, and kt2 were calculated from the data in Fig. 7B and are 11.4 ± 0.3 μM, 8.7 s−1, and 0.38 ± 0.08 s−1, respectively, for the ShpM66A-apoHtsA reaction and 170 ± 8 μM, 120 s−1, and 2.5 ± 0.2 s−1, respectively, for the ShpM153A-apoHtsA reaction (Table I). Interestingly, the values of Kd, kt1, and kt2 in the ShpM66A reaction are all smaller than those in the ShpM153A reaction. These results show that replacement of Met66 with Ala causes Shp to bind to HtsA more tightly but, at the same time, makes it more difficult for Shp to transfer its hemin to apoHtsA
FIGURE 7. The hemin binding site and its solvent exposure in the crystal structure of Shp180.

(A) A ribbon representation of Shp180 structure showing hemin binding. The backbone, hemin group, iron, and Met axial ligands are blue lines and ribbons, grey sticks, a red ball, and yellow sticks, respectively. (B and C) Space-filling blowups of the hemin binding site, showing the solvent exposure of the porphyrin ring on the Met66 and Met153 sides. The colors of the different components are the same as those in panel A. The representations were derived from the crystal structure of Shp180 (18).
ShpM153H, but not ShpM66H, transfers its hemin to apoHtsA. The transfer process is a single exponential process, indicating that the M153H replacement does not alter the kinetic mechanism from that seen for wild-type Shp. However, the mutation decreases the dissociation equilibrium constant (Kd) for formation of the Shp-apoHtsA complex by 7-fold and decreased the intra-complex transfer rate constant by 12-fold. These results show that bis-Met coordination in Shp is more efficient than His-Met coordination for hemin transfer from Shp to apoHtsA.
DISCUSSION
We have been studying the Shp/HtsA system as a model to understand heme transfer from one protein to another. We previously examined the kinetic mechanism for the Shp-to-HtsA heme and hemin transfer with the wild-type proteins (13). In this study, the extensive spectral and kinetic characterizations of the axial mutants of Shp, in combination with the high resolution structure for Shp180, have further advanced our understanding of the hemin transfer mechanism and helped elucidate molecular details of the reaction. The heme binding domain of Shp contains residues 30–180 and is functionally active. The axial Met153 residue of Shp is critical for its relatively high affinity for hemin, whereas the other axial residue, Met66, destabilizes the hemin binding. Nonetheless, both Met66 and Met153 are critical for rapid hemin transfer. More significantly, kinetic characterization of the Shp mutant-apoHtsA reactions has allowed us to detect intermediates during hemin transfer and propose a novel mechanism of simultaneous attack on both sides of bound hemin in Shp by ligand side chains from apoHtsA.
Truncated Shp180 has the same EPR and UV-visible spectral properties as full-length Shp. Its structure exhibits a immunoglobulin-like-β-sandwich fold and is similar to the structures reported for the S. aureus heme-uptake proteins IsdC and IsdA (22, 23). Shp180 has a well-defined hemin binding site with two Met axial ligands (Fig. 7) and has retained the ability to transfer both hemin and heme to apoHtsA by the activated ternary complex mechanism that is observed for the full-length Shp-apoHtsA reactions.
However, the affinity of Shp180 for apoHtsA is 2-fold less than that of full length Shp and the rate of internal transfer in the Shp180-apoHtsA complex is ~10-fold slower. Thus, although the heme-binding domain is functionally active, the C-terminal region does play a role in enhancing the speed of the heme transfer reaction. An understanding of structural cause of this enhancement will require determination of the structure of the full length protein, which so far we have been unable to crystallize.
ShpM66A and ShpM153A share similar spectral features with apolar distal histidine mutants (H64V, H64L) of human and sperm whale metmyoglobin (20), including a broadened and blue-shifted Soret peak in the 395–405 nm region and a high spin, charge-transfer band in the 600–650 nm region. The multiple derivative peaks in the g=6 region of the EPR spectra of the ferric forms of these Shp mutants are also very similar to those observed for H64V and H64L metmyoglobin (20), which have been shown by crystallography to be water-free, pentacoordinate hemin complexes (21). Thus, oxidized ShpM66A and ShpM153A are almost certainly in pentacoordinate hemin-Met form with only a small portion of water-hemin-Met coordination.
The Met153-Fe coordination bond confers the high affinity of Shp for hemin. Replacement of Met153 with Ala increases the rate of hemin dissociation by 700-fold and decreases the overall affinity of Shp for hemin by two orders of magnitude. The Met153 side of bound hemin in the crystal structure of Shp180 is less exposed to solvent than the Met66 side, which is only partially covered by a three-turn α helix (Fig. 7B and 7C). This interpretation is further supported by the observation that imidazole can coordinate to the heme iron of ShpM66A, but not ShpM153A (data not shown).
Bis-Met coordination in Shp appears to be the result of selective evolutionary pressure to increase the speed of hemin transfer. The affinity of ShpM66A for hemin is 3-fold higher than that of wild-type Shp. Replacement of Met66 with His prevents hemin from being transferred to apoHtsA. The EPR spectrum of ShpM66H is virtually identical to that of aquometmyoglobin and its affinity for hemin is very high, on the order of that of native sperm whale myoglobins (19). Utilizing Met66 as an axial ligand appears to destabilize the bound cofactor and facilitate its transfer. Although oxidized ShpM153H exhibits a spectrum characteristic of a low spin hemichrome, the value of ktransfer for hemin transfer from ShpM153H to apoHtsA is only one twelfth of that in the wild-type Shp/HtsA reaction. Thus, bis-Met coordination in Shp facilitates rapid hemin transfer. Bis-Met coordination has only been found previously in bacterioferritin (24), and its physiological role in this protein is unknown.
Heme and hemin transfer from wild-type Shp to apoHtsA is a concerted process and only a single kinetic phase is observed (13). In contrast, hemin transfer from M66A and M153A Shp to apoHtsA shows two distinct kinetic phases and an intermediate that appears to be a true ternary Shp-hemin-HtsA complex with one axial ligand provided by each protein. Although a change in mechanism of hemin transfer due to loss of an axial ligand was not unexpected, the slowing of the overall exchange process was a surprise.
The distinct spectral intermediates in the reactions of apoHtsA with ShpM66A and ShpM153A are the loss of the 600-nm band, suggesting ligation to form a hexacoordinate hemin complex (25). The simplest interpretation of the intermediate is binding of an axial ligand side chain from apoHtsA to the sixth coordination position of hemin still bound to Shp to form a true ternary complex. A second feature in both mutant reactions is that loss of the A600 peak is the first fast phase. These results are significant because they indicate that both sides of the bound hemin in Shp can be attacked by the axial residues in apoHtsA after forming the initial holoShp-apoHtsA complex. The decay of the intermediate is observed by a further red shift and intensification of the Soret, α, and β bands, resulting in the spectrum of the hexacoordinate holoHtsA product. Thus, we propose that the two axial bonds in the HtsA product are sequentially formed in the transfer reactions of these mutants. The intermediates are Met153-Fe(III)hemin-X and Met66-Fe(III)hemin-Y ternary complexes, where X and Y are the axial ligands donated by HtsA (Fig. 8A and 8B). Preliminary mutagenesis results suggest that X and Y in HtsA are Met79 and His229 (Lei et al., unpublished data)
FIGURE 8. Proposed schemes for the reactions of the wild-type and mutant Shp proteins with apoHtsA.

M66 and M153 are the axial ligands in Shp, X and Y the axial ligands, Met79 and His229, in HtsA, and the short lines to hemin represent the axial bonds. The two axial bonds of the HtsA product are formed at about same time in the wild-type Shp reaction but are sequentially formed in the M66A and M153A mutant reactions with a Shp-hemin-HtsA ternary complex intermediate with one ligand from each protein.
The features of the mutant Shp reactions have led us to propose a “plug-in” mechanism for the concerted one step, internal hemin transfer process carried out by wild-type holoShp. There are two possible interpretations of the one step transfer reaction: 1) the displacement of the two Shp Met ligands is truly simultaneous or 2) the displacement of the first Shp Met ligand is rate-limiting. In both interpretations, the two new coordination bonds are formed at effectively the same time (Fig. 8C) and would give equivalent kinetic results.
The two axial ligands of apoHtsA must be close to the two axial positions of hemin in the holoShp-apoHtsA binary complex. This idea implies that either the empty heme pocket in apoHtsA pre-exists or is induced quickly after the binary complex is formed. This view also requires that the HtsA axial side chains easily slide into the exposed Shp heme binding site to “pull or pry” the cofactor out of Shp. Thus, our initial interpretation can be called a “plug-in” mechanism for transferring the hemin actively from one protein to another. The bound hemin in Shp180 has significant exposure to solvent on both Met153 and Met66 sides (Fig. 7). Thus, simultaneous ligand displacement by adjacent apoHtsA ligands in a Shp-HtsA complex is structurally feasible by sliding movements of the amino acid side chains across both sides of the hemin plane.
Both ShpM66A and ShpM153A appear to have no ligand or partial water coordination at the mutated axial positions. This open pentacoordinate geometry appears to facilitate insertion of the first apoHtsA ligand to generate a discrete and kinetically stable intermediate. The intermediates for ShpM153A and ShpM66A decay to holoHtsA at rates of 2 and 0.4 s−1, respectively, which are ~20 to 100-fold slower than ktransfer for full length Shp. The underlying cause for the appearance of the intermediate and the slowing of the second ligand displacement reaction is unclear. One possible reason is that the first displaced Met in the wild-type Shp reaction sterically clashes with the ApoHtsA axial ligand, destabilizing the ternary complex intermediate but at the same time facilitating transfer by “pushing” the hemin into the binding pocket of apoHtsA (Fig. 8C). The association rate constants of hemin binding to apoShpM66A and apoShpM153A are 4 and 14 times greater, respectively, than that for hemin binding to wild-type apoShp. This result suggests that the heme pocket in wild-type Shp is more sterically restricted and perhaps designed to help eject non-coordinated heme from its active site once the HtsA ligands have been inserted and to destabilize ternary complexes that slow the net rate of transfer. The Met-to-Ala replacements appear to relieve this steric pressure. In addition, the lack of a sixth ligand or weaker coordination with water should strengthen the remaining Fe(III)-Met bond, making its displacement more difficult and probably requiring the binding of the first HtsA ligand to the unoccupied coordination site. This latter phenomenon appears to occur for the ShpM66A/apoHtsA reaction.
The findings in this work suggest a unique “plug-in” mechanism for hemin transfer from Shp to HtsA. The key features are insertion of the axial ligands of apoHtsA on both sides of the partially exposed hemin in Shp and subsequent, simultaneous displacement of the two axial bonds to pull the hemin from Shp to HtsA. Although the mechanism needs to be verified, these ideas may serve as an initial frame work for examining a variety of other clinically relevant heme transfer processes in pathogenic bacteria, including the HasA/HasR and hemoglobin/ShuA systems in which hemin transfer has been biochemically demonstrated and the S. aureus Isd system, in which hemin transfer remains undocumented.
Acknowledgments
This work was supported by grants AI057347 (BL), GM27659 (DMD), HL47020 (JSO), GM35649 (JSO), GM55807 (MF), and GM08349 (RA) from the National Institutes of Health, P20 RR-020185 (BL) from the National Center for Research Resources, C-0612 (JSO) from the Robert A. Welch Foundation, and US Department of Agriculture National Research Initiative/Competitive Grants Programs grant 2004-35204-14637 and Formula Funds, and the Montana State University Agricultural Experimental Station. We thank Tyler Nygaard and Maki Fukumura for technical support.
Footnotes
The abbreviations used are: Heme, Fe(II)-protoporphyrin IX complex; hemin, Fe(III)-protoporphyrin IX complex; HtsA, lipoprotein component of heme-specific ABC transporter HtsABC of Streptococcus pyogenes; apoHtsA, metalloporphyrin-free HtsA; holoHtsA, HtsA with bound heme or hemin; Shp, streptococcal heme-binding protein; apoShp, metalloporphyrin-free Shp; holoShp, Shp with bound heme or hemin; ShpM66A and ShpM153A, the alanine replacement mutants of the methionine residues at positions 66 and 153 of Shp, respectively; Shp180, the heme-binding domain of Shp containing amino acids 30-180.
“The atomic coordinates for the crystal structure of Shp180 are available in the Protein Data Bank (http://www.rcsb.org/pdb/home) under pdb2Q7A (18).”
References
- 1.Stojiljkovic I, Hantke K. Mol Microbiol. 1994;13:719–732. doi: 10.1111/j.1365-2958.1994.tb00465.x. [DOI] [PubMed] [Google Scholar]
- 2.Drazek ES, Hammack CA, Schmitt MP. Mol Microbiol. 2000;36:68–84. doi: 10.1046/j.1365-2958.2000.01818.x. [DOI] [PubMed] [Google Scholar]
- 3.Lei B, Liu M, Voyich JM, Prater CI, Kala SV, DeLeo FR, Musser JM. Infect Immun. 2003;71:5962–5969. doi: 10.1128/IAI.71.10.5962-5969.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Griffiths E, Williams P. In: Iron and infection: molecular, physiological and clinical aspects. Bullen JJ, Griffiths E, editors. John Wiley & Sons, Inc.; Chichester, United Kingdom: 1999. pp. 87–212. [Google Scholar]
- 5.Burkhard KA, Wilks A. J Biol Chem. 2007;282:15126–15136. doi: 10.1074/jbc.M611121200. [DOI] [PubMed] [Google Scholar]
- 6.Izadi-Pruneyre N, Huche F, Lukat-Rodgers GS, Lecroisey A, Gilli R, Rodgers KR, Wandersman C, Delepelaire P. J Biol Chem. 2006;281:25541–25550. doi: 10.1074/jbc.M603698200. [DOI] [PubMed] [Google Scholar]
- 7.Lei B, Smoot LM, Menning H, Voyich JM, Kala SV, Deleo FR, Musser JM. Infect Immun. 2002;70:4494–4500. doi: 10.1128/IAI.70.8.4494-4500.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mazmanian SK, Skaar EP, Gaspar AH, Humayun M, Gornicki P, Jelenska J, Joachmiak A, Missiakas DM, Schneewind O. Science. 2003;299:906–909. doi: 10.1126/science.1081147. [DOI] [PubMed] [Google Scholar]
- 9.Torres VJ, Pishchany G, Humayun M, Schneewind O, Skaar EP. J Bacteriol. 2006;188:8421–8429. doi: 10.1128/JB.01335-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Letoffe S, Nato F, Goldberg ME, Wandersman C. Mol Microbiol. 1999;33:546–555. doi: 10.1046/j.1365-2958.1999.01499.x. [DOI] [PubMed] [Google Scholar]
- 11.Liu M, Lei B. Infect Immun. 2005;73:5086–5092. doi: 10.1128/IAI.73.8.5086-5092.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nygaard TK, Liu M, McClure MJ, Lei B. BMC Microbiol. 2006;6:82. doi: 10.1186/1471-2180-6-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nygaard TK, Blouin GC, Liu M, Fukumura M, Olson JS, Fabian M, Dooley DM, Lei B. J Biol Chem. 2006;281:20761–20771. doi: 10.1074/jbc.M601832200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moore GR, Pettigrew GW. Cytochrome c: Evolutionary, structural and physiochemical aspects. Springer-Verlag; Berlin: 1990. [Google Scholar]
- 15.Eaton WA, Hofrichter J. Meth Enzymol. 1981;76:175–261. doi: 10.1016/0076-6879(81)76126-3. [DOI] [PubMed] [Google Scholar]
- 16.Deniau C, Gilli R, Izadi-Pruneyre N, Letoffe S, Delepierre M, Wandersman C, Briand C, Lecroisey A. Biochemistry. 2003;42:10627–10633. doi: 10.1021/bi030015k. [DOI] [PubMed] [Google Scholar]
- 17.Liu X, Olczak T, Guo H-C, Dixon DW, Genco CA. Infect Immun. 2006;74:1222–1232. doi: 10.1128/IAI.74.2.1222-1232.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aranda R, IV, Worley CE, Liu M, Bitto E, Cates MS, Olson JS, Lei B, Phillips GN., Jr The Protein Data Bank. 2007 doi: 10.2210/pdb2Q7A/pdb. www.rcsb.org/pdb/home. [DOI] [PMC free article] [PubMed]
- 19.Hargrove MS, Singleton EW, Quillin ML, Ortiz LA, Phillips GN, Jr, Olson JS, Mathews AJ. J Biol Chem. 1994;269:4207–4214. doi: 10.2210/pdb1mgn/pdb. [DOI] [PubMed] [Google Scholar]
- 20.Ikeda-Saito M, Hori H, Andersson LA, Prince RC, Pickering IJ, George GN, Sanders CR, 2nd, Lutz RS, McKelvey EJ, Mattera R. J Biol Chem. 1992;267:22843–22852. [PubMed] [Google Scholar]
- 21.Quillin ML, Arduini RM, Olson JS, Phillips GN., Jr J Mol Biol. 1993;234:140–155. doi: 10.1006/jmbi.1993.1569. [DOI] [PubMed] [Google Scholar]
- 22.Sharp KH, Schneider S, Cockayne A, Paoli M. J Biol Chem. 2007;282:10625–10631. doi: 10.1074/jbc.M700234200. [DOI] [PubMed] [Google Scholar]
- 23.Grigg JC, Vermeiren CL, Heinrichs DE, Murphy ME. Mol Microbiol. 2007;63:139–149. doi: 10.1111/j.1365-2958.2006.05502.x. [DOI] [PubMed] [Google Scholar]
- 24.Andrews SC, Le Brun NE, Barynin V, Thomson AJ, Moore GR, Guest JR, Harrison PM. J Biol Chem. 1995;270:23268–23274. doi: 10.1074/jbc.270.40.23268. [DOI] [PubMed] [Google Scholar]
- 25.Moreira LM, Poli AL, Costa-Filho AJ, Imasato H. Biophys Chem. 2006;124:62–72. doi: 10.1016/j.bpc.2006.05.030. [DOI] [PubMed] [Google Scholar]