

Abstract
Embryonic knockdown of candidate dyslexia susceptibility gene (CDSG) homologs in cerebral cortical progenitor cells in the rat results in acute disturbances of neocortical migration. In the current report we investigated the effects of embryonic knockdown and overexpression of the homolog of DCDC2, one of the CDSGs, on the postnatal organization of the cerebral cortex. Using a within-litter design, we transfected cells in rat embryo neocortical ventricular zone around E15 with either 1) small hairpin RNA (shRNA) vectors targeting Dcdc2, 2) a DCDC2 overexpression construct, 3) Dcdc2 shRNA along with DCDC2 overexpression construct, 4) an overexpression construct comprised of the C Terminal domain of DCDC2, or 5) an overexpression construct comprised of the DCX Terminal domain of DCDC2. RNAi of Dcdc2 resulted in pockets of heterotopic neurons in the periventricular region. Approximately 25% of the transfected brains had hippocampal pyramidal cell migration anomalies. Dcdc2 shRNA-transfected neurons migrated in a bimodal pattern, with approximately 7% of the neurons migrating a short distance from the ventricular zone, and another 30% migrating past their expected lamina. Rats transfected with Dcdc2 shRNA along with the DCDC2 overexpression construct rescued the periventricular heterotopia phenotype, but did not affect the percentage of transfected neurons that migrate past their expected laminar location. There were no malformations associated with any of the overexpression constructs, nor was there a significant laminar disruption of migration. These results support the claim that knockdown of Dcdc2 expression results in neuronal migration disorders similar to those seen in the brains of dyslexics.
Keywords: neuronal migration, heterotopias, RNAi, malformation, cerebral cortex
Linkage analysis has revealed a number of gene intervals conferring susceptibility to developmental dyslexia—a language-based learning disability affecting 4–10% of the population. Recently, candidate dyslexia susceptibility genes (CDSGs) have been proposed at some of these intervals, including MRPL19 and C2ORF3 on Chr 2 (Anthoni et al., 2007), ROBO1 on Chr 3 (Hannula-Jouppi et al., 2005), DCDC2 and KIAA0319 on Chr 6 (Francks et al., 2004, Cope et al., 2005, Meng et al., 2005, Paracchini et al., 2006, Schumacher et al., 2006), DYX1C1 on Chr 15 (Taipale et al., 2003, Brkanac et al., 2007, Marino et al., 2007) but see also (Scerri et al., 2004, Bellini et al., 2005, Marino et al., 2005). We have previously demonstrated that embryonic knockdown of Dcdc2, Kiaa0319, or Dyx1c1 function in the rat disrupts the process of neuronal migration to the cerebral cortex, as assessed during the prenatal period (Meng et al., 2005, Paracchini et al., 2006, Wang et al., 2006). Recent evaluation of the postnatal consequences of embryonic knockdown of Dyx1c1 function in rats revealed the presence of a variety of neocortical malformations, including molecular layer ectopias and periventricular heterotopias (Rosen et al., 2007).
The potential role of these genes in neuronal migration is intriguing when considered in light of previous evidence demonstrating the presence of neuronal migration disorders in the brains of dyslexics (Galaburda and Kemper, 1979, Galaburda et al., 1985, Humphreys et al., 1990), which consist of nests of neurons and glia in the molecular layer (ectopias), intracortical laminar dysplasias, and occasional instances of focal microgyria. In addition, there is a reported increased incidence of developmental dyslexia in patients with periventricular nodular heterotopias (Chang et al., 2005, Sokol et al., 2006). Taken together, these results suggest that disruption of the function of any of these CDSGs may underlie the anatomic phenotype of this disorder.
Of the CDSGs currently reported, the strongest support has been given for DCDC2 and KIAA0319 on Chr 6 (Francks et al., 2004, Cope et al., 2005, Meng et al., 2005, Harold et al., 2006, Paracchini et al., 2006, Schumacher et al., 2006). In the original report identifying DCDC2 as a candidate dyslexia susceptibility gene, in utero electroporation of small hairpin RNA (shRNA) targeted against the rat homolog of this gene was shown to disrupt neuronal migration when assessed 4–7 days after transfection (Meng et al., 2005). What is not known is the postnatal phenotype of this embryonic knockdown. Here we examine the brains of postnatal rats where Dcdc2 was either knocked down (using shRNA) or overexpressed during cerebral cortical development. We find that knockdown, but not overexpression, of Dcdc2 results in neocortical malformations in the cerebral cortex.
EXPERIMENTAL PROCEDURES
In Situ Hybridization
In order to better interpret the knockdown and overexpression findings, we first determined the expression of Dcdc2 in the prenatal brain by in situ hybridization. We obtained time-mated pregnant females (Charles River Laboratory, Wilmington, MA) and sacrificed the litters on E15, E17 or E19. Three embryos from each litter were immediately frozen and they were cut in either the horizontal, sagittal, or coronnal plane on a cryostat at 18 μm, and the slides were processed for in situ hybridization of Dcdc2 as described below.
The cDNA prepared from frontal, parietal, and occipital lobes of human embryonic brain (20 weeks, Biochain Institute, Hayward, CA) was amplified with respective forward (ATGAGCGGCAGCAGCGCCAGG) and reverse primers (CTAAGCCACGGCAGCATAGTCC) for 35 cycles. All fragments were then cloned into t vector (Invitrogen, Carlsbad, CA) and sequenced to verify Dcdc2 amplification. Rat embryonic and postnatal brain cDNAs were synthesized from total RNA and amplified with the primers (Forward = ATGAACGGTCCCAGCCCCAGG; Reverse = CTATGCCACAGCAGAAGAGGCTT) to rat Dcdc2. The amplified DNA was gel-purified, cloned, and sequence verified to be Dcdc2. Nonradioactive in situ hybridizations were done by UB-In Situ (Natick, MA), as previously described (Berger and Hediger 2001), using a digoxigenin-labeled cRNA probe. The antisense and sense probes were obtained from the polymerase chain reaction (PCR) products, amplified from rat E14 brain cDNA, and cloned in pGEMT-Easy flanking T7 and SP6 promoters. Two probes, one from the first 400 bp generated from PCR primer pairs and the second full-length cDNA yielded similar results.
In Utero Electroporation
In utero electroporation was performed at the University of Connecticut and all procedures were approved by the Institutional Animal Care and Use Committee of that institution.
Five pregnant Wistar rats were obtained (Charles River Laboratory) and each litter was assigned to one of five conditions: Dcdc2 shRNA, Rescue, DCDC2 Overexpression, DCDC2-DCX Domain Overexpression, or DCDC2-C Terminal Domain Overexpression (see Table 1). Within each litter, pups were randomly assigned to receive one of two treatments: Treatment 1 was the “experimental” construct (shRNA or Overexpression) and treatment 2 was a “control” construct (scrambled shRNA, Rescue, or fluorescent protein only). This design was essential for the analysis of migrational distance as it controlled for between-litter variation in gestational age. In utero electroporation of plasmid DNA was performed at E15 as described previously (Bai et al., 2003, Rosen et al., 2007). Concentration of GFP and RFP plasmids were 0.5 μg/μL, the shRNA was 1.5 μg/μL, and overexpression plasmids were 2.0 μg/μL.
Table 1.
Summary of litter treatments (N used for migration analysis/Total N).
| Litter | Group | Treatment 1 | Treatment 2 |
|---|---|---|---|
| 1 | Dcdc2 shRNA | pU6shRNA-Dcdc2A +
pCAGGS-GFP (5/5) |
pU6shRNA-Dcdc2 scram +
pCAGGS-mRFP + pCAGGS-GFP (2/2) |
| 2 | Rescue | pU6shRNA-Dcdc2A +
pCAGGS-GFP (3/3) |
pU6shRNA-Dcdc2A +
PCAGGS-DCDC2-GFP + pCAGGS-mRFP (3/4) |
| 3 | DCDC2 Overexpression | pCAGGS-DCDC2-GFP +
pCAGGS-mRFP (4/5) |
pCAGGS-GFP
(2/2) |
| 4 | DCDC2 DCX Domain Overexpression | pCAGGS-DCDC2-DCX-GFP +
pCAGGS-mRFP (3/3) |
pCAGGS-GFP
(5/5) |
| 5 | DCDC2 C Terminal Domain Overexpression | pCAGGS-DCDC2-CTerm-GFP +
pCAGGS-mRFP (3/5) |
pCAGGS-GFP
(4/4) |
Plasmids
For the Dcdc2 shRNA condition, plasmids encoding shRNA (pU6shRNA-Dcdc2A) and plasmids encoding enhanced green fluorescent protein (GFP) (pCAGGS-GFP) were co-transfected into the ventricular zone (VZ). Littermates were co-transfected with plasmids encoding a scrambled version of the shRNA fused with GFP (pU6shRNA-Dcdc2 scram along with plasmids encoding monomeric red fluorescent protein (RFP) (pCAGGS-RFP) and plasmid encoding GFP. In the Rescue condition, subjects were co-transfected with pU6shRNA-Dcdc2A, a fusion construct coding for the human DCDC2 protein with GFP (pCAGGS-DCDC2-GFP), and pCAGGS-RFP. Littermates were transfected with pCAGGS-GFP. Pups in the DCDC2 overexpression group were co-transfected with pCAGGS-DCDC2-GFP and pCAGGS-RFP, and their littermates with pCAGGS-GFP. The DCDC2 DCX Domain overexpression group was transfected with pCAGGS-DCDC2 DCX domain-GFP plasmids and pCAGGS-RFP, and the remaining pups in the litter received pCAGGS-GFP. In the DCDC2 C-Terminal Domain overexpression group, pups were co-transfected with pCAGGS-DCDC2 C-Terminal-GFP pCAGGS-RFP, and paired littermates were transfected with pCACGS-GFP. Previous research indicated that co-transfection is highly efficient (Rosen et al., 2007).
Histology
At P21, animals were deeply anesthetized (Ketamine/Xylazine 10:1, 100 mg/mL) and sacrificed by transcardial perfusion with 0.9% saline followed by 4% paraformaldehyde. The brains were removed from the skull and were coronally sectioned at 80 μm on a vibratome. Sections were then mounted and coverslipped with VECTASHIELD Mounting Medium (Vector Labs, CA) and visualized under fluorescence for the presence of GFP and/or RFP. One series of every tenth section was stained for Nissl substance using Thionin. One adjacent series of free-floating sections was processed for immunohistochemical detection of GFP (Chemicon, 1:200) using ABC protocols. Adjacent series in some brains were processed for immunofluorescence detection of Cux-1 (CDP (M-222), Santa Cruz Biotechnology, CA, 1:1000) —a transcription factor that labels supragranular neurons in the cerebral cortex (Nieto et al., 2004)—with Alexa Fluor 594 secondary antibody (Invitrogen, CA, 1:200).
Analysis
In situ quantification
Individual sense and antisense sections from horizontally prepared brains (1 each from E15, E17, and E19 rats) were imaged with monochrome digital camera (Insight, Diagnostic Instruments, Sterling Heights, MI) on a light box (Aristo Grid Lamp Products, Waterbury, CT) and interfaced via firewire to Macintosh G4 computer (Apple Computer, Cupertino, CA). Each antisense section image and its corresponding sense section were captured using SPOT software (Diagnostic Instruments) with common exposure settings. Using ImageJ <http://rsb.info.nih.gov/ij/>, optical density values were measured for the combined cortical plate and ventricular zone. A total of 9–13 sections were measured for each brain. The average difference in optical density between sense and antisense images were computed and expressed as a percent of sense density.
Postnatal Analysis
All analyses of postnatal brains were performed blind with respect to condition. Nissl-stained sections were surveyed for the presence of neocortical and/or hippocampal malformations, and the location noted. Quantitative analysis of migrational distance was conducted as previously described (Rosen et al., 2007). Briefly, the location of immunohistochemically labeled cells was charted in the immunohistochemically stained series using Neurolucida (MBF Biosciences, Williston, VT). These were then imported into Canvas X (ACD Systems, Miami, FL), and a counting rectangle subdivided into 10 equal-size bins was scaled to extend from the white matter to the pial surface. The number of labeled cells within each decile was manually counted and recorded. For each brain, the rectangle was arbitrarily placed in the transfected hemisphere from the 4–8 sections. The percentage of labeled cells in each decile was determined for each animal, and the mean value across all animals within each condition was determined. Differences in the distribution of migrated neurons across deciles were assessed using repeated measures analysis of variance.
Image Processing
Fluorescent images were obtained on a confocal microscope (Zeiss LSM 510 Meta, Carl Zeiss, Inc., Thornwood, NY). Photomicrographs were adjusted for exposure and sharpened (unsharp mask filter) using Adobe Photoshop (Adobe Inc., San Jose, CA). Some images were acquired using the Virtual Slice Module of Neurolucida. Image montages were created in Canvas X.
RESULTS
Dcdc2 is ubiquitously expressed at a modest level in the forebrain
In situ hybridization of Dcdc2 in embryonic rat embryos revealed relatively ubiquitous expression in the forebrain during development (Fig. 1A–F). Overall, expression is modest throughout the brain. Quantification of expression in the combined cortical plate and ventricular zone demonstrated an approximately 2 fold increase in optical density in the antisense sections at E15. In comparison, there was an approximately 0.6–fold increase in optical density at E17 and E 19. These results are mostly compatible with those of Reiner et al (2006), who found limited expression of Dcdc2A in the brains of mice at E14.5.
Figure 1.

In situ hybridization of Dcdc2 in embryonic rat brains. Photomontages of in situ hybridization of Dcdc2 antisense and sense probes in E15 (A,B), E17 (C,D), and E19 (E,F) rat embryos. The expression of Dcdc2 is relatively ubiquitous with a modest increase in E19 in the ventricular zone, striatum, and cortical plate. Bar in all panels = 1 mm.
Periventricular heterotopias are present only in Dcdc2 shRNA transfected subjects
For the purposes of qualitative analysis, the Dcdc2 shRNA treated rats from litters 1 and 2 were combined. Examination of the Nissl-stained sections revealed what we term periventricular heterotopias (PVH), which reflects both nodular heterotopias and heterotopias in the white matter, in 7 of 8 Dcdc2 shRNA-treated rats (Fig 2A, B, E, ,F). PVH consisted of large collections of neurons within the white matter, or at the cortical/white matter border that were visible on Nissl- and immunohistochemically-stained sections.
Figure 2.
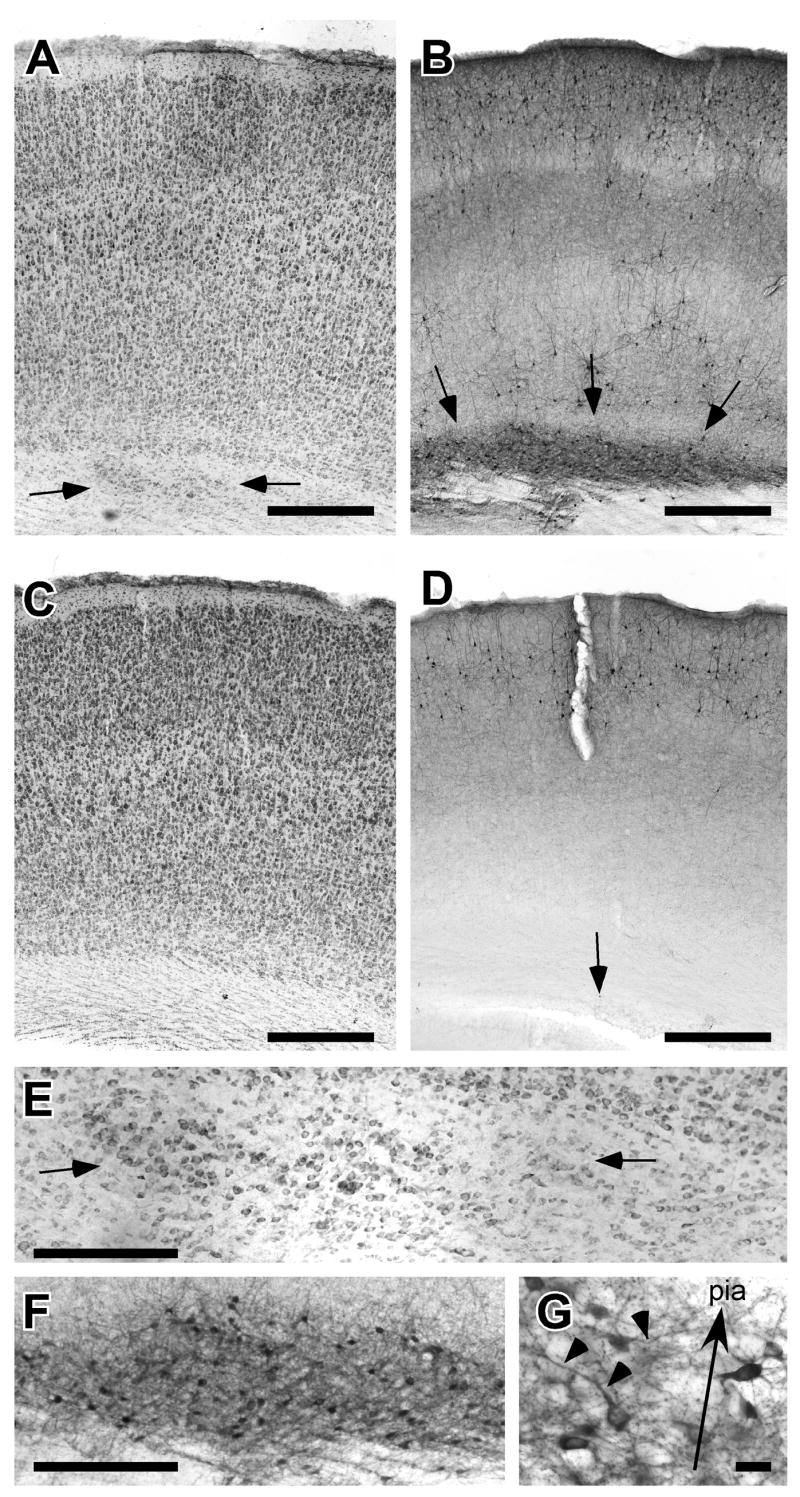
Periventricular heterotopias (PVH) in a rat embryonically transfected with shRNA targeted against Dcdc2. A. Photomicrograph of cerebral cortex of Nissl-stained section illustrating region of PVH (arrows). This animal was embryonically transfected with Dcdc2 shRNA + GFP. B. Photomicrograph of section adjacent to Panel A immunohistochemically stained for GFP. Transfected neurons are located within the PVH. C. Photomicrograph of cerebral cortex of Nissl-stained section of a rat from the “rescue” condition. This animal was embryonically cotransfected with Dcdc2 shRNA + GFP along with a human DCDC2 protein overexpression plasmid, and shows no evidence of PVH. D. Photomicrograph of section adjacent to Panel C immunohistochemically stained for GFP. There is a solitary transfected neuron in Layer 6 (arrow), but no evidence of PVH. Bar for A–D = 500 μm. E. High power photomicrograph of PVH illustrated in panel A (arrows). F. High power photomicrograph of GFP-positive neurons in PVH. In comparison with Panel E, note that not all neurons in the PVH are transfected. Bar for E, F = 100 μm. G. High power photomicrograph of box in panel F. Note that transfected neurons in the PVH have neuronal morphology, but are misoriented. Arrow indicates direction of pial surface, arrowheads denote misoriented apical dentrites. Bar = 25 μm.
In order to determine the specificity of the RNAi for Dcdc2 we performed experiments in which the Dcdc2 shRNA was co-transfected with a plasmid encoding human DCDC2 (pCAGGS-DCDC2-GFP). Human DCDC2 nucleotide sequence does not match rat Dcdc2 sequence in the region targeted by the Dcdc2 shRNA and therefore is not susceptible to RNAi. Of the rats simultaneously transfected with Dcdc2 shRNA and the human DCDC2 overexpression construct, 1 out of 4 had a small (<10) collection of hetertopic neurons clustered together on one section (3 of these neurons were GFP-positive). The remainder had no obvious malformations (Fig 2 C, D). This suggests that overexpressing the human DCDC2 protein in Dcdc2 shRNA-treated rats rescued the PVH phenotype, and indicates that the effects of the RNAi are not due to off target effects. None of the rats in other treatment groups had PVH.
In the Dcdc2 shRNA condition, only a subset of the neurons in the PVH were immunopositive for GFP, indicating that there were large numbers of ectopic neurons that had not been transfected with Dcdc2 shRNA. Because the efficiency of co-transfection is nearly 100% (Rosen et al., 2007), this suggests that there are non cell-autonomous effects of Dcdc2 shRNA transfection. Those cells that were immunopositive for GFP were clearly neuronal in morphology, but their normal radial orientation was disturbed (Fig. 2E, F, G).
Immunostaining for Cux-1, a transcription factor uniquely expressed in supragranular layers (Nieto et al., 2004), revealed large numbers of Cux-1 positive neurons in the PVH (Fig 3). Of the shRNA-transfected neurons throughout the cerebral cortex, most were Cux-1 positive. The presence of numerous non-transfected Cux-1 positive neurons in the PVH again supports the contention that there are non-cell autonomous effects of Dcdc2 shRNA transfection.
Figure 3.

Confocal microscopy of the laminar specific transcription factor Cux-1 in the brain of rats embryonically transfected with shRNA targeted against Dcdc2. Top row illustrates high density of Cux-1 immunoreactive neurons in layer 2/3 as expected. Neurons transfected with Dcdc2 shRNA + GFP are co-labeled with Cux-1 (arrows). Bar = 25 μm. Middle row illustrates a single neuron in layer 5 that was transfected with Dcdc2 shRNA + GFP and is co-labeled with Cux-1. Bar = 25 μm. Bottom row is large PVH that contains numerous Cux-1 positive neurons (arrows and arrowheads), only a small subset of which are co-labeled with GFP (arrows). Bar = 50 μm.
Malformations of the Hippocampus
There were hippocampal malformations in 2 of 8 Dcdc2 shRNA treated rats (see Fig. 4A, B). These malformations consist of cells originally from the pyramidal layer localized in the stratum radiatum and stratum oriens (see Fig. 4A,B), and were not associated with the injection site. Comparison between immunostained and adjacent Thionin-stained sections revealed that only a small percentage of the ectopic neurons stained positively for GFP (Fig. 4A′). As with the PVH discussed above, this suggested non-cell autonomous effects in the hippocampus of Dcdc2 shRNA transfection. Of the remaining rats in the experiment, there were 3 (2 GFP controls and 1 Dcdc2 DCX domain overexpression group) that exhibited more modest malformations of the hippocampus (<5 mismigrated neurons), but they were associated with the injection site and were likely artifactual.
Figure 4.

Hippocampal malformations in rats embryonically transfected with shRNA targeted against Dcdc2. A. Photomicrograph of Nissl-stained section of rat embryonically transfected with Dcdc2 shRNA + GFP. There is a periventricular heterotopia (white arrow) as well as a malformation of the hippocampus (arrows). Bar = 250 μm. A′ High power photomicrograph of hippocampal malformation. Arrows are for orientation with A. Bar = 100 μm. B and B′. Section adjacent to A and A′ immunohistochemically stained for GFP. Note that only a small subset of neurons in the malformation are GFP-positive. Bar for B = 250 μm, B′ = 100 μm.
Dcdc2 shRNA-transfected neurons migrate in a bimodal pattern
Migration distance analysis was performed within each group using a repeated measure ANOVA with Treatment (1 vs. 2) as the between, Deciles as the within, and the percent of neurons within each decile as the dependent measure. In Group 1 (Fig. 5A,B), there was a significant Treatment × Decile interaction (F9.45 = 3.9, P < .05), indicating that the laminar dispersion of Dcdc2 shRNA-transfected neurons significantly differed from those of neurons transfected with the mutant form of Dcdc2. Further analysis revealed that there were significantly more Dcdc2 shRNA-transfected neurons in the lower four deciles (F3,15 = 4.9, P < .05) and a significantly greater number of these neurons that migrated past the expected lamina (F9.45 = 3.9, P < .05).
Figure 5.

Patterns of neuronal migration to the cerebral cortex in each of the five experimental groups. First column contains plots of GFP-positive neurons in the cerebral cortex of rats exposed to Treatment 1 (the “experimental” condition). The second column contains plots from Treatment 2 (“control” condition). The last column contains histograms representing the percent of neurons contained within each of the deciles ranging from the white matter to the pial surface. Analysis reveals significant differences in the pattern of neuronal migration only between A and B, with there being significantly greater number of neurons in both the lower and upper deciles in the Dcdc2 shRNA group as compared to the control condition.
In Group 2, the Dcdc2 shRNA + pCAGGS-DCDC2-GFP condition, there was no significant difference in the distribution of transfected neurons between the treatments (F6,30 < 1, NS). This suggested that the treatment of shRNA-transfected neurons with the DCDC2 overexpression construct failed to rescue this migration phenotype. Although there appeared to be a mild arrest of migration in animals transfected with either the DCDC2 overexpression construct (Fig 5E,F) or the C terminal domain (Fig 5I,J), there were no significant differences in laminar dispersion of the transfected neurons between Groups 3, 4, or 5 (F9.36 = 1.3, NS; F9.54 < 1, NS; F9.44 = 1.9, NS; respectively)(Fig 5E–J).
DISCUSSION
Embryonic knockdown of Dcdc2 function is associated with neocortical malformations
Previous work showed that the embryonic knockdown of Dcdc2 function in neocortical progenitor cells by transfection with shRNA resulted in a disturbance in neuronal migration when assessed 4–7 days post transfection. Here, we demonstrate that this disruption of neuronal migration results in malformations that can be seen in the postnatal cerebral cortex. Specifically, we found periventricular heterotopias in the brains of animals transfected with shRNA targeted against Dcdc2. These heterotopias, which are apparent in Nissl-stained sections, are located at the neocortical/white matter border, and are composed of both transfected and non-transfected neurons. Moreover, large numbers of these non-transfected neurons were Cux-1 positive, suggesting that they were neurons destined for supragranular layers whose migration was halted by non-cell autonomous effects of Dcdc2 shRNA treatment. Co-transfection of Dcdc2 shRNA with the DCDC2 overexpression construct mostly rescued this phenotype, as there was only one small collection of heterotopic neurons noted in this group. There were no PVH in any of the rats from the three overexpression conditions. Taken together, this supports the claim that PVH are the result of the knockdown of Dcdc2 function.
Dcdc2 and neuronal migration
The specific function of Dcdc2 in neuronal migration has yet to be elucidated, but analysis of its protein structure provides some clues. DCDC2 is one of an eleven-member group of proteins distinguished by the presence of tandem or single dcx domains. DCX, the first gene of this family to be characterized, was identified after the discovery of mutations in a gene that caused double cortex syndrome and lissencephaly in humans (Allen et al., 1998, des Portes et al., 1998). The dcx domain appears critical for binding to and stabilizing microtubules and is regulated by phosphorylation (Gleeson et al., 1999, Graham et al., 2004, LoTurco, 2004, Reiner et al., 2004, Schaar et al., 2004). More recently another member of the DCX family, Dclk, has been shown to genetically interact with Dcx in mice, and functional knockdown of either of these genes results in the interruption of normal neuronal migration in neocortex. Interestingly, two functioning copies of Dcx and Dclk appear to be necessary both for growth of axons across the corpus callosum as well as for neuronal migration in cerebral cortex (Deuel et al., 2006, Koizumi et al., 2006, Friocourt et al., 2007). A comparison of the biochemical and cellular functions of proteins in the Dcx family found that Dcdc2 exhibits the same functional features shown by Dclk and Dcx (Coquelle et al., 2006), and analysis suggests that DCX, DCDC2, and DCLK are the most conserved genes in this superfamily (Reiner et al., 2006).
RNAi of DCX and Dcdc2 leads to qualitatively different impairments in migration, however, indicating that their roles in migration are distinct. Whereas RNAi of DCX leads to large continuous subcortical band heterotopia, RNAi of Dcdc2 leads to smaller isolated PVH. DCDC2 may act earlier in the migration path for migrating cells than does DCX because PVH occurs nearer to the ventricles than are subcortical band heterotopia. Another striking difference between DCX and DCDC2 RNAi is that DCX RNAi does not lead to cells that overmigrate, and instead causes a general impairment of all migration to upper layers (Bai et al., 2003, Ramos et al., 2006). This difference suggests that DCDC2, unlike DCX, also functions in the normal termination of migration.
Overexpression of DCDC2 does not affect neuronal migration
The initial report identifying DCDC2 as a candidate dyslexia susceptibility gene did not offer evidence as to the functional consequences of the specific polymorphisms linked to the behavioral phenotype (Meng et al., 2005). It could be, therefore, that the DCDC2 gene variants result in either overexpression or knockdown of the DCDC2 protein. The results reported here confirm the initial reports that knockdown of Dcdc2 expression via shRNA in neocortical progenitor cells disrupts neuronal migration to the cerebral cortex (Meng et al., 2005). Overexpression of DCDC2, however, does not significantly impair neuronal migration. Thus, there were no cerebral cortical malformations in rats embryonically transfected with plasmids encoding either the full DCDC2 protein, or the C terminal or DCX domain. Although overexpression of the DCDC2 protein or the C Terminal domain appeared to mildly arrest migration (see Fig E,F, I, J), there was not a statistically significant disruption of laminar disposition comparable to that seen in following embryonic transfection with Dcdc2 shRNA. Taken together, these results do not provide support for the role of DCDC2 overexpression in the neuronal migration. This does not discount, however, the possibility that DCDC2 overexpression may affect other anatomic, physiologic, or behavioral phenotypes. These possibilities are being addressed in ongoing experiments.
Laminar organization is disrupted following Dcdc2 shRNA transfection
Migration distance analysis revealed that Dcdc2 shRNA-transfected neurons migrated in a bimodal pattern, with peaks at the white matter border and upper laminae. Those neurons in the upper laminae migrated past their expected location when compared to their control littermates. This is a similar pattern to that seen following Dyx1c1 shRNA transfections, and is discussed in detail elsewhere (Rosen et al., 2007). In the current experiment, however, we found that there was no significant difference in the laminar disposition of neurons between rats transfected with Dcdc2 shRNA and those co-transfected with Dcdc2 shRNA and the DCDC2 overexpression construct. Specifically, while a few neurons that remained along the white matter border with the cerebral cortex in the “rescue” condition, the majority of the neurons migrated past the expected lamina.
The lack of rescue of this migration phenotype could indicate that “overmigration” is the result to an off-target effect of the Dcdc2 shRNA. Or, it could be the case that the human DCDC2 overexpression construct was not effective because of the slight differences in sequence between the human and rat. This is unlikely, however, since, as we have shown, this construct successfully rescued the malformation phenotype. Another explanation could be that both the knockdown and overexpression constructs have the same effects on the migration distance phenotype. This does not appear to be the case, however, as transfection with the DCDC2 overexpression construct alone did not produce an overmigration phenotype, and instead transfected neurons migrated to slightly lower laminae (Fig 5 E,F). On the other hand, there were far fewer surviving transfected neurons following Dcdc2 shRNA + DCDC2 overexpression treatment, when compared to their littermates who were transfected solely with Dcdc2 shRNA. This raises the possibility that co-transfection in this case was particularly toxic to the cells, and that those neurons that survive comprise a special population that is atypical. The stoichiometry of the knockdown and overexpression constructs are not known at this time, and it could be that the timing of their expression in the cell may prove important in understanding this phenomenon. At the very least, we cannot exclude the possibility that this overmigration phenotype is the consequence of some as yet undefined off-target effect of Dcdc2 shRNA treatment.
Comparison to knockdown of other candidate dyslexia susceptibility genes
Previously, we have demonstrated that embryonic knockdown of the candidate dyslexia genes Kiaa0319 (Paracchini et al., 2006) and Dyx1c1 (Wang et al., 2006, Rosen et al., 2007) disrupted neuronal migration. In the case of both of these genes, transfected neurons were severely delayed in their migration out of the ventricular zone when assessed 4–7 days post transfection (Paracchini et al., 2006, Wang et al., 2006). In adulthood, we found that the disruption of neocortical migration by knockdown of Dyx1c1 function caused a variety of malformations in the forebrain similar to those reported here. For both Dyx1c1 and Dcdc2, the majority of shRNA-transfected brains examined had PVH that contained transfected and non-transfected neurons. Approximately 25% of the brains in both groups were noted to have remarkably similar hippocampal malformations. In addition, the bimodal pattern of migration of transfected neurons was similar for both Dyx1c1 and Dcdc2 shRNA groups. In both cases, approximately 7–15% of the transfected neurons failed to migrate past the white matter/neocortical border, and the peak locations of the neurons that did migrate were superficial to their expected lamina (but see above). On the other hand, there were phenotypic differences between the brains of rats embryonically transfected with Dyx1c1 or Dcdc2 shRNA. In the case of the former, there were clusters of mostly non-transfected neurons in the molecular layer of the neocortex, which were not associated with the injection site. There were no such molecular layer ectopias in the Dcdc2 shRNA-transfected brains.
The phenotypic similarities between Dyx1c1 and Dcdc2 shRNA brains could indicate that they share cellular and/or molecular pathways that are important for neuronal migration. There is no evidence as yet to directly link the function of these two genes, but previous reports suggest that Dyx1c1 is localized in the cytoplasm along microtubules (Wang et al., 2006), which is also the site where Dcdc2 is hypothesized to be localized. On the other hand, it could be that the phenotypes shared between these two genes are the result of a non-cell autonomous disruption of neuronal migration, while those that are unique are the result of the specific, and differential, effects of the knockdown of the gene in question. If this were the case, one would hypothesize that PVH, hippocampal malformations, and the bimodal distribution of transfected neurons would be seen following knockdown of function of any number of neuronal migration genes. As mentioned above, embryonic knockdown of Kiaa0319 via RNAi disrupts neuronal migration when assessed 4–7 days post transfection (Paracchini et al., 2006), and preliminary examination of these brains postnatally reveals all three phenotypes (unpublished observations). On the other hand, embryonic knockdown of Dcx does not produce PVH, nor is there an over-migration phenotype (Bai et al., 2003, Ramos et al., 2006). Dissection of the cellular and molecular consequence of the knockdown of these neuronal migration genes will help elucidate these issues.
Dyslexia candidate susceptibility genes and neuronal migration disorders
Post mortem examination of the brains of individuals with developmental dyslexia has revealed neocortical malformations (Galaburda and Kemper, 1979, Galaburda et al., 1985, Humphreys et al., 1990). We and others have previously shown that animals with induced or spontaneously occurring malformations of the cerebral cortex have profound changes in other aspects of cerebral anatomy (Jacobs et al., 1999, Rosen et al., 2001), connectivity (Giannetti et al., 1999, Giannetti et al., 2000, Jenner et al., 2000, Rosen et al., 2000), physiology (Luhmann and Raabe, 1996, Frenkel et al., 2000, Gabel and LoTurco, 2001, Gabel and LoTurco, 2002, Jacobs and Prince, 2005), and behavior (Fitch et al., 1994, Fitch et al., 1997, Peiffer et al., 2002, Peiffer et al., 2004). Recent reports demonstrate that embryonic knockdown of any of three CDSG homologs—Dyx1c1, Kiaa0319, and Dcdc2—by transfection with shRNA disrupts neuronal migration in rats (Meng et al., 2005, Paracchini et al., 2006, Wang et al., 2006). More recently, we have shown that embryonic knockdown of the candidate dyslexia susceptibility gene Dyx1c1 in neocortical progenitor cells results in malformations of the cerebral cortex similar to those seen in developmental dyslexia (Rosen et al., 2007). Moreover, these animals exhibit behavioral deficits in rapid auditory processing that are reminiscent of those reported in language impaired individuals and in animals with induced cortical malformations (Threlkeld et al., 2007). In the current experiment, we report that knockdown of function of the candidate dyslexia susceptibility gene homolog Dcdc2 also results in neuronal migration anomalies that resemble those found in humans with developmental dyslexia, including laminar dysplasias (Galaburda et al., 1985) and periventricular nodular heterotopias (Chang et al., 2005, Chang et al., 2007). Thus, of the currently identified dyslexia candidate susceptibility genes whose functions have been investigated, all appear to play a role in neural development, specifically in neuronal migration. Taken together, these results support the link between neuronal migration disorders and developmental dyslexia. Future research will consider the long term anatomic, connectional, physiological, and behavioral consequences of these genetically induced malformations.
Acknowledgments
This work was supported, in part, by HD20806 from NIH.
ABBREVIATIONS
- PVH
Periventricular Heterotopia
- E
Embryonic
- P
Postnatal
- shRNA
small hairpin RNA
- CDSG
candidate dyslexia candidate gene
- GFP
enhanced green fluorescent protein
- RFP
monomeric red fluorescent protein
Footnotes
TJB, YW, VJP, and AJV contributed equally to this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allen KM, Gleeson JG, Shoup SM, Walsh CA. A YAC contig in Xq22.3–q23, from DXS287 to DXS8088, spanning the brain- specific genes doublecortin (DCX) and PAK3. Genomics. 1998;52:214–218. doi: 10.1006/geno.1998.5424. [DOI] [PubMed] [Google Scholar]
- Anthoni H, Zucchelli M, Matsson H, Muller-Myhsok B, Fransson I, Schumacher J, Massinen S, Onkamo P, Warnke A, Griesemann H, Hoffmann P, Nopola-Hemmi J, Lyytinen H, Schulte-Korne G, Kere J, Nothen MM, Peyrard-Janvid M. A locus on 2p12 containing the co-regulated MRPL19 and C2ORF3 genes is associated to dyslexia. Hum Mol Genet. 2007;16:667–677. doi: 10.1093/hmg/ddm009. [DOI] [PubMed] [Google Scholar]
- Bai J, Ramos RL, Ackman JB, Thomas AM, Lee RV, LoTurco JJ. RNAi reveals doublecortin is required for radial migration in rat neocortex. Nat Neurosci. 2003;6:1277–1283. doi: 10.1038/nn1153. [DOI] [PubMed] [Google Scholar]
- Bellini G, Bravaccio C, Calamoneri F, Donatella Cocuzza M, Fiorillo P, Gagliano A, Mazzone D, del Giudice EM, Scuccimarra G, Militerni R, Pascotto A. No evidence for association between dyslexia and DYX1C1 functional variants in a group of children and adolescents from Southern Italy. J Mol Neurosci. 2005;27:311–314. doi: 10.1385/jmn:27:3:311. [DOI] [PubMed] [Google Scholar]
- Brkanac Z, Chapman NH, Matsushita MM, Chun L, Nielsen K, Cochrane E, Berninger VW, Wijsman EM, Raskind WH. Evaluation of candidate genes for DYX1 and DYX2 in families with dyslexia. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. 2007 doi: 10.1002/ajmg.b.30471. Epub ahead of print:n/a. [DOI] [PubMed] [Google Scholar]
- Chang BS, Katzir T, Liu T, Corriveau K, Barzillai M, Apse KA, Bodell A, Hackney D, Alsop D, Wong S, Walsh CA. A structural basis for reading fluency: White matter defects in a genetic brain malformation. Neurology. 2007;69:2146–2154. doi: 10.1212/01.wnl.0000286365.41070.54. [DOI] [PubMed] [Google Scholar]
- Chang BS, Ly J, Appignani B, Bodell A, Apse KA, Ravenscroft RS, Sheen VL, Doherty MJ, Hackney DB, O’Connor M, Galaburda AM, Walsh CA. Reading impairment in the neuronal migration disorder of periventricular nodular heterotopia. Neurology. 2005;64:799–803. doi: 10.1212/01.WNL.0000152874.57180.AF. [DOI] [PubMed] [Google Scholar]
- Cope N, Harold D, Hill G, Moskvina V, Stevenson J, Holmans P, Owen MJ, O’Donovan MC, Williams J. Strong evidence that KIAA0319 on chromosome 6p is a susceptibility gene for developmental dyslexia. Am J Hum Genet. 2005;76:581–591. doi: 10.1086/429131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coquelle FM, Levy T, Bergmann S, Wolf SG, Bar-El D, Sapir T, Brody Y, Orr I, Barkai N, Eichele G, Reiner O. Common and Divergent Roles for Members of the Mouse DCX Superfamily. Cell Cycle. 2006;5 doi: 10.4161/cc.5.9.2715. [DOI] [PubMed] [Google Scholar]
- des Portes V, Pinard JM, Billuart P, Vinet MC, Koulakoff A, Carrie A, Gelot A, Dupuis E, Motte J, Berwald-Netter Y, Catala M, Kahn A, Beldjord C, Chelly J. A novel CNS gene required for neuronal migration and involved in X- linked subcortical laminar heterotopia and lissencephaly syndrome. Cell. 1998;92:51–61. doi: 10.1016/s0092-8674(00)80898-3. [DOI] [PubMed] [Google Scholar]
- Deuel TA, Liu JS, Corbo JC, Yoo SY, Rorke-Adams LB, Walsh CA. Genetic interactions between doublecortin and doublecortin-like kinase in neuronal migration and axon outgrowth. Neuron. 2006;49:41–53. doi: 10.1016/j.neuron.2005.10.038. [DOI] [PubMed] [Google Scholar]
- Fitch RH, Brown CP, Tallal P, Rosen GD. Effects of sex and MK-801 on auditory-processing deficits associated with developmental microgyric lesions in rats. Behav Neurosci. 1997;111:404–412. doi: 10.1037//0735-7044.111.2.404. [DOI] [PubMed] [Google Scholar]
- Fitch RH, Tallal P, Brown C, Galaburda AM, Rosen GD. Induced microgyria and auditory temporal processing in rats: A model for language impairment? Cereb Cortex. 1994;4:260–270. doi: 10.1093/cercor/4.3.260. [DOI] [PubMed] [Google Scholar]
- Francks C, Paracchini S, Smith SD, Richardson AJ, Scerri TS, Cardon LR, Marlow AJ, MacPhie IL, Walter J, Pennington BF, Fisher SE, Olson RK, DeFries JC, Stein JF, Monaco AP. A 77-kilobase region of chromosome 6p22.2 is associated with dyslexia in families from the United Kingdom and from the United States. Am J Hum Genet. 2004;75:1046–1058. doi: 10.1086/426404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenkel M, Sherman GF, Bashan KA, Galaburda AM, LoTurco JJ. Neocortical ectopias are associated with attenuated neurophysiological responses to rapidly changing auditory stimuli. NeuroReport. 2000;11:575–579. doi: 10.1097/00001756-200002280-00029. [DOI] [PubMed] [Google Scholar]
- Friocourt G, Liu JS, Antypa M, Rakic S, Walsh CA, Parnavelas JG. Both doublecortin and doublecortin-like kinase play a role in cortical interneuron migration. J Neurosci. 2007;27:3875–3883. doi: 10.1523/JNEUROSCI.4530-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabel LA, LoTurco JJ. Electrophysiological and morphological characterization of neurons within neocortical ectopias. J Neurophysiol. 2001;85:495–505. doi: 10.1152/jn.2001.85.2.495. [DOI] [PubMed] [Google Scholar]
- Gabel LA, LoTurco JJ. Layer I ectopias and increased excitability in murine neocortex. J Neurophysiol. 2002;87:2471–2479. doi: 10.1152/jn.2002.87.5.2471. [DOI] [PubMed] [Google Scholar]
- Galaburda AM, Kemper TL. Cytoarchitectonic abnormalities in developmental dyslexia; a case study. Ann Neurol. 1979;6:94–100. doi: 10.1002/ana.410060203. [DOI] [PubMed] [Google Scholar]
- Galaburda AM, Sherman GF, Rosen GD, Aboitiz F, Geschwind N. Developmental dyslexia: Four consecutive cases with cortical anomalies. Ann Neurol. 1985;18:222–233. doi: 10.1002/ana.410180210. [DOI] [PubMed] [Google Scholar]
- Giannetti S, Gaglini P, Di Rocco F, Di Rocco C, Granato A. Organization of cortico-cortical associative projections in a rat model of microgyria. NeuroReport. 2000;11:2185–2189. doi: 10.1097/00001756-200007140-00024. [DOI] [PubMed] [Google Scholar]
- Giannetti S, Gaglini P, Granato A, Di Rocco C. Organization of callosal connections in rats with experimentally induced microgyria. Childs Nerv Syst. 1999;15:444–448. doi: 10.1007/s003810050435. discussion 449–450. [DOI] [PubMed] [Google Scholar]
- Gleeson JG, Lin PT, Flanagan LA, Walsh CA. Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron. 1999;23:257–271. doi: 10.1016/s0896-6273(00)80778-3. [DOI] [PubMed] [Google Scholar]
- Graham ME, Ruma-Haynes P, Capes-Davis AG, Dunn JM, Tan TC, Valova VA, Robinson PJ, Jeffrey PL. Multisite phosphorylation of doublecortin by cyclin-dependent kinase 5. The Biochemical journal. 2004;381:471–481. doi: 10.1042/BJ20040324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannula-Jouppi K, Kaminen-Ahola N, Taipale M, Eklund R, Nopola-Hemmi J, Kaariainen H, Kere J. The axon guidance receptor gene ROBO1 is a candidate gene for developmental dyslexia. PLoS Genet. 2005;1:e50. doi: 10.1371/journal.pgen.0010050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harold D, Paracchini S, Scerri T, Dennis M, Cope N, Hill G, Moskvina V, Walter J, Richardson AJ, Owen MJ, Stein JF, Green ED, O’Donovan MC, Williams J, Monaco AP. Further evidence that the KIAA0319 gene confers susceptibility to developmental dyslexia. Mol Psychiatry. 2006;11:1085–1091. 1061. doi: 10.1038/sj.mp.4001904. [DOI] [PubMed] [Google Scholar]
- Humphreys P, Kaufmann WE, Galaburda AM. Developmental dyslexia in women: Neuropathological findings in three cases. Ann Neurol. 1990;28:727–738. doi: 10.1002/ana.410280602. [DOI] [PubMed] [Google Scholar]
- Jacobs KM, Mogensen M, Warren E, Prince DA. Experimental microgyri disrupt the barrel field pattern in rat somatosensory cortex. Cereb Cortex. 1999;9:733–744. doi: 10.1093/cercor/9.7.733. [DOI] [PubMed] [Google Scholar]
- Jacobs KM, Prince DA. Excitatory and inhibitory postsynaptic currents in a rat model of epileptogenic microgyria. J Neurophysiol. 2005;93:687–696. doi: 10.1152/jn.00288.2004. [DOI] [PubMed] [Google Scholar]
- Jenner AR, Galaburda AM, Sherman GF. Connectivity of ectopic neurons in the molecular layer of the somatosensory cortex in autoimmune mice. Cereb Cortex. 2000;10:1005–1013. doi: 10.1093/cercor/10.10.1005. [DOI] [PubMed] [Google Scholar]
- Koizumi H, Tanaka T, Gleeson JG. Doublecortin-like kinase functions with doublecortin to mediate fiber tract decussation and neuronal migration. Neuron. 2006;49:55–66. doi: 10.1016/j.neuron.2005.10.040. [DOI] [PubMed] [Google Scholar]
- LoTurco J. Doublecortin and a tale of two serines. Neuron. 2004;41:175–177. doi: 10.1016/s0896-6273(04)00006-6. [DOI] [PubMed] [Google Scholar]
- Luhmann HJ, Raabe K. Characterization of neuronal migration disorders in neocortical structures. 1. Expression of epileptiform activity in an animal model. Epilepsy Res. 1996;26:67–74. doi: 10.1016/s0920-1211(96)00041-1. [DOI] [PubMed] [Google Scholar]
- Marino C, Citterio A, Giorda R, Facoett iA, Menozzi G, Vanzin L, Lorusso M, Nobile M, Molteni M. Association of short-term memory with a variant within DYX1C1 in developmental dyslexia. Genes Brain Behav. 2007 doi: 10.1111/j.1601-183X.2006.00291.x. ePub ahead of print. [DOI] [PubMed] [Google Scholar]
- Marino C, Giorda R, Luisa Lorusso M, Vanzin L, Salandi N, Nobile M, Citterio A, Beri S, Crespi V, Battaglia M, Molteni M. A family-based association study does not support DYX1C1 on 15q21.3 as a candidate gene in developmental dyslexia. Eur J Hum Genet. 2005;13:491–499. doi: 10.1038/sj.ejhg.5201356. [DOI] [PubMed] [Google Scholar]
- Meng H, Smith SD, Hager K, Held M, Liu J, Olson RK, Pennington BF, DeFries JC, Gelernter J, O’Reilly-Pol T, Somlo S, Skudlarski P, Shaywitz SE, Shaywitz BA, Marchione K, Wang Y, Paramasivam M, LoTurco JJ, Page GP, Gruen JR. DCDC2 is associated with reading disability and modulates neuronal development in the brain. Proc Natl Acad Sci U S A. 2005;102:17053–17058. doi: 10.1073/pnas.0508591102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto M, Monuki ES, Tang H, Imitola J, Haubst N, Khoury SJ, Cunningham J, Gotz M, Walsh CA. Expression of Cux-1 and Cux-2 in the subventricular zone and upper layers II–IV of the cerebral cortex. J Comp Neurol. 2004;479:168–180. doi: 10.1002/cne.20322. [DOI] [PubMed] [Google Scholar]
- Paracchini S, Thomas A, Castro S, Lai C, Paramasivam M, Wang Y, Keating BJ, Taylor JM, Hacking DF, Scerri T, Francks C, Richardson AJ, Wade-Martins R, Stein JF, Knight JC, Copp AJ, LoTurco JJ, Monaco AP. The chromosome 6p22 haplotype associated with dyslexia reduces the expression of KIAA0319, a novel gene involved in neuronal migration. Hum Mol Genet. 2006;15:1659–1666. doi: 10.1093/hmg/ddl089. [DOI] [PubMed] [Google Scholar]
- Peiffer AM, Friedman JT, Rosen GD, Fitch RH. Impaired gap detection in juvenile microgyric rats. Brain Res Dev Brain Res. 2004;152:93–98. doi: 10.1016/j.devbrainres.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Peiffer AM, Rosen GD, Fitch RH. Sex differences in rapid auditory processing deficits in ectopic BXSB/MpJ mice. NeuroReport. 2002;13:2277–2280. doi: 10.1097/00001756-200212030-00021. [DOI] [PubMed] [Google Scholar]
- Ramos RL, Bai J, LoTurco JJ. Heterotopia formation in rat but not mouse neocortex after RNA interference knockdown of DCX. Cereb Cortex. 2006;16:1323–1331. doi: 10.1093/cercor/bhj074. [DOI] [PubMed] [Google Scholar]
- Reiner O, Coquelle FM, Peter B, Levy T, Kaplan A, Sapir T, Orr I, Barkai N, Eichele G, Bergmann S. The evolving doublecortin (DCX) superfamily. BMC Genomics. 2006;7:188. doi: 10.1186/1471-2164-7-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner O, Gdalyahu A, Ghosh I, Levy T, Sapoznik S, Nir R, Sapir T. DCX’s phosphorylation by not just another kinase (JNK) Cell Cycle. 2004;3:747–751. [PubMed] [Google Scholar]
- Rosen GD, Bai J, Wang Y, Fiondella CG, Threlkeld SW, LoTurco JJ, Galaburda AM. Disruption of neuronal migration by targeted RNAi knockdown of Dyx1c1 results in neocortical and hippocampal malformations. Cereb Cortex. 2007;17:2562–2572. doi: 10.1093/cercor/bhl162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen GD, Burstein D, Galaburda AM. Changes in efferent and afferent connectivity in rats with cerebrocortical microgyria. J Comp Neurol. 2000;418:423–440. [PubMed] [Google Scholar]
- Rosen GD, Windzio H, Galaburda AM. Unilateral induced neocortical malformation and the formation of ipsilateral and contralateral barrel fields. Neuroscience. 2001;103:931–939. doi: 10.1016/s0306-4522(01)00044-6. [DOI] [PubMed] [Google Scholar]
- Scerri TS, Fisher SE, Francks C, MacPhie IL, Paracchini S, Richardson AJ, Stein JF, Monaco AP. Putative functional alleles of DYX1C1 are not associated with dyslexia susceptibility in a large sample of sibling pairs from the UK. J Med Genet. 2004;41:853–857. doi: 10.1136/jmg.2004.018341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaar BT, Kinoshita K, McConnell SK. Doublecortin microtubule affinity is regulated by a balance of kinase and phosphatase activity at the leading edge of migrating neurons. Neuron. 2004;41:203–213. doi: 10.1016/s0896-6273(03)00843-2. [DOI] [PubMed] [Google Scholar]
- Schumacher J, Anthoni H, Dahdouh F, Konig IR, Hillmer AM, Kluck N, Manthey M, Plume E, Warnke A, Remschmidt H, Hulsmann J, Cichon S, Lindgren CM, Propping P, Zucchelli M, Ziegler A, Peyrard-Janvid M, Schulte-Korne G, Nothen MM, Kere J. Strong Genetic Evidence of DCDC2 as a Susceptibility Gene for Dyslexia. Am J Hum Genet. 2006;78:52–62. doi: 10.1086/498992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokol DK, Golomb MR, Carvahlo KS, Edwards-Brown M. Reading impairment in the neuronal migration disorder of periventricular nodular heterotopia. Neurology. 2006;66:294. doi: 10.1212/01.wnl.0000204246.83103.7d. author reply 294. [DOI] [PubMed] [Google Scholar]
- Taipale M, Kaminen N, Nopola-Hemmi J, Haltia T, Myllyluoma B, Lyytinen H, Muller K, Kaaranen M, Lindsberg PJ, Hannula-Jouppi K, Kere J. A candidate gene for developmental dyslexia encodes a nuclear tetratricopeptide repeat domain protein dynamically regulated in brain. Proc Natl Acad Sci U S A. 2003;100:11553–11558. doi: 10.1073/pnas.1833911100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Threlkeld SW, McClure MM, Bai J, Wang Y, LoTurco JJ, Rosen GD, Fitch RH. Developmental disruptions and behavioral impairments in rats following in utero RNAi of Dyx1c1. Brain Res Bull. 2007;71:508–514. doi: 10.1016/j.brainresbull.2006.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Paramasivam M, Thomas A, Bai J, Kaminen N, Kere J, Voskul J, Rosen G, Galaburda A, LoTurco J. Dyx1c1 functions in neuronal migration in developing neocortex. Neuroscience. 2006;143:515–522. doi: 10.1016/j.neuroscience.2006.08.022. [DOI] [PubMed] [Google Scholar]