

Abstract
A yeast two-hybrid screen using the last 28 amino acids of the cytoplasmic domain of the neural cell adhesion molecule L1 identified RanBPM as an L1-interacting protein. RanBPM associates with L1 in vivo and the N-terminal region of RanBPM (N-RanBPM), containing the SPRY domain, is sufficient for the interaction with L1 in a glutathione S-transferase pulldown assay. L1 antibody patching dramatically changes the subcellular localization of N-RanBPM in transfected COS cells. Overexpression of N-RanBPM in COS cells reduces L1-triggered extracellular signal-regulated kinase 1/2 activation by 50% and overexpression of N-RanBPM in primary neurons inhibits L1-mediated neurite outgrowth and branching. These data suggest that RanBPM is an adaptor protein that links L1 to the extracellular signal-regulated kinase/MAPK pathway
Keywords: adhesion molecule adaptor, axon extension, Ig superfamily
The neural cell adhesion molecule L1 is important for a variety of developmental processes, including neurite outgrowth (Lagenaur and Lemmon 1987), axon fasciculation (Kunz et al. 1998), myelination (Haney et al. 1999) and migration of neuronal precursors (Lindner et al. 1983). Mutations in the human L1 gene cause a number of X-linked disorders, such as X-linked hydrocephalus, agenesis of the corpus callosum and spastic paraplegia type I (Kenwrick et al. 1996; Fransen et al. 1997; Kamiguchi et al. 1998a). Furthermore, L1 knockout mice exhibit several phenotypes similar to human patients with L1 mutations, such as hypoplasia of the corticospinal tract and corpus callosum, hypoplasia of the cerebellar vermis and hydrocephalus (Dahme et al. 1997; Cohen et al. 1998; Fransen et al. 1998).
The extracellular domain of L1, composed of six Ig domains and five fibronectin type III repeats, is capable of binding to a variety of ligands, including L1 itself, other members of the Ig superfamily, integrins, neuropilin and several extracellular matrix components (for review, see Haspel and Grumet 2003). L1 can bind to ligands in trans to mediate adhesion or bind to ligands in cis to function as coreceptors. There is compelling evidence that the L1 cytoplasmic domain (L1CD) is crucial for L1 function. The L1CD is highly conserved and mutations in it cause Mental Retardation Aphasia Shuffling Gait and Adducted Thumbs (MASA) syndrome (Fransen et al. 1997). The L1CD is phosphorylated by several kinases and phosphorylation appears to regulate L1 function (Wong et al. 1996). L1 is known to activate the extracellular signal-regulated kinase (ERK) pathway and it has been suggested that ERK activation is involved in L1-mediated neurite outgrowth and migration (Schaefer et al. 1999; Schmid et al. 2000). To date, three proteins, ankyrin, adaptor protein-2 (AP-2) and ezrin, have been shown to interact with the L1CD but their relationship to ERK activation is unclear.
The ankyrin-binding site on the L1CD (amino acids 1204–1229) couples L1 to the underlying actin cytoskeleton. The interaction with ankyrin seems to mediate the stationary behavior of L1 and may play a critical role in the regulation of L1-mediated adhesion and migration (Gil et al. 2003). L1 can bind to the clathrin adaptor AP-2 through the YRSL motif and this interaction is critical for clathrin-mediated L1 endocytosis (Kamiguchi et al. 1998b). AP-2-mediated L1 endocytosis is critical for L1 recycling at the growth cone (Kamiguchi and Lemmon 2000), sorting of L1 to axons in dorsal root ganglion neurons and L1 transcytosis in hippocampal neurons (Kamiguchi and Lemmon 1998; Wisco et al. 2003). The L1CD also binds to ezrin, a member of the ezrin, radixin and moesin family of membrane–cytoskeleton-linking proteins (Dickson et al. 2002), through the YRSL motif and the juxtamembrane region (Cheng et al. 2005). This interaction provides a link between L1 and the actin cytoskeleton and plays a critical role in the regulation of neurite branching (Cheng et al. 2005).
As no interactor with the L1CD has a clear relationship to ERK activation, we sought to identify additional L1 binding proteins by performing a yeast two-hybrid screen. We chose the last 28 amino acids of the L1CD as bait because we have previously shown that this region is phosphorylated (Schaefer et al. 1999) but no protein interactions have been reported for this region. We identified RanBPM as an L1-interacting protein. RanBPM was originally cloned because it interacts with RAN, a Ras-like small GTPase, that functions as a carrier in nuclear–cytoplasmic exchange (Nakamura et al. 1998). Subsequently, a number of studies have identified RanBPM as a binding partner with several unrelated proteins, such as the hepatocyte growth factor (HGF) receptor Met (Wang et al. 2004), integrin lymphocyte function-associated antigen-1 (LFA-1) (Denti et al. 2004) and serine/threonine kinase Mirk/Dyrk (Zou et al. 2003). RanBPM has also been shown to associate directly with the guanine nucleotide exchange factor Sos and to stimulate Ras/ERK (Wang et al. 2002). It also regulates the transcriptional activity downstream of several receptors (Rao et al. 2002; Wang et al. 2002; Denti et al 2004).
We have demonstrated that L1 and RanBPM interact both in vitro and in vivo. The N-terminus of RanBPM was sufficient for the interaction with L1. In transfected cells, L1 and RanBPM colocalized in the plasma membrane and antibody-induced L1 patching caused redistribution of RanBPM with substantial colocalization with L1. Overexpression of the N-terminal fragment of RanBPM decreased L1-induced ERK activation by twofold in COS cells and partially inhibited L1-mediated neurite outgrowth in cerebellar neurons. These data suggest that RanBPM serves as an adaptor in L1-mediated signaling involved in neurite growth.
Materials and methods
Materials
All cell culture reagents were from Gibco (Carlsbad, CA, USA). The Nucleofector transfection kit was from Amaxa (Cologne, Germany). All chemicals were from Sigma (St Louis, MO, USA). Glutathione-Sepharose 4B beads, pGEX-4T-1 vector and anti-glutathione S-transferase (GST) were from Amersham Biosciences (Piscataway, NJ, USA). Protease inhibitors and anti-HA were purchased from Roche (Indianapolis, NJ, USA). Polyclonal anti-ERK2 (sc-154) and monoclonal anti-phosphorylated ERK were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The rabbit anti-human L1 antibody, rabbit anti-rat L1 antibody and monoclonal anti-L1 antibody 74-5H7 were produced in the laboratory (Schaefer et al. 2002). The anti-RanBPM antibody (5M) was a kind gift from Dr T Nishimito (Kyushu University, Japan). The monoclonal anti-RanBPM antibody was a kind gift from Dr E. Bianchi (Institut Pasteur, Paris, France).
Yeast two-hybrid screen
A cDNA encoding the last 28 amino acids of the L1CD was fused to sequences encoding the GAL4-DNA binding domain in the pGBKT7 vector (Clontech, Palo Alto, CA, USA) to make the vector pGBKT7-L1CD28. The bait lacked any intrinsic transcription activation activity or toxicity to the host strain. A pre-transformed adult human brain two-hybrid prey cDNA library in the Y187 yeast strain (Clontech) was screened by mating with AH 109 transformed with the bait construct pGBKT7-L1CD-28. Positive clones were selected on SD/-Leu/-Trp/-Ade/-His/15 mM 3-aminotriazole. Plasmid DNAs from positive clones were isolated and used to reverify the interaction. To map the binding region, various combinations of the bait vector pAS2-L1CD or pGBKT7-L1CD28 and the prey vector pACT2-tRanBPM, pACT2-N-terminal region of RanBPM (N-RanBPM) or pACT2-C-terminal region of RanBPM were cotransformed into AH109 and grown on low stringency plates (SD/-Leu/-Trp/-His/5 mM 3-aminotriazole) to test the interaction.
Plasmid construction
The pcDNA3-human L1 vector (Wong et al. 1995) and the pAS2-L1CD vector (Kamiguchi et al. 1998b) were described previously. The mammalian expression vector pcDN A3-N-RanBPM encodes amino acids 117–354 of RanBPM with an HA tag at the N-terminus. The yeast prey vectors pACT2-N-RanBPM and pACT2-C-terminal region of RanBPM encode amino acids 117–354 and 355–729 of RanBPM, respectively. The GST-tagged bacterial recombinant expression vector pGEX-N-RanBPM was generated by inserting codons 117–354 of RanBPM into pGEX-4T-1.
Glutathione S-transferase pull-down assay
pGEX-N-RanBPM was transformed into BL21 cells and induced with 1 mM Isopropyl-β-D-thiogalactopyranosid (IPTG) for 4 h at 37°C. GST fusion proteins were purified using glutathione-Sepharose 4B beads. Equal amounts of GST or GST-N-RanBPM (~100 μg protein on 100 μL 50% slurry of beads) were incubated with glutathione-Sepharose 4B beads for 2 h at 4°C and then washed twice with the binding buffer (20 mM HEPES, 50 mM NaCl, 0.1% Tween-20, pH 7.4). Immunoaffinity-purified mouse L1 (50 μL) (see below) was added to the beads and incubated overnight at 4°C. The beads were washed three times with the binding buffer. Bound proteins were eluted with 30 μL sodium dodecyl sulfate (SDS) sample buffer and resolved by SDS–polyacrylamide gel electrophoresis (PAGE) (7.5% gel) followed by western blotting with anti-GST (1: 2000) and anti-rat L1 (1: 1000).
L1 affinity purification
The procedure was performed as described previously (Schaefer et al. 1999) with slight modifications. Briefly, brains from adult mice were homogenized in 50 mM Tris, pH 7.5, 1 mM pervanadate, 0.32 M sucrose and EDTA-free complete protease inhibitors (Roche). The homogenates were separated by ultracentrifugation on a sucrose gradient for 45 min at 58 400 g. The plasma membrane layer was collected and then centrifuged for 30 min at 150 000 g to pellet the membranes. The plasma membrane pellet was solubilized in 50 mM Tris, pH 7.5, with 1% Triton-X-100 and centrifuged for 45 min at 150 000 g to remove the insoluble material. The solubilized membrane fraction was run through the 74-5H7 column overnight at 4°C. Bound proteins were eluted with 0.1 M diethylamine (pH 11.2) and immediately neutralized with Tris (2 M, pH 4). Peak fractions were dialysed with 50 mM Tris, pH 7.5, overnight at 4°C and then resolved on SDS–PAGE followed by silver staining to verify the band patterns. To test the interaction between L1 and RanBPM, 1 μg of immunoaffinity-purified mouse L1 was resolved on 7.5% SDS-PAGE followed by western blotting with rabbit anti-RanBPM (1: 2000) and mouse anti-RanBPM (1: 1000).
Western blot analysis
Samples were mixed with SDS sample buffer with 2-mercaptoethanol, boiled for 5 min and then separated by SDS–PAGE. The proteins were transferred to Immobilon-P membrane for immunobloting. For chemiluminescent western blots, membranes were blocked with 10% bovine serum albumin in Tris-buffered saline. Horseradish peroxidase-coupled secondary antibodies (Jackson Immuno-Research, West Grove, PA, USA) and SuperSignal West Pico chemiluminescence kit (Pierce, Rockford, IL, USA) were used for detection. For western blots with the Odyssey Infrared Fluorescence Imager (LI-COR, Lincoln, NE, USA), the membrane was blocked with 5% non-fat dry milk in phosphate-buffered saline. IRDye 800-(Rockland, Gilbertsville, PA, USA) or Alexa 680- (Molecular Probes, Eugene, OR, USA) coupled secondary antibodies were used.
Cell culture and transfection
COS-7 cells were grown in Dulbecco’s minimum essential medium supplemented with 10% fetal bovine serum and GlutaMAX. Transient transfection was performed using the Nucleofector electroporation device according to the manufacturer's protocol (program A24). Cells were grown for 48 h after transfection.
Immunofluorescence
For immunofluorescence staining, transfected COS-7 cells were grown on glass coverslips for 48 h. Cells were fixed with 4% paraformaldehyde and permeabilized in phosphate-buffered saline with 0.03% Triton-X-100. Cells were incubated with the corresponding primary antibodies (1: 1000 rabbit anti-hL1 for the pcDNA3-human L1 vector or 2 μg/mL anti-HA for the pcDNA3-N-RanBPM) followed by incubation with secondary antibodies (1: 200 Alexa 488 anti-mouse IgG and 1: 200 Texas Red-X anti-rabbit IgG). In the experiments involving antibody patching, COS-7 cells were cotransfected with pcDNA3-human L1 and pcDNA3-N-RanBPM/pFLAG-RanBPM and grown for 48 h after transfection. Live cells were incubated with 1: 100 heat-inactivated rabbit anti-human L1 antibody for 10 min at 37°C. Cells were then fixed, permeabilized and stained with anti-HA antibodies, followed by incubation with Alexa 488 goat anti-mouse IgG and Texas Red-X anti-rabbit IgG. Images were acquired with a confocal laser microscope (LSM 510; Zeiss, Oberkochen, Germany) using an Argon and HeNe laser and a 100× Plan Neofluor (N.A. 1.3) oil objective.
Extracellular signal-regulated kinase activation
COS-7 cells were transfected with the pcDNA3-human L1 vector alone or cotransfected with the pcDNA3-human L1 vector and the pcDNA3-HA-N-RanBPM vector. Transfected cells were plated at a density of 1 × 106 cells/35-mm dish. At 24 h after transfection, cells were serum starved for 16–20 h. The cells were then treated with heat-inactivated rabbit anti-human L1 antibody (1: 100) for various periods. After the treatment, cells were lysed in 100 μL lysis buffer/35-mm dish (50 mM Tris, 150 mM NaCl, pH 7.2, 1% Triton X-100 and EDTA-free Complete protease inhibitors). Lysates were collected and centrifuged at 16 000 g for 5 min to remove the insoluble material. The supernatant fluid was collected and mixed with sample buffer. Samples were boiled for 5 min and resolved on SDS–PAGE (10% gels) followed by western blotting. To detect ERK activation, two-color western blot was performed with rabbit anti-total ERK2 (1: 500) and mouse anti-phosphorylated ERK (1: 500) followed by incubation with Alexa 680 goat anti-mouse IgG (1: 5000) and IRDye goat anti-rabbit IgG (1: 5000). Another two-color western blot was performed with rabbit anti-human L1 (1: 5000) and mouse anti-HA (1 μg/mL) followed by incubation with Alexa 680 goat anti-mouse IgG (1: 5000) and IRDye goat anti-rabbit IgG (1: 5000). To perform the quantitative analysis, the integrated intensity for the ERK1/2 band on the original scan was quantified with the Odyssey software (LI-COR). ERK activation was calculated by dividing the integrated intensity of the band in the treated samples by the integrated intensity of the band in the untreated samples. Similar results were obtained in triplicate experiments.
Neurite outgrowth analysis
The preparation of cerebellar neuron cultures, L1 substrates and electroporation was described previously (Cheng and Lemmon 2004). Briefly, post-natal day 8 mouse cerebella were dissected and dissociated into single cells by trypsin and deoxynuclease treatment. Cells were transfected with the Nucleofector electroporation device (program G13) and plated onto L1-coated coverslips. After 48 h, cells were fixed, permeabilized and incubated with anti-HA antibodies to detect N-RanBPM-expressing neurons. Images were acquired with a Spot CCD camera RT slider (Diagnostic Instruments, Sterling Heights, MI, USA) coupled to a DLMB microscope (Leica, Nussloch, Germany) with a 20× objective. Images were analysed with Neurolucida (MicroBrightField Inc., Williston, VT, USA) and figures prepared with Photoshop 7 (Adobe, San Jose, CA, USA). Quantification of neurite outgrowth was performed as described by Cheng and Lemmon (2004).
Results
Identification of RanBPM as a L1-interacting protein by yeast two-hybrid screening
We performed a yeast two-hybrid screen on a pre-transformed adult human brain cDNA library using the last 28 amino acids of the L1CD as bait. Twenty-five positive clones were identified after screening 4 × 107 clones. DNA sequencing revealed that seven of the positive clones encoded a partial sequence of RanBPM (GeneBank Accession no. AB055311). The longest sequence of the seven library clones was ~2.8 kb in length and encoded amino acids 117–729 of RanBPM (Fig. 1). Independent cotransformation of the bait construct pGBKT7-L1CD28 or pAS2-L1CD (complete L1CD fused with the GAL4 activation domain) and the candidate clone pACT2-RanBPM into yeast further confirmed the interaction. RanBPM interacted with both the last 28 amino acids of the L1CD and the complete L1CD (Fig. 2a). The selectivity of this interaction was demonstrated by lack of interaction between the last 28 amino acids of the L1CD and SV40 large T antigen (Fig. 2a).
Fig. 1.
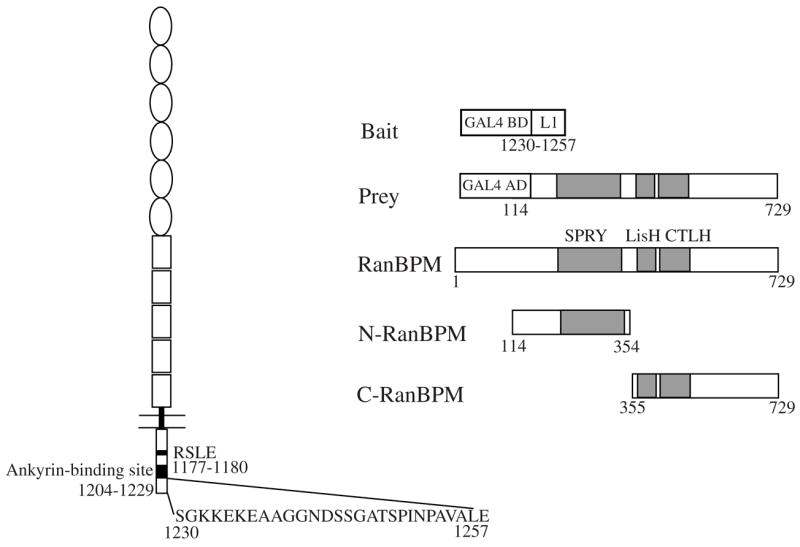
Identification of RanBPM as an L1-interacting protein by yeast two-hybrid screen. The structures of L1, bait protein with last 28 amino acids of the L1 cytoplasmic domain, longest prey clone isolated in the screen, complete RanBPM protein, N-terminal region of RanBPM (N-RanBPM) and C-terminal region of RanBPM (C-RanBPM) construct are shown.
Fig. 2.
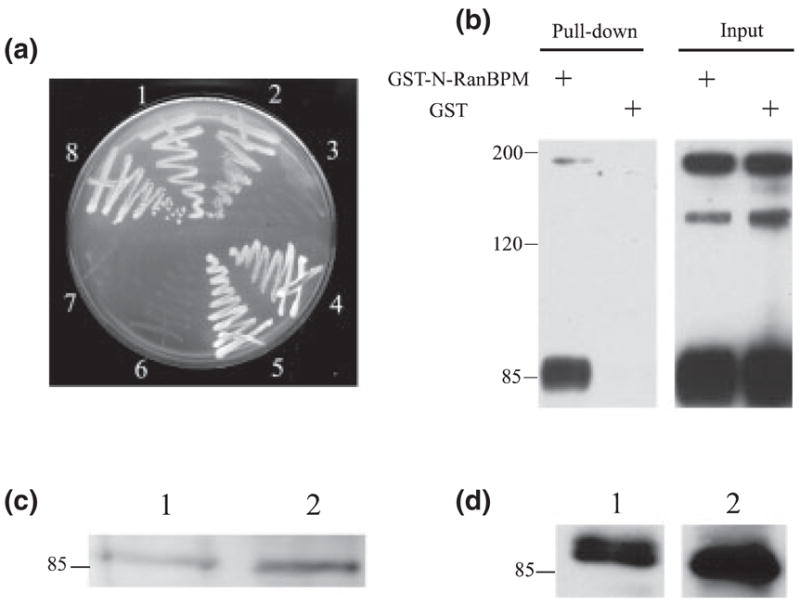
RanBPM interacts with L1 in vitro and in vivo, (a) Yeast AH109 cells were cotransformed and selected on a SD/-Trp/-Leu/-His/5 mM 3-aminotriazole plate. (1–3) AH109 yeast strain was cotransformed with pGBKT7-L1 cytoplasmic domain (L1CD)28 and the following GAL4 prey vectors: (1) pACT2-RanBPM; (2) pACT2-N-terminal region of RanBPM (N-RanBPM) and (3) pACT2-C-terminal region of RanBPM (C-RanBPM). (4–6) AH109 strain was cotransformed with pAS2-L1CD and the following GAL4 prey vectors: (4) pACT2-RanBPM; (5) pACT2-N-RanBPM and (6) pACT2-C-RanBPM. (7) Cotransformation of pGBKT7-L1CD28 and large T antigen-AD as negative control. (8) Cotransformation of p53-DBD and large T antigen-AD as positive control, (b) Glutathione S-transferase (GST)-N-RanBPM binds to purified L1 in GST pull-down assay. GST or GST-N-RanBPM was immobilized on beads and incubated with purified L1. L1 and an L1 fragment (85 kDa) containing the L1CD were detected on beads immobilized with GST-N-RanBPM but not on GST alone. (c and d) RanBPM was detected in L1 purified from brain. (1) Brain lysates as a positive control; (2) purified L1 from mouse brain. (c) Mouse anti-RanBPM; (d) rabbit anti-RanBPM.
N-terminal region of RanBPM containing the SPRY domain is sufficient for the interaction with L1
RanBPM contains a SPRY domain (dual-specificity kinase splA and ryanodine receptor domain; amino acids 212–333), a LisH domain (Lissencephaly type-1-like homology motif, amino acids 364–397) and a CTLH domain (C-terminal to LisH motif, amino acids 403–459). The function of the SPRY domain is still unclear but several studies have indicated that it is a protein–protein interaction domain (Rao et al. 2002; Wang et al. 2004). We speculated that the SPRY domain might therefore be involved in the interaction with L1. To test our hypothesis, two prey vectors were constructed. pACT2-N-RanBPM encodes amino acids 117–334 of RanBPM, containing the complete SPRY domain. pACT2-C-terminal region of RanBPM encodes amino acids 335–729 of RanBPM, containing the LisH, CTLH and the remaining C-terminal part. In the yeast two-hybrid assay, N-RanBPM interacted with both the last 28 amino acids of the L1CD and the complete L1CD, while C-terminal region of RanBPM interacted with neither (Fig. 2a). These results suggest that sequences in N-RanBPM are critical for the interaction with L1. To test this interaction using native L1, a GST fusion protein with the N-RanBPM was purified, immobilized on glutathione beads and used in a GST pull-down assay. Immunoaffinity-purified L1 from mouse brain bound to GST-N-RanBPM but not to GST alone (Fig. 2b). Due to protease cleavage, the L1 preparation contained full-length L1 (~200 kDa), an amino-terminal extracellular fragment (~135 kDa) and a C-terminal fragment containing the L1CD (~85 kDa) (Grumet et al. 1984). Full-length L1 and the 85-kDa C-terminal bound to GST-N-RanBPM but the 135-kDa extracellular fragment did not, consistent with the notion that GST-N-RanBPM binds to the L1CD. Results from both the yeast two-hybrid and the GST pull-down assays suggest that N-RanBPM, containing the SPRY domain, is sufficient for the interaction with L1.
RanBPM interacts with L1 in vivo
To investigate whether L1 and RanBPM interact in vivo, L1 and L1-associated proteins were purified from mouse brain and detected with two different antibodies against RanBPM, a rabbit antibody (Nishitani et al. 2001) and a mouse monoclonal antibody (Denti et al. 2004). Both antibodies detected a 90–100-kDa band in immunoaffinity-purified L1 samples (Fig. 2c, lane 2 and Fig. 2d, lane 2) in the range of the reported RanBPM molecular weight (Nakamura et al. 1998). As a positive control, the same antibody also detected a band of similar molecular weight in the brain lysate samples (Fig. 2c, lane 1 and Fig. 2d, lane 1). This strongly suggests that RanBPM associates with L1 in vivo.
Colocalization of L1 and N-terminal region of RanBPM after antibody-induced patching of L1
The biochemical studies above argue that L1 interacts with RanBPM. Colocalization of L1 and RanBPM in cells would lend support to this hypothesis. Preliminary studies indicated that HA-tagged, full-length RanBPM in pcDNAS gave poor expression in COS-7 cells but that HA-tagged N-RanBPM was expressed at acceptable levels. These studies also indicated that the N-RanBPM was concentrated along the plasma membrane (Fig. 3a) and that L1 and N-RanBPM showed good colocalization. Colocalization of two membrane-associated molecules in the plasma membrane does not prove that they interact so we hypothesized that altering the distribution of L1 would also redistribute RanBPM if they interact. Therefore, antibody-induced patching of L1 was performed on COS-7 cells cotransfected with L1 and HA-tagged N-RanBPM. We performed L1 patching assays with a short antibody incubation time (10 min at 37°C and no secondary antibody), which is sufficient to induce L1 patching as well as some internalization (Long et al. 2001). Confocal microscopic inspection revealed that most of the cells exhibited a patchy distribution of L1 (Fig. 3e) and a redistribution of N-RanBPM into the patches while some RanBPM remained at the perimeter of the cells. There is extensive colocalization between the L1 and N-RanBPM clusters (Fig. 3f), suggesting that L1 interacts with N-RanBPM and L1 can influence the distribution of N-RanBPM.
Fig. 3.
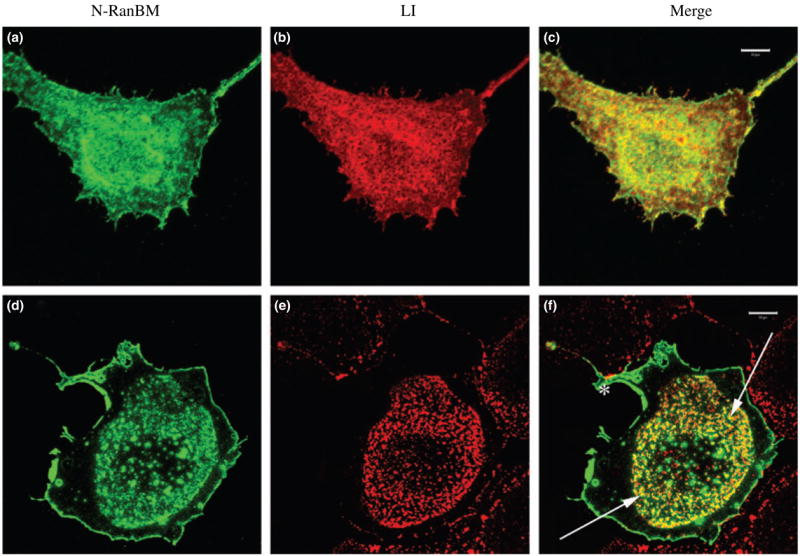
Colocalization of L1 and N-terminal region of RanBPM (N-RanBPM) after antibody-induced patching of L1. These confocal images represent Z-stacks through the cells, (a–c) COS-7 cells cotransfected with HA-tagged N-RanBPM (a) and L1 (b). Cells were fixed and stained without patching, (c) N-RanBPM (green) showed extensive colocalization with L1 (red). Bar, 10 μrn. (d–f) COS-7 cells cotransfected with HA-tagged N-RanBPM (d) and L1 (e). In this field only one cell expressed N-RanBPM while adjacent cells expressed some L1. Cells were patched with Ranti-L1 for 10 min before fixation. L1 (red) exhibited a patchy distribution in the center of the cell and N-RanBPM (green) was partially colocalized with L1 patches indicated with arrows. Some RanBPM remained at the perimeter of the cell (asterisk) and is not associated with L1. Bar, 10 μrn.
N-terminal region of RanBPM inhibits L1-mediated MAPK activation
Previous studies demonstrated that RanBPM directly interacts with Sos to activate Ras and induce ERK phosphorylation (Wang et al. 2002). Also, overexpression of full-length RanBPM led to the constitutive activation of the Ras-ERK pathway (Wang et al. 2002). As L1 cross-linking is known to activate ERK (Schaefer et al. 1999; Schmid et al. 2000; Loers et al. 2005), we hypothesized that RanBPM might function as an adaptor to mediate L1-induced ERK activation. To test this hypothesis, we performed L1 cross-linking with polyclonal anti-human L1 antibodies on both COS-7 cells transfected with L1 alone and COS-7 cells cotransfected with L1 and N-RanBPM. In COS-7 cells transfected with L1 alone, L1 cross-linking triggered a strong activation of ERK, as previously described in NIH 3T3 cells (Schaefer et al. 1999). Maximum activation was observed within 10 min of stimulation (4.55-fold increase in ERK activity, n = 3, SD, 0.28) and the activation went down to the basal level after 30 min of stimulation (Fig. 4). In cells cotransfected with L1 and N-RanBPM, antibody cross-linking also triggered the activation of ERK after 10 min of treatment but the activation was weaker compared with cells transfected with L1 alone (2.00-fold increase in ERK activity, n = 3, SD, 0.08). This was not due to a difference in L1 expression level, as shown by the blot probed with anti-L1 (Fig. 4). Quantitative analysis demonstrated that N-RanBPM reduced the L1-induced ERK activation by twofold. This suggests that RanBPM is involved in L1-induced ERK activation.
Fig. 4.
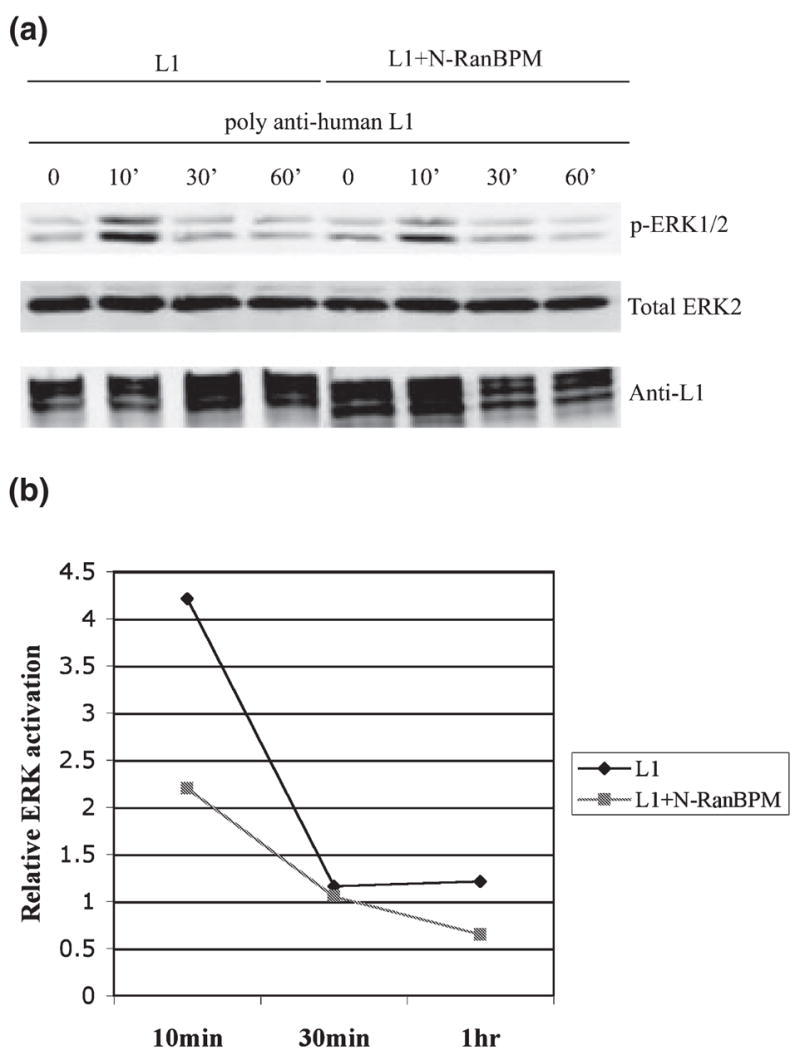
N-terminal region of RanBPM (N-RanBPM) inhibits extracellular signal-regulated kinase (ERK) activation induced by L1 cross-linking. (a) Serum-starved COS-7 cells transfected with L1 alone or cotransfected with L1 + N-RanBPM were treated with polyclonal anti-L1 for various time periods. ERK activation was detected with anti-phosphorylated ERK. The blot with total ERK2 was to ensure equal loading and the blot with anti-L1 was to ensure equal L1 expression level between samples, (b) Densitometric analysis of the relative ERK activation change induced by antibody cross-linking. A summary of three experiments is shown. Error bars, SD.
N-terminal region of RanBPM reduces L1-mediated neurite outgrowth
As L1-mediated neurite outgrowth involves MAPK activation (Schmid et al. 2000), we speculated that RanBPM might regulate L1-mediated neurite outgrowth through the MAPK pathway. We transfected the N-RanBPM vector pcDNA3-N-RanBPM into wild-type cerebellar neurons and grew transfected cells on an L1 substrate (Fig. 5b). As a positive control, we also transfected Enhanced Green Fluorescent Protein (EGFP) vector into neurons (Fig. 5a). EGFP-transfected neurons had an average neurite length of 240 μm after 48 h. In comparison, the N-RanBPM-expressing neurons had an average longest neurite length of 206 μm, a 14% decrease. This decrease was significant at the p < 0.02 level and is representative of three experiments (Fig. 5d). On the neurite length distribution curve (Fig. 5c), there was a significant shortening of neurites in the N-RanBPM-expressing neurons compared with EGFP-expressing neurons. Interestingly, when we examined total neurite length, as opposed to single longest neurite, the effect appeared larger with a reduction from 366 to 300 μm, a 18% reduction that was significant at the p < 0.0002 level. This is entirely accounted for by a reduction in secondary branching. While the number of primary neurites in N-RanBPM-treated cells was virtually identical to that in EGFP-transfected neurons (2.14 ± 0.06 vs. 2.13 ± 0.04, respectively, mean ± SEM), the number of secondary branches was reduced by 53% (0.91 ± 0.15 vs. 1.93 ± 0.21, p < 0.002). This result supports the idea that N-RanBPM functions as a dominant-negative adaptor to inhibit the L1-induced activation of the ERK pathway and thus reduces L1-mediated neurite outgrowth, possibly by having its most significant effect on branching.
Fig. 5.
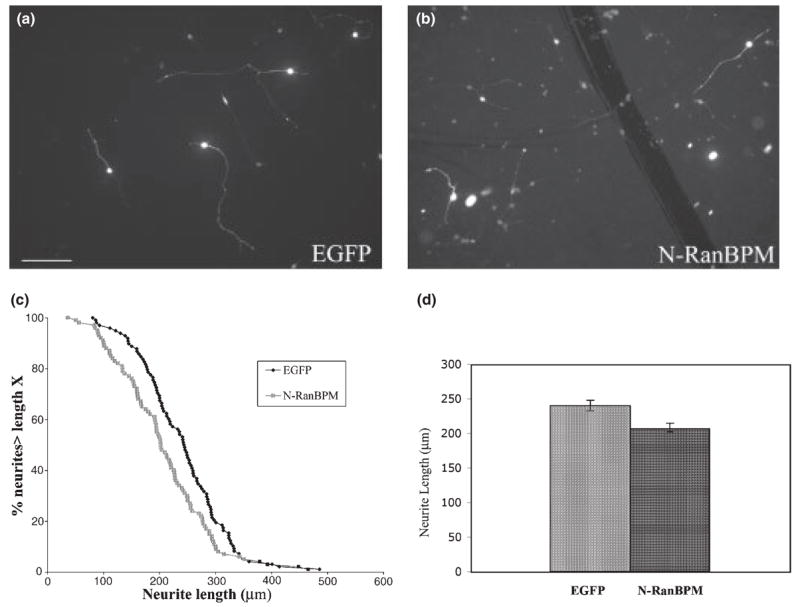
N-terminal region of RanBPM (N-RanBPM) inhibits L1-mediated neurite outgrowth, (a and b) Cerebellar neurons transfected with EGFP (a) or N-RanBPM (b) were grown on L1 substrate. Cultures were fixed and stained after 48 h. Bar, 100 μm. (c) The distribution curve of neurite length. The percentage of neurons with neurites longer than any given length was plotted against neurite length, (d) Bar graph showing mean neurite length ±SEM for neurons expressing EGFP or N-RanBPM.
Discussion
Using yeast two-hybrid screening, we have identified RanBPM as an L1-binding protein. RanBPM interacts with L1 in vitro and in vivo. The N-terminal region of RanBPM, containing the SPRY domain, is sufficient for the interaction with L1. We also demonstrate that N-RanBPM can inhibit L1-induced MAPK activation and L1-mediated neurite outgrowth, suggesting that RanBPM is an adaptor protein that participates in L1-induced MAPK activation.
Previous studies have demonstrated that the RanBPM SPRY domain is sufficient for interaction with Met and the serine/threonine kinase Mirk/Dyrk1B (Wang et al. 2002; Zou et al. 2003). Our study also demonstrates that the N-RanBPM containing the complete SPRY domain is sufficient for the interaction with L1 in the GST pull-down and yeast two-hybrid assays. Denti et al. (2004) have demonstrated that RanBPM can be phosphorylated both constitutively and in response to stress. Osmotic shock induced a shift towards hyperphosphorylated forms of RanBPM. Pre-treatment of cells with p38 kinase inhibitor blocks this phosphorylation, suggesting that the p38 kinase may phosphorylate RanBPM in response to stress. The phosphorylation site predicted by Scansite (http://www.scansite.mit.edu) identifies a p38 kinase phosphorylation site at Thr320, inside the SPRY domain; this site is conserved across species, including human, mouse, Xenopus, Caenorhabditis elegans and yeast (Wang et al. 2002). Therefore, the p38 kinase phosphorylation site inside the SPRY domain may be physiologically relevant and may regulate protein interactions with RanBPM in response to stress.
Although RanBPM was first identified as a centrosomal protein, Denti et al. (2004) have demonstrated, using a monoclonal antibody against RanBPM, that at least a fraction of RanBPM associates with the plasma membrane. Similarly, we found that HA-tagged N-RanBPM can be localized to the plasma membrane, based on confocal microscopy. The plasma membrane localization of RanBPM, together with RanBPM’s role in the ERK pathway and its interaction with several transmembrane proteins (including integrin LFA-1, HGF receptor Met and L1), suggest that RanBPM can function as an adaptor protein for transmembrane proteins on the cell surface.
L1 is known to associate with components of the ERK/MAPK pathway in vivo (Schaefer et al. 1999). In L1 immunoprecipitates from rat brain, Raf-1, ERK2 (Schaefer et al. 1999) and MAPK/ERK Kinase (MEK)-1 (VL, unpublished data) can be detected. Furthermore, L1 cross-linking can activate ERK (Schaefer et al. 1999; Schmid et al. 2000). However, it is not clear how L1 activates the ERK pathway after L1 cross-linking. The direct association of RanBPM with L1 and the ability of RanBPM to recruit Sos and stimulate Ras activation (Wang et al. 2002) suggest that RanBPM may be the adaptor protein for L1 to activate MAPK. We demonstrate that the ERK1/2 activation in COS cells coexpressing L1 and N-RanBPM is weaker than that in COS cells expressing only L1. Quantitative analysis shows a twofold difference in ERK activation. This suggests that N-RanBPM acts as a dominant-negative RanBPM to block L1-induced MAPK activation. The underlying mechanism is still unclear. It is possible that N-RanBPM can compete with endogenous RanBPM to bind L1 but fails to recruit Sos and thus cannot activate ERK efficiently. However, Maness and associates have argued that ras is not involved in the L1 activation of ERK (Schmid et al. 2000) based on pharmacological studies. It is not clear whether this is due to a difference in cell types or whether RanBPM can influence erk independent of Sos and ras.
Overexpressing N-RanBPM reduces neurite growth to a small but statistically significant degree (14%). One could question whether this decrease is biologically relevant. However, we have recently demonstrated that deletion of 110 of 114 amino acids of the L1CD decreases neurite length on L1 substrates by 17% (Cheng et al. 2005), which is remarkable given the very high conservation of the L1CD. It is, in fact, similar to the effect of Overexpressing N-RanBPM on neurite length. This suggests that loss of interaction with RanBPM accounts for most of the effects of the L1CD on neurite length and RanBPM is a regulator of neurite length. Other studies have shown that the MAPK pathway is required for L1-mediated neurite outgrowth, as the MEK inhibitors PD98059 and U0126 can block L1-stimulated neurite outgrowth (Schmid et al. 2000; Watanabe et al. 2004; Loers et al. 2005). In our study, N-RanBPM overexpression decreases neurite length by 14%, presumably by decreasing ERK activation. The less potent effect of N-RanBPM overexpression, compared with the MEK inhibitors, is probably because the N-RanBPM inhibits L1-induced ERK activation by 50% but the MEK inhibitors can block ERK activation completely. Nonetheless, both studies suggest that L1-mediated neurite outgrowth requires activation of the MAPK pathway and RanBPM is involved in L1-mediated neurite outgrowth through its role in MAPK signaling. A more refined analysis revealed that N-RanBPM had a slightly larger effect on overall neurite growth (an 18% reduction in total neurite length) that is probably explained by a large (50%) reduction in secondary branch formation. In unpublished time-lapse studies we have found that secondary branching is not common in our cerebellar cultures until about 48 h have elapsed. Earlier studies on erk and src inhibitors of L1-mediated neurite growth (Schmid et al. 2000; Watanabe et al. 2004; Loers et al. 2005) have used conditions (relatively short culture times, 24 h or less) and analysis methods (total neurite growth or percent cells with neurites) that would not have uncovered a role for erk and src in neurite branching. Clearly this needs to be investigated further.
RanBPM associates with the plasma membrane and can bind to numerous transmembrane proteins, including the integrin LFA-1 and the HGF receptor Met as well as the neural cell adhesion molecule L1. The emerging view is that RanBPM may serve as a scaffolding protein to regulate signal transduction. Furthermore, it has been suggested that L1 can interact with integrins in cis (Silletti et al. 2000). The ability of RanBPM to interact with both L1 and integrins suggests that RanBPM may play a role in the ‘cross-talk’ between L1 and β1 integrin. The ubiquitous expression pattern of RanBPM and its interaction with multiple membrane receptors or cell adhesion molecules place RanBPM in an ideal position to integrate different signaling pathways.
Acknowledgments
We thank Dr T. Nishimito (Kyushu University, Japan) for providing the rabbit anti-RanBPM antibody and Dr E. Bianchi for providing the monoclonal anti-RanBPM antibody. We are grateful to Ken Henry and Tom Newpher for reagents and helpful discussions.
Abbreviations used
- AP-2
Adaptor Protein-2
- EGFP
Enhanced Green Fluorescent Protein
- ERK
extracellular signal-regulated kinase
- GST
glutathione S-transferase
- HA
hemagglutinin peptide
- HGF
Hepatocyte Growth Factor
- IPTG
Isopropyl-β-D-thiogalactopyranosid
- L1CD
L1 cytoplasmic domain
- LFA-1
lymphocyte function-associated antigen-1
- MASA
Mental Retardation Aphasia Shuffling Gait and Adducted Thumbs
- MEK
MAPK/ERK kinase
- PAGE
polyacrylamide gel electrophoresis
- SDS
sodium dodecyl sulfate
References
- Cheng L, Lemmon V. Pathological missense mutations of neural cell adhesion molecule L1 affect neurite outgrowth and branching on an L1 substrate. Mol Cell Neurosci. 2004;27:522–530. doi: 10.1016/j.mcn.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Cheng L, Itoh K, Lemmon V. L1-mediated branching is regulated by two ezrin-radixin-moesin (ERM)-binding sites, the RSLE region and a novel juxtamembrane ERM-binding region. J Neurosci. 2005;25:395–403. doi: 10.1523/JNEUROSCI.4097-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen NR, Taylor JS, Scott LB, Guillery RW, Soriano P, Furley AJ. Errors in corticospinal axon guidance in mice lacking the neural cell adhesion molecule L1. Curr Biol. 1998;8:26–33. doi: 10.1016/s0960-9822(98)70017-x. [DOI] [PubMed] [Google Scholar]
- Dahme M, Bartsch U, Martini R, Anliker B, Schachner M, Mantei N. Disruption of the mouse L1 gene leads to malformations of the nervous system. Nat Genet. 1997;17:346–349. doi: 10.1038/ng1197-346. [DOI] [PubMed] [Google Scholar]
- Denti S, Sirri A, Cheli A, Rogge L, Innamorati G, Putignano S, Fabbri M, Pardi R, Bianchi E. RanBPM is a phosphoprotein that associates with the plasma membrane and interacts with the integrin LFA-1. J Biol Chem. 2004;279:13 027–13 034. doi: 10.1074/jbc.M313515200. [DOI] [PubMed] [Google Scholar]
- Dickson TC, Mintz CD, Benson DL, Salton SR. Functional binding interaction identified between the axonal CAM L1 and members of the ERM family. J Cell Biol. 2002;157:1105–1112. doi: 10.1083/jcb.200111076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransen E, Van Camp G, Vits L, Willems PJ. L1-associated diseases: clinical geneticists divide, molecular geneticists unite. Hum Mol Genet. 1997;6:1625–1632. doi: 10.1093/hmg/6.10.1625. [DOI] [PubMed] [Google Scholar]
- Fransen E, Van Camp G, D’Hooge R, Vits L, Willems PJ. Genotype-phenotype correlation in L1 associated diseases. J Med Genet. 1998;35:399–404. doi: 10.1136/jmg.35.5.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil OD, Sakurai T, Bradley AE, Fink MY, Cassella MR, Kuo JA, Felsenfeld DP. Ankyrin binding mediates L1CAM interactions with static components of the cytoskeleton and inhibits retrograde movement of L1CAM on the cell surface. J Cell Biol. 2003;162:719–730. doi: 10.1083/jcb.200211011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumet M, Hoffman S, Chuong CM, Edelman GM. Polypeptide components and binding functions of neuronglia cell adhesion molecules. Proc Natl Acad Sci USA. 1984;81:7989–7993. doi: 10.1073/pnas.81.24.7989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haney CA, Sahenk Z, Li C, Lemmon VP, Roder J, Trapp BD. Heterophilic binding of L1 on unmyelinated sensory axons mediates Schwann cell adhesion and is required for axonal survival. J Cell Biol. 1999;146:1173–1184. doi: 10.1083/jcb.146.5.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haspel J, Grumet M. The L1CAM extracellular region: a multi-domain protein with modular and cooperative binding modes. Front Biosci. 2003;8:s1210–s1225. doi: 10.2741/1108. [DOI] [PubMed] [Google Scholar]
- Kamiguchi H, Lemmon V. A neuronal form of the cell adhesion molecule L1 contains a tyrosine-based signal required for sorting to the axonal growth cone. J Neurosci. 1998;18:3749–3756. doi: 10.1523/JNEUROSCI.18-10-03749.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiguchi H, Lemmon V. Recycling of the cell adhesion molecule L1 in axonal growth cones. J Neurosci. 2000;20:3676–3686. doi: 10.1523/JNEUROSCI.20-10-03676.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiguchi H, Hlavin ML, Lemmon V. Role of L1 in neural development: what the knockouts tell us. Mol Cell Neurosci. 1998a;12:48–55. doi: 10.1006/mcne.1998.0702. [DOI] [PubMed] [Google Scholar]
- Kamiguchi H, Long KE, Pendergast M, Schaefer AW, Rapoport I, Kirchhausen T, Lemmon V. The neural cell adhesion molecule L1 interacts with the AP-2 adaptor and is endocytosed via the clathrin-mediated pathway. J Neurosci. 1998b;18:5311–5321. doi: 10.1523/JNEUROSCI.18-14-05311.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenwrick S, Jouet M, Donnai D. X linked hydrocephalus and MASA syndrome. J Med Genet. 1996;33:59–65. doi: 10.1136/jmg.33.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz S, Spirig M, Ginsburg C, Buchstaller A, Berger P, Lanz R, Rader C, Vogt L, Kunz B, Sonderegger P. Neurite fasciculation mediated by complexes of axonin-1 and Ng cell adhesion molecule. J Cell Biol. 1998;143:1673–1690. doi: 10.1083/jcb.143.6.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagenaur C, Lemmon V. An L1-like molecule, the 8D9 antigen, is a potent substrate for neurite extension. Proc Natl Acad Sci USA. 1987;84:7753–7757. doi: 10.1073/pnas.84.21.7753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner J, Rathjen FG, Schachner M. L1 mono- and polyclonal antibodies modify cell migration in early postnatal mouse cerebellum. Nature. 1983;305:427–430. doi: 10.1038/305427a0. [DOI] [PubMed] [Google Scholar]
- Loers G, Chen S, Grumet M, Schachner M. Signal transduction pathways implicated in neural recognition molecule L1 triggered neuroprotection and neuritogenesis. J Neurochem. 2005;92:1463–1476. doi: 10.1111/j.1471-4159.2004.02983.x. [DOI] [PubMed] [Google Scholar]
- Long KE, Asou H, Snider MD, Lemmon V. The role of endocytosis in regulating L1-mediated adhesion. J Biol Chem. 2001;276:1285–1290. doi: 10.1074/jbc.M006658200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura M, Masuda H, Horii J, Kuma K, Yokoyama N, Ohba T, Nishitani H, Miyata T, Tanaka M, Nishimoto T. When overexpressed, a novel centrosomal protein, RanBPM, causes ectopic microtubule nucleation similar to gamma-tubulin. J Cell Biol. 1998;143:1041–1052. doi: 10.1083/jcb.143.4.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishitani H, Hirose E, Uchimura Y, Nakamura M, Umeda M, Nishii K, Mori N, Nishimoto T. Full-sized RanBPM cDNA encodes a protein possessing a long stretch of proline and glutamine within the N-terminal region, comprising a large protein complex. Gene. 2001;272:25–33. doi: 10.1016/s0378-1119(01)00553-4. [DOI] [PubMed] [Google Scholar]
- Rao MA, Cheng H, Quayle AN, Nishitani H, Nelson CC, Rennie PS. RanBPM, a nuclear protein that interacts with and regulates transcriptional activity of androgen receptor and glucocorticoid receptor. J Biol Chem. 2002;277:48 020–48 027. doi: 10.1074/jbc.M209741200. [DOI] [PubMed] [Google Scholar]
- Schaefer A, Kamei Y, Kamiguchi H, Wong EV, Rapoport I, Kirchhausen T, Beach CM, Landreth G, Lemmon SK, Lemmon V. L1 endocytosis is controlled by a phosphorylation-dephosphorylation cycle stimulated by outside-in signaling by L1. J Cell Biol. 2002;157:1223–1232. doi: 10.1083/jcb.200203024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer AW, Kamiguchi H, Wong EV, Beach CM, Landreth G, Lemmon V. Activation of the MAPK signal cascade by the neural cell adhesion molecule L1 requires L1 internalization. J Biol Chem. 1999;274:37 965–37 973. doi: 10.1074/jbc.274.53.37965. [DOI] [PubMed] [Google Scholar]
- Schmid RS, Pruitt WM, Maness PF. A MAP kinase-signaling pathway mediates neurite outgrowth on L1 and requires Src-dependent endocytosis. J Neurosci. 2000;20:4177–4188. doi: 10.1523/JNEUROSCI.20-11-04177.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silletti S, Mei F, Sheppard D, Montgomery AM. Plasmin-sensitive dibasic sequences in the third fibronectin-like domain of L1-cell adhesion molecule (CAM) facilitate homomultimerization and concomitant integrin recruitment. J Cell Biol. 2000;149:1485–1502. doi: 10.1083/jcb.149.7.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Li Z, Messing EM, Wu G. Activation of Ras/Erk pathway by a novel MET-interacting protein RanBPM. J Biol Chem. 2002;277:36 216–36 222. doi: 10.1074/jbc.M205111200. [DOI] [PubMed] [Google Scholar]
- Wang D, Li Z, Schoen SR, Messing EM, Wu G. A novel MET-interacting protein shares high sequence similarity with RanBPM, but fails to stimulate MET-induced Ras/Erk signaling. Biochem Biophys Res Commun. 2004;313:320–326. doi: 10.1016/j.bbrc.2003.11.124. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Yamazaki M, Miyazaki H, Arikawa C, Itoh K, Sasaki T, Maehama T, Frohman MA, Kanaho Y. Phospholipase D2 functions as a downstream signaling molecule of MAP kinase pathway in L1-stimulated neurite outgrowth of cerebellar granule neurons. J Neurochem. 2004;89:142–151. doi: 10.1111/j.1471-4159.2004.02308.x. [DOI] [PubMed] [Google Scholar]
- Wisco D, Anderson ED, Chang MC, Norden C, Boiko T, Folsch H, Winckler B. Uncovering multiple axonal targeting pathways in hippocampal neurons. J Cell Biol. 2003;162:1317–1328. doi: 10.1083/jcb.200307069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong EV, Cheng G, Payne HR, Lemmon V. The cytoplasmic domain of the cell adhesion molecule L1 is not required for homophilic adhesion. Neurosci Lett. 1995;200:155–158. doi: 10.1016/0304-3940(95)12100-i. [DOI] [PubMed] [Google Scholar]
- Wong EV, Schaefer AW, Landreth G, Lemmon V. Involvement of p90rsk in neurite outgrowth mediated by the cell adhesion molecule L1. J Biol Chem. 1996;271:18 217–18 223. doi: 10.1074/jbc.271.30.18217. [DOI] [PubMed] [Google Scholar]
- Zou Y, Lim S, Lee K, Deng X, Friedman E. Serine/threonine kinase Mirk/Dyrk1B is an inhibitor of epithelial cell migration and is negatively regulated by the Met adaptor Ranbinding protein M. J Biol Chem. 2003;278:49 573–49 581. doi: 10.1074/jbc.M307556200. [DOI] [PubMed] [Google Scholar]