

Abstract
Apoptosis is a significant factor in cardiac dysfunction and graft failure in cardiac rejection. In this study, we examined potential signaling molecules responsible for caspase 3 activation in a model of acute cardiac allograft rejection. The roles of reactive oxygen species (ROS) and nitric oxide (NO) were determined in untreated allografts and allograft recipients treated with either cyclosporine (CsA), α-phenyl-t-butylnitrone (PBN, a spin-trapping agent), vitamin C (VitC), Mn(III)tetrakis (1-methyl-4-pyridyl)porphyrin); MnTmPyP, a superoxide dismutase (SOD) mimetic), or l-(1-iminoethyl)lysine) (l-NIL), an inhibitor of inducible NO synthase (iNOS) enzyme activity. Graft tissue was taken for measuring superoxide radical production, Western blotting, and direct measurement of caspase 3 activity. Activation of caspase 3 in untreated allografts was revealed by the appearance of cleaved caspase 3 from pro-caspase 3 by Western blotting and functional caspase 3 catalytic activity. CsA or PBN inhibited iNOS expression and caspase 3 activity. VitC and MnTmPyP did not alter iNOS expression or decrease NO levels but did inhibit caspase 3 activity. In contrast, l-NIL completely inhibited the increase in NO production without altering iNOS expression and inhibited caspase 3 activity. The prevention of TUNEL staining by MnTmPyP and l-NIL confirmed downstream effects of superoxide and NO on apoptosis. These studies indicate that both superoxide and NO (precursors of peroxynitrite formation) play a significant role in caspase 3 activation in cardiac allograft rejection. Antioxid. Redox Signal. 10, 1031–1039.
Introduction
Early studies suggest a correlation in time of the expression of inducible nitric oxide synthase (iNOS) and apoptosis of cardiac myocytes in acute cardiac rejection (30). Considering that postmitotic cardiac myocytes have a limited capacity to regenerate compared with embryonic and neonatal cardiac myocytes, the death of cardiac myocytes via apoptosis in acute cardiac allograft rejection becomes a significant factor leading to contractile dysfunction and graft failure.
The signaling molecule(s) responsible for apoptosis in acute cardiac rejection are incompletely understood. By far, the majority of the information obtained to date in studies of acute cardiac rejection arises from nonquantitative assessments of apoptosis by using such techniques as DNA fragmentation, TUNEL staining, Hoechst staining, or nuclear imaging of annexin V (9, 15, 13, 29, 30). One of the pivotal points in the apoptotic cascade is the convergence of the activation of intrinsic and extrinsic apoptotic pathways at the level of activation of caspase 3. Thus, caspase 3 activation is considered to be essential to execution of apoptosis. Furthermore, the determination of the proteolytic activity of caspase 3 can be used as a quantitative measure of functional apoptotic activity in biologic tissue. However, to date, reports of the quantitative measuring of caspase 3 catalytic activity in acute cardiac allograft rejection are extremely limited.
The role of NO in apoptosis is complex owing to proapoptotic and antiapoptotic activity, depending on experimental conditions (11). Thus, no consensus determines when NO turns apoptosis on or off in any cell or tissue type, let alone the heart (2). In one study, nitric oxide (NO) derived from iNOS of recipients was implicated as contributing to activation of caspase 3 in iNOS+/+ vs. iNOS−/− mice (12). However, in this case, absence of iNOS in recipients does not eliminate the contributions of NO derived from iNOS upregulation within cardiac myocytes of the donor. In a second study, treatment with zinc chloride also decreased caspase 3 catalytic activity in a rat model of acute cardiac allograft rejection (14). The mechanism of this action is currently unknown, but complex formation between zinc and caspase 3 has been suggested to explain why zinc is a potent inhibitor of caspase 3 (22). Collectively, it is clear that the mechanisms of caspase 3 activation in acute cardiac allograft rejection are not yet fully understood.
In the present study, we examined functional apoptosis in acute cardiac allograft rejection by using caspase 3 catalytic activity. To determine possible signaling molecules, we compared caspase 3 catalytic activity in graft tissue from untreated allograft recipients or with allograft recipients treated with a variety of agents that scavenge reactive oxygen species or inhibit bulk NO production to determine a role of these factors. Additional studies were performed to assess downstream effects of these treatments on apoptosis by using TUNEL staining.
Methods
Transplantation
All animal procedures were approved by the local institutional animal care and use committee. All animals received humane care in compliance with the “Guide for the Care and Use of Laboratory Animals.” Lewis (Lew:RT11) and Wistar-Furth (WF:RT1u) rat strains (Harlan Labs, Indianapolis, IN) were chosen for genetic disparity at both the major and minor histocompatibility loci for donor-to-recipient combination of Lewis → Lewis (for isografts) or Wistar-Furth → Lewis (for allografts) rats. Isogeneic or allogeneic heterotopic cardiac transplantation was performed aseptically in pentobarbital (50 mg/kg, i.p.) anesthetized animals as previously described in our laboratory (3). Donor hearts were arrested in cold University of Wisconsin preservation solution and transplanted into anesthetized (50 mg/kg pentobarbital, i.p.) recipient animals (total cold and warm ischemic time, <35 min).
Untreated allografts served as controls. To assess a role of alloimmune activation, some recipients received cyclosporine (CsA). To assess a role of reactive oxygen, some recipients received the antioxidant vitamin, vitamin C, or the nonspecific free radical scavenger and spin-trap agent, α-phenyl-t-butylnitrone (PBN). To specify the role of superoxide anion radical, other recipients received the cell-permeable superoxide dismutase (SOD) mimetic agent, Mn(lll)tetrakis (1-methyl-4-pyridyl)porphyrins (MnTmPyP). To determine a role of inducible nitric oxide synthase (iNOS), other recipients received l-(1-iminoethyl)lysine (l-NIL). Treatment doses and routes of administration included CsA (2.5 mg/kg, i.p.); vitamin C (1,200 mg/kg, orally by gavage); PBN (150 mg/kg, i.p.); MnTmPyP (5 mg/kg, i.p.); and l-NIL (1 and 60 μg/ml in drinking water). Drug treatments were not given to donor animals but rather to recipient animals starting on the day of transplantation after revascularization and completing the surgical transplantation of the donor graft until harvesting of grafts on postoperative day 6.
Graft survival was monitored twice daily for presence or absence of palpable activity and was confirmed on direct inspection after laparotomy. In other studies, grafts were arrested in pentobarbital-anesthetized animals at postoperative day 6 and flushed with cold University of Wisconsin solution, minced, and portions were frozen in liquid N2 for Western blotting and caspase 3 activity measurements. Other portions of the graft were taken and evaluated for histologic rejection, as described later. In some cases, plasma was also taken to document changes in NO metabolite levels by using a Griess reaction assay.
Histologic rejection scoring
Tissue was fixed in 4% phosphate-buffered formalin. Paraffin-embedded sections were stained with hematoxylin and eosin. Histologic rejection was scored blinded by using criteria established by the International Society for Heart and Lung Transplantation, as modified to a linear score system to allow statistical analysis, as described previously (25, 31).
Western blot
Frozen tissue was processed and samples electrophoresed on 7.5% SDS polyacrylamide gels and transferred to Nytran membranes. Blots were probed with 1:2,000 dilution of rabbit anti-iNOS (Santa Cruz Biotechnology, Santa Cruz, CA) and visualized by using a 1:5,000 dilution of donkey anti-rabbit IgG horseradish peroxidase conjugate and enhanced chemiluminescence. For caspase-3 Western blotting, samples were electrophoresed on 12% SDS polyacrylamide gels and transferred to Nytran membranes. The membranes were probed with 1:500 dilution of goat anti-caspase-3 (Santa Cruz Biotechnology) and visualized as described earlier. Blots were stripped and reprobed with a 1:100 dilution of β-actin for protein-loading controls.
Superoxide production
For the SOD mimetic study, heart tissue (∼100 mg) was homogenized in Krebs–Henseleit buffer containing 10 mM HEPES. Tubes containing homogenate or buffer were incubated in the dark without and with 100 μM MnTmPyP for 30 minutes. The amount of chemiluminescence after addition of 20 μM lucigenin attributed to superoxide production was determined by the difference without and with MnTmPyP after subtraction of the background. Values were calculated as relative light units and normalized to homogenate protein levels.
Caspase 3 activity
Caspase 3 activity (n = 4 to 6 in each group) was performed by using a commercial kit (Sigma Aldrich, St. Louis, MO). The assay is based on the hydrolysis of the peptide substrate, acetyl-ASP-Glu-Val-p-nitroanilide by caspase 3 to release p-nitroaniline, which is detected spectrophotometrically at 405 nm by using a plate reader. Protease activity was determined in units of micromolar p-nitroaniline/min/mg protein.
TUNEL staining
Apoptosis was measured with the TUNEL assay by using ApopTag technology (Chemicon International, Temecula, CA) according to the manufacturer's instructions and as previously established in our laboratories (19). Sections were counterstained for visualization of apoptotic nuclei. Apoptotic nuclei were counted from at least four section fields for each graft sample, averaged, and the mean for each experimental group determined.
Data analysis
All values are expressed as mean ± SEM. Statistical analysis was performed with one-way analysis of variance with a Student–Newman–Keuls test for multiple comparisons of group means or with Student's t test for comparisons between two group means. Statistical significance was set at p < 0.05.
Results
Histologic rejection scores were significantly (p < 0.01) increased in untreated allografts compared with isograft controls. In the first series, allograft recipients were treated with CsA to show that caspase 3 activation was due to alloimmune activation. We previously showed that cardiac allografts survived to at least 117 days when given continuously at this dose (10) and that the increase in IFN-γ and CD3 mRNA in a mixed splenocyte culture was prevented in cells taken from CsA-treated rats at posttransplant day 6 and stimulated ex vivo without added CsA (24). The latter indicates that the dose used effectively blocks both cytokine gene expression and lymphocyte activation. We also showed that all of the drug-treatment regimens used for this study showed significantly (p < 0.01 each) decreased histologic rejection compared with untreated allografts (Table 1).
Table 1.
Histologic Rejection Scores of Cardiac Grafts for Experimental Groups at Posttransplant Day 6
| Groups | Rejection score |
|---|---|
| Isograft | 0.9 ± 0.2 |
| Untreated allograft | 5.2 ± 0.3 |
| CsA | 2.4 ± 0.3* |
| PBN | 3.2 ± 0.4* |
| VitC | 3.7 ± 0.2* |
| MnTmPyP | 4.0 ± 0.3* |
| l-NIL | 2.6 ± 0.2* |
Histologic score values represent the mean ± SEM of n = 6–8 for each group.
p < 0.01 vs. untreated allografts.
In addition to decreasing graft rejection, CsA also prevented the increase in iNOS protein expression in immunoblots of allograft homogenates (Fig. 1A). This regimen also prevented the increase in plasma NO metabolites (Fig. 1B). Caspase 3 protease activity was significantly (p < 0.01) increased in allografts vs. isograft controls (Fig. 1C). This increase was prevented in allograft recipients treated with CsA.
FIG. 1.

(A) Western blot of graft homogenates showing the upregulation of immunoreactive iNOS in rejecting cardiac allografts and the prevention by treatment with CsA. (B) Increase in plasma NO metabolites in allografts (allo) compared with isograft (iso) controls and prevention by treatment with CsA (n = 5 to 9 each). (C) Increase in caspase 3 protease activity in allografts (n = 6) compared with isograft controls (n = 5) and reversal by treatment with CsA (n = 3). ‡p < 0.01 vs. isografts and CsA-treated allografts.
To evaluate a general role of reactive oxygen species, allograft recipients were treated with the nonspecific radical scavenger and spin-trap agent, PBN (mechanism of actions shown in Fig. 2A). Treatment with PBN inhibited the increase in iNOS protein expression (Fig. 2B) and increased plasma NO metabolites above the value seen in untreated allografts (Fig. 2C). PBN effectively blocked the increased in caspase 3 protease activity seen in allografts (Fig. 2D). In another strategy, we used intervention with the natural antioxidant vitamin, VitC. In contrast to that with PBN, treatment with VitC did not prevent the increase in iNOS protein expression (Fig. 3A) or the increase in plasma NO metabolite levels typically seen in unreated allografts (Fig. 3B). Similar to the findings with PBN, VitC blocked the increased in caspase 3 protease activity seen in allografts (Fig. 3C).
FIG. 2.

(A) Diagram showing the radical trapping action of PBN. (B) Increase in iNOS protein expression normalized to β-actin protein loading in allografts and prevention by treatment with PBN (n = 3–4 each). (C) Increase in plasma NO metabolites and effects of PBN. (D) Increase in caspase 3 protease activity in allografts compared with isografts is prevented by PBN (n = 3–6 each). *p < 0.05 vs. isograft and PBN; ‡p < 0.01 vs. isograft; ‡p < 0.01 vs. allograft.
FIG. 3.

(A) Upregulation of iNOS protein in allografts vs. undetectable in isograft controls (not shown) was unaltered by treatment with antioxidant VitC. (B) Increase in plasma NO metabolites in untreated allografts and allografts treated with VitC (n = 5–6 each). (C) Increase in caspase 3 protease activity in rejecting allografts is prevented by treatment with VitC (n = 5–6 each). *p < 0.05 vs. isograft and VitC; ‡p < 0.01 vs. isograft.
To evaluate the specific role of superoxide anion radical, a subset of allograft recipients were treated with the cell-permeable SOD mimetic, MnTmPyP. This treatment did not alter the increase in iNOS expression (Fig. 4A) but slightly potentiated the increase in plasma NO metabolite levels (Fig. 4B) while decreasing the chemiluminescence signal indicating superoxide production (Fig. 4C). Treatment with MnTmPyP prevented the increase in caspase 3 protease activity (Fig. 4D).
FIG. 4.

(A) Expression of iNOS expression was not altered by treatment with the SOD mimetic, MnTmPyP (n = 3–4 each). (B) Increases in plasma NO metabolites were seen in untreated allografts and in allografts treated with MnTmPyP (n = 5–7 each). (C) Superoxide production as determined by the proportion of lucigeninenhanced chemiluminescence inhibitable by treatment with MnTmPyP was increased in allografts vs. isograft controls (n = 6 each). (D) Increase in caspase 3 protease activity was prevented by treatment with MnTmPyP (n = 5–6 each). *p < 0.05 vs. isograft and MnTmPyP; ‡p < 0.01 vs. isograft; (‡)p < 0.01 vs. allograft.
To evaluate a role of NO derived from iNOS, allograft recipients were treated with the iNOS inhibitor, l-NIL. Figure 5A and B shows the immunoblot and densitometry of the cleaved or activated caspase-3 (i.e., 17-kDA protein), which is increased in allografts compared with isograft controls. Treatment with the highest dose of l-NIL prevented both the increase in cleaved caspase 3 protein and the increase in caspase 3 protease activity (Fig. 5C). In contrast, treatment with a low dose of l-NIL (not shown) had little effect on the caspase-3 activation shown in Fig. 5B. Treatment with l-NIL did not alter iNOS protein levels (Fig. 6A). However, consistent with its expected mode of action, treatment with l-NIL was effective in inhibiting NO production, reflected by its ability to block the increase in NO metabolites in plasma back to isograft control levels (Fig. 6B).
FIG. 5.
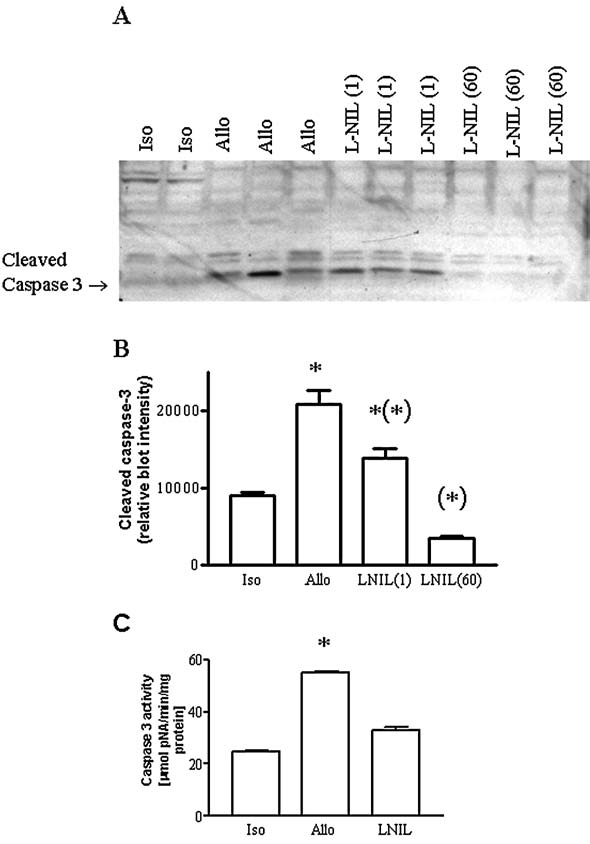
(A) Western blot, and (B) densitometry showing the increase in detection of cleaved, activated caspase 3 in individual allografts (n = 3) compared with individual isografts controls (n = 2) and inhibition by treatment with 60 μg/ml l-NIL (n = 3) but not a 1 μg/ml dose (n = 3). (C) Increase in caspase 3 protease activity was prevented by treatment with the higher dose of l-NIL (n = 5–6 each). *p < 0.05 vs. isograft and l-NIL. *p < 0.05 vs. allograft.
FIG. 6.

(A) Treatment with l-NIL had no capacity to change iNOS protein levels. (B) Increase in plasma NO metabolites was, however, blocked by treatment with l-NIL (n = 4–6 each). *p < 0.05 vs. allografts; ‡p < 0.01 vs. isografts and l-NIL.
To show the actions of various drug treatments on apoptosis, we performed additional studies by using the TUNEL staining technique. The results showed TUNEL-positive apoptotic cells in untreated allografts (Fig. 7, panels A and B). Treatment with either CsA or PBN decreased TUNEL staining and significantly (p < 0.01) inhibited the number of apoptotic cells compared with untreated allografts, whereas VitC did not produce inhibition of TUNEL staining or numbers of apoptotic cells (Fig. 7). Furthermore, TUNEL staining and numbers of increased apoptotic cells were abolished by treatment with either MnTmPyP or l-NIL (Fig. 7).
FIG. 7.
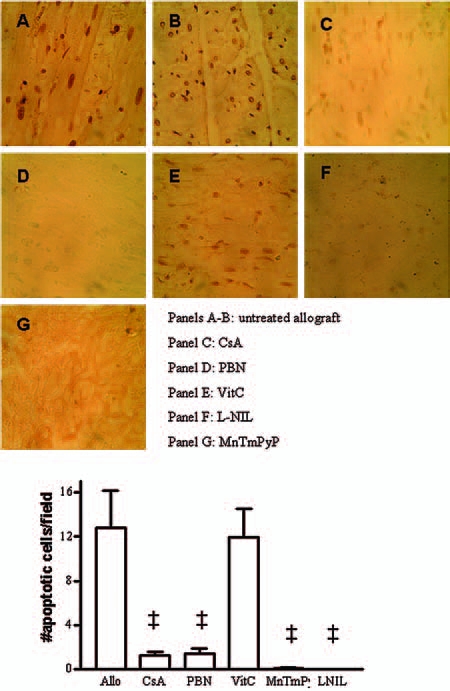
TUNEL staining (A–G) and number of apoptotic cells in cardiac allografts was inhibited or completely abolished by treatment with either CsA, PBN, MnTmPyP, or l-NIL but not VitC (n = 4 each). ‡p < 0.01 each vs. untreated allografts. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article at www.liebertonline.com/ars).
Discussion
Loss of cardiac myocytes due to apoptosis is considered to be a significant factor contributing, in part, to graft dysfunction and failure in acute cardiac rejection. A better understanding of the pathways leading to apoptosis may provide avenues to improve graft function and prevent graft failure. We used a variety of interventions, each shown to decrease histologic rejection of cardiac grafts. The new findings are that this study for the first time showed that both reactive oxygen and reactive nitrogen species contribute as signaling molecules causing activation of caspase 3 protease and apoptosis in acute cardiac allograft rejection.
Generally, it is believed that inflammatory cytokines such as TNF-α or IFN-γ, which are known to be produced during acute allograft rejection, are able to cause apoptosis of isolated cardiac myocytes in culture. Reactive oxygen species are known to be increased after exposure of cardiac myocytes to cytokines in culture and are also believed to play a significant role in graft failure in cardiac-transplant rejection. In this context, antioxidants such as N-acetylcysteine and dithiothreitol and a NOS inhibitor were shown to inhibit DNA cleavage and staining for apoptosis in neonate rat cardiac myocytes stimulated ex vivo with a combination of TNF-α, IL-1β, and IFN-γ (31).
Surprisingly, no subsequent studies to our knowledge have evaluated signaling molecules of apoptosis, such as the role of reactive oxygen species, by using either qualitative or quantitative end points under in vivo conditions of acute cardiac rejection. A role of reactive oxygen species in our model of acute cardiac rejection was implicated in the initial portions of this study by the findings that the antioxidant, vitamin C, or free radical spin-trap agent, PBN, prevented activation of caspase 3. Neither of these two agents has previously been evaluated to prevent cardiac apoptosis after inflammatory stimuli or alloimmune activation in myocardium in vivo. In the case of PBN, this treatment was associated with a downregulation in iNOS, whereas no changes in iNOS were seen with vitamin C administration. Collectively, these studies indicate for the first time that reactive oxygen species play a role in caspase 3 activation in acute cardiac allograft rejection. The role of reactive oxygen supports our previous study that used another antioxidant, a vitamin E analogue, showing attenuation of caspase-3 activation in this model (17).
The findings with vitamin C were conflicting owing to blockade of caspase 3 activation, yet without effect to limit TUNEL staining. We surmise that this could be explained by caspase 3–dependent and caspase 3–independent pathways for producing apoptosis. The action of vitamin C in biology is complex, and it displays both antioxidant and prooxidant effects (7, 16), which may be due, in part, to the presence or absence of free iron, which determines its beneficial and detrimental actions in biologic systems under inflammatory conditions.
More specifically to address the role of reactive oxygen species (specifically superoxide anion radicals), we performed additional studies by using a cell-permeable SOD mimetic, MnTmPyP. This intervention prevented activation of caspase 3 but without having any action on iNOS expression. This suggests evidence for the first time that the superoxide anion radical plays a significant role in caspase 3 activation in acute cardiac allograft rejection.
Another portion of the study was to determine the role of reactive nitrogen species, specifically the role of NO derived from iNOS on caspase 3 activation. A role of iNOS in cytokine-induced apoptosis in isolated rat neonate cardiac myocytes was implicated previously (1), but the role of iNOS in apoptosis in acute allograft rejection is incompletely understood. In a previous mouse cardiac transplant model, nitric oxide (NO) derived from iNOS of recipients was implicated as contributing to activation of caspase 3 in iNOS+/+ vs. iNOS−/− mice (12). A limitation of this study is that the absence of iNOS in recipients does not eliminate the contributions of bulk NO in the graft due to NO derived from iNOS upregulation occurring within cardiac myocytes per se of the donor. Indeed, apoptosis could be eliminated in acute cardiac allograft rejection only if iNOS was deleted in both donor and recipient mice (29).
In an earlier study using pyrimidylimidazole-based iNOS inhibitors, a decreased number of TUNEL-positive apoptotic cells was observed by this treatment strategy, suggesting a role for iNOS (31). But this intervention decreased iNOS protein expression as well suggesting an additional impact of this iNOS inhibitor secondary to decreases in inflammatory signaling. Downregulation of iNOS protein by these inhibitors has been confirmed in HEK 293 cells as well (13).
In the present study, we used the iNOS inhibitor, l-NIL, at a dose that decreased histologic rejection. We showed that the increase in functional caspase 3 protease activity in cardiac allografts was prevented by treatment with l-NIL. This treatment ablated the increase in plasma NO levels but had no effect on iNOS protein levels, suggesting that this intervention inhibited NO production by iNOS but not its expression. Therefore, we conclude that NO derived from iNOS plays a significant role as a signaling molecule for caspase 3 activation in acutely rejecting cardiac allografts. These findings were consistent with the other data showing that treatment with l-NIL also blocked the allograft-induced apoptosis.
It is interesting that MnTmPyP and l-NIL individually blocked caspase 3 activity, suggesting a role of both NO and superoxide in the development of apoptosis in acute cardiac rejection. Thus, increases in superoxide could decrease NO levels by reacting with NO to form peroxynitrite. In this context, we found that MnTmPyP increased plasma NO levels, and that this prevented activation of caspase 3 activity. TUNEL staining also confirmed effects of MnTmPyP to abolish downstream effects of caspase 3 activation on apoptosis. This shows that apoptosis is not likely due to a direct effect of NO but rather to peroxynitrite as the signaling molecule. Peroxynitrite is formed from the reaction of NO and superoxide, producing a potent oxidizing and nitrating species. This has potential significance in that addition of peroxynitrite to normal isolated rat neonate cardiac myocytes causes apoptosis (1). That peroxynitrite formation occurred in our models was shown previously by the detection of nitrotyrosine, a footprint of peroxynitrite, in rejecting cardiac allografts (18, 24). Consistent with this are other previous studies from our laboratory showing that treatment with a peroxynitrite-decomposition catalyst inhibited the increase in poly(ADP)ribose seen in acutely rejecting cardiac allografts in vivo (25). In this regard, a downstream effect of caspase 3 is to cleave poly(ADP)ribose synthase, thereby increasing levels of poly(ADP)ribose. Thus, it is possible that both inhibition of NO and superoxide by these interventions could act upstream to prevent peroxynitrite formation, which in turn could mediate activation of caspase 3 proteolytic activity and the subsequent effects of apoptosis in acute cardiac allograft rejection.
With one exception, all treatment regimens produced near or complete blockade of caspase 3 activation or TUNEL staining under conditions in which each regimen inhibited histologic rejection scores. However, the effect on rejection scores was not completely inhibited by every drug intervention. This could be explained by the fact that apoptosis is only one facet of the overall process of graft rejection and that the histologic scoring by definition includes aspects, such as grades of inflammatory cell infiltration, hemorrhage, and necrosis, that are not considered in the apoptosis analysis (28). In this context, it was reported previously in a cardiac rejection model that a greater correlation exists between apoptotic myocytes and left ventricular pressure development than with histologic rejection scores (21).
Pathways of caspase 3 activation in other posttransplant model systems
As indicated earlier, this is the first study to examine the role of NO and superoxide in signaling caspase 3 activation in acute cardiac allograft rejection. Interestingly, it was recently shown that SOD1 overexpression inhibited caspase 3 activity as well as caspase 9 activity but not caspase 8 activity at 4 h of reperfusion in a model of ischemic reperfusion injury in isogeneic cardiac transplants (32). These findings suggest a role of superoxide produced in ischemia–reperfusion preferentially on caspase 9–dependent activation in cardiac transplants. The conventional paradigm would be that activation of caspase 3 via caspase 8 and caspase 9 would reflect alloimmune-mediated (CD8, Fas-Fas ligand, TNF) vs. free radical–mediated (superoxide) pathways, respectively. It is important to underscore that the focus of the study was on prolonged ischemia with cold saline storage conditions rather than traditional organ-preserving solutions to study the effects on apoptosis after revascularization by using isogeneic cardiac transplants and its latent effects on chronic vasculopathy. The study did not address the separate issue of the effect on alloimmune activation–induced caspase 3 activation in transplanted hearts, so it is difficult to relate these results with the new findings in our model because of the dissimilarities in design.
We believe that the impact of ischemia–reperfusion injury and caspase 3 activation via reactive oxygen-mediated caspase 8 activation would not be contextually relevant to our model. The reasons are that we used short ischemic times and cold University of Wisconsin preserving solutions. Under our conditions, no obvious signs of ischemia–reperfusion injury appear in the posttransplant period. This was shown by findings in our cardiac allografts that inactivation of cardiac aconitase activity, which is very sensitive to inhibition by superoxide and well known to be inactivated after ischemia–reperfusion in heart, was not found until posttransplant day 4 (23). We also found no significant increase in iNOS protein expression in Western blots of cardiac allografts before posttransplant day 4 (26). Also, we previously reported that NF-κB is not activated at both 2 and 24 h of revascularization in our model of acute cardiac allograft rejection (27). This indicates a negligible role of reactive oxygen-induced ischemia–reperfusion injury in our model. Thus, it is unlikely that ROS-mediated apoptosis as a result of revascularization could be expected if this redox-sensitive transcription factor were not also activated. Finally, it is important to note that all of our drug treatments commenced long after revascularization and surgeries had been completed. Therefore, our interventions do not affect caspase 3 activation and apoptosis due to ischemia–reperfusion per se, yet they were clearly able to block apoptosis in allografts.
Only one published study examined caspase 8 and caspase 9 activation in any model of acute cardiac rejection (33). This study, conducted in rabbits, showed equivalent increases in both caspase 8 and 9, which would argue against a preferential caspase 8 pathway in acute rejection via alloimmune responses (i.e., CD8, Fas-Fas ligand, TNF). Another study showed that CD8 depletion did not change activation of either caspase 3 or 8 in hepatic allograft rejection (20). Finally, in a model of acute cardiac rejection (4), genetic mutations of Fas and Fas ligand that resulted in nonfunctional Fas/FasL-mediated pathways caused no change in the extent of apoptosis, leading the authors to conclude that the Fas-mediated pathway (i.e., caspase 8–mediated pathway) is not essential for acute cardiac rejection.
Collectively, based on these findings and those derived from other experimental models (5, 6, 34), the issue that reactive oxygen is solely a mediator of caspase 9 activation cannot be supported, as caspase 8 is also activated. Likewise, interrupting Fas-mediated activation is now known to inhibit both caspase 8 and caspase 9 activation. Thus, growing evidence suggests that considerable cross-talk occurs between the traditional understood pathways of activation of caspase 8 and caspase 9 by reactive oxygen species. Because of these limitations and the concept of nontraditional pathways of caspase activation, it is clear that future studies are warranted to unravel the various complex pathways leading to apoptosis in cardiac transplants, including the role of ischemic storage times, acute rejection, and chronic rejection.
Acknowledgments
This work was supported by NIH grant HL078937.
Abbreviations
CsA, cyclosporin A; iNOS, inducible nitric oxide synthase; l-NIL, l-(1-iminoethyl)lysine (l-NIL); MnTmPyP, Mn(lll)tetrakis (1-methyl-4-pyridyl)porphyrin; NO, nitric oxide; PBN, α-phenyl-t-butylnitrone; SOD, superoxide dismutase; VitC, vitamin C.
References
- 1.Arstall MA. Sawyer DB. Fukazawa R. Kelly RA. Cytokine-mediated apoptosis in cardiac myocytes: the role of inducible nitric oxide synthase induction and peroxynitrite generation. Circ Res. 1999;85:829–840. doi: 10.1161/01.res.85.9.829. [DOI] [PubMed] [Google Scholar]
- 2.Brüne B. Nitric oxide: NO apoptosis or turning it on? Cell Death Differ. 2003;10:864–869. doi: 10.1038/sj.cdd.4401261. [DOI] [PubMed] [Google Scholar]
- 3.Cooper M. Lindholm P. Pieper G. Seibel R. Moore G. Nakanishi A. Dembny K. Komorowski R. Johnson C. Adams M. Roza A. Myocardial nuclear factor-κB activity and nitric oxide production in rejecting cardiac allografts. Transplantation. 1998;66:838–844. doi: 10.1097/00007890-199810150-00005. [DOI] [PubMed] [Google Scholar]
- 4.Djamli A. Odorico JS. Fas-mediated cytotoxicity is not required for rejection of murine nonvascularized heterotopic cardiac allografts. Transplantation. 1998;66:1793–1801. doi: 10.1097/00007890-199812270-00038. [DOI] [PubMed] [Google Scholar]
- 5.Feldenberg LR. Thevananther S. del Rio M. de Leon M. Devarajan P. Partial ATP depletion induces Fas- and caspase-mediated apoptosis in MDCK cells. Am J Physiol Renal Physiol. 1999;276:F837–F846. doi: 10.1152/ajprenal.1999.276.6.F837. [DOI] [PubMed] [Google Scholar]
- 6.Gómez-Lechón MJ. Serralta A. Donato MT. Jiménez N. O'Connor E. Castell JV. Mir J. The immunosuppressant drug FK506 prevents Fas-induced apoptosis in human hepatocytes. Biochem Pharmacol. 2004;68:2427–2433. doi: 10.1016/j.bcp.2004.08.028. [DOI] [PubMed] [Google Scholar]
- 7.Hiroi M. Ogihara T. Hirano K. Hasegawa M. Morinobu T. Tamai H. Niki E. Regulation of apoptosis by glutathione redox state in PC12 cells exposed simultaneously to iron and ascorbic acid. Free Radic Biol Med. 2005;38:1057–1072. doi: 10.1016/j.freeradbiomed.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 8.Ing DJ. Zang J. Dzau VJ. Webster KA. Bishopric NH. Modulation of cytokine-induced cardiac myocyte apoptosis by nitric oxide, Bak and Bcl-xL. Circ Res. 1999;84:21–33. doi: 10.1161/01.res.84.1.21. [DOI] [PubMed] [Google Scholar]
- 9.Josien R. Müschen M. Gilbert E. Douillard P. Heslan JM. Soulillou JP. Cuturi MC. Fas ligand, tumor necrosis factor-α expression, and apoptosis during allograft rejection and tolerance. Transplantation. 1998;66:887–893. doi: 10.1097/00007890-199810150-00013. [DOI] [PubMed] [Google Scholar]
- 10.Khanna AK. Plummer MS. Hilton G. Pieper GM. Ledbetter S. Anti-transforming growth factor antibody at low but not high doses limits cyclosporine-mediated nephrotoxicity without altering rat cardiac allograft survival: potential of therapeutic applications. Circulation. 2004;110:3822–3829. doi: 10.1161/01.CIR.0000150400.15354.7D. [DOI] [PubMed] [Google Scholar]
- 11.Kim YM. Bombeck CA. Billiar TA. Nitric oxide as a bifunctional regulator of apoptosis. Circ Res. 1999;84:253–256. doi: 10.1161/01.res.84.3.253. [DOI] [PubMed] [Google Scholar]
- 12.Koglin J. Granville DJ. Glysing-Jensen T. Mudgett JS. Cathry CM. McManus BM. Russell ME. Attenuated acute cardiac rejection in NOS2−/− recipients correlates with reduced apoptosis. Circulation. 1999;99:836–842. doi: 10.1161/01.cir.99.6.836. [DOI] [PubMed] [Google Scholar]
- 13.Kolodziejski PJ. Koo JS. Eissa NT. Regulation of inducible nitric oxide synthase by rapid cellular turnover and cotranslational down-regulation by dimerization inhibitors. Proc Natl Acad Sci U S A. 2004;101:18141–18146. doi: 10.1073/pnas.0406711102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kown MH. Van der Steenhoven T. Blankenberg FG. Hoyt G. Berry GJ. Tait JF. Strauss HW. Robbins RC. Zinc-mediated reduction of apoptosis in cardiac allografts. Circulation. 2000;102(suppl III):III228–III232. doi: 10.1161/01.cir.102.suppl_3.iii-228. [DOI] [PubMed] [Google Scholar]
- 15.Liu Y. Son NH. Szabolcs MJ. Ma N. Sciacca RR. Albala A. Edwards N. Cannon PJ. Effects of inhibition of poly(adenosine diphosphate-ribose) synthase on acute cardiac allograft rejection. Transplantation. 2004;78:668–674. doi: 10.1097/01.tp.0000131662.01491.2e. [DOI] [PubMed] [Google Scholar]
- 16.Naidu KA. Vitamin C in human health and disease is still a mystery: an overview. Nutr J. 2003 2003 Aug 21;2:7. doi: 10.1186/1475-2891-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nguyen TK. Nilakantan V. Felix CC. Khanna AK. Pieper GM. Beneficial effect of α-tocopheryl succinate in rat cardiac transplants. J Heart Lung Transplant. 2006;25:707–715. doi: 10.1016/j.healun.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 18.Nilakantan V. Halligan NLN. Nguyen TK. Hilton G. Khanna AK. Roza AM. Johnson CP. Adams MB. Griffith OW. Pieper GM. Post-translational modification of manganese superoxide dismutase in acutely rejecting cardiac transplants: role of inducible nitric oxide synthase. J Heart Lung Transplant. 2005;24:1591–1599. doi: 10.1016/j.healun.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 19.Nilakantan V. Hilton G. Maenpaa C. Van Why SK. Pieper GM. Johnson CP. Shames BD. Favorable balance of anti-oxidant/pro-oxidant systems and ablated oxidative stress in Brown Norway rats in renal ischemia-reperfusion injury. Mol Cell Biochem. 2007;304:1–11. doi: 10.1007/s11010-007-9480-z. [DOI] [PubMed] [Google Scholar]
- 20.Oguru Y. Martinez O. Villanueva JC. Tait J. Strauss HW. Higgins JPT. Tanaka K. Ewquivel CO. Blankenberg FG. Krams SM. Apoptosis and allograft rejection in the absence of CD8+ cells. Transplantation. 2001;71:1827–1834. doi: 10.1097/00007890-200106270-00020. [DOI] [PubMed] [Google Scholar]
- 21.Oshima K. Sen L. Cui G. Tung T. Sacks BM. Arellano-Kruse A. Laks H. Localized interleukin-10 gene transfer induces apoptosis of alloreactive T cells via Fas/FasL pathway, improves function, and prolongs survival of cardiac allografts. Transplantation. 2002;73:1019–1026. doi: 10.1097/00007890-200204150-00002. [DOI] [PubMed] [Google Scholar]
- 22.Perry DK. Smyth MJ. Stennicke HR. Salvesen GS. Duriez P. Poirier GG. Hannun YA. Zinc is a potent inhibitor of the apoptotic protease, caspase 3: a novel target for zinc in the inhibition of apoptosis. J Biol Chem. 1997;272:18530–18533. doi: 10.1074/jbc.272.30.18530. [DOI] [PubMed] [Google Scholar]
- 23.Pieper GM. Halligan NLN. Hilton G. Konorev EA. Felix CC. Roza AM. Adams MB. Griffith OW. Non-heme iron protein: a potential target of nitric oxide in acute cardiac allograft rejection. Proc Natl Acad Sci U S A. 2003;100:3125–3130. doi: 10.1073/pnas.0636938100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pieper GM. Khanna AK. Kampalath BN. Felix CC. Hilton G. Johnson CP. Adams MB. Roza AM. Inhibition of nitrosylation, nitration and lymphocyte proliferation and gene expression in acute and delayed cardiac allograft rejection by an orally active dithiocarbamate. J Cardiovasc Pharmacol. 2004;43:522–530. doi: 10.1097/00005344-200404000-00007. [DOI] [PubMed] [Google Scholar]
- 25.Pieper GM. Nilakantan V. Chen M. Zhou J. Khanna AK. Henderson HD., Jr Johnson CP. Roza AM. Szabó C. Protective mechanisms of a metalloporphyrinic peroxynitrite decomposition catalyst, WW85, in rat cardiac transplants. J Pharmacol Exp Ther. 2005;314:53–60. doi: 10.1124/jpet.105.083493. [DOI] [PubMed] [Google Scholar]
- 26.Pieper GM. Nilakantan V. Halligan NLN. Khanna AK. Hilton G. Vásquez-Vivar J. Nitric oxide formation in acutely rejecting cardiac allografts correlates with GTP cyclohydrolase activity. Biochem J. 2005;391:541–547. doi: 10.1042/BJ20050575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pieper GM. Olds C. Hilton G. Lindholm PF. Adams MB. Roza AM. Antioxidant treatment inhibits activation of myocardial nuclear factor κB and inhibits nitrosylation of myocardial heme protein in cardiac transplant rejection. Antioxid Redox Signal. 2001;3:81–88. doi: 10.1089/152308601750100542. [DOI] [PubMed] [Google Scholar]
- 28.Pieper GM. Roza AM. Adams MB. Hilton G. Johnson C. Felix CC. Kampalath B. Darkes M. Wanggui Y. Cameron B. Fricker SP. A ruthenium (III) polyaminocarboxylate complex: a novel nitric oxide scavenger, enhances graft survival and decreases nitrosylated heme protein in models of acute and delayed cardiac transplant rejection. J Cardiovasc Pharmacol. 2002;39:441–448. doi: 10.1097/00005344-200203000-00016. [DOI] [PubMed] [Google Scholar]
- 29.Szabolcs MJ. Ma N. Athan E. Zhong J. Ming M. Sciacca RR. Huseman J. Albala A. Cannon PJ. Acute cardiac allograft rejection in nitric oxide synthase-2−/− and nitric oxide synthase-2+/+ mice: effects of cellular chimeras on myocardial inflammation and cardiomyocyte damage and apoptosis. Circulation. 2001;103:2514–2520. doi: 10.1161/01.cir.103.20.2514. [DOI] [PubMed] [Google Scholar]
- 30.Szabolcs M. Michler RE. Yang X. Aji W, Roy D, Athan E, Sciacca RR, Minanov OP, and Cannon PJ. Apoptosis of cardiac myocytes during cardiac allograft rejection: relation to induction of nitric oxide synthase. Circulation. 1996;94:1665–1673. doi: 10.1161/01.cir.94.7.1665. [DOI] [PubMed] [Google Scholar]
- 31.Szabolcs MJ. Sun J. Ma N. Albala A. Sciacca RR. Philips GB. Parkinson J. Edwards N. Cannon PJ. Effects of selective inhibitors of nitric oxide synthase-2 dimerization on acute cardiac allograft rejection. Circulation. 2002;106:2392–2396. doi: 10.1161/01.cir.0000034719.08848.26. [DOI] [PubMed] [Google Scholar]
- 32.Tanka M. Mokhtari GK. Terry RD. Balsam LB. Lee KH. Kofidis T. Tsao PS. Robbins RC. Overexpression of human copper/zinc superoxide dismutase (SOD1) suppresses ischemia-reperfusion injury and subsequent development of graft coronary artery disease in murine cardiac grafts. Circulation. 2004;110(suppl II):II200–II206. doi: 10.1161/01.CIR.0000138390.81640.54. [DOI] [PubMed] [Google Scholar]
- 33.Tung TC. Oshima K. Cui G. Laks H. Sen L. Dual upregulation of Fas and Bax promotes alloreactive T cell apoptosis in IL-10 gene targeting of cardiac allografts. Am J Physiol Heart Circ Physiol. 2003;285:H964–H973. doi: 10.1152/ajpheart.00976.2002. [DOI] [PubMed] [Google Scholar]
- 34.Zhuang S. Simon G. Peroxynitrite-induced apoptosis involves activation of multiple caspases in HL-60 cells. Am J Physiol Cell Physiol. 2000;279:C341–C351. doi: 10.1152/ajpcell.2000.279.2.C341. [DOI] [PubMed] [Google Scholar]