

Abstract
The effect of the non-selective, 1-aminoindan-1,5-dicarboxylic acid (AIDA), and selective (3,4-dihydro-2H-pyrano[2,3-b]quinolin-7-yl)-(cis-4- methoxycyclohexyl) methanone (JNJ16259685), metabotropic glutamate subtype 1 (mGlu1) receptor antagonists, on rat sciatic nerve chronic constrictive injury (CCI)- induced hyperalgesia, allodynia, spinal dorsal horn apoptosis, and gliosis was examined at 3 and 7 days post-injury. RT-PCR analysis showed increased expression of bax, apoptotic protease-activating factor-1 (apaf-1), nestin, GFAP, and caspase-7 mRNA in the dorsal horn spinal cord by 3 days post-CCI. At 7 days post-CCI, only over-expression of bcl-2, nestin and GFAP mRNA was observed. Administration of AIDA reduced thermal hyperalgesia and mechanical allodynia at 3 and 7 days post-CCI; administration of JNJ16259685 reduced thermal hyperalgesia at 3 and 7 days post-CCI, but not mechanical allodynia. AIDA decreased the mRNA levels of bax, apaf-1, GFAP and caspase-7 genes. JNJ16259685 increased the mRNA levels of bcl- 2 and GFAP gene, and decreased APAF-1 and caspases-7 genes. Inhibiting mGlu1 receptors also reduced TUNEL-positive profiles and immunohistochemical reactivity for caspase-7. We report here that despite inhibiting CCI-induced over-expression of pro-apoptotic genes in the spinal cord dorsal horn, the selective mGlu1 receptor antagonist JNJ16259685 exerted only a slight and transient allodynic effect. Moreover, JNJ16259685, but not the non-selective AIDA, increased astrogliosis which may account for its decreased analgesic efficacy. This study provides evidence that the contemporary and partial blockade of group I and likely ionotropic glutamate receptors may be a more suitable therapy than selective blockade of mGlu1 subtype receptors condition to decrease neuropathic pain symptoms.
Keywords: neuropathic pain, metabotropic glutamate receptor, apoptosis, caspase-7
Introduction
Neuropathic pain is caused by an injury or dysfunction in the peripheral or central nervous system [1, 2, 3]. In neuropathic pain conditions, noxious stimuli are perceived as more painful (hyperalgesia), and normal, harmless stimuli may elicit pain (allodynia) [1]. The mechanisms that centrally control such spread of pain stem from neurochemical and functional changes, and therefore, neuropathic pain should be considered a neuropathological condition. Evidence for this classification is offered by several animal models of chronic pain showing that a sustained release of glutamate, cytokines and neurotrophic factors (i.e. neurokinins, BDNF, TNF-alpha, etc.) induces sensitisation at both dorsal root ganglion neurons and second-order neurons in the dorsal horn [4, 5, 6]. Indeed, there is evidence that synaptic facilitation requires the release of neurokinins, glutamate, and tropomyosin-related kinase (TRK) family of neurotrophin receptors. Spinal N-methyl-D-aspartate (NMDA) glutamate receptor contributes to triggering intracellular signals that induce long lasting effects at the transcriptional level [3, 7, 8, 9, 10, 11, 12].
Moreover, not only central sensitisation, but also a disinhibitory process associated either with excitotoxic-induced neuronal death [2, 10, 13, 14] or with a rearrangement in the pathophysiology of the endogenous antinociceptive pathway [15], has been suggested to be required for the induction of dorsal horn circuitry sensitization. Morphological studies suggested the occurrence of neuronal apoptosis in the spinal cord following peripheral nerve insult [16, 17]. Nevertheless, there is also evidence that astrocytes undergo apoptosis in the spinal cord of neuropathic rat [18]. We have shown that the occurence of glutamate-dependent apoptosis may be an early and rapid event, as pro-apoptotic bax and bcl-xS mRNA levels increased in the spinal cord of rats within 2–3 days after sciatic nerve chronic constrictive injury (CCI) [19, 20]. Many studies have highlighted the involvement of metabotropic glutamate (mGlu) receptors in nociception control [21, 22, 23, 24, 25, 26]. Group I metabotropic glutamate receptors (mGluRs) (mGlu1 and -5) have been implicated in the processes of central sensitization and persistent nociception [27]. Treatment with selective antagonists for mGlu1 and mGlu5 receptors attenuated the development of mechanical allodynia and decreased extracellular glutamate in the spinal cord [28, 29]. We have previously shown that mGlu5 receptor blockade was transiently anti-allodynic and reduced spinal cord apoptosis in neuropathic rats [20], although an analysis of terminal caspase activation was not carried out in that study.
Caspases, a family of cysteine proteases, play a critical role in the execution phase of apoptosis and are responsible for many of the biochemical and morphological changes associated with apoptosis [30]. Procaspase-9 has been proposed as an initiator caspase that activates the effector caspases-3, -6, and -7 [31]. This apoptotic step is implicated in motorneuron degeneration produced by mutant superoxide dismutase-1 [32], in models of peripheral neuropathy [33], Alzheimer disease [34, 35, 36], Huntington disease [37], and neuropathic pain, suggesting a critical role for these proteases in numerous neurological conditions. To our knowledge, however, there is no study aimed at identifying the possible relationship between mGlu1 receptors, the activation of specific spinal caspases and the development of hyperalgesia/allodynia in neuropathic pain models.
In this study, we have addressed the issue of whether the blockade of mGlu1 receptors might modify early over-expression of pro-apoptotic and gliosis genes in the spinal cord dorsal horn, together with allodynia and hyperalgesia development by CCI of the sciatic nerve.
Materials and Methods
Animals
Male Wistar rats (Harlan, Udine, Italy) (250–300 g) were housed 3 per cage under controlled illumination (12:12 h light:dark cycle; light on 06.00 h) and environmental conditions (room temperature 20–22°C, humidity 55–60%) for at least 1 week before the commencement of experiments. Rat chow and tap water were available ad libitum. Behavioural testing was performed before surgery to establish a baseline for comparison with post-surgical values. CCI and sham rats were assessed for thermal and mechanical hyperalgesia before being used for further study in other tests. Rats were divided into multiple groups:
Naïve rats (n = 5);
Sham-operated rats treated for 3 days with vehicle (n = 5) or 1-aminoindan-1,5-dicarboxylic acid (AIDA) (2 mg/Kg i.p.) (n = 5); or 3,4-dihydro-2H-pyrano[2,3-b]quinolin-7-yl)-(cis-4-methoxycyclohexyl) methanone (JNJ16259685) (5 mg/Kg i.p.) (n = 5);
CCI rats treated for 3 days with vehicle (n = 5) or AIDA (2 mg/Kg i.p.) (n = 5); or JNJ16259685 (5 mg/Kg i.p.) (n = 5);
Sham-operated rats treated for 7 days with vehicle (n = 5) or AIDA (2 mg/Kg i.p.) (n = 5); or JNJ16259685 (5 mg/Kg i.p.) (n = 5);
CCI rats treated for 7 days with vehicle (n = 5) or AIDA (2 mg/Kg i.p.) (n = 5); or JNJ16259685 (5 mg/Kg i.p.) (n = 5).
In this study, a minimal still effective dose of AIDA (2 mg/kg) has been chosen based on previous evidence that it does not affect motor coordination [38] and poorly interacts with other glutamate receptor subtypes. Moreover, JNJ16259685 has been used because it is about 6000 times more potent than CPCCOEt [39].
The experimental procedures were approved by the Animal Ethics Committee of the Second University of Naples. Animal care was in compliance with the IASP and European Community (E.C. L358/1 18/12/86) guidelines on the use and protection of animals in experimental research. All efforts were made to minimise animal suffering and the number of animals used.
Drugs
The mGlu1 glutamate receptor antagonist AIDA (Tocris, Bristol, UK) was dissolved in saline with NaOH. Few drops of NaOH were supplemented until the solution was clear. The mGlu1 glutamate receptor antagonist JNJ16259685 (Johnson and Johnson Pharmaceutical Research, Janssen Pharmaceutica, Beerse, Belgium) was dissolved in Ringer solution. Vehicle was saline solution for AIDA, and the Ringer solution for JNJ16259685.
Sciatic nerve chronic constriction injury
Mononeuropathy was induced according to the method of Bennet and Xie (1988). Rats were anaesthetised with sodium pentobarbital (60 mg/kg, i.p.). The sciatic nerve was exposed at mid-thigh and freed of connective tissue, four chromic gut ligatures (4–0) were loosely tied around the nerve. Sham rats were anaesthetised, the sciatic nerve was exposed at the same level, but not ligated.
Nociceptive Behaviour
Thermal hyperalgesia was evaluated by using a Plantar Test Apparatus (Ugo Basile, Varese, Italy). On the day of the experiment each animal was placed in a plastic cage (22cm × 17cm × 14cm; length × width × height) with a glass floor. After a 30 min habituation period, the plantar surface of the hind paw was exposed to a beam of radiant heat through the glass floor. The radiant heat source consisted of an infrared bulb (Osram halogen-bellaphot bulb; 8 V, 50 W). A photoelectric cell detected light reflected from the paw and turned off the lamp when paw movement interrupted the reflected light. The paw withdrawal latency was automatically displayed to the nearest 0.1 sec; the cut-off time was 30 s in order to prevent tissue damage.
Mechanical allodynia was measured by using Dynamic Plantar Anesthesiometer (Ugo Basile, Varese, Italy). Rats were allowed to move freely in one of the two compartments of the enclosure positioned on the metal mesh surface. Rats were adapted to the testing environment before any measurements were taken. After that the mechanical stimulus was delivered to the plantar surface of the hindpaw of the rat from below the floor of the test chamber by an automated testing device. A steel rod (2 mm) was pushed with electronical ascending force (0–30 g in 10 sec). When the animal withdrew its hindpaw, the mechanical stimulus was automatically withdrawn and the force recorded to the nearest 0.1 g. Nociceptive responses for thermal and mechanical sensitivity were expressed as thermal paw withdrawal latency (PWL) in seconds and mechanical paw withdrawal threshold (PWT) in grams.
Sham and CCI rats were treated with AIDA or JNJ16259685 (AIDA: 2 mg/kg i.p., twice a day; JNJ16259685: 5 mg/Kg i.p.). Each rat served as its own control, the responses being measured both before and after surgical procedures.
RNA extraction and RT-PCR
Total RNA was extracted from homogenized dorsal sides of lumbar L4–L5 spinal cord using an RNA Tri-Reagent (Molecular Research Center Inc., Cincinnati, OH) according to the manufacturer’s protocol. The extracted RNA was subjected to DNase I treatment at 37° C for 30 min. The total RNA concentration was determined by UV spectrophotometer. The mRNA levels of the genes under analysis were measured by RT-PCR amlification, as previously reported [40]. RT minus controls were carried out to check potential genomic DNA contamination. These RT minus controls were performed without using the reverse transcriptase enzyme in the reaction mix Sequences for the rat mRNAs from GeneBank (DNASTAR INC., Madison, WI) were used to design primer pairs for RT-PCRs (OLIGO 4.05 software, National Biosciences Inc., Plymouth, MN). Each RT-PCR was repeated at least four times to achieve best reproducibility data. A semiquantitative analysis of mRNA levels was carried out by the “Gel Doc 2000 UV System” (Bio-Rad, Hercules, CA). The measured mRNA levels were normalised with respect to hypoxanthine-guanine phosphoribosyltransferase (HPRT), chosen as housekeeping gene. The HPRT gene expression did not change in several experimental conditions [41].To our knowledge there is no molecular evidence for variation in HPRT mRNA-levels in CCI neuropathic pain model. The gene expression values were expressed as arbitrary units ±SE. Amplification of genes of interest and HPRT were performed simultaneously.
Immunohistochemistry
The animals were perfused transcardially with 150 ml of saline solution (0.9% NaCl), and 250 ml of Bouin’s fixative (picric acid, 40% formaldehyde, acetic acid glacial). Lumbar spinal cord at the L4–L5 level was taken out and kept in the fixative for 24 hours followed by 75% ethanol for another 24 hours. The tissue was then dehydrated in graded ethanols and embedded in paraffin resin. Serial 5 µm sections of the lumbar segments of the spinal cord were sliced. Sections were deparaffinized by further passages in Citric Solvent (Fisher Scientific, Pittsburgh, PA) and in isopropanol. Citrate antigen retrieval was performed by placing slides in citrate buffer (0.1 M citric acid monohydrate, and 0.1 M sodium citrate, Sigma) in a water filled steamer for 20 min. After cooling, endogenous peroxidase activity was quenched by incubating sections for 15 min in 0.3% hydrogen peroxide in PBS and non-specific antibody binding was inhibited by incubation for 30 min in blocking solution (1% BSA, 0.2% powdered skim milk, 0.3% Triton-X 100 in PBS). Primary antibodies were diluted in PBS blocking buffer and slides were incubated overnight at 4°C in primary antibodies to rabbit anti-GFAP (1:10,000; DAKO, CA) and rabbit anti-caspase-7 (1:100; Cell Signaling, CA). The rabbit anti-cleaved caspase-7 antibody detects endogenous levels of the large fragment of caspases-7 resulting from cleavage at aspartic acid 198. The antibody does not cross-react with full length inactive caspases-7 or with other caspases. HRP-labelled secondary antibodies (1:1,000; Jakson Immunoresearch Laboratories, West Grove, PA) specific to the IgG species used as a primary antibody and fluorescein labeled tyramide amplification (TSA; Perkin Elmer Life Science Products, Boston, MA) were used to locate the specific antigens in each section. Sections were counterstained with bisbenzimide (Hoechst 33258) and mounted with PBS:glycerol (1:1). For GFAP-optical detection, streptavidin-labeled secondary antibodies (1:200; Molecular Probes, Eugene, OR) specific to the IgG species used as a primary antibody and DAB reaction were used to locate the specific antigens in each section. Sections were counterstained with Mayer’s hematoxyline, dehydraded in graded alcohols, fixed in xylene, and mounted with Entellan (EMS, Hatfield, PA). The staining was visualized using an optical microscope (Leica DMLB, Cambridge, UK). Positive profiles were quantified within the selected areas (498.7 mm2) in the dorsal horn by using digital system IM500 Leika.
Fluorescently labelled sections were viewed with a Zeiss Axioskop (Zeiss, Germany) epifluorescence microscope and the images were captured with a Zeiss Axiocam and Axiovison software.
We used different secondary antibodies for GFAP immunostaining in order to better clarify whether JNJ16259685 treatment was able to affect astrocyte morphology and activation rather than a quantification of increased positive cell number.
For Caspase-7 fluorescence-staining, digitised images were obtained from 5 non consecutive sections per every L4–L5 lumbar spinal cord (total sections per group = 25), using an instrument connected to an Axioskop Zeiss microscope. Quantification of numbers of Caspase-7 profiles in the superficial dorsal horn was performed with Neurolucida Software (MicroBrightField, Colchester, VT) using a 20X plan apochromat objective by an observer blind to the treatment. The areas to be analysed were outlined using NIH Image Software, and the integrate optical density (IOD) of staining in each outlined area was measured. Next, measurements from corresponding areas of the dorsal horn were compared. Staining on the control side always resembled that in sham animals; thus, a percentage difference in the IOD was calculated as 100 X (IOD CCI – IOD control)/IOD control. This measure is positive when the staining on the CCI side exceeds that on the control side and is negative when the opposite is the case. For GFAP staining quantitative assessment was carried out by determining the intensity within fixed area of the dorsal horn. A square measuring 104 µm2 was placed onto areas of the lateral, central, and medial dorsal horn. The intensity was recorded within these areas.
Detection of DNA cleavage
To analyse fragmented DNA, terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labelling (TUNEL) was performed using an in situ cell death detection kit (Roche Molecular Biochemicals, Mannheim, Germany). The kit contains terminal deoxynucleotidyl transferase, which catalyses polymerisation of fluorescein dUTP to free 3′-OH DNA ends in a template-independent manner. Incorporated fluorescein is detected by sheep anti-fluorescein Fab fragments, conjugated with horseradish peroxidase (POD). After substrate reaction, the slides were counterstained with haematoxylin. TUNEL-positive cells were identified directly by light microscopic examination. Positive cells in five non consecutive sections (10 µm), randomly selected from each L4–L5 lumbar spinal cord, were quantified by an experimenter blinded to the treatment and expressed as means ±SE per group.
Statistics
Behavioural data are represented as means ± SE. ANOVA, followed by Sheffe’s post hoc test, was used to determine the statistical significance between mean behavioural difference scores pre- and post-surgery in sciatic nerve-constricted and sham-operated animals. Histological data are shown as the means ± SE. ANOVA and Dunnett’s post hoc test was used to determine the statistical significance between groups. Molecular data are shown as means ± SE. ANOVA, followed by Student-Neuman-Keuls post hoc test was used to determine the statistical significance between groups. P<0.05 was considered statistically significant.
Results
AIDA prevents thermal hyperalgesia and mechanical allodynia by 3 and 7 days CCI, JNJ16259685 prevents thermal hyperalgesia, but not mechanical allodynia
Post-surgery mean difference scores for thermal hyperalgesia were significantly lower for the CCI sciatic nerve groups at 3 and 7 days (Fig. 1 A, B) than sham rats The increased sensitivity on the CCI sides was not present on the contralateral sides (data not shown). Similarly, sciatic nerve CCI rats showed mechanical allodynia only on the ipsilateral sides of the nerve ligature at 3 and 7 days post-surgery (Fig. 1 C, D). Administration of AIDA, a preferential mGlu1 receptor antagonist, was effective in preventing the appearance of thermal hyperalgesia and mechanical allodynia at both 3 and 7 days post-CCI (Fig. 1 A, C). Administration of JNJ16259685, a selective mGlu1 receptor antagonist, prevented the appearance of thermal hyperalgesia at both 3 and 7 days post-CCI (Fig. 1B), but not mechanical allodynia (Fig. 1D).
Figure 1.
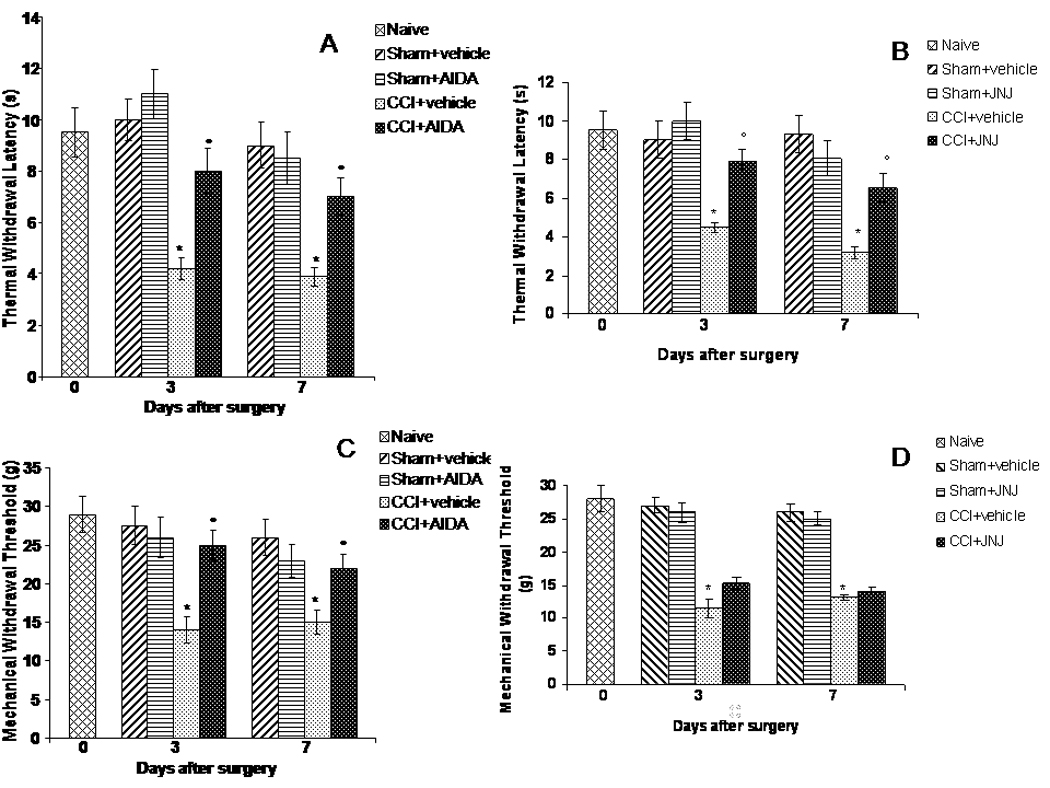
Effects of AIDA and JNJ16259685 on reflex withdrawal responses to noxious thermal and mechanical stimuli in CCI rats. Data represent the mean of thermal PWL (sec ± s.e.) and the mechanical PWT (g ± s.e.), for thermal hyperalgesia and mechanical allodynia, respectively, of 5 randomly selected rats for each histogram. The onset of CCI-induced thermal hyperalgesia was evaluated in ipsilateral (A, B) and contralateral (data not shown) sides.
Post-surgery mean difference scores for thermal hyperalgesia were significantly lower for the CCI sciatic nerve groups at 3 and 7 days (Fig. 1 A, B). The increased sensitivity on the CCI sides was not present on the contralateral sides. Similarly, sciatic nerve CCI rats showed mechanical allodynia only the ipsilateral sides of the nerve ligature at 3 and 7 days post-surgery (Fig. 1 C, D). Administration of AIDA (2 mg/Kg i.p. twice, daily), a preferential mGlu1 receptors antagonist, was effective in preventing the appearance of thermal hyperalgesia and mechanical allodynia at both 3 and 7 days post-CCI (Fig. 1 A, C). Administration of JNJ16259685 (5 mg/Kg i.p., daily), a selective mGlu1 receptors antagonist, prevented the appearance of thermal hyperalgesia at both 3 and 7 days post-CCI (Fig. 1B), but not mechanical allodynia (Fig. 1D). Vehicle is ringer solution (0.9 % NaCl). *p< 0.05 vs sham operated rats or control rats, °p< 0.05 vs CCI rats.
AIDA prevents the over-expression of bax, APAF-1 and caspase-7 genes, and increases the expression of bcl-2 gene by 3 days CCI
The semiquantitative analysis of mRNA levels measured by RT-PCR amplification, at the L4–L5 dorsal spinal cord of rats, showed an increase in the pro-apoptotic bax gene but no change in the anti-apoptotic bcl-2 gene 3 days after sciatic nerve CCI (Table 1). Indeed, at this stage the bax/bcl-2 ratio was significantly increased (mean ± SE of arbitrary units: 1.89 ± 0.17 vs 0.40 ± 0.08 in CCI with respect to the sham rats, respectively) in CCI rats. Seven days post-CCI, no change in the bax/bcl-2 ratio was observed (mean ± SE of arbitrary units: 0.30 ± 0.06 vs 0.40 ± 0.08 in CCI with respect to the sham rats, respectively) (Table 1). In sham rats, AIDA increased the expression of pro-apoptotic bax after 3 days of treatment, but not after 7 days. This fact could be due because the spinal cord mGlu receptors may potentially have a constitutive, tonic neurotrophic role, as we demonstrated in our previous work [20]. In fact, a number of factors (e.g. exposure time to drugs or ambient glutamate, NMDA receptor functional overactivation, function of astrocytes clearing extracellular glutamate and production of neurotoxic/neuroprotective factors) must be considered when examining the dual role (neurotoxicity and neuroprotection) that group I mGlu receptors play in the CNS. Moreover, AIDA also increased baseline expression of the anti-apoptotic bcl-2 after 3 days of treatment (Table 1). In CCI rats, AIDA prevented bax over-expression and increased the anti-apoptotic bcl-2 expression 3 days by sciatic nerve injury (Table 1).
Table 1.
The mRNA levels (mean±SE) of the genes under analysis measured by RT-PCR amplification are reported. Each RT-PCR was repeated at least four times. The semiquantitative analysis of mRNA levels was carried out by the “Gel Doc 2000 UV System” (Bio-Rad, Hercules, CA). The measured mRNA levels were normalised with respect to HPRT (housekeeping gene) and gene expression values were expressed as arbitrary units ± SE.
| Gene | Sham | Sham/AIDA 3 days | CCI 3 days | CCI/AIDA 3 days | Sham/AIDA 7 days | CCI 7 days | CCI/AIDA 7 days |
|---|---|---|---|---|---|---|---|
| bax/HPRT | 0.17±0.03 | 0.85±0.12* | 0.68±0.13* | 0.26±0.04° | 0.08±0.01 | 0.21±0.02 | 0.04±0.01*° |
| bcl-2/HPRT | 0.43±0.05 | 0.73±0.031 | 0.36±0.04 | 0.51±0.05° | 0.42±0.03 | 0.71±0.04* | 0.25±0.02*° |
| nestin/HPRT | 1.25±0.07 | 2.11±0.17* | 5.31±0.37* | 3.59±0.18*° | 2.14±0.19* | 2.74±0.27* | 1.54±0.15° |
| GFAP/HPRT | 5.21±0.31 | 3.77±0.34* | 13.02±0.60* | 5.22±0.36° | 2.41±0.14* | 11.04±1.21* | 4.85±0.33° |
| APAF-1/HPRT | 0.63±0.11 | 1.04±0.17* | 1.62±0.32* | 0.93±0.06° | 0.54±0.08 | 0.98±0.13* | 0.83±0.14 |
| caspase-7/HPRT | 1.02±0.06 | 1.15±0.07 | 1.64±0.08* | 1.17±0.05 | 0.99±0.06 | 0.83±0.09 | 1.04±0.04 |
p<0.05 vs the corresponding sham,
p<0.05 vs the CCI AIDA untreated rats, as analysed by ANOVA, followed by Student-Neuman-Keuls test.
To check activation of the apoptosome complex, expression of the apoptotic protease-activating factor-1 (APAF-1) was analyzed. The mRNA levels of APAF-1 increased strongly 3 days after CCI (mean ± SE of arbitrary units: 1.62 ± 0.32 vs 0.63 ± 0.11 in CCI with respect to the sham rats, respectively) (Table 1). Consistently, the expression level of the apoptotic executor gene caspase-7 was also increased 3 days following nerve injury (mean ± SE of arbitrary units: 1.64±0.08vs 1.02±0.06 in CCI with respect to the sham rats, respectively) (Table 1). Treatment with AIDA did not modify baseline APAF-1 or caspase-7 mRNA levels after 3 and 7 days of treatment; however, it reduced expression of both genes 3 days following CCI. AIDA inhibited the APAF-1 over-expression in 7 day CCI spinal cord (mean ± SE of arbitrary units: 0.93 ± 0.06 vs 1.62 ± 0.32 in CCI + treated and CCI rats, respectively). In sham rats, 3 days-treatment with AIDA increased the baseline level of APAF-1 mRNA expression (mean ± SE of arbitrary units: 1.04 ± 0.17 vs 0.63 ± 0.11) in treated and sham rats, respectively) (Table 1); however, this effect was not apparent after 7 days of treatment. For all genes examined, sham and naïve rat spinal cord showed similar levels of expression.
AIDA prevents the incidence of TUNEL-positive profiles in the dorsal horn spinal cord by 3 days post-CCI
Consistent with biomolecular data, sciatic nerve CCI induced a marked increase in the incidence of TUNEL-positive profiles by 3 days post-CCI on the side ipsilateral to the nerve injury (means ± SE: 12 ± 2.5 profiles) (Fig. 2). Treatment with AIDA in CCI, but not in sham rats, caused a significant decrease in the number of TUNEL-positive profiles in the dorsal horn spinal cord at 3 (means ± SE: 4 ± 0.8 profiles) (Fig. 2), but not 7 days after CCI (data not shown).
Figure 2.
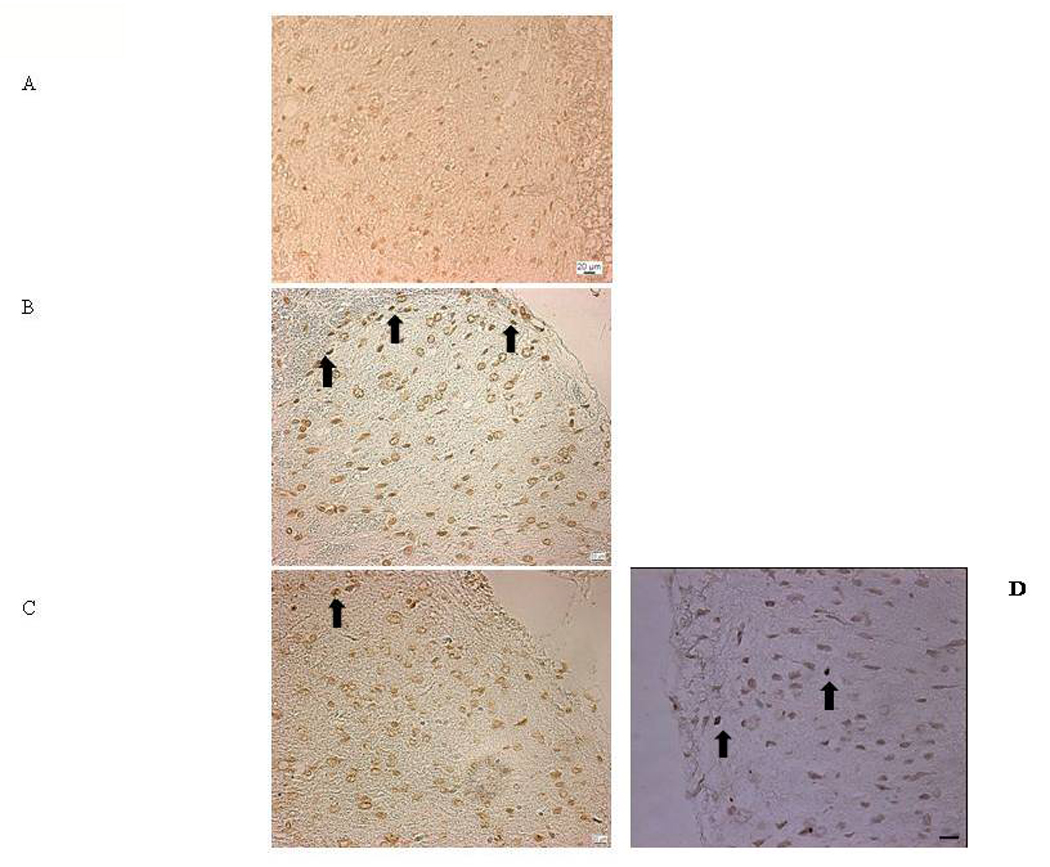
TUNEL-positive profiles in dorsal horn spinal cord of rats 3 days after sciatic nerve CCI. CCI induced a marked increase in the incidence of TUNEL-positive profiles by 3 days post-surgery in the ipsilateral CCI (B), as compared to sham-operated rats (A), as shown by arrows. AIDA treatment caused a reduction of the TUNEL-positive profiles by 3 days post-CCI (C). The same effect was observed after JNJ16259685 treatment (D). Scale bars: 20 µm.
JNJ16259685 increases expression of bcl-2 mRNA by 3 and 7 days CCI, without affecting bax gene over-expression, and prevents the over-expression of APAF-1 and caspases-7 genes
In CCI rats, JNJ16259685 treatment increased the anti-apoptotic bcl-2 expression at 3 and 7 days by sciatic nerve injury (Table 2), without affecting pro-apoptotic bax over-expression. The bax/bcl-2 ratio was decreased both at 3 days (mean ± SE of arbitrary units: 0.54 ± 0.09 vs 1.89 ± 0.13 in CCI/treated and CCI rats, respectively) and 7 days by sciatic nerve CCI (mean ± SE of arbitrary units: 0.13 ± 0.09 vs 0.30 ± 0.07 in CCI/treated and CCI rats, respectively).
Table 2.
The mRNA levels (mean±SE) of the genes under analysis measured by RT-PCR amplification are reported. Each RT-PCR was repeated at least four times. The semiquantitative analysis of mRNA levels was carried out by the “Gel Doc 2000 UV System” (Bio-Rad, Hercules, CA). The measured mRNA levels were normalised with respect to HPRT (housekeeping gene) and gene expression values were expressed as arbitrary units ± SE.
| Gene | Sham | CCI 3 days | CCI/JNJ16259685 3 days | CCI 7 days | CCI/JNJ16259685 7 days |
|---|---|---|---|---|---|
| bax/HPRT | 0.17±0.03 | 0.68±0.13* | 0.67±0.20* | 0.21±0.02 | 0.18±0.02 |
| bcl-2/HPRT | 0.43±0.05 | 0.36±0.04 | 1.25±0.18*° | 0.71±0.04* | 1.34±0.27*° |
| GFAP/HPRT | 5.21±0.31 | 13.02±0.60* | 19.79±0.35° | 11.04±1.21* | 5.83±0.18° |
| APAF-1/HPRT | 0.63±0.11 | 1.62±0.32* | 0.53±0.02° | 0.98±0.13* | 0.69±0.10° |
| caspase-7/HPRT | 1.02±0.06 | 1.64±0.08* | 0.84±0.04° | 0.83±0.09 | 1.30±0.06* |
p<0.05 vs the corresponding sham,
p<0.05 vs the CCI JNJ16259685 untreated rats, as analysed by ANOVA, followed by Student-Neuman-Keuls test. JNJ16259685 did not affect behavioural analysis in sham rats. For this reason we did not perform other experiments on JNJ16259685 treatment in sham rats.
JNJ16259685 inhibited the APAF-1 and caspase-7 gene over-expression in 3 days CCI spinal cord (APAF-1: mean ± SE of arbitrary units: 0.53 ± 0.2 vs 1.62 ± 0.32 in CCI/treated and CCI rats, respectively), (caspase-7: mean ± SE of arbitrary units: 0.84 ± 0.04 vs 1.64 ± 0.08 in CCI/treated and CCI rats, respectively). In 7 days CCI spinal cord, JNJ16259685 inhibited the APAF-1 gene over-expression (APAF-1: mean ± SE of arbitrary units: 0.69 ± 0.10 vs 0.98 ± 0.13 in CCI/treated and CCI rats, respectively), but not caspase-7 gene expression which was not modified at this time as compared to the sham operated rats (caspase-7: mean ± SE of arbitrary units: 1.30 ± 0.06 vs 0.83 ± 0.09 in CCI/treated and CCI rats, respectively)
JNJ16259685 prevents the incidence of TUNEL-positive profiles in the dorsal horn spinal cord by 3 days post-CCI
Treatment with JNJ16259685 in CCI, but not in sham rats, caused a significant decrease in the number of TUNEL-positive profiles in the dorsal horn spinal cord at 3 (means ± SE: 6 ± 0.7 profiles) (Fig. 2), but not 7 days after CCI (data not shown).
AIDA prevents the over-expression of nestin and GFAP genes by 3 and 7 days CCI
To check for the presence of gliosis, changes in the expression of GFAP and nestin reactive astrocytosis marker genes were analyzed. The mRNA levels of the GFAP gene were increased at 3 and 7 days post-CCI (Table 1). Similarly, nestin mRNA levels were increased at 3 and 7 days after CCI (Table 1). These molecular data are consistent with the changes in the size and shape of the astrocyte cell bodies (reactive gliosis) in the superficial laminae ipsilateral to CCI that we observed previously [20] and confirmed in this study (fig. 3). In sham rats, AIDA increased the baseline expression of nestin and reduced GFAP (mean ±SE of arbitrary units: 3.77 ±0.34 vs 5.21 ±0.31 in treated and untreated sham rats, respectively) after 3 days of treatment (Table 1). These changes were still apparent in sham rats after 7 days of treatment with AIDA (table 1). In CCI rats, AIDA completely abolished GFAP over-expression and also decreased nestin levels at 3 days CCI as compared to CCI untreated rats (table 1). Moreover, AIDA normalized both nestin and GFAP mRNA levels 7 days after CCI (table 1).
Figure 3.
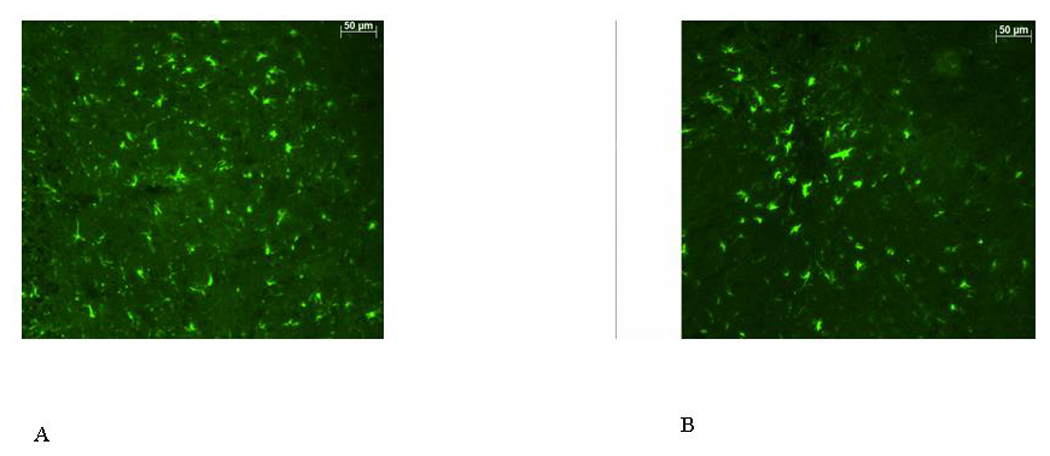
AIDA partially reduced reactive gliosis in the lumbar spinal cord of rats with CCI. GFAP immunoreactivity in 5 µm-thick nonadjacent sections from L4–L5 lumbar spinal cord from 7 days sciatic nerve CCI rats before (A) and after (B) AIDA treatment. Astrocyte morphology was characterized by changes in cell sizes after 7 days CCI. This reactive gliosis was attenuated by AIDA. Scale bar: 50 µm
Treatment with AIDA produced a reduction of GFAP immunoreactivity in the superficial dorsal horn of the spinal cord 7 days after CCI (GFAP immunoreactivity in arbitrary units ± SEM: 60.60 ± 1.89 vs 44.70 ± 1.44, in CCI-rats and CCI-AIDA treated rats, respectively; P<0.003) (Fig. 3B).
JNJ16259685 increases the expression of GFAP gene by 3 days CCI
JNJ16259685 increased the mRNA levels of GFAP gene at 3 days by sciatic nerve CCI (mean ± SE of arbitrary units: 19.79 ± 0.35 vs 13.02 ± 0.60 in CCI/treated and CCI rats, respectively)
Treatment with JNJ16259685 produced a change in GFAP positive-cell morphology in the superficial dorsal horn of the spinal cord 7 days after CCI (Fig. 4).
Figure 4.

JNJ16259685 increased reactive gliosis in the lumbar spinal cord of rats with CCI. GFAP immunoreactivity in 5 µm-thick nonadjacent sections from L4–L5 lumbar spinal cord from 7 days sciatic nerve CCI rats before (A) and after (B) JNJ16259685 treatment. Astrocyte morphology was characterized by changes in cell sizes after 7 days CCI. Scale bar: 50 µm
CCI increases caspase-7 immunoreactivity
We investigated the location of the active form of caspase-7 expression in the dorsal spinal cord following sciatic nerve injury and after treatment with AIDA or JNJ16259685 by using immunohistochemistry. As shown in fig. 5, constitutive immunohistochemical reactivity for the active form of caspase-7 was found in the superficial laminae of dorsal horn of the spinal cord of sham or naïve rats. However, a strong immunohistochemical reactivity for the active form of caspase-7 was observed at 3 (Fig. 5B), and 7 days after CCI (data not shown). The IOD of caspase-7 immunostaining was increased by 38±6% in 3 days CCI-rats vs sham rats. Seven days CCI-rats showed increased IOD of caspases-7 immunostaining by 20±5% vs sham rats. At this time point, caspase-7 enzyme activity was still present, although caspases-7 mRNA levels did not increase. The antibody used is direct toward the active form of Caspase-7, and this evidence may suggest that 7 days CCI Caspase-7 is still active in the spinal cord, with a possible role in pain maintenance. Consistent with RT-PCR analysis, AIDA reduced the staining for caspase-7 immunoreactivity in the superficial laminae of the spinal cord at 3 days post-CCI (Fig. 5C)
Figure 5.
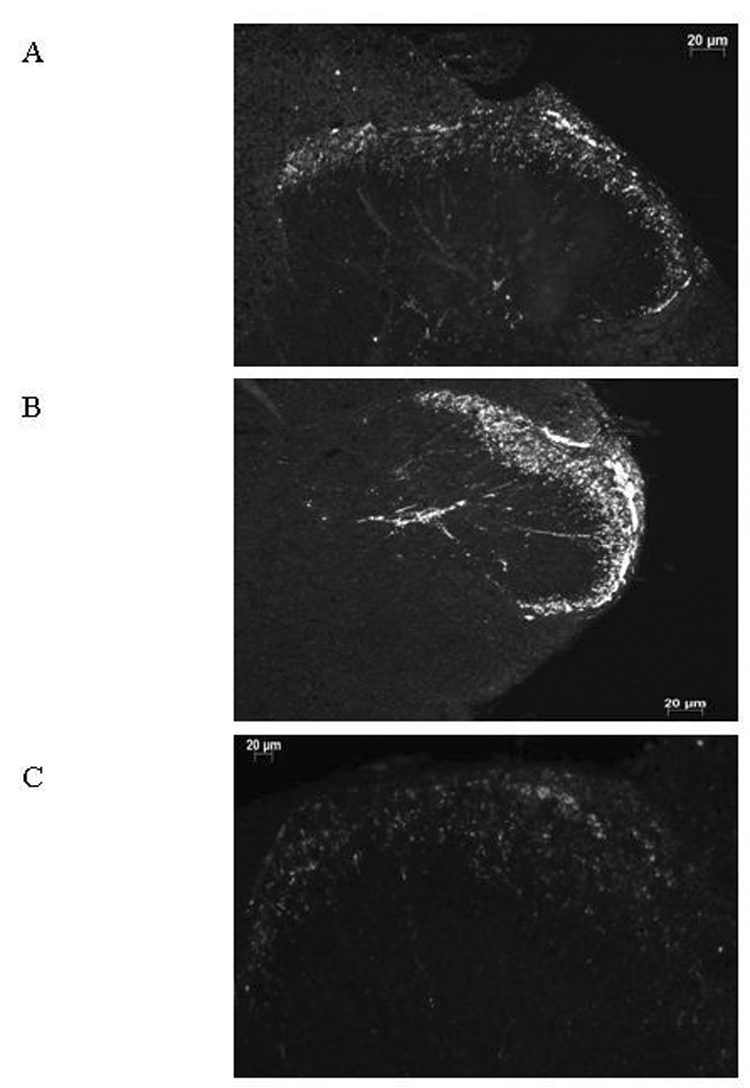
Caspase immunostaining. Caspase-7 immunostaining in lamina II spinal cord of rats 3 days after sciatic nerve CCI. Strong immunohistochemical reactivity for caspase-7 was found in the dorsal horn cells of the spinal cord at 3 days after CCI (A: sham rat; B: CCI-rat). At 3 days post-CCI, AIDA treatment resulted in reduction staining for caspase-7 immunoreactivity in the superficial laminae of the spinal cord (C). Scale bar: 20 µm.
Discussion
It has been shown that increased expression of group I mGlu receptors parallels the development of thermal hyperalgesia and mechanical allodynia, and this change has been suggested to contribute to the development and maintenance of chronic central pain syndrome [22, 24, 29, 43]. This study provides molecular evidence that, in the model of sciatic nerve ligature in the rat [40], neuropathic pain is associated with marked activation of caspase-7 together with its transient increase in expression, and apoptosis in the superficial laminae of the spinal cord.
In our previous work [19] we found that a massive neuro-protective machinery might be activated in the spinal cord dorsal horn of neuropathic rats. Fourteen days after injury we observed no sign of an active apoptotic process, as determined by TUNEL and Hoechst analysis. In addition, in other our previous work [20] we showed a possible dual role for bcl-2 family gene after sciatic nerve CCI. In the first few days post-CCI, the bax/bcl-2 ratio increased massively favouring a pro-apoptotic condition. We found that apoptosis occurred transiently within the substantia gelatinosa 3 days post-CCI. These bcl-2 family genes showed an inversed pattern of expression at later stages. Thus, there was a significant lowering in bax/bcl-2 ratio over time as a consequence of increased expression of anti-apoptotic bcl-2 gene. This fact could be the same in the case of caspases-7 gene expression found in the current study. Apoptosis and neuropathic pain develop together just in the early phase of induction and maintenance of pain. Afterward, the apoptotic process appears to be slowed and then blocked. This fact could be due to the own neuroprotection mechanisms of the nervous system [20].
We have shown in a previous study that the selective blockade of mGlu5 receptors with MPEP had anti-hyperalgesic and acute anti-allodynic effects in neuropathic rats [20]. In the current study, we show that the selective mGlu1 receptor antagonist JNJ16259685 is able to prevent thermal hyperalgesia, but not the development of mechanic al allodynia in rats. On the other hand, we show that the preferential mGlu1 receptor antagonist AIDA prevents the development of the mechanical allodynia in rats, suggesting that mGlu1, opposite to mGlu5 receptor [20], appears to be less involved in mediating abnormal mechanical responses after nerve injury. It is worthy of note that AIDA may also antagonize NMDA and AMPA ionotropic receptors [44], and that group I mGlu receptor agonists produce potentiation of the responses to NMDA and AMPA receptors [45, 46]. Given that, it is likely that combined involvement of group I metabotropic and ionotropic glutamate receptors takes part in neuropathic pain development and manteinance [47]. Our current study confirms this hypothesis. The fact that JNJ16259685 is able to increase bcl-2 expression, without affecting pro-apoptotic bax expression, might mean that blockade of mGlu1 receptors could be protective only in the early phase of neuropathic pain induction. Stimulation of these receptors up-regulates c-junand c-fosgene expression [48], in this way activating multiple downstream signalling regulating several functions including programmed cell death.
In addition to confirming the over-expression of the pro-apoptotic bax gene [20, 42], we found that APAF-1 gene expression was significantly increased in the first 2–3 days following CCI. This protein has a key role in the transactivation of the downstream effectors caspases-3 and -7 [32, 49, 50, 51, 52, 53]. Neuronal apoptosis is typically mediated by caspases, and up-regulation of caspase gene expression and protein activity have been reported in rodent models of neurological diseases [32, 54, 55]. Caspase signalling pathways may also contribute to pain-related behaviour in several models of neuropathies [33].
We have previously shown caspases-3 activation in a mouse model of CCI neuropathic pain [48]. To confirm a role for caspase signalling pathways in pain-related behaviour associated with sciatic nerve CCI, we quantified expression and localization of these proteases in the dorsal horn in neuropathic rats. We found activation of caspase-7 in the superficial laminae of the spinal cord 3 days after sciatic nerve CCI. Consistent with our observation, a few previous reports have shown highly localized expression of caspase-7 in other brain injury paradigms [36, 56].
These findings suggest that specific effector caspases may play selective roles in neurological diseases, as well as in cancer cell lines [36, 57]. It has been shown that neuronal death can proceed in the absence of caspase-3 [58] or by activation of caspase-6 and caspase-7 [59]. Caspase-7 is present in the brain and its transient (2–4 days) up-regulation and activation after traumatic brain injury has been reported [56]. Although caspase-3 and caspase-7 are both effector caspases, caspase-7 has a different preferred enzymatic site (specific substrates) as compared to caspase-3 [60]. Therefore, caspase-7 could have either a complementary or even independent role relative to caspase-3 [56]. Separate from their role as apoptotic inducers, caspases may be involved in other neural functions including synaptic plasticity. For example, caspase-3 has been found constitutively active in neurons [61], and there is evidence that caspase-3 may be involved in the long-term potentiation (LTP) phenomenon since some LTP key players are caspase-3 substrates [61]. In addition, it has been shown that blocking caspase activity is able to prevent trans-synaptic neuronal apoptosis and the loss of inhibition in lamina II of the dorsal horn after peripheral nerve injury [62]. Given that, we can not exclude a novel role for caspase-7 in controlling neuro-adaptive processes, such as learning and memory [63]. In this report, we have found constitutive activation of caspase-7 in the outer laminae of spinal cord of neuropathic rats suggesting that it may plan non-apoptotic roles in the nervous system. Our result showing that JNJ16259685 is able to block caspases-7 gene expression only at 3 days post-injury enhances this statement.
We have previously shown that in neuropathic rats the cells that over-express pro-apoptotic bax gene and undergo apoptosis are neurons [20]. It is likely, therefore, that also in this study the cells over-expressing caspase-7 may be neurons; however, we can not exclude caspase-7 over-expression in astrocytes [56].
Opposite to AIDA, the blockade of mGlu1 receptors with JNJ16259685, while having the same anti-apoptotic effect, triggered over-expression of gliosis associated genes. These data confirm an active involvement of astrocytes, which have high expression of mGlu1 receptors, in the maintenance of pain [64, 65]. Astrocytes, through their mGlu1 receptors, can maintain neuronal homeostasis by controlling the extracellular glutamate levels [66]. Activation of spinal cord astrocytes participates in neuronal plasticity, as well as in many neurodegenerative diseases [67]. Activated glia enhance pain transmission by releasing several neuroactive substances (e.g. proinflammatory/pro-allodynic cytokines, glutamate, reactive oxygen species, nitric oxide) [68, 69], which may serve as a driving force for triggering and maintaining pain [6, 68, 70]. There are also evidence to support a role of spinal astrocytes in maintaining chronic pain. In particular, c-Jun N-terminal kinase (JNK) is activated persistently in spinal astrocytes in a neuropathic pain condition produced by spinal nerve ligation. This activation is required for the maintenance of neuropathic pain [71]. Thus, the presence of activated glia could explain the mechanical allodynia that is still present with JNJ16259685 treatment. Besides astrocytes, activated spinal cord microglia show altered morphology, gene expression and function, and have an ongoing role in pain-associated behaviours [72, 73, 74]. p38 MAPK is activated in spinal microglia after nerve injury and contributes importantly to neuropathic pain development and maintenance [75]. It has been recently demonstrated that disinhibition of neurons in the superficial spinal dorsal horn, via microglia - neuron signaling leading to disruption of chloride homeostasis, is a potential cellular substrate for neuropathic pain [76]. The fact that we found here a decrease in reactive astrocytes with AIDA, together with a reduction in mechanical allodynia, might suggests a pivotal role for these cells in pain processing and maintenance. Spinal cord microglia involved in the nociceptive pathway could potentially offer a novel target for pain treatment
In conclusion, this study provides evidence that the contemporary and partial blockade of group I and likely ionotropic glutamate receptors may be a more suitable therapy than selective blockade of mGlu1 subtype receptors to decrease neuropathic pain symptoms. In the early phase of pain induction, caspase-7 could be a novel therapeutic target to reduce the pain-associated apoptotic process.
Acknowledgments
This study was supported by grant from MIUR Italy (PRIN 2005). Dr. Kevin A. Roth was supported by NIH Grant (R01 NS35107).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bonica JJ. Advances in Pain Research and Therapy. New York: Raven Press; 1970. pp. 141–166. [Google Scholar]
- 2.Zimmermann M. Pathobiology of neuropathic pain. Eur J Pharmacol. 2001;429:23–37. doi: 10.1016/s0014-2999(01)01303-6. [DOI] [PubMed] [Google Scholar]
- 3.Siniscalco D, de Novellis V, Rossi F, Maione S. Neuropathic Pain: is the end of suffering starting in the gene therapy? Curr Drug Targets. 2005;6(1):75–80. doi: 10.2174/1389450053344966. [DOI] [PubMed] [Google Scholar]
- 4.Sandkuhler J, Benrath J, Brechtel C, Ruscheweyh R, Heinke B. Synaptic mechanisms of hyperalgesia. Prog. Brain Res. 2000;129:81–100. doi: 10.1016/S0079-6123(00)29007-9. [DOI] [PubMed] [Google Scholar]
- 5.Willis WD. Role of neurotransmitters in sensitization of pain responses. In: Sorg BA, Bell IR, editors. Spinal cord plasticity in chemical intolerance. Vol. 933. Ann NY: Acad Sci.; 2001. pp. 142–156. [DOI] [PubMed] [Google Scholar]
- 6.Ledeboer A, Sloane EM, Milligan ED, Frank MG, Mahony JH, Maier SF, Watkins LR. Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain. 2005;115:71–83. doi: 10.1016/j.pain.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 7.Sher GD, Mitchell D. Intrathecal N-methyl-D-aspartate induces hyperexcitability in rat dorsal horn convergent neurones. Neurosci. Lett. 1990;119:199–202. doi: 10.1016/0304-3940(90)90833-u. [DOI] [PubMed] [Google Scholar]
- 8.Cumberbatch MJ, Herrero JF, Headley PM. Exposure of rat spinal neurones to NMDA, AMPA and kainate produces only short-term enhancements of responses to noxious and non-noxious stimuli. Neurosci. Lett. 1994;181:98–102. doi: 10.1016/0304-3940(94)90569-x. [DOI] [PubMed] [Google Scholar]
- 9.Dickenson AH, Chapman V, Green GM. The pharmacology of excitatory and inhibitory amino acid-mediated events in the transmission and modulation of pain in the spinal cord. Gen. Pharmacol. 1997;28:633–638. doi: 10.1016/s0306-3623(96)00359-x. [DOI] [PubMed] [Google Scholar]
- 10.Mayer DJ, Mao J, Holt J, Price D. Cellular mechanisms of neuropathic pain, morphine tolerance, and their interactions. Proc. Natl. Acad. Sci. USA. 1999;96:7731–7736. doi: 10.1073/pnas.96.14.7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martin WJ, Malmberg AB, Basbaum AI. PKCgamma contributes to a subset of the NMDA-dependent spinal circuits that underlie injury-induced persistent pain. J. Neurosci. 2001;21:5321–5327. doi: 10.1523/JNEUROSCI.21-14-05321.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fang L, Wu J, Lin Q, Willis WD. Calcium-calmodulin-dependent protein kinase II contributes to spinal cord central sensitisation. J. Neurosci. 2002;22:196–4204. doi: 10.1523/JNEUROSCI.22-10-04196.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mao J, Price DD, Zhu J, Lu J, Mayer DJ. The inhibition of nitric oxide-activated poly (ADP-ribose) synthetase attenuates transsynaptic alteration of spinal cord dorsal horn neurons and neuropathic pain in the rat. Pain. 1997;72:355–366. doi: 10.1016/s0304-3959(97)00063-8. [DOI] [PubMed] [Google Scholar]
- 14.Sugimoto T, Bennett GJ, Kajander KC. Transsynaptic degeneration in the superficial dorsal horn after sciatic nerve injury: effects of a chronic constriction injury, transection, and strychnine. Pain. 1990;42:205–213. doi: 10.1016/0304-3959(90)91164-E. [DOI] [PubMed] [Google Scholar]
- 15.Vanegas H, Schaible HG. Descending control of persistent pain: inhibitory or facilitatory? Brain Res Rev. 2004;46:295–309. doi: 10.1016/j.brainresrev.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 16.Kawamura T, Akira T, Watanabe M, Kagitani Y. Prostaglandin E1 prevents apoptotic cell death in superficial dorsal horn of rat spinal cord. Neuropharmacology. 1997;36:1023–1030. doi: 10.1016/s0028-3908(97)00096-8. [DOI] [PubMed] [Google Scholar]
- 17.Whiteside GT, Munglani R. Cell death in the superficial dorsal horn in a model of neuropathic pain. J Neurosci Res. 2001;64:168–173. doi: 10.1002/jnr.1062. [DOI] [PubMed] [Google Scholar]
- 18.Polgár E, Hughes DI, Arham AZ, Todd AJ. Loss of neurons from laminas I-III of the spinal dorsal horn is not required for development of tactile allodynia in the spared nerve injury model of neuropathic pain. J Neurosci. 2005;25:6658–6666. doi: 10.1523/JNEUROSCI.1490-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maione S, Siniscalco D, Galderisi U, de Novellis V, Uliano R, Di Bernardo G, Berrino L, Cascino A, Rossi F. Apoptotic genes expression in the lumbar dorsal horn in a model neuropathic pain in rat. NeuroReport. 2002;13:101–106. doi: 10.1097/00001756-200201210-00024. [DOI] [PubMed] [Google Scholar]
- 20.de Novellis V, Siniscalco D, Galderisi U, Fuccio C, Nolano M, Santoro L, Cascino A, Roth KA, Rossi F, Maione S. Blockade of glutamate mGlu5 receptors in a rat model of neuropathic pain prevents early over-expression of pro-apoptotic genes and morphological changes in dorsal horn lamina II. Neuropharmacology. 2004;46:468–479. doi: 10.1016/j.neuropharm.2003.10.014. [DOI] [PubMed] [Google Scholar]
- 21.Neugebauer V, Lucke T, Schaible HG. Requirement of metabotropic glutamate receptors for the generation of inflammation-evoked hyperexcitability in rat spinal cord neurons. Eur J Neurosci. 1994;7:1179–1186. doi: 10.1111/j.1460-9568.1994.tb00616.x. [DOI] [PubMed] [Google Scholar]
- 22.Fisher K, Fundytus ME, Cahill CM, Coderre TJ. Intrathecal administration of the mGluR compound, (S)-4CPG, attenuates hyperalgesia and allodynia associated with sciatic nerve constriction injury in rats. Pain. 1998;1:59–66. doi: 10.1016/S0304-3959(98)00082-7. [DOI] [PubMed] [Google Scholar]
- 23.Jia H, Rustioni A, Valtschanoff JG. Metabotropic glutamate receptors in superficial laminae of the rat dorsal horn. J Comp Neurol. 1999;4:627–642. [PubMed] [Google Scholar]
- 24.Maione S, Oliva P, Marabese I, Palazzo E, Rossi F, Berrino L, Filippelli A. Periaqueductal gray matter metabotropic glutamate receptors modulate formalin-induced nociception. Pain. 2000;(1–2):183–189. doi: 10.1016/s0304-3959(99)00269-9. [DOI] [PubMed] [Google Scholar]
- 25.de Novellis V, Mariani L, Palazzo E, Vita D, Marabese I, Scafuro M, Rossi F, Maione S. Periaqueductal grey cb1 cannabinoid and metabotropic glutamate subtype5 receptors modulate changes in rostral ventromedial medulla neuronal activities induced by subcutaneous formalin in the rat. Neuroscience. 2005;134:269–281. doi: 10.1016/j.neuroscience.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 26.Marabese I, Rossi F, Palazzo E, de Novellis V, Starowicz K, Cristino L, Vita D, Gatta L, Guida F, Di Marzo V, Rossi F, Maione S. Periaqueductal gray metabotropic glutamate receptor subtype 7 and 8 mediate opposite effects on amino acid release, rostral ventromedial medulla cell activities, and thermal nociception. J Neurophysiol. 2007;98:43–53. doi: 10.1152/jn.00356.2007. [DOI] [PubMed] [Google Scholar]
- 27.Chiechio S, Copani A, Melchiorri D, Canudas AM, Storto M, Calvani M, Nicolai R, Nicoletti F. Metabotropic receptors as targets for drugs of potential use in the treatment of neuropathic pain. J Endocrinol Invest. 2004;27 6 Suppl:171–176. [PubMed] [Google Scholar]
- 28.Mills CD, Xu GY, McAdoo DJ, Hulsebosch CE. Involvement of metabotropic glutamate receptors in excitatory aminoacid and GABA release following spinal cord injury in rat. J Neurochem. 2001;79:835–848. doi: 10.1046/j.1471-4159.2001.00630.x. [DOI] [PubMed] [Google Scholar]
- 29.Mills CD, Johnson KM, Hulsebosch CE. Group I metabotropic glutamate receptors in spinal cord injury: roles in neuroprotection and the development of chronic central pain. J Neurotrauma. 2002;19:23–42. doi: 10.1089/089771502753460213. [DOI] [PubMed] [Google Scholar]
- 30.Cohen GM. Caspase: the executioners of apoptosis. Biochem J. 1997;(Pt 1):1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Srinivasula SM, Ahmad M, Fernandes-Alnemri T, Alnemri ES. Autoactivation of procaspase-9 by Apaf-1 mediated oligomerization. Mol Cell. 1998;7:949–957. doi: 10.1016/s1097-2765(00)80095-7. [DOI] [PubMed] [Google Scholar]
- 32.Guegan C, Vila M, Rosoklija G, Hays AP, Przedborski S. Recruitement of the mitochondrial-dependent apoptotic pathway in amyotrophic lateral sclerosis. J. Neurosci. 2001;17:6569–6576. doi: 10.1523/JNEUROSCI.21-17-06569.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Joseph EK, Levine JD. Caspase signalling in neuropathic and inflammatory pain in the rat. Eur. J. Neurosci. 2004;20:2896–2902. doi: 10.1111/j.1460-9568.2004.03750.x. [DOI] [PubMed] [Google Scholar]
- 34.Selznick LA, Holtzman DM, Han BH, Gökden M, Srinivasan AN, Johnson EM, Jr, Roth KA. In SituImmunodetection of Neuronal Caspase-3 Activation in Alzheimer Disease. J. Neuropath. Exp. Neurol. 1999;58:1020–1026. doi: 10.1097/00005072-199909000-00012. [DOI] [PubMed] [Google Scholar]
- 35.Stadelmann C, Deckwerth TL, Srinivasan A, Bancher C, Bruck W, Jellinger K, Lassmann H. Activation of caspase-3 in single neurons and autophagic granules of granulovacuolar degeneration in Alzheimer’s disease. Evidence for apoptotic cell death. Am J Pathol. 1999;155:1459–1466. doi: 10.1016/S0002-9440(10)65460-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pompl NP, Yemul S, Xiang Z, Ho L, Haroutunian V, Purohit D, Mohs R, Pasinetti GM. Caspase gene expression in the brain as a function of the clinical progression of Alzheimer disease. Arch. Neurol. 2003;60:369–376. doi: 10.1001/archneur.60.3.369. [DOI] [PubMed] [Google Scholar]
- 37.Hermel E, Gafni J, Propp SS, Leavitt BR, Wellington CL, Young JE, Hackam AS, Logvinova AV, Peel AL, Chen SF, Hook V, Singaraja R, Krajewski S, Goldsmith PC, Ellerby HM, Hayden MR, Bredesen DE, Ellerby LM. Specific caspase interactions and amplification are involved in selective neuronal vulnerabilità in Huntington’s disease. Cell Death Differ. 2004;4:424–438. doi: 10.1038/sj.cdd.4401358. [DOI] [PubMed] [Google Scholar]
- 38.Kłodzińska A, Tatarczyńska E, Stachowicz K, Chojnacka-Wójcik E. The anxiolytic-like activity of AIDA (1-aminoindan-1,5-dicarboxylic acid), an mGLu 1 receptor antagonist. J Physiol Pharmacol. 2004;55(1 Pt 1):113–126. [PubMed] [Google Scholar]
- 39.Lavreysen H, Wouters R, Bischoff F, Nóbrega Pereira S, Langlois X, Blokland S, Somers M, Dillen L, Lesage AS. JNJ16259685, a highly potent, selective and systemically active mGlu1 receptor antagonist. Neuropharmacology. 2004;47:961–972. doi: 10.1016/j.neuropharm.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 40.Bennet JG, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- 41.Galderisi U, Di Bernardo G, Cipollaro M, Peluso G, Cascino A, Cotrufo R, Melone MA. Differentiation and apoptosis of neuroblastoma cells: role of N-myc gene product. J Cell Biochem. 1999;73:97–105. doi: 10.1002/(sici)1097-4644(19990401)73:1<97::aid-jcb11>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 42.Siniscalco D, Fuccio C, Giordano C, Ferraraccio F, Palazzo E, Luongo L, Rossi F, Roth KA, Maione S, de Novellis V. Role of reactive oxygen species and spinal cord apoptotic genes in the development of neuropathic pain. Pharmacol. Res. 2007;55:158–166. doi: 10.1016/j.phrs.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 43.Fundytus ME, Yashpal K, Chabot JG, Osborne MG, Lefebvre CD, Dray A, Henry JL, Coderre TJ. Knockdown of spinal metabotropic glutamate receptor 1 (mGluR1) alleviates pain and restores opioid efficacy after nerve injury in rats. Br. J. Pharmacol. 2001;132:354–367. doi: 10.1038/sj.bjp.0703810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Salt TE, Turner JP, Kingston AE. Evaluation of agonists and antagonists acting at group I metabotropic glutamate receptors in the thalamus in vivo. Neuropharmacology. 1999;38:1505–1510. doi: 10.1016/s0028-3908(99)00081-7. [DOI] [PubMed] [Google Scholar]
- 45.Jones MW, Headley PM. Interactions between metabotropic and ionotropic glutamate receptor agonists in the rat spinal cord in vitro. Neuropharmacology. 1995;34:1025–1031. doi: 10.1016/0028-3908(95)00055-b. [DOI] [PubMed] [Google Scholar]
- 46.Ugolini A, Corsi M, Bordi F. Potentiation of NMDA and AMPA responses by group I mGluR in spinal cord motoneurons. Neuropharmacology. 1997;36:1047–1055. doi: 10.1016/s0028-3908(97)00103-2. [DOI] [PubMed] [Google Scholar]
- 47.Zhang L, Lu Y, Chen Y, Westlund KN. Group I metabotropic glutamate recptor antagonists block secondary thermal hyperalgesia in rats with knee joint inflammation. JPET. 2002;300:149–156. doi: 10.1124/jpet.300.1.149. [DOI] [PubMed] [Google Scholar]
- 48.Miglio G, Varsaldi F, Dianzani C, Fantozzi R, Lombardi G. Stimulation of group I metabotropic glutamate receptors evokes calcium signals and c-jun and c-fos gene expression in human T cells. Biochem Pharmacol. 2005;70:189–199. doi: 10.1016/j.bcp.2005.04.038. [DOI] [PubMed] [Google Scholar]
- 49.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytocrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 50.Finucane DM, Bossy-Wetzel E, Waterhouse NJ, Cotter TG, Green DR. Bax-induced caspase activation and apoptosis via cytocrome c release from mitochondria is inhibitable by Bcl-xL. J Biol Chem. 1999;274:2225–2233. doi: 10.1074/jbc.274.4.2225. [DOI] [PubMed] [Google Scholar]
- 51.Putcha GV, Deshmukh M, Johnson EM., Jr Bax translocation is a critical event in neuronal apoptosis: regulation by neuroprotectants, BCL-2, and caspase. J Neurosci. 1999;19:7476–7485. doi: 10.1523/JNEUROSCI.19-17-07476.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Slee EA, Harte MT, Kluck RM, Wolf BB, Casiano CA, Newmeyer DD, Wang HG, Reed JC, Nicholson DW, Alnemri ES, Green DR, Martin SJ. Ordering the cytocrome c-induced caspase cascade: hierarchical activation of caspase-2, -3, -6, -7, -8, and –10 in a caspase-9-dependent manner. J Cell Biol. 1999;144:281–292. doi: 10.1083/jcb.144.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zou H, Li Y, Liu X, Wang X. An APAF-1 cytocrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem. 1999;274:11549–11556. doi: 10.1074/jbc.274.17.11549. [DOI] [PubMed] [Google Scholar]
- 54.Yakovlev AG, Faden AI. Caspase-dependent apoptotic pathways in CNS injury. Mol. Neurobiol. 2001;(1–3):131–144. doi: 10.1385/MN:24:1-3:131. [DOI] [PubMed] [Google Scholar]
- 55.Furuya T, Hayakawa H, Yamada M, Yoshimi K, Hisahara S, Miura M, Mizuno Y, Mochizuki H. Caspase-11 mediates inflammatory dopaminergic cell death in the 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine mouse model of Parkinson’s Disease. J Neurosci. 2004;8:1865–1872. doi: 10.1523/JNEUROSCI.3309-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Larner SF, McKinsey DM, Hayes RL, Wang KKW. Caspase 7: increased expression and activation after traumatic brain injury in rats. J Neurochem. 2005;94:97–108. doi: 10.1111/j.1471-4159.2005.03172.x. [DOI] [PubMed] [Google Scholar]
- 57.Lee AT, Azimahtol HL, Tan AN. Styrylpyrone derivative (SPD) induces apoptosis in a caspase-7-dependent manner in the human breast cancer cell line MCF-7. Cancer Cell Int. 2003;3:16–23. doi: 10.1186/1475-2867-3-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Young C, Roth KA, Klocke BJ, West T, Holtzman DM, Labruyere J, Qin YQ, Dikranian K, Olney JW. Role of caspase-3 in ethanol-induced developmental neurodegeneration. Neurobiol Dis. 2005;20:608–614. doi: 10.1016/j.nbd.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 59.Chee JL, Guan XL, Lee JY, Dong B, Leong SM, Ong EH, Liou AK, Lim TM. Compensatory caspase activation in MPP+-induced cell death in dopaminergic neurons. Cell Mol Life Sci. 2005;62:227–238. doi: 10.1007/s00018-004-4413-4. [DOI] [PubMed] [Google Scholar]
- 60.Riedl SJ, Fuentes-Prior P, Renatus M, Kairies N, Krapp S, Huber R, Salvesen GS, Bode W. Structural basis for the activation of human procaspase-7. PNAS. 2001;98:14790–14795. doi: 10.1073/pnas.221580098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gulyaeva NV. Non-apoptotic functions of caspase-3 in nervous tissue. Biochemistry (Mosc) 2003;68:1171–1180. doi: 10.1023/b:biry.0000009130.62944.35. [DOI] [PubMed] [Google Scholar]
- 62.Scholz J, Broom DC, Youn DH, Mills CD, Kohno T, Suter MR, Moore KA, Decosterd I, Coggeshall RE, Woolf CJ. Blocking caspase activity prevents transsynaptic neuronal apoptosis and the loss of inhibition in lamina II of the dorsal horn after peripheral nerve injury. J. Neurosci. 2005;25:7317–7323. doi: 10.1523/JNEUROSCI.1526-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stepanichev MY, Kudryashova IV, Yakovlev AA, Onufriev MV, Khaspekov LG, Lyzhin AA, Lazareva NA, Gulyaeva NV. Central administration of a caspase inhibitor impairs shuttle-box performance in rats. Neuroscience. 2005;136:579–591. doi: 10.1016/j.neuroscience.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 64.Agrawal SK, Theriault E, Fehlings MG. Role of group I metabotropic glutamate receptors in traumatic spinal cord white matter injury. J Neurotrauma. 1998;15:929–941. doi: 10.1089/neu.1998.15.929. [DOI] [PubMed] [Google Scholar]
- 65.Catania MV, Bellomo M, Di Giorgi-Gerevini V, Seminara G, Giuffrida R, Romeo R, De Blasi A, Nicoletti F. Endogenous activation of group-I metabotropic glutamate receptors is required for differentiation and survival of cerebellar Purkinje cells. J Neurosci. 2001;21:7664–7673. doi: 10.1523/JNEUROSCI.21-19-07664.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Floyd CL, Rzigalinski BA, Sitterding HA, Willoughby KA, Ellis EF. Antagonism of group I metabotropic glutamate receptors and PLC attenuates increases ininositol trisphosphate and reduces reactive gliosis in strain-injured astrocytes. J Neurotrauma. 2004;21:205–216. doi: 10.1089/089771504322778668. [DOI] [PubMed] [Google Scholar]
- 67.Barbeito LH, Pehar M, Cassina P, Vargas MR, Peluffo H, Viera L, Estevez AG, Beckman JS. A role for astrocytes in motor neuron loss in amyotrophic lateral sclerosis. Brain Res Rew. 2004;47:263–274. doi: 10.1016/j.brainresrev.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 68.Watkins L, Milligan ED, Maier SF. Glial activation: a driving force for pathological pain. Trends Neurosci. 2001;24:450–455. doi: 10.1016/s0166-2236(00)01854-3. [DOI] [PubMed] [Google Scholar]
- 69.Watkins L, Milligan ED, Maier SF. Glia: a novel drug discovery target for clinical pain. Nat Rev Drug Discov. 2003;2:973–985. doi: 10.1038/nrd1251. [DOI] [PubMed] [Google Scholar]
- 70.Ohtori S, Takahashi K, Moriya H, Myers RR. TNF-alpha and TNF-alpha receptor type 1 upregulation in glia and neurons after peripheral nerve injury: studies in murine DRG and spinal cord. Spine. 2004;29:1082–1088. doi: 10.1097/00007632-200405150-00006. [DOI] [PubMed] [Google Scholar]
- 71.Ji RR, Kawasaki Y, Zhuang ZY, Wen YR. Decosterd I. Possible role of spinal astrocytes in maintaining chronic pain sensitization: review of current evidence with focus on bFGF/JNK pathway. Neuron Glia Biol. 2006;2:259–269. doi: 10.1017/S1740925X07000403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Inoue K, Tsuda M, Koizumi S. ATP- and adenosine-mediated signaling in the central nervous system: chronic pain and microglia: involvement of the ATP receptor P2X4. J Pharmacol Sci. 2004;94:112–114. doi: 10.1254/jphs.94.112. [DOI] [PubMed] [Google Scholar]
- 73.Tsuda M, Inoue K, Salter MW. Neuropathic pain and spinal microglia: a big problem from molecules in “small” glia. Trends Neurosci. 2005;28:101–107. doi: 10.1016/j.tins.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 74.Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci. 2007;10:1361–1368. doi: 10.1038/nn1992. [DOI] [PubMed] [Google Scholar]
- 75.Ji RR, Suter MR. MAPK, microglial signaling, and neuropathic pain. Mol Pain. 2007;3(33):38. doi: 10.1186/1744-8069-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Keller AF, Beggs S, Salter MW, De Koninck Y. Transformation of the output of spinal lamina I neurons after nerve injury and microglia stimulation underlying neuropathic pain. Mol Pain. 2007;3:27. doi: 10.1186/1744-8069-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]