

Abstract
The morphogenesis of organs as diverse as lungs, teeth and hair follicles is initiated by a downgrowth from a layer of epithelial stem cells1,2. During follicular morphogenesis, stem cells form this bud structure by changing their polarity and cell–cell contacts. Here we show that this process is achieved through simultaneous receipt of two external signals: a Wnt protein to stabilize β-catenin, and a bone morphogenetic protein (BMP) inhibitor to produce Lef1. β-Catenin then binds to, and activates, Lef1 transcription complexes that appear to act uncharacteristically by downregulating the gene encoding E-cadherin, an important component of polarity and intercellular adhesion. When either signal is missing, functional Lef1 complexes are not made, and E-cadherin downregulation and follicle morphogenesis are impaired. In Drosophila, E-cadherin can influence the plane of cell division and cytoskeletal dynamics3. Consistent with this notion, we show that forced elevation of E-cadherin levels block invagination and follicle production. Our findings reveal an intricate molecular programme that links two extracellular signalling pathways to the formation of a nuclear transcription factor that acts on target genes to remodel cellular junctions and permit follicle formation.
During skin development, signals from adjacent epithelial and mesenchymal cells instruct select ectodermal cells to form hair follicle buds. In turn, each bud signals to a small group of underlying mesenchymal cells to condense1,2. Once the bud proliferates to form a larger bulb (matrix), it encases this dermal condensate (papilla), and further differentiates into the cells of the hair shaft (Fig. 1a)1. Recent evidence suggests that Wnt signalling is involved in this process at a time that correlates with bud-specific patterns of upregulation of P-cadherin and downregulation of E-cadherin4–9. Cadherins form the transmembrane core of adherens junctions (AJs) by bridging to α-catenin and the cytoskeleton through β-catenin, a protein which on its own is prone to degradation10,11. β-Catenin’s degradation machinery is transiently suppressed by Wnt signalling. This renders β-catenin a new-found stability and function, binding to and activating members of the Lef1/Tcf family of DNA binding proteins11–13. Whether there is underlying functional significance to β-catenin’s link between adhesion and transcription is an issue that we now address.
Figure 1.
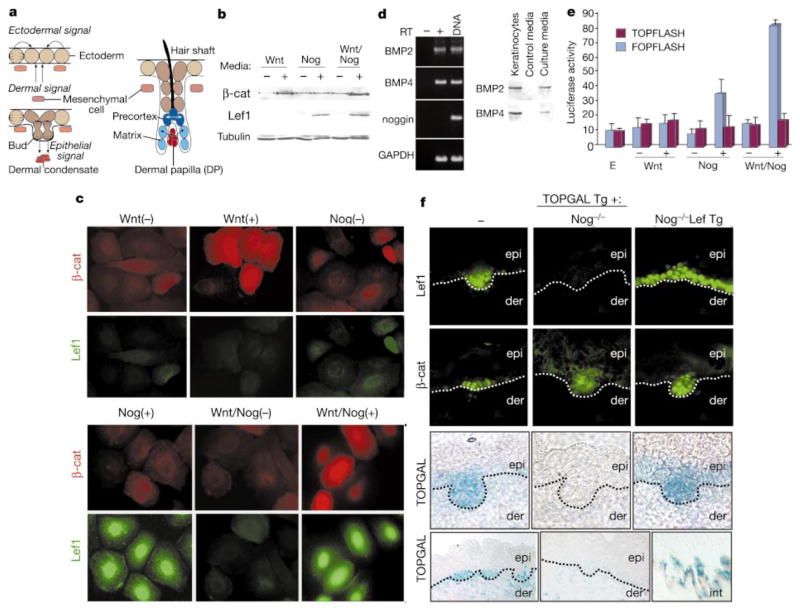
Wnt and noggin induce a transcriptionally competent Lef1 complex that is absent in Nog −/− mice. a, Cell–cell signalling (denoted by arrows) in developing follicles: noggin is expressed by mesenchyme and Wnts by ectoderm. b, SDS–PAGE immunoblots showing Wnt3a-induced β-catenin upregulation and noggin-induced Lef1 induction in lysates from keratinocytes treated with control (−) or conditioned (+) media.
c, Immunofluorescence of keratinocytes, revealing nuclear Wnt3a-induced β-catenin and noggin-induced Lef1. d, RT–PCR and immunoblot analyses revealing BMP2 and BMP4, but not noggin, in keratinocytes. e, Effect of Wnt3a and/or noggin on TOPFLASH (test) and FOPFLASH (control) in transiently transfected keratinocytes. E, low calcium media.
f, Dependency of nuclear β-catenin, Lef1 and TOPGAL expression on noggin in vivo. E16.5 skins from the knockout/transgenic (Tg) mice as indicated at top. Note that in epithelial buds lacking noggin, neither Lef1 nor TOPGAL are expressed and β-catenin is diffuse; these features are rescued on the K14-Lef1 transgenic background. Dotted lines denote mesenchymal–epithelial boundaries between epidermis (epi) and dermis (der). Bottom row are low-magnification views; lower right is of Nog −/− intestinal epithelium, which uses Tcf4, rather than Lef1, to regulate Wnt signalling7. For b, c, f, antibodies or TOPGAL activity assays are denoted at left.
Wnts are expressed in ectodermal buds14,15, and are prime candidates to stabilize β-catenin at these sites. We confirmed this by testing the ability of the canonical skin Wnt3a to generate nuclear β-catenin in mouse keratinocytes. Keratinocytes exposed to Wnt3a-conditioned media displayed an ~7 times increase in β-catenin, as judged by immunoblot and densitometry analysis (Fig. 1b). This increase was paralleled by accumulation of β-catenin in ~85 ± 5% of the nuclei of treated cells (Fig. 1c).
Wnt-treated cultures did not express appreciable Lef1, suggesting the need for additional signalling molecules to induce a DNA binding protein for β-catenin activation. As bud formation in vivo requires a mesenchymal cue1, we searched for candidates expressed by developing dermal condensates. Epithelial cells and mesenchymal cells within follicle buds express BMP2 and BMP4 (refs 16, 17), while only mesenchymal cells express their inhibitor, noggin18. Polymerase chain reaction with reverse transcription (RT–PCR) and western analyses revealed that in vitro, keratinocytes express BMPs, but not noggin (Fig. 1d). When exposed to noggin-conditioned media, Lef1 was induced, and localized to the nucleus (Fig. 1b, c).
Together, Wnt and noggin promoted transactivation of the TOPFLASH reporter gene, which regulated luciferase expression through Lef/Tcf binding sites in the TOP promoter12 (Fig. 1e). When the Tcf/Lef1 binding sites were mutated (FOPFLASH), promoter activity was abolished, as it was when noggin was absent and BMPs were present. Additionally, although some stabilization of cytoplasmic β-catenin occurred naturally in keratinocyte cultures, transactivation was clearly enhanced by Wnt3a. Thus, in vitro, noggin and Wnt3a appeared to act in concert to generate transcriptionally competent Lef1 complexes.
Noggin and Wnts also appeared to be essential for this process in vivo (Fig. 1f). In skin from embryonic day (E) 16.5, nuclear Lef1 and β-catenin could be seen in most follicles except for the mature guard follicles developing at E13.5 independently of Lef1. Lef1-positive hair buds also expressed β-galactosidase under the control of the TOP promoter7. In contrast, noggin-null (Nog−/−) mice lacked two-thirds of E16.5 buds18, and those that formed exhibited cytoplasmic β-catenin and little or no Lef1 or TOPGAL activity (Fig. 1f). When Nog−/−/TOPGAL mice were mated on a background of K14-Lef15 to force Lef1 expression, nuclear β-catenin and TOPGAL expression were restored in ~80% of the E16.5 buds (Fig. 1f). This said, follicles were not restored to wild-type levels, indicating that some of the altered genes and morphology associated with Nog−/− follicles are not under control of Lef1/β-catenin.
When cells of embryonic epidermis (epi) reorient to form an epithelial bud, they switch from E-cadherin to P-cadherin based AJs (Fig. 2a, b). This timing coincided with Lef1 and TOPGAL activation (Fig. 2c, d). At the mesenchymal–epithelial interface, the cadherin switch persisted throughout follicle development, and was still in Lef1-positive, stem cell progeny of adult follicles (Fig. 2e–g). As in buds, adult hair precursor cells were not only E-cadherin dim and P-cadherin bright, but also active for nuclear Lef1, β-catenin and TOPGAL7,19. These results were interesting in light of findings by Shimamura et al.20, who noted that in developing mouse brain, E-cadherin was absent wherever Wnt-1 was expressed. A correlation between reduced E-cadherin and elevated nuclear β-catenin has also been observed in vitro and in human cancers21.
Figure 2.
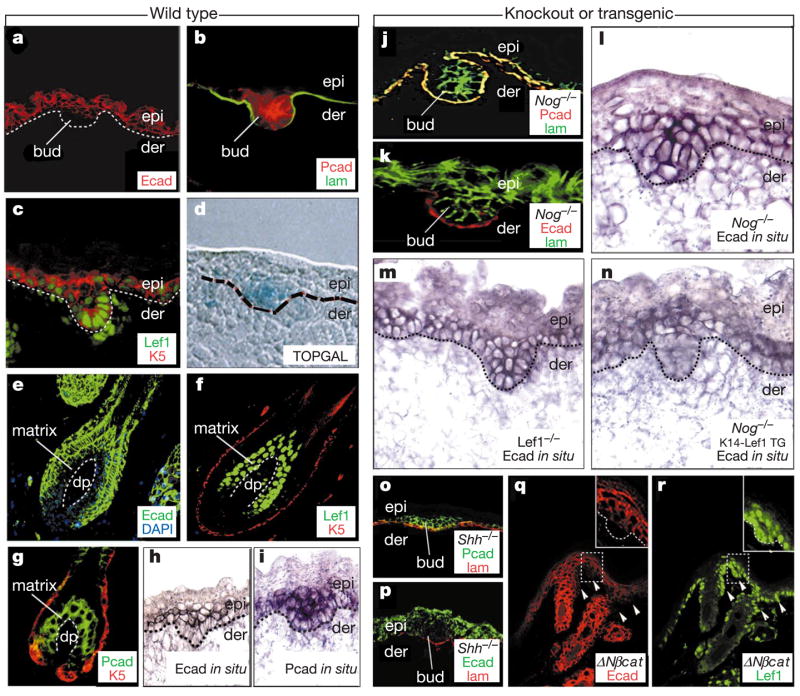
During follicle morphogenesis and cycling, skin stem cells change cadherin expression in a fashion dependent upon noggin and Wnt signalling. Wild-type skins from TOPGAL reporter mice at E16.5 (a–d, h, i) or adult (e–g) (left) or mutant E16.5 skins from the genetic backgrounds indicated (j–r) (right) were processed for double immunofluorescence, TOPGAL (β-galactosidase) activity or in situ hybridizations, using the markers indicated (colour coding denotes secondary antibodies). Wherever noggin and Wnt signalling were active, E-cadherin was downregulated, and nuclear Lef1 was present, indicating β-catenin activated Lef1 complexes. This was true even for Shh-null skin (o, p), arrested at the bud stage, and for the Lef1-positive epithelial invaginations (arrowheads) of K14-ΔNβcat transgenic (Tg) skin, expressing stable β-catenin (q, r). Without noggin, few buds formed and E-cadherin remained high (j–m), but this was rescued by K14-Lef1 (n). lam, laminin 5 antibody, demarcating the epithelial–mesenchymal boundary (elsewhere delineated by dotted lines); K5 antibody, marking the basal layer of epidermis and outer root sheath of the hair follicle. epi, embryonic epidermis; der, dermis; DP, dermal papilla.
In situ hybridization revealed that the cadherin switch was regulated at the messenger RNA level (Fig. 2h, i). Both cadherin promoters harbour multiple sequence motifs corresponding to the optimal Lef1/Tcf binding site, and the E-cadherin promoter has been shown to bind Lef122. Although P-cadherin appeared unaffected by a Nog-null or Lef1-null background, E-cadherin mRNA and protein failed to be downregulated (Fig. 2j–m), a feature which was restored when mice were bred on the K14-Lef1 background (Fig. 2n). These data reveal the E-cadherin gene as a candidate for negative repression by noggin-induced Lef1.
To further assess the extent to which E-cadherin downregulation correlated specifically with follicle downgrowth, we analysed E16.5 skin null for sonic hedgehog (Shh), which when absent impairs follicle development soon after Lef1/β-catenin activation14. Arrested Shh−/− hair buds were positive for P-cadherin, and suppressed E-cadherin at both protein (shown) and mRNA (not shown) levels (Fig. 2o, p). Taken together, these data place noggin involvement earlier than Shh, and specifically required for E-cadherin downregulation.
A priori, the negative effects of Lef1 on E-cadherin mRNA expression could be independent of β-catenin, a view consistent with how Tcf/Lef family members are known to repress target genes12. To test this possibility, we examined E-cadherin expression in skin from a mouse expressing a constitutively stable β-catenin. In interfollicular epidermis, the transgene (K14ΔNβ-catenin) elicits de novo follicle-like downgrowths6, which display evidence of Wnt-responsive gene transactivation on a TOPGAL background7. E-cadherin downregulation was consistently observed at Lef1-positive sites of K14ΔNβ-catenin induced epithelial invaginations (Fig. 2q, r). A complementary study on Wnt-1-null mice reported a broadening of the E-cadherin pattern in developing brain tissue20. Our findings now uncover a dual and atypical importance of both stabilized β-catenin and Lef1 in repressing E-cadherin expression.
To begin to address whether the changes we observed in E-cadherin mRNA expression are reflected at the transcriptional level, we first cloned 6.5 kilobases (kb) of 5′ murine E-cadherin gene sequence, and engineered a mutation in the promoter that had previously been shown by gel shift assays to bind recombinant Lef122. When tested in primary mouse keratinocytes, both wild-type and mutant promoters yielded comparable β-galactosidase reporter gene expression, indicating that the Lef1/Tcf site is not required for promoter activity (Fig. 3a).
Figure 3.
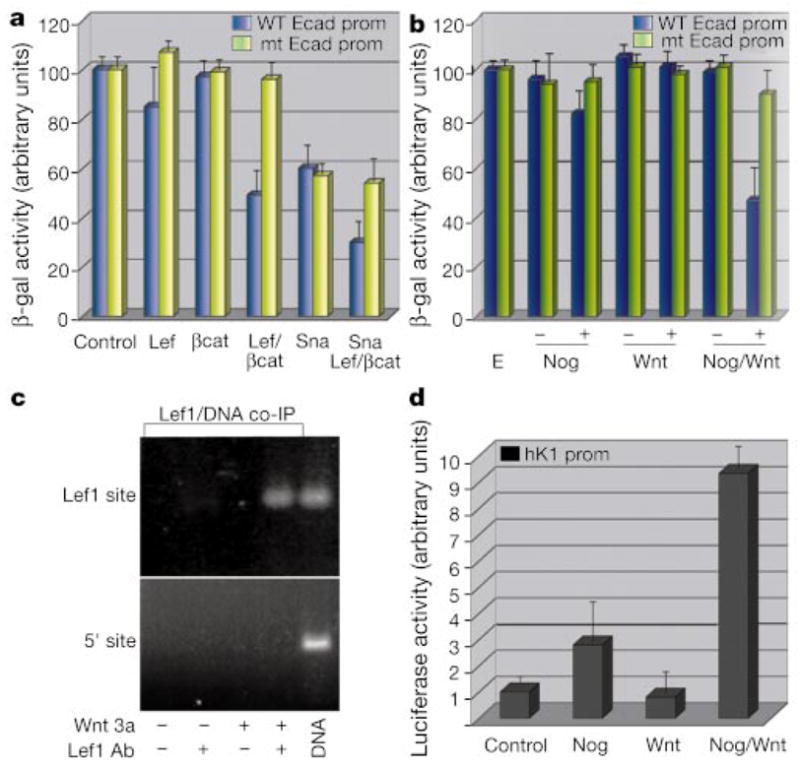
Lef1 and β-catenin transcriptionally downregulate E-cadherin in vitro. a, b, d, Promoter activity assays on extracts from keratinocytes treated without (a) or with (b, d) control (E) or conditioned media as indicated. Cells were co-transfected with E-cadherin-β-galactosidase (WT (wild-type) or mt (mutant) Lef1-site; a, b) or HK1-luciferase (d)19 ± K14-expression vectors encoding Lef (Lef1), βcat (ΔNβcat), Sna (Snail), or control (empty vector). c, Chromatin IP analyses of Lef1-expressing keratinocytes ± Wnt-conditioned medium. Fragmented, crosslinked DNA from chromatin immunoprecipitated with Lef1 or control antibody was subjected to PCR with primers encompassing the known Lef1 site or control sequence in the E-cadherin promoter. DNA, genomic control.
Transfection of either stable β-catenin or Lef1 on their own did not appreciably affect the activity of the E-cadherin promoter. In contrast, Lef1 and stabilized β-catenin in combination markedly suppressed E-cadherin promoter activity in a manner dependent upon the Lef1/Tcf binding site (Fig. 3a). Similar repression was observed when noggin and Wnt 3a together were added to the keratinocyte cultures (Fig. 3b).
To test whether Wnt-activated Lef1 complexes bind to the endogenous E-cadherin promoter, we conducted chromatin immunoprecipitation analyses (ChIP) on Lef1-expressing keratinocytes. Only when these cells were exposed to Wnt3A were we able to specifically precipitate 120-base-pair (bp) chromatin fragments of the E-cadherin promoter containing the Lef1 binding site (Fig. 3c). In contrast to the repressive effects on the E-cadherin promoter, Wnt and noggin together activated the murine HK1-hair keratin promoter, previously identified as a bona fide Lef1/β-catenin responsive target in keratinocytes (Fig. 3d)19.
Our findings suggest that Lef1’s ability to function as repressor and activator in the same Wnt-treated cells depends upon the context of responsive promoter elements. Located 3′ from the Lef1 motif in the E-cadherin promoter is an E-box sequence, which in various cell lines serves as the binding and regulatory site for the Snail family of transcriptional repressors23,24. Mutation of the Lef1 binding site did not interfere with Snail’s ability to repress the E-cadherin promoter (Fig. 3a). Moreover, these two sites together functioned additively to repress wild-type E-cadherin promoter activity by up to 70% of its normal activity in keratinocytes (Fig. 3a).
Despite the presence of functional in vitro binding sites for Snail and Lef1, the E-cadherin promoter might still be regulated in vivo by indirect pathways involving these factors. Irrespective of mechanism, the independent repressor action of Snail was intriguing given that the large guard hair follicles that develop on both the noggin-null and Lef1-null genetic backgrounds still exhibited E-cadherin downregulation (not shown). These data reveal E-cadherin down-regulation as a common thread among the waves of follicle morphogenesis and expose the existence of multiple mechanisms to govern this downregulation.
To test the functional importance of E-cadherin downregulation in hair follicle morphogenesis, we engineered transgenic mice expressing elevated levels of an epitope-tagged E-cadherin. Previous studies have shown that the addition of a carboxy-terminal tag does not interfere with E-cadherin’s ability to form intercellular junctions25. Several of the newborn animals harbouring the K14-Ecad-herin-HA transgene were sickly, and were killed shortly after birth. Immunofluorescence analyses of frozen newborn skin sections revealed a mosaic pattern of anti-HA negative (Fig. 4a) and positive (Fig. 4b) domains. Signs of the characteristic E-cadherin down-regulation were seen in the non-transgenic (Tg −) epithelium, but were not detected in transgene positive (Tg +) areas. Haematoxylin and eosin staining confirmed that the most notable morphological difference between Tg − and Tg + regions was the paucity of hair follicles in Tg + skin (Fig. 4c, d).
Figure 4.
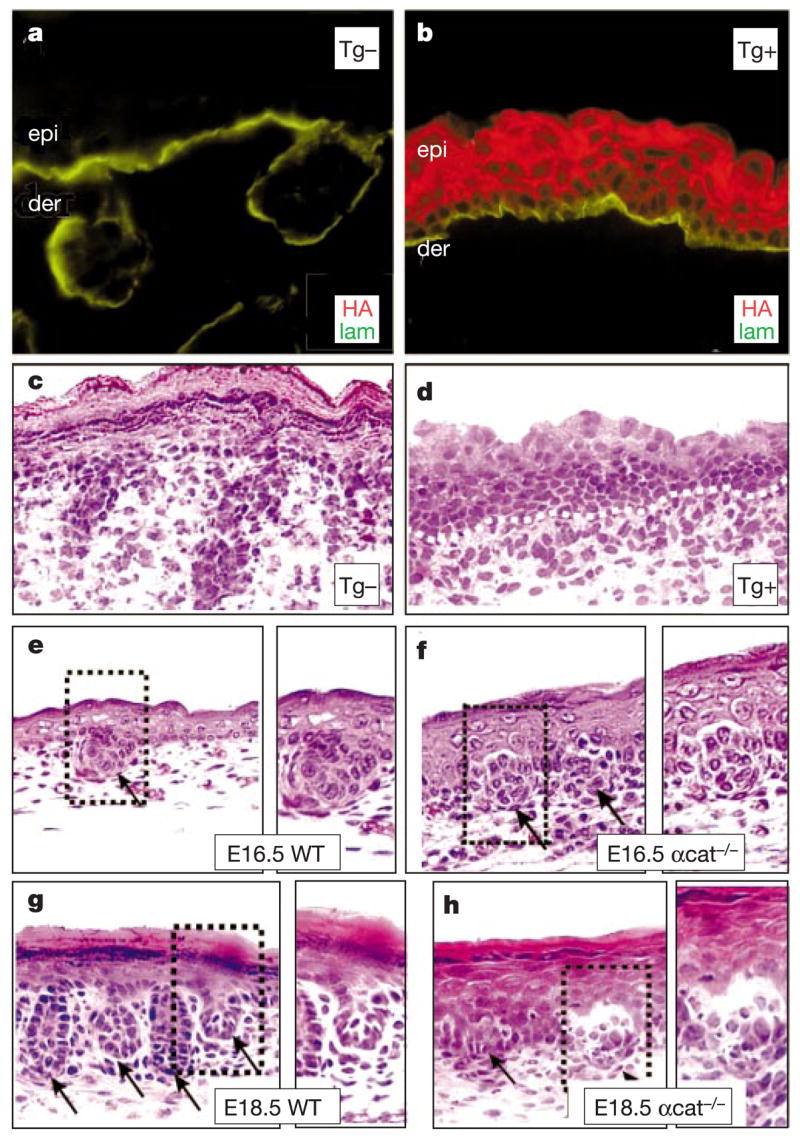
Levels of adherens junction proteins influence follicle morphogenesis. a–d, Immunofluorescence and haematoxylin-eosin staining of frozen back-skin sections from a representative newborn mouse mosaic for the K14-Ecadherin-HA transgene. Transgene-negative (Tg −) and positive (Tg +) regions were identified by anti-HA (HA), and anti-laminin 5 (lam) delineated the basement membrane. e–h, Haematoxylin-eosin staining of back-skin sections from wild-type and α-catenin K14-Cre conditional-null (αcat −/−) embryos. Boxed areas are shown at higher magnification to right of each frame. Note that in α-catenin-null skin, follicle buds (arrows) begin to form at E16.5, but cellular disorganization arrests their downgrowth by E18.5.
Skin conditionally null for a-catenin (by K14-Cre) also lacked proper AJs26. At E16.5, developing placodes were visible (Fig. 4e, f), but they were severely arrested by birth (Fig. 4g, h). Correspondingly, sebaceous glands failed to develop. These findings demonstrate the deleterious consequences of too few as well as too many AJ proteins in hair follicle morphogenesis, and provide compelling evidence that dynamic changes in AJ gene expression may be a key step in morphogenesis. In this regard, it seems particularly relevant that either complete loss or overexpression of E-cadherin can also impair formation of intestinal epithelia and mammary glands27,28. In contrast, a reduction in AJs is characteristic of epithelial cancers, whose cellular masses bear some resemblance to aberrant bud formation21. The extent to which these effects go beyond simple adhesion is at present unknown.
Our findings shed new light on the molecular mechanism underlying the early steps of epithelial bud development, a process that is anticipated to have implications that extend beyond the hair follicle. Inverse correlations between E-cadherin and Wnt signalling have been noted in various tissues and organs, during development and in cancers. In some human epithelial cancers, E-cadherin gene mutations seem to promote not only malignant invaginations but also Wnt signalling, as judged by the detection of nuclear β-catenin in the tumour tissue21. Our studies now provide evidence for a converse mechanism, namely nuclear β-catenin/Lef1 mediated suppression of E-cadherin expression during normal follicle development. The downregulation of E-cadherin may in turn perpetuate the Wnt signalling pathway by increasing the pool of transcriptionally competent β-catenin10.
Although Lef/Tcf factors are known to function as repressors, β-catenin involvement is typically associated with converting these proteins from repressors to activators12. In this regard, our in vitro results on the E-cadherin promoter are surprising, as they suggest a role for β-catenin in mediating the repressive effects of Lef1 on the E-cadherin promoter. One possibility is that the atypical context of the Lef1 binding site in the E-cadherin promoter may recruit other cofactors, such as Snail, to endow β-catenin stimulated Lef1 with this unusual repressor activity. Although we have shown that the Snail and Lef1 binding sites can function independently, Lef1’s ability to bend DNA still affords a means of bringing these complexes into contact. Additionally, β-catenin’s ability to seemingly act through both chromatin-dependent12 and independent13 mechanisms to modulate Lef1’s action hints at unexplored avenues for how these negative activities might be coordinated in vivo. But our results do not preclude the possibility that in vivo, the ability of Lef1 and β-catenin to downregulate E-cadherin mRNA expression may be indirect, perhaps through transcriptionally activating one of the E-box repressors, such as Snail, that bind to this promoter29. Once transcriptionally downregulated, E-cadherin’s relatively short half-life and multiple modes of posttranslational regulation10 may facilitate the ensuing changes that accompany follicle morphogenesis.
We have found that in noggin-regulated waves of Lef1-expressing follicles, Wnts appear to collaborate by stabilizing β-catenin so that it can bind and activate a transcription factor that participates in finely balancing the levels of E-cadherin to sculpt the epithelial bud. The sources of noggin and Wnt offer an additional level of regulation in the process. Whereas canonical Wnts have been found in skin epithelium14,15, noggin is mesenchymal18, and BMPs and their surface receptors are expressed by both mesenchyme and epithelium16,17. Our findings suggest a model in which embryonic skin epithelial stem cells require simultaneous inputs of stimulatory and inhibitory signals from multiple neighbouring cell types for the purpose of producing an activated transcription factor able to remodel AJ gene expression and form a follicle bud (see Supplementary Information). Such a mechanism furnishes an exquisite level of governance by converging signals to specify stem cell lineage.
Methods
Plasmid construction
TOPFLASH and FOPFLASH were gifts (H. Clevers). The E-cadherin promoter was generated by PCR with primers (Celera Genomics database) and a BAC clone as template. Mutation of the Lef1 binding site (−242 to −233) was achieved using the QuickChange Site-Directed Mutagenesis Kit (Stratagene). Promoter fragments were subcloned into pGL3-basic (Promega; luciferase) or pNASSβ(M) (β-galactosidase)7. HA-tagged Snail cDNA (A. Garcia de Herreros) was subcloned into the BamHI/NotI sites of the K14-cassette6. E-cadherin-HA cDNA was generated by PCR amplification of pKS + UM plasmid (R. Kemler) with a forward primer at the XhoI site (3′ end), and a reverse primer containing the HA tag, a stop codon and XbaI site. E-cadherin-HA localized to intercellular junctions in keratinocytes.
Nuclear Lef1 and β-catenin generated by noggin and Wnt3a conditioned media
Keratinocytes from newborn mouse skin were cultured in low calcium E-media7, and then either treated for (1) 12 h with control-media (Nog(−)) or conditioned-media (Nog(+)) from a noggin-secreting cell line (R. Harland), or (2) 5 h with control-media (Wnt(−)) or conditioned-media (Wnt(+)) from a Wnt3A-secreting cell line (S. Takada). In some cases, 7 h noggin/control-media was replaced with Nog/Wnt conditioned or Nog(−)/Wnt(−) media for 5 h. In all cases, EGTAwas added to 5 mM and control/conditioned media was at a final dilution of 1:5.
For harvesting, cells were washed twice with PBS and lysed in RIPA buffer (1% Triton X100, PBS, 10 mM EDTA, 150 mM NaCl, 1% sodium deoxycholate, 0.1% SDS, protease inhibitors). After determining protein concentrations (BCA, Pierce) for equal loadings, SDS–PAGE and immunoblot analyses were performed with mouse anti-tubulin (Sigma), rabbit anti-Lef1 (UC72), and mouse anti-β-catenin (Sigma). HRP-conjugated secondary-antibodies were followed by enhanced chemiluminescence (Amersham).
BMP/Noggin expression analyses
RNAs were purified by Trizol-extraction of keratinocytes (Invitrogen) and spin-column chromatography (Qiagen). Reverse transcription was performed with a Superscript kit (Invitrogen). BMP2, BMP4, noggin, and GAPDH mRNAs were detected by RT–PCR. For BMP immunodetection, 1 ml of spent-keratinocyte or control media were precipitated with two volumes of acetone, while keratinocytes were pelleted directly by centrifugation. Samples were subjected to 10% SDS–PAGE and immunoblotting with anti-BMPs (R&D Systems).
Transfections
FUGENE6 (Roche) was used to transfect newborn skin keratinocytes with expression vectors. For β-galactosidase reporters, CMV-luciferase was used to control for transfection efficiency, and for luciferase reporters, CMV-β-galactosidase was used. After 24 h, cells were treated with Wnt and/or noggin media as described. β-Galactosidase activity was measured with the Galacto-Lite-Assay Kit (Tropix Inc.) and luciferase activity by the Dual-Luciferase Kit (Promega). For standardizations, the activity in the transfected lysate of control cells was assigned an arbitrary value.
Animals
noggin and Shh null mice (A. McMahon) and Lef1 null mice (R. Grosschedl) were gifts. α-Catenin conditional null, TOPGAL, K14-Lef1 and K14-ΔNβcat animals have been described5–7,27. K14-EcadherinHA transgenic mice were generated in the laboratory.
In situ hybridization
Digoxygenin-labelled probes were synthesized with the DIG-RNA labelling kit and detected with Anti-Digoxigenin AP (both from Roche). Antisense E-cadherin cRNA was generated from T7 RNA polymerase transcription of a BglII-XhoI extracellular domain fragment that had been subcloned into pCRII (Invitrogen) and linearized with HindIII. Sense probe was generated by XhoI digestion and Sp6 RNA polymerase transcription. 3′UTR P-cadherin cRNAs were generated by subcloning a 379 bp PCR product into pCRII. Probes were applied to 15-μm sections of frozen, optimal cutting temperature (OCT) compound-embedded tissue and processed as described7.
Immunohistochemistry and tissue analysis
Sections (10 μm) of frozen embryos or derived-keratinocytes were fixed in 4% paraformaldehyde and processed for indirect immunofluorescence. Primary antibodies used were: rabbit anti-Lef1 (UC54), rat anti-E-cadherin (M. Takeichi), rat anti-P-cadherin (Zymed), guinea pig anti-K5, rat anti-HA (Roche) and rabbit anti-laminin5 (8LN5; R Burgeson). Secondary FITC (fluorescein isothiocyanate) or Texas Red conjugated antibodies (Jackson Labs) were diluted 1:100. For detection of nuclear β-catenin, whole embryos were fixed overnight in 4% PFA, dehydrated, embedded in paraffin, and rehydrated before probing with mouse anti-β-catenin antibody (Clone 15B8, Sigma). Expression of TOPGAL was determined by X-gal staining of frozen embryo sections (7.5 μm) embedded in OCT and fixed in 0.1% glutaraldehyde7. Histological analysis of embryonic skin was performed by staining with haematoxylin and eosin (Richard Allan Scientific).
ChIP and PCR
Protein-DNA complexes were crosslinked in whole cells with 1% formaldehyde, followed by sonication to fragment genomic DNA to a mid-range of 600 bp (protocols from H. Singh). DNA from anti-Lef1 immunoprecipitation was subjected to PCR using Taq-polymerase (Promega) and primers specific for a 123-bp sequence encompassing the Lef1 binding site (Lef1 site; 5′CAAAGAAAATAAAAACATAAGAAAC3′; 5′TCCTATTCCACGGTCGTTCG3′)22 and a site ~2 kb upstream (5′ site; 5′AGCACCTCTATAGATGAGGC3′, 5′TACTAAGGC-CAAAACAATCACTG3′). PCR was performed with 40 cycles of denaturation at 94 °C for 30 s, annealing at 55 °C for 45 s, 72 °C for 30 s. PCR products were separated on 1.5% agarose gels and visualized with ethidium bromide.
Acknowledgments
We thank L. Degenstein and J. Fan for transgenic technology; B. Merrill and M. Perez-Moreno for comments on the manuscript; W. Lowry and H. Rhee for sharing experimental expertise; M. Takeichi and R. Burgeson for antibodies; A. McMahon and R. Grosschedl for knockout mice; R. Kemler, A. Garcia de Herreros and H. Clevers for cDNAs and vectors; R. Harland and S. Takada for cell lines. C.J. is supported by the Helen Hay Whitney foundation. R.D. was a graduate student at the University of Chicago, where a portion of this work was conducted. This work was supported by the National Institutes of Health.
Footnotes
Supplementary Information accompanies the paper on Nature’s website
Competing interests statement The authors declare that they have no competing financial interests.
References
- 1.Hardy MH. The secret life of the hair follicle. Trends Genet. 1992;8:55–61. doi: 10.1016/0168-9525(92)90350-d. [DOI] [PubMed] [Google Scholar]
- 2.Hogan BL. Morphogenesis. Cell. 1999;96:225–233. doi: 10.1016/s0092-8674(00)80562-0. [DOI] [PubMed] [Google Scholar]
- 3.Jan Y-N, Jan L-Y. Asymmetric cell division in the Drosophila nervous system. Nature Rev Neurosci. 2001;2:772–779. doi: 10.1038/35097516. [DOI] [PubMed] [Google Scholar]
- 4.van Genderen C, et al. Development of several organs that require inductive epithelial-mesenchymal interactions is impaired in LEF-1-deficient mice. Genes Dev. 1994;8:2691–2703. doi: 10.1101/gad.8.22.2691. [DOI] [PubMed] [Google Scholar]
- 5.Zhou P, Byrne C, Jacobs J, Fuchs E. Lymphoid enhancer factor 1 directs hair follicle patterning and epithelial cell fate. Genes Dev. 1995;9:700–713. doi: 10.1101/gad.9.6.700. [DOI] [PubMed] [Google Scholar]
- 6.Gat U, DasGupta R, Degenstein L, Fuchs E. De novo hair follicle morphogenesis and hair tumors in mice expressing a truncated β-catenin in skin. Cell. 1998;95:605–614. doi: 10.1016/s0092-8674(00)81631-1. [DOI] [PubMed] [Google Scholar]
- 7.DasGupta R, Fuchs E. Multiple roles for activated LEF/TCF transcription complexes during hair follicle development and differentiation. Development. 1999;126:4557–4568. doi: 10.1242/dev.126.20.4557. [DOI] [PubMed] [Google Scholar]
- 8.Huelsken J, Vogel R, Erdmann B, Cotsarelis G, Birchmeier W. β-catenin controls hair follicle morphogenesis and stem cell differentiation in the skin. Cell. 2001;105:533–545. doi: 10.1016/s0092-8674(01)00336-1. [DOI] [PubMed] [Google Scholar]
- 9.Hirai Y, Nose A, Kobayashi S, Takeichi M. Expression and role of E- and P-cadherin adhesion molecules in embryonic histogenesis. II. Skin morphogenesis. Development. 1989;105:271–277. doi: 10.1242/dev.105.2.271. [DOI] [PubMed] [Google Scholar]
- 10.Gumbiner BM. Regulation of cadherin adhesive activity. J Cell Biol. 2000;148:399–404. doi: 10.1083/jcb.148.3.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Daniels DL, Eklof-Spink K, Weis WI. β-catenin molecular plasticity and drug design. Trends Biochem Sci. 2001;26:672–678. doi: 10.1016/s0968-0004(01)01952-1. [DOI] [PubMed] [Google Scholar]
- 12.Brantjes H, Barker N, van Es J, Clevers H. TCF Lady Justice casting the final verdict on the outcome of Wnt signalling. Biol Chem. 2002;383:255–261. doi: 10.1515/BC.2002.027. [DOI] [PubMed] [Google Scholar]
- 13.Chan S-K, Struhl G. Evidence that Armadillo transduces Wingless by mediating nuclear export or cytosolic activation of pangolin. Cell. 2002;111:265–280. doi: 10.1016/s0092-8674(02)01037-1. [DOI] [PubMed] [Google Scholar]
- 14.St-Jacques B, et al. Sonic hedgehog signaling is essential for hair development. Curr Biol. 1998;8:1058–1068. doi: 10.1016/s0960-9822(98)70443-9. [DOI] [PubMed] [Google Scholar]
- 15.Reddy S, et al. Characterization of Wnt gene expression in developing and postnatal hair follicles and identification of Wnt5a as a target of Sonic hedgehog in hair follicle morphogenesis. Mech Dev. 2001;107:69–82. doi: 10.1016/s0925-4773(01)00452-x. [DOI] [PubMed] [Google Scholar]
- 16.Kratochwil K, Dull M, Farinas I, Galceran J, Grosschedl R. Lef1 expression is activated by BMP-4 and regulates inductive tissue interactions in tooth and hair development. Genes Dev. 1996;10:1382–1394. doi: 10.1101/gad.10.11.1382. [DOI] [PubMed] [Google Scholar]
- 17.Kulessa H, Turk G, Hogan BL. Inhibition of BMP signaling affects growth and differentiation in the anagen hair follicle. EMBO J. 2000;19:6664–6674. doi: 10.1093/emboj/19.24.6664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Botchkarev VA, et al. Noggin is a mesenchymally derived stimulator of hair-follicle induction. Nature Cell Biol. 1999;1:158–164. doi: 10.1038/11078. [DOI] [PubMed] [Google Scholar]
- 19.Merrill BJ, Gat U, DasGupta R, Fuchs E. Tcf3 and Lef1 regulate lineage differentiation of multipotent stem cells in skin. Genes Dev. 2001;15:1688–1705. doi: 10.1101/gad.891401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shimamura K, Hirano S, McMahon AP, Takeichi M. Wnt-1-dependent regulation of local E-cadherin and α N-catenin expression in the embryonic mouse brain. Development. 1994;120:2225–2234. doi: 10.1242/dev.120.8.2225. [DOI] [PubMed] [Google Scholar]
- 21.Conacci-Sorrell M, Zhurinsky J, Ben-Ze’ev A. The cadherin-catenin adhesion system in signaling and cancer. J Clin Invest. 2002;109:987–991. doi: 10.1172/JCI15429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huber O, et al. Nuclear localization of β-catenin by interaction with transcription factor LEF-1. Mech Dev. 1996;59:3–10. doi: 10.1016/0925-4773(96)00597-7. [DOI] [PubMed] [Google Scholar]
- 23.Cano A, et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nature Cell Biol. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- 24.Batlle E, et al. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nature Cell Biol. 2000;2:84–89. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- 25.Adams CL, Chen YT, Smith SJ, Nelson WJ. Mechanisms of epithelial cell-cell adhesion and cell compaction revealed by high-resolution tracking of E-cadherin-green fluorescent protein. J Cell Biol. 1998;142:1105–1119. doi: 10.1083/jcb.142.4.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vasioukhin V, Bauer C, Degenstein L, Wise B, Fuchs E. Hyperproliferation and defects in epithelial polarity upon conditional ablation of α-catenin in skin. Cell. 2001;104:605–617. doi: 10.1016/s0092-8674(01)00246-x. [DOI] [PubMed] [Google Scholar]
- 27.Hermiston ML, Wong MH, Gordon JI. Forced expression of E-cadherin in the mouse intestinal epithelium slows cell migration and provides evidence for nonautonomous regulation of cell fate in a self-renewing system. Genes Dev. 1996;10:985–996. doi: 10.1101/gad.10.8.985. [DOI] [PubMed] [Google Scholar]
- 28.Boussadia O, Kutsch S, Hierholzer A, Delmas V, Kemler R. E-cadherin is a survival factor for the lactating mouse mammary gland. Mech Dev. 2002;115:53–62. doi: 10.1016/s0925-4773(02)00090-4. [DOI] [PubMed] [Google Scholar]
- 29.Kolligs FT, et al. ITF-2, a downstream target of the Wnt/TCF pathway, is activated in human cancers with β-catenin defects and promotes neoplastic transformation. Cancer Cell. 2002;1:145–155. doi: 10.1016/s1535-6108(02)00035-1. [DOI] [PubMed] [Google Scholar]