

Abstract
Elevated expression of Bmi-1 is associated with many cancers including breast cancer. Here we examined the oncogenic potential of Bmi-1 in MCF10A cells, a spontaneously immortalized, nontransformed strain of human mammary epithelial cells (HMECs). Bmi-1 overexpression alone in MCF10A cells did not result in oncogenic transformation. However, Bmi-1 co-overexpression with activated H-Ras (RasG12V) resulted in efficient transformation of MCF10A cells in vitro. Although early passage H-Ras expressing MCF10A cells were not transformed, late passage H-Ras expressing cells exhibited features of transformation in vitro. Early and late passage H-Ras expressing cells also differed in levels of expression of H-Ras, and Ki-67, a marker of proliferation. Subsets of early passage H-Ras expressing cells exhibited high Ras expression and were negative for Ki-67, whereas most late passage H-Ras expressing cells expressed low levels of Ras and were Ki-67 positive. Injection of late passage H-Ras expressing cells in SCID mice formed carcinomas with leiomatous, hemangiomatous, and mast cell components; these tumors were quite distinct from those induced by late passage cells co-overexpressing Bmi-1 and H-Ras, which formed poorly differentiated carcinomas with spindle cell features. Bmi-1 and H-Ras co-overexpression in MCF10A cells also induced features of epithelial-to-mesenchymal transition (EMT). Importantly, Bmi-1 inhibited senescence and permitted proliferation of cells expressing high levels of Ras. Examination of various growth regulatory pathways suggested that Bmi-1 overexpression together with H-Ras promotes HMEC transformation and breast oncogenesis by deregulation of multiple growth regulatory pathways by p16INK4a-independent mechanisms.
Keywords: Bmi-1, HMECs, Breast Cancer, Senescence, p53, H-Ras
Introduction
Polycomb group (PcG) of proteins play an important role as epigenetic gene silencers during development (1). In addition to their role in development, these proteins were recently reported to be overexpressed in various human cancers such as malignant lymphomas and various solid tumors (2) . In particular, Bmi-1 oncogene is overexpressed in a number of malignancies such as mantle cell lymphoma (3), B-cell non-Hodgkin's lymphoma (4), myeloid leukemia (5), non-small cell lung cancer (6), colorectal cancer (7), breast and prostate cancers (8, 9). In addition, we have recently reported that Bmi-1 is also overexpressed in head and neck cancers, in particular nasopharyngeal carcinoma and oral cancer (10, 11). Apart from its role in oncogenesis, Bmi-1 has been shown to be required for self-renewal of hematopoietic stem cells and neuronal stem cells (12-15). In addition, recently it was shown that Bmi-1 regulates self-renewal of normal and cancer stem cells in breast, and that modulation of Bmi-1 expression in mammosphere-initiating cells alters mammary development in a humanized non obese diabetic-severe combined immunodeficient mouse model (16, 17).
The polycomb proteins which are overexpressed in tumors, such as Bmi-1 and EZH2, appear to deregulate cell cycle progression (2). Recent studies using in vivo mouse and in vitro cell culture models have shown that Bmi-1 regulates the expression of INK4A/ARF locus, which encodes two important tumor suppressors p16INK4A and p19ARF (p14ARF in human) (18, 19). By downregulating p16INK4A and ARF, Bmi-1 can potentially regulate p16-pRb and p53-p21 pathways of senescence (20). Indeed, overexpression of Bmi-1 bypasses senescence in human and rodent fibroblasts, HMECs, nasopharyngeal epithelial cells and normal oral keratinocytes (11, 18, 19, 21, 22). Along these lines, we have recently reported that Bmi-1 downregulation by another PcG protein Mel-18, and Bmi-1 knockdown using an RNA interference (RNAi) approach induces premature senescence via upregulation of p16INK4A (23). Apart from regulating INK4a/ARF locus, Bmi-1 can also regulate cell proliferation and oncogenesis via INK4a/ARF-independent pathways. For example, Bmi-1 overexpression leads to immortalization of the 76N strain of HMECs via activation of telomerase (21). Like most HMECs, 76N cells do not express p16INK4a, and Bmi-1 does not appear to regulate p14ARF expression in these cells (21). In addition, we recently reported that in normal human oral keratinocytes (NHOK), and skin keratinocytes, Bmi-1 does not downregulate p16INK4A, suggesting the possible role of other unidentified targets of Bmi-1 that are involved in cell proliferation (10, 24).
Our recent data suggests that independent of its effect on p16INK4A, Bmi-1 regulates AKT activity in MCF10A and MCF7 cells (25). It is thought that the precursor cells for breast cancer are p16INK4A -negative due to promoter methylation and silencing (26), suggesting that overexpression of Bmi-1 in p16INK4A negative tumors may contribute to oncogenesis via 16INK4A-independent growth regulatory pathways. Here, we examined the oncogenic potential of Bmi-1 in an immortal but untransformed HMEC line MCF10A, which does not express p16INK4A and p14ARF due to deletion of INK4A/ARF locus (27, 28). In addition to p16INK4A, this cell line also does not express p15INK4B (27). Using in vitro cell culture and in vivo mouse models, we show that overexpression of Bmi-1 alone is not sufficient for oncogenic transformation of immortal HMECs, which lack p16INK4a. However, the combined overexpression of the G12V mutant of H-Ras and Bmi-1 was able to transform HMECs in culture as determined by transformation assays. Furthermore, orthotopic injection of cells co-overexpressing Bmi-1 and activated H-Ras resulted in the formation of poorly differentiated and invasive tumors in severe combined immunodeficient (SCID) mice.
Materials and Methods
Cells, cell culture, expression vectors, retrovirus production and infection of HMECs
MCF10A and MCF10A-derived cell lines were cultured as described (21). A retroviral vector overexpressing Bmi-1 has been described earlier (21, 23). A retroviral vector pMSCV-Ras expressing H-Ras G12V mutant was constructed by subcloning cDNA of H-Ras from pcDNA3.1 obtained from UMR cDNA Resource Center (University of Missouri, MO). Stable cell lines expressing gene(s) of interest were generated by infection of the retroviral vector(s) expressing a particular gene and selecting cells in either puromycin, G418 or hygromycin as described (21, 23). The retroviruses were produced by transient transfection of the retroviral vector together with pIK packaging plasmid into tsa 54 packaging cell line as described (21, 23).
Antibodies, Western blot analysis, immunostaining, Matrigel, soft-agar and wound healing assays
Bmi-1 was detected using either F6 mouse monoclonal antibody (mAb) from Upstate Cell Signaling Solutions (Charlottesville, VA), or 1H6B10G7 mAb from Zymed (S. San Francisco, CA). For the analysis of AKT pathway, phospho-AKT 1/2/3 (Ser-473) (sc-7985-R), AKT-1 (B-1, sc-5298) and AKT-2 (F-7, sc-5270) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Among other antibodies, CDK4 (C-22, sc260), Cyclin D1 (A-12, sc-8396), H-Ras (F-235, sc-29), p21 (F-5, sc-6246), p53 (DO-1, sc-126), p53-Ser-15 (sc-11764-R), PUMA (FL-193, sc-28226), Bax (6A7, sc-23959), ERK (C-16, sc-93), phospho-ERK (E-4, sc-7383), PRAK (A-7, sc-46667), QM (C-17, sc- 798) antibodies were obtained from Santa Cruz Biotechnology, Santa Cruz, CA. p53-Ser-37 rabbit polyclonal Ab was obtained from Cell Signaling Technology (Beverly, MA). Vimentin, Fibronectin and E-Cadherin mAbs were obtained from BD Transduction Laboratories (Franklin Lake, NJ). For immunohistochemistry, a rabbit polyclonal Ab against Vimentin was obtained from Abcam (Cambridge, MA). β-Actin and α-Smooth Muscle Actin (α-SMA) mAbs were obtained from Sigma-Aldrich (St. Louis, MO). α-Tubulin mAb was obtained from Developmental Studies Hybridoma Bank (University of Iowa, IA). For Ki-67 and H-Ras co-immunostaining, Alexa Fluor 488 conjugated Ki-67 (Cat# 558616, BD Biosciences, San Jose, CA) and a Ras mAb, (Cat# 610001, BD Biosciences, San Jose, CA) were used. Induction of oncogene induced senescence (OIS) was determined using staining for senescence-associated beta galactosidase (SA-β-Gal) marker as described (29).
To determine the AKT and ERK activity in synchronized cells, MCF10A cells were growth factor deprived using D3 medium (30) for 48 hrs and stimulated for 40 min by addition of D medium, which contains 12.5 ng/ml epidermal growth factor (EGF) (30). Western blot analyses of total cell extracts using antibodies that detect various proteins were performed as described (21, 23). Immunostaining for EMT markers such as E-Cadherin, Fibronectin and Vimentin, and soft-agar, Matrigel and wound healing assays were performed as described (25, 31).
Mice and in vivo experiments
For mammary fat pad injection experiments, four cohorts of ten SCID mice each were used. Each cohort was injected in the right axillary mammary fat pad with 1×106 cells from each cell line. Tumor growth was measured weekly by caliper, and mice were euthanized by CO2 asphyxiation once tumors reached 2 cm in diameter, or until mice became clinically ill. All animal work was performed following NIH guidelines under an approved animal protocol.
Necropsy, histopathology, histochemistry and immunohistochemistry
At necropsy examination, tumor tissue, brain, lung, heart, liver, spleen, and kidney were collected and fixed in 4% paraformaldehyde and routinely processed into paraffin blocks from which 4 μm sections were cut and stained with hematoxylin and eosin (H&E). Selected sections were also stained using Masson's Trichrome and Geimsa methods. For immunohistochemical analysis, following deparaffinization, rehydration, antigen retrieval, and quenching of endogenous peroxidase activity, polyclonal and monoclonal primary antibodies were applied. Negative controls were obtained by substitution of the primary antibody with buffer solution. Sections for vimentin staining were pretreated by microwaving in sodium citrate buffer, sections for CD31 were pretreated by microwaving in EDTA, and sections for α-SMA were not subjected to antigen retrieval. Following washing, the labeled avidin-biotinylated enzyme complex technique was employed for amplification of primary antibody binding. For visualization, 3, 3-diaminobenzidine was applied and counterstained with Mayer's hematoxylin. The sections were dehydrated through graded alcohols, immersed in xylene, and mounted with coverslips. Selected areas were photographed using an Olympus DP70 (Olympus) digital camera.
Results
Bmi-1 overexpression does not lead to transformation of HMECs
Although Bmi-1 is known to be overexpressed in many cancers including breast cancer, its exact role in oncogenesis is unclear. To define the role of Bmi-1 in breast cancer progression, we overexpressed Bmi-1 in MCF10A, a non-tumorigenic but immortal HMEC cell line (Fig. 1A). Next, we examined the oncogenic potential of MCF10A cells overexpressing Bmi-1. Consistent with recent observation that 4 or more oncogenic events are required for the in vitro transformation of HMECs (32), Bmi-1 overexpressing MCF10A cells did not form colonies in soft-agar indicating that Bmi-1 is insufficient to cause transformation of immortal HMECs, which also do not express p16INK4A and p14ARF tumor suppressors. Similar results were obtained using Bmi-1 overexpressing 76N HMECs (Supplementary Fig. S1), which do not express p16INK4A tumor suppressor and are immortal (21).
Figure 1.
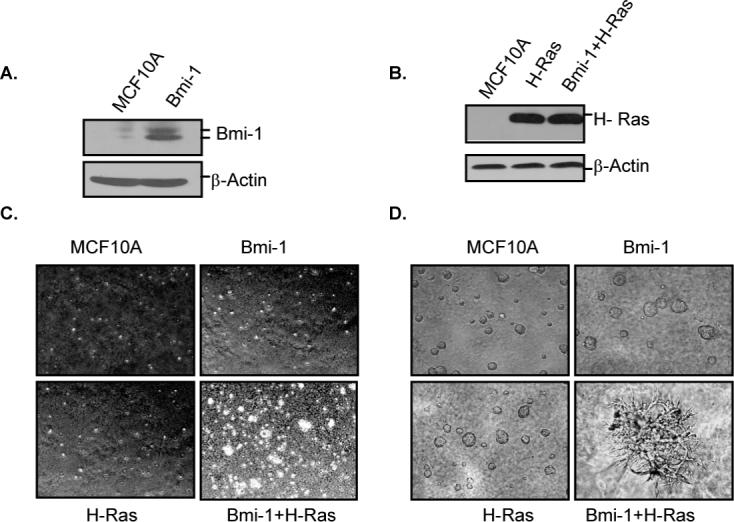
Bmi-1 and H-Ras co-overexpression transforms HMECs. A, Bmi-1 overexpressing MCF10A cells were generated by stable overexpression of Bmi-1, and cells (as indicated) were analyzed for Bmi-1 overexpression by western blot analysis. B, H-Ras was introduced in control MCF10A and MCF10A-Bmi-1 cells, and cells were analyzed for H-Ras expression by western blot analysis. Cells after Ras selection were considered at passage 1. C, MCF10A and MCF10A cells expressing H-Ras alone, Bmi-1 alone or Bmi-1 together with H-Ras (as indicated), at passage two (after Ras selection) were analyzed under light microscope for anchorage-independent growth using soft-agar assays, and photographed (4×). D, MCF10A and MCF10A-derived cells (as indicated) at passage 2 were analyzed for acini formation using Matrigel assays and photographed (6×).
Overexpression of H-Ras together with Bmi-1 transforms MCF10A cells via deregulation of multiple growth regulatory pathways
Next, we overexpressed a constitutively active mutant G12V of H-Ras (33) in control MCF10A and Bmi-1 overexpressing MCF10A cells (Fig. 1B). The pool populations of cells expressing H-Ras (MCF10A-H-Ras), Bmi-1 (MCF10A-Bmi-1) or both Bmi-1 and H-Ras (MCF10A-Bmi-1+H-Ras) were studied for transformed phenotype using soft agar and Matrigel assays (Fig. 1C and 1D). The soft-agar assay indicated that cells expressing either Bmi-1 or H-Ras alone did not exhibit anchorage-independent growth. However, cells co-overexpressing both Bmi-1 and H-Ras readily formed colonies in soft-agar (Fig. 1C). Bmi-1 and H-Ras co-overexpression in 76N cells also led to colony formation in soft-agar (Supplementary Fig. S1). Thus, our data indicate that Bmi-1 and HRas co-overexpression leads to transformation of HMECs, however, neither Bmi-1 nor H-Ras alone are sufficient to transform HMECs. To further confirm the in vitro transformation potential of MCF10A-derived cells, Bmi-1, H-Ras and Bmi-1+H-Ras expressing cells were seeded in Matrigel. After one week, cells were assessed for growth in matrigel. The results indicated that control MCF10A, MCF10A-Bmi-1 and MCF10A-H-Ras cells formed normal spherical acini, while MCF10A- Bmi-1+H-Ras cells formed large irregular branched structures indicative of transformed phenotype of seeded cells (Fig. 1D).
We have recently shown that Bmi-1 overexpression leads to activation of AKT/PKB (25), which is also a downstream target of H-Ras. The other known downstream effectors of H-Ras include extracellular signal-regulated kinases ERK1 and ERK2. To determine the mechanism of Bmi-1 and H-Ras induced transformation of HMECS, we analyzed MCF10A and MCF10A derived cells for the expression of AKT and ERK kinases. The cells were starved for 48 hrs and then EGF containing D-medium was added for 40 min. The activity of ERK and AKT kinases was determined by Western blot analysis.
The results indicated that control MCF10A and MCF10A-Ras cells had very little or no basal phospho-AKT (pAKT) expression, while MCF10A-Bmi-1 and MCF10A-Bmi-1+H-Ras cells expressed significant amount of activated AKT (pAKT) even under EGF starved conditions (Fig. 2A). AKT activity was induced in all cells after EGF addition, however, the induction of AKT activity was more noticeable in Bmi-1+H-Ras expressing cells. On the other hand, ERK activity was constitutively high in H-Ras and Bmi-1+H-Ras expressing cells regardless of EGF (Fig. 2A). It was induced after addition of EGF in MCF10A and MCF10A-Bmi-1 cells (Fig. 2A). These results suggested that Bmi-1 and H-Ras could transform HMECs by activating AKT and ERK kinases.
Figure 2.

Various growth regulatory pathways are dysregulated in cells co-overexpressing Bmi-1 and H-Ras. All cells were analyzed at passage 2 after Ras selection and/or mock infection. A, Western blot analysis of phospho-AKT, total AKT (AKT1 and AKT2), phospho-ERK and total ERK in control MCF10A and MCF10A-derived cells (as indicated). Western blot analysis using β-actin served as a loading control. B, Western blot analysis of p53, pRb, CDK4 and Cyclin D1 in asynchronously growing MCF10A and MCF10A-derived cells (as indicated). β-actin and QM are loading controls.
Next, we determined the expression of cyclin D1 and CDK4, as the overexpression of these two cell cycle regulatory proteins has been linked to breast cancer progression (34, 35). Our results indicated that compared to control cells, Bmi-1 or H-Ras overexpression upregulated Cyclin D1, while Bmi-1 and H-Ras co-overexpression upregulated CDK4 as well as Cyclin D1 expression in MCF10A cells (Fig. 2B). We also noticed a higher molecular weight form of CDK4 in Bmi-1 and H-Ras co-expressing cells, which may represent increased phosphorylation of CDK4 in these cells. We also determined the expression of pRb and p53 tumor suppressors in control and MCF10A-derived cells. Since, MCF10A cells are p16INK4A negative and contained high hyperphosphorylated pRb, no significant differences were found between different forms of pRb in control MCF10A and MCF10A-derived cells (Fig. 2B). On examining p53 expression, we found that MCF10A-H-Ras cells contained slightly higher p53 protein levels, while MCF10A-Bmi-1 and MCF10A-Bmi-1+H-Ras cells showed downregulation of p53 (Fig. 2B). Thus, the notable differences between H-Ras alone and Bmi-1+H-Ras overexpressing cells are- upregulation of AKT, upregulation of CDK4, and downregulation of p53 in Bmi-1+H-Ras expressing cells. Collectively, our data indicate that Bmi-1 together with H-Ras overexpression leads to activation of ERK and AKT, upregulation of Cyclin D1 and CDK4 expression, and downregulation of p53.
H-Ras expressing late passage (LP) HMECs cells exhibit a transformed phenotype
It has been reported in the literature that in some instances, H-Ras overexpression alone can lead to transformation of MCF10A cells, while other reports suggest the opposite (36-40). In our case, the early passage H-Ras expressing cells were clearly not transformed. These early passage cultures of cells were also heterogeneous and exhibited mixed morphologies with some enlarged senescent cells and some small normal proliferating cells. The late passage (>5 passages) (LP) culture of H-Ras expressing cells, on the other hand exhibited more uniform morphology with most cells proliferating. We considered whether these LP cells have undergone selection for rapidly proliferating cells and that during this selection may have acquired transformed properties.
To probe this hypothesis, MCF10A-Bmi-1+H-Ras (LP) and MCF10A-H-Ras (LP) cells were plated on soft-agar and allowed to form colonies for 10−14 days. The results indicated that similar to Bmi-1 and H-Ras co-overexpressing cells, MCF10A- H-Ras (LP) cells formed colonies in soft-agar indicating that H-Ras (LP) cells have also undergone transformation (Fig. 3A). The late passage Bmi-1 overexpressing MCF10A cells still did not make any colonies in soft-agar, indicating that Bmi-1 expression alone is not sufficient to cause transformation of HMECs even after extensive passaging in culture. Transformed phenotype of LP H-Ras cells was also confirmed by Matrigel assays. These assays indicated that LP H-Ras and H-Ras+Bmi-1 MCF10A cells form very disorganized, highly branched structures, which ultimately connect through branches and appear like a big sieved structure (Fig. 3B).
Figure 3.

Late passage (LP) H-Ras expressing MCF10A cells exhibit transformed features. All MCF10A-derived cells were analyzed at passage 8. A, Control MCF10A, and MCF10A-derived LP cells (as indicated) were grown in soft-agar to determine anchorage-independent growth potential of these cells. Cells were photographed (4×) at day 14. B, 3-Dimensional (3-D) growth of MCF10A-H-Ras (LP) and MCF10A-Bmi-1+H-Ras (LP) was analyzed using Matrigel assays as described in materials and methods. Cells in matrigel were photographed (10×) at day 7. C, EMT phenotype of MCF10A and MCF10A-derived LP cells was analyzed by immunostaining using antibodies specific for E-cadherin, vimentin, and fibronectin (as indicated). To visualize nuclei cells were stained with DAPI, and immunostained cells were visualized and photographed using Zeiss LSM510 UV META confocal microscope (60×). D, The migration potential of MCF10A and MCF10A-derived cells was determined by wound healing assay over 15 hrs time period as described in Materials and Methods. Cells were photographed using a light microscope (4×).
Bmi-1 expression together with H-Ras induces EMT in HMECs
When examining the morphology of MCF10A derived cells, we noticed that cells expressing both H-Ras and Bmi-1 exhibited fibroblastic morphology, while control and Bmi-1 or H-Ras alone expressing cells exhibited epithelial morphology. The fibroblastic morphology of MCF10A-Bmi-1+H-Ras cells suggested the induction of an EMT phenotype in these cells. To confirm this, we examined control MCF10A and MCF10A derived cells for the presence of EMT markers by immunostaining and Western blot analysis (Fig. 3D). The results indicated that control MCF10A and MCF10A-Bmi-1(LP) cells expressed E-Cadherin, a cell junction protein characteristic of epithelial cells, while MCF10A-H-Ras (LP) and MCF10A-Bmi-1+H-Ras (LP) lost the expression of E-Cadherin expression. On the other hand, MCF10A-H-Ras (LP) and MCF10A-Bmi-1+H-Ras (LP) cells expressed fibroblastic markers such as Vimentin and Fibronectin (Fig. 3C). Similar results were obtained using Western blot analysis using antibodies specific for E-Cadherin, Fibronectin, and Vimentin (Supplementary Fig. S2). These data indicate that Bmi-1 and H-Ras co-overexpression induces a strong EMT phenotype. The late passage MCF10A-H-Ras cells also exhibited an EMT phenotype, although these cells appeared less fibroblastic compared to MCF10A-Bmi-1+H-Ras cells.
As H-Ras and Bmi-1+H-Ras expressing cells exhibited EMT phenotype, which is closely linked to migration and invasion, we performed a wound healing assay to determine the migratory potential of these cells. The control MCF10A, and Bmi-1, H-Ras and Bmi-1+H-Ras overexpressing MCF10A cells were grown to 80% confluence, starved in D3 medium for 48 hrs. A wound was made in the middle of culture dish containing near-confluent cells and the cells were stimulated with EGF containing D medium for 15 hrs. Cells were photographed at 0 hr, before adding D medium and at 15 hr, after stimulating with D medium. The results indicated that MCF10A-Bmi-1+H-Ras cells have the highest migration potential and that these cells filled the wound quickly compared to other cells (Fig. 3D). H-Ras expressing MCF10A cells also showed moderate migratory potential, although much less compared to MCF10A-Bmi-1+H-Ras cells (Fig. 3D). These cells tend to undergo cell death during migration. Control MCF10A cells showed no migration, while Bmi-1 expressing cells only exhibited a minimal migration (Fig. 3D). Thus, our data suggest that Bmi-1 and H-Ras co-overexpressing cells have acquired migration and invasion potential typical of highly transformed HMECs.
Expression level of H-Ras determines proliferation in H-Ras expressing MCF10A cells
The differential ability of MCF10A-H-Ras (EP) and MCF10A-H-Ras (LP) cells to undergo transformation could be related to the different levels of Ras expression, which in turn may determine proliferation in these cells. To examine this possibility, we determined expression of H-Ras by Western blot analysis in control MCF10A, and MCF10A-derived EP and LP cells, and performed Ras and Ki-67 co-immunostaining in these cells (Fig. 4). The Western blot analysis of control, EP and LP cells of H-Ras, Bmi-1, and Bmi-1+H-Ras indicated that H-Ras (EP) cells expressed high level of Ras, whereas H-Ras (LP) cells expressed low level of H-Ras (Fig. 4A). On the other hand, Bmi-1+H-Ras (LP) cells expressed a high level of H-Ras (Fig. 4A, 4B). Bmi-1+H-Ras (EP) cells and H-Ras (EP) cells expressed similar levels of Ras (Fig. 4A, 4B). Since EP cultures are heterogeneous with cells expressing variable levels of Ras, it is possible that cells expressing Ras above a certain threshold level are not proliferating and at increasing number of population doublings, there may be selection for cells that express lower level of Ras, which permits continued proliferation. Accordingly, H-Ras (LP) cells will have lower levels of Ras. Consistent with this hypothesis, on a single cell basis, we observed that in H-Ras EP cultures, most cells with high Ras stained negative for Ki-67, a proliferation marker, whereas cells with low Ras stained positive for Ki-67 (Fig. 4C, D). On the other hand, H-Ras (LP) culture mostly contained cells with low Ras, which stained positive with Ki-67 ((Fig. 4C, D). The percentage of low Ras expressing cells, which were Ki-67 positive was also high in MCF10A-Bmi-1+H-Ras (EP) culture, although some cells in this culture also expressed high Ras, which were positive for Ki-67 (Fig. 4C, D). Importantly, most Bmi-1+H-Ras (LP) cells expressed high Ras, which stained positive for Ki-67, indicating that Bmi-1 permits proliferation of these cells despite high Ras (Fig. 4C, D). In all cultures, variable percentages of low Ras expressing cells were Ki-67 negative. Because of growth asynchrony in culture, such cells may not be proliferating at the time of staining.
Figure 4.

Expression level of H-Ras determines proliferation in MCF10A cells overexpressing H-Ras. A, H-Ras expression in MCF10A control and MCF10A-derived early passage (EP) (passage 2 after Ras selection) and late passage (LP) cells (passage 8) was determined by Western blot analysis as described in Materials and Methods. B, To determine the relative expression of H-Ras in MCF10A and MCF10A-derived cells, its signal in each lane was quantified by densitometric analysis using ImageJ1.3 software (NIH, Bethesda, MD), and normalized to α-tubulin signal. C, H-Ras and Ki-67 co-immunostaining was performed to determine proliferation in MCF10A-derived EP (passage 2) and LP (passage 8) cells. MCF10A cells were used as control, which do not express detectable Ras but are Ki-67 positive under our experimental conditions. Representative photos (60×) of co-staining in each cell line (as indicated) are shown. D, quantification of Ras and Ki-67 expressing cells in MCF10A-derived EP (passage 2) and LP (passage 8) culture of H-Ras, and Bmi-1+H-Ras cells. Co-staining was performed in triplicates and total of 100−200 stained cells were counted in multiple fields.
MCF10A cells expressing H-Ras, and Bmi-1 + H-Ras form histologically distinct tumors in vivo
In order to address the contributory role of Bmi-1 on tumor progression, MCF10A, MCF10A-Bmi-1 (LP), MCF10A-H-Ras (LP), MCF10A-Bmi-1+H-Ras (LP) cells were injected into the mammary fat pad. As expected, MCF10A control cells did not produce tumors in vivo. Injection of MCF10A+Bmi-1 cells also did not result in tumor formation even after 60 days, indicating that overexpression of Bmi-1 alone is not sufficient for neoplastic transformation of mammary epithelial cells in vivo. In contrast, MCF10A-H-Ras and MCF10A-Bmi-1+H-Ras cells produced progressively enlarging tumors in the mammary fat pad. Grossly, these tumors were strikingly different (Fig. 5A); MCF10A-Bmi-1+H-Ras cells formed tumors that were solid, firm, and irregular, whitish-tan on cut surface with well differentiated vasculature. In contrast, tumors formed by MCF10A-H-Ras cells were variably hemorrhagic and often cystic, composed predominantly of large thin cysts filled with clotted and/or unclotted blood (Fig. 5A). Tumor growth rates when measured by tumor volume were not significantly different between these two groups; however, this was due to the general volume of tumors being similar, rather than the actual tumor mass which was significantly less in the tumors formed by Bmi-1+H-Ras cells.
Figure 5.

Gross morphology, histopathology, and immunohistochemistry of tumors originating from xenografts. A, Gross morphology of tumors resulting from injection of MCF10A-H-Ras (LP) and MCF10A-Bmi-1+H-Ras cells (LP) (as indicated). Left: tumors induced by MCF10A-H-Ras. Right: tumors induced by MCF10A-Bmi-1+H-ras cells. B, Histopathology of tumors resulting from injection of MCF10A-H-Ras (left) and MCF10A-Bmi-1+H-Ras (right) cells. Left: tumors induced by MCF10A-H-Ras cells were composed of variable populations of poorly- to well-differentiated endothelial cells forming haphazard vascular channels (52), spindle shaped cells resembling smooth muscle (middle), and multiple variable sized clusters of poorly- to well-differentiated mast cells (bottom). Right: tumors induced by MCF10A-Bmi-1+H-Ras cells were composed of a homogeneous population of sheets and intersecting bundles of poorly differentiated spindle cells (top) that infiltrated adjacent adipose tissue and bone (middle). Cells were poorly-differentiated with large pleomorphic nuclei and frequent mitoses (bottom). C, Histochemical and immunohistochemical staining of tumors induced by MCF10A-H-Ras (left) and MCF10A-Bmi-1+H-Ras (right) cells. Left: tumors induced by MCF10A-H-Ras were multifocally immunoreactive for antibodies against α-SMA and CD31; mast cell clusters were diffusely positive with Geimsa staining for mast cell granules, and tumors were diffusely negative for collagen by Masson's Trichrome staining. Right: Tumors induced by MCF10A-H-Ras+Bmi-1 cells were diffusely negative for α-SMA and CD31 except for the presence of intratumoral capillaries (arrowheads), diffusely negative with Geimsa staining except for occasional resident mast cells (arrowhead), and showed very little collagen production with Masson's Trichrome stain. D, Kaplan-Meier survival curve. Whereas MCF10A and MCF10A-Bmi-1 xenografted mice did not develop tumors and survived throughout the course of the study, mice xenografted with MCF10A-H-Ras and MCF10A-Bmi-1+H-Ras had decreased survival following the development of palpable tumors. MCF10A-H-Ras xenografted mice had significantly decreased survival compared to MCF10A-Bmi-1+H-Ras mice (p<0.002).
Histologically, MCF10A-H-Ras tumors consisted of variable populations of poorly to fairly well-differentiated smooth muscle, variably cystic irregular vascular spaces lined by poorly- to fairly well-differentiated endothelial cells, and multifocal clusters and nests of poorly- to well-differentiated mast cells (Fig. 5B). In contrast, MCF10A-Bmi-1+H-Ras tumors were composed of streams and bundles of poorly-differentiated spindle-shaped cells with scant, faintly eosinophilic fibrillar cytoplasm embedded in scant eosinophilic stroma, large round to oval hyperchromatic nuclei with multiple prominent nucleoli, and numerous mitotic figures (approximately 2−3/hpf) (Fig. 5B). These cells often infiltrated into the surrounding fat pad, effacing normal ducts and adipose tissue, and in one case, infiltrating and destroying the cortical bone of a subjacent rib and invading and effacing the bone marrow (Fig. 5B).
MCF10A-H-Ras tumors were multifocally immunoreactive to antibodies to α-SMA and CD31 (PECAM), illustrating the smooth muscle and hemangiomatous components of these tumors (Fig. 5C). Geimsa staining for mast cell granules confirmed the multifocal mast cell clusters of varying differentiation in the MCF10A-H-Ras tumors, whereas Masson's Trichrome staining illustrated no collagen production in these tumors (Fig. 5C). MCF10A-Bmi-1+H-Ras tumors were diffusely negative for α-SMA and CD31 except for pre-existing intratumoral capillaries supplying the tumors that were immunoreactive to CD31 (Fig. 5C). Geimsa staining confirmed the absence of mast cells in these tumors except for a rare mature resident mast cell, and Masson's Trichrome staining confirmed that these tumors are composed of spindle cells with scant collagen production, more suggestive of a myogenic phenotype than a fibrosarcomatous one (Fig. 5C). Both MCF10A-H-Ras and MCF10A-Bmi-1+H-Ras tumors were diffusely immunoreactive to antibodies to cytokeratin and vimentin (Supplementary Fig. S3). Animals with tumors formed by MCF10A-H-Ras cells were often very hemorrhagic, resulting in early morbidity due to anemia rather than tumor burden in contrast to mice bearing tumors formed by MCF10A-H-Ras+Bmi-1 cells, which as a group lived longer with tumors than MCF10A-H-Ras tumor bearing mice (Fig. 5D).
MCF10A-H-Ras (LP) and MCF10A-Bmi-1+H-Ras (LP) cells display partially defective p53 phosphorylation and attenuated induction of p53 target genes in response to DNA damage
H-Ras is known to cause premature senescence or oncogene induced senescence (OIS) in primary cells, which is mediated by p16INK4A and p53-p21 pathways of senescence (41-43). Using SA-β-Gal marker, we noticed senescence induction in a significant number (40−50%) of MCF10A cells by H-Ras overexpression at early passages (Supplementary Fig. S4A), although these cells do not express p16IKN4A, suggesting that the partial OIS in MCF10A cells may depend on p53 and its target genes. Consistent with partial OIS, early passage MCF10A-Ras cells also showed slower growth compared to vector control MCF10A and MCF10A cells co-overexpressing H-Ras and Bmi-1 (Supplementary Fig. S2B).
The senescent cells in MCF10A-H-Ras and MCF10A-Bmi-1+H-Ras cells were progressively lost, and rapidly proliferating cells were selected in later passages. We hypothesized that the selection of rapidly proliferating cells in LP cultures of MCF10A-H-Ras, and MCF10ABmi-1+H-Ras cells may depend on a defect in p53 pathway in these cells. To examine this hypothesis, we determined p53 expression in control MCF10A, MCF10A-Bmi-1 (LP), MCF10A-H-Ras (LP) and MCF10A-Bmi-1+H-Ras (LP) cells. The results indicated that unlike in early passage MCF10A-H-Ras cells ((Fig. 2B), p53 was downregulated in MCF10A-H-Ras (LP) cells (Fig. 6A). Downregulation of p53 in MCF10A-H-Ras (LP) cells was similar to MCF10A-Bmi-1+H-Ras early passage (Fig. 2B), or MCF10A-Bmi-1+H-Ras (LP) cells (Fig. 6A). To determine the mechanism of p53 downregulation and its possible significance with respect to transformed phenotype of MCF10A-H-Ras (LP) and MCF10A-Bmi-1+H-Ras (LP) cells, we further studied the p53 pathway in these cells.
Figure 6.

Analysis of p53 pathway in control MCF10A and MCF10A-derived late passage cells. All cells except parental MCF10A cells, passage 9 cells were used for the analysis. A, Upper panel-Western blot analysis of Bmi-1, H-Ras and p53 in control MCF10A and MCF10A-derived (LP) cells (as indicated) was performed as described in Fig. 2. Lower panel- Densitometric analysis of signals (of p53 and H-Ras) present in each lane was performed, normalized to corresponding α-tubulin signal, and plotted to determine the expression levels of p53 and H-Ras as indicated. B, Analysis of DNA damage response in MCF10A and MCF10A-derived LP cells. The cells were treated with CPT for indicated amount of time, harvested and analyzed by western blot analysis for total p53, phosphorylated p53 (Ser-15 and Ser-37), p53 target genes (p21, PUMA, Bax and PIG3), PRAK and BCL2. β-Actin was used as a loading control.
MCF10A control and MCF10A- derived LP cells were treated with the DNA damaging agent Camptothecin (CPT) (500 nM) for the indicated amount of time (Fig. 6B). Next, the expression of p53, phospho-p53 and p53 target genes was studied by Western blot analysis. Recently, it has been reported that H-Ras induces senescence via p38-regulated/activated protein kinase (PRAK)-mediated phosphorylation of p53 at Ser-37 residue, and that PRAK mediated induction of Ser-37 phosphorylation of p53 is lost during H-Ras mediated oncogenesis (42). Hence, we also determined PRAK expression in MCF10A and MCF10A-derived cells.
The results indicated that although MCF10A-H-Ras (LP) and MCF10A-Bmi-1+H-Ras (LP) cells had overall low p53 compared to control MCF10A and MCF10A-Bmi-1 cells, p53 remained inducible by CPT in all four set of cells, although the induced levels of p53 was still low in MCF10A-H-Ras (LP) and MCF10A-Bmi-1+H-Ras (LP) cells (Fig. 6B, Supplementary Figure S5). Further analysis of phospho-p53 indicated that MCF10A-H-Ras (LP) and MCF10A-Bmi-1+H-Ras (LP) cells were partially defective in phosphorylation of p53 at Ser-15 and Ser-37 residues (Fig. 6B,Supplementary Figure S5). Quantification of Western blot data showed reduced phosphorylation of p53 at Ser-15 in both MCF10A-H-Ras (LP) and MCF10A-Bmi-1+H-Ras (LP) cells at 4 hr and 8 hr time points, whereas the basal levels of p53 Ser-15 were similar in all MCF10A-derived cells (Supplementary Figure S5). Ser-37 phosphorylation was also compromised in MCF10A-H-Ras and MCF10A-Bmi-1+H-Ras cells. Neither of these cell lines showed any induction of Ser-37 phosphorylation of p53 by CPT treatment (Supplementary Figure S5).
Since, it has been reported that PRAK mediates Ser-37 phosphorylation of p53 induced by H-Ras, we hypothesized that PRAK may be lost during selection of rapidly proliferating cells in H-Ras overexpressing cells in culture. To examine this possibility, we determined PRAK expression in these cells by Western blot analysis. The results indicated that regardless of DNA damage, PRAK expression is not lost in control or H-Ras overexpressing cells (Fig. 6B). Interestingly, PRAK expression was upregulated in H-Ras expressing cells (Fig. 6B). The upregulation of PRAK is consistent with the notion that PRAK is a H-Ras target, which acts negatively to suppress H-Ras induced proliferation (44). Nonetheless, it seems that this PRAK-mediated negative feedback regulation of H-Ras mediated proliferation is lost in H-Ras overexpressing MCF10A cells, which may have allowed these cells to undergo transformation in culture.
Next, we studied the induction of p21 and PUMA (p53 upregulated modulator of apoptosis), two well known transcriptional targets of p53 (45, 46), which are associated with p53 functions such as apoptosis and senescence. Our results indicated that both p21 and PUMA induction by CPT is partially compromised in H-Ras overexpressing MCF10A cells (Fig. 6B, Supplementary Figure S5), and p21 induction was more compromised in MCF10A-Bmi-1+H-Ras cells. Attenuated response of these targets of p53 is consistent with defective phosphorylation at Ser-15 and Ser-37 residues. We also examined expression of Bax and PIG3 (p53 inducible gene 3), two other known targets of p53 (45). Analysis of these two genes indicated that Bax is expressed at very low levels and is inducible in control MCF10A cells. However, H-Ras expressing MCF10A cells had higher levels of Bax, which were not inducible by DNA damage (Fig. 6B, Supplementary Figure S5). Interestingly, among all four cell types, MCF10A-H-Ras (LP) cells expressed high BCL2, which may be related to transformed properties of these cells. PIG3, which usually has a delayed kinetics of induction by p53 (47), was not inducible in any of the cell types within the time frame used in our experiments. However, compared to MCF10A control cells, PIG3 was downregulated in MCF10A-H-Ras, MCF10A-Bmi-1 and MCF10A-Bmi-1+H-Ras cells, with the latter cells showing the most dramatic downregulation of PIG3 (Fig. 6B).
Discussion
Several recent studies have suggested that PcG proteins, in particular EZH2 and Bmi-1 are overexpressed in human cancers. Recent elegant studies have clearly shown that oncogenic transformation of human cells is a multi-step process (48). It is very likely that overexpression of EZH2 or Bmi-1 alone is not sufficient to cause transformation of human cells. To gain an insight into breast cancer progression, here we examined the transformation potential of Bmi-1 oncoprotein in immortalized HMECs. Although immortalized HMECs that we studied lack p16INK4A and p14-ARF, Bmi-1 expression still provides an oncogenic signal in these cells by the activation of PI3KAKT pathway (25). However, the oncogenic signal provided by Bmi-1 alone does not appear to be sufficient to cause transformation of HMECs, despite these cells being immortal and lacking p16INK4A , p14ARF and p15INK4B (27). This observation underscores the stringency of transformation in HMECs. Nonetheless, Bmi-1 overexpression is frequently observed in invasive breast tumors (8, 9, 25), suggesting the involvement of additional oncogenic events during breast cancer progression in such tumors.
To understand the genetic basis of these presumptive additional oncogenic events, we overexpressed a constitutively active mutant G12V of H-Ras (33) in Bmi-1 overexpressing MCF10A cells. G12V mutant of H-Ras promotes proliferation and oncogenesis via activation of mitogen-activated protein kinase (MAPK) kinase (MEK)/MAPK and the phosphoinositide 3-kinase (PI-3K)/AKT pathways. However, the activation of these pathways and their outcome is cell-type specific. For example, in primary cells, activation of these pathways lead to induction of OIS, while in immortalized cells with compromised p53-p21 and/or p16INK4A pathways, H-Ras G12V may promote proliferation. Our reasoning behind using H-Ras G12V in these assays was based on its relevance to breast cancer, and its reported use in oncogenic assays involving HMECs (32). Although, the direct mutational activation of H-Ras is rare in breast cancer, its hyperactivation by persistent growth factor signaling caused by EGFR and HER2/neu overexpression occurs in a proportion of breast cancers (49, 50).
OIS caused by G12V mutant of H-Ras may require both functional p16INK4A and p53. In MCF10A cells, which have functional p53, we initially noticed the appearance of a heterogeneous culture with approximately 40−50% cells exhibiting senescent morphology upon H-Ras overexpression. Consistent with partial senescence induction, our Western blot data also indicated upregulation of p53 protein in these cells. Senescence acts as a strong barrier to oncogenesis (20), hence the initial induction of senescence in a proportion of MCF10A cells by H-Ras indicates an anti-oncogenic response. As expected, these early passage cells were not transformed by soft-agar and Matrigel assays. However, late passage culture, which were much more homogenous and did not contain cells with senescent morphology, displayed transformed phenotype in Matrigel and soft-agar assays. Ras and Ki-67 co-staining data also suggest that early passage culture of MCF10A-H-Ras are more heterogeneous in terms of Ras expression, while the late passage culture of these cells are homogenous in terms of Ras expression. Importantly, only low Ras expressing cells tend to be Ki-67 positive suggesting that low Ras permits proliferation, while high Ras blocks proliferation, possibly via OIS. This differential effect of Ras on proliferation explains the emergence of low Ras expressing culture at late passages.
The H-Ras overexpression in Bmi-1 overexpressing MCF10A cells caused senescence only in a minority of cells. When compared to H-Ras overexpressing culture, homogenous culture with proliferating cells appeared much more rapidly from MCF10A-Bmi-1+H-Ras cultures. These data indicate that to some extent, Bmi-1 can overcome H-Ras induced OIS, even in p16INK4A negative cells, presumably via p16INK4a/ARF-independent targets of Bmi-1. The homogenous culture that rapidly emerged from Bmi-1+H-Ras expressing cells continued to express high Ras. Most cells in this culture were Ki-67 positive despite expressing high Ras, suggesting that in the presence of Bmi-1, cells can tolerate high Ras, and thus there is no selection for cells expressing low Ras.
Analysis of Ras effector pathways suggest that activation of PI3K-AKT pathway may also play a role in Bmi-1 and H-Ras induced transformation of HMECs. Although, after EGF stimulation, compared to control MCF10A cells, early passage H-Ras expressing cells exhibited higher phospho-AKT, EGF starved MCF10A-H-Ras had no phospho-AKT, while Bmi-1 and Bmi-1+H-Ras expressing MCF10A cells contained significant level of phospho-AKT even under growth factor starved conditions. On the other hand, early passage MCF10A-H-Ras and MCF10A-Bmi-1+H-Ras cells exhibited constitutively high ERK kinase activity, which was inducible but not constitutive in control MCF10A and MCF10A-Bmi-1 cells. Collectively, our data suggest that Bmi-1 induces AKT activity, which is further augmented by H-Ras co-overexpression, and that H-Ras constitutively activates ERK kinases in MCF10A cells.
On examination of Ser-37 and Ser-15 phosphorylation of p53 in response to DNA damage, we found that Ser-37 phosphorylation of p53 is significantly low and not inducible in both late passage H-Ras, and Bmi-1+H-Ras expressing cells. However, PRAK expression is not affected in these cells. These data suggest that the kinase activity of PRAK responsible for Ser-37 phosphorylation is lost in H-Ras overexpressing MCF10A cells. In addition, these cells also had much lower induction of Ser-15 phosphorylated p53, suggesting a possible defect in other p53 activating kinases such as ATM. A detailed analysis of various p53 phosphorylating kinases in late passage MCF10A-H-Ras and MCF10A-Bmi-1+H-Ras remains to be elucidated. Nevertheless, our data clearly indicate that these late passage H-Ras and Bmi-1+H-Ras expressing cells have defects in p53 phosphorylating pathways, which results in attenuation of induction of p53 targets such as p21 and PUMA. This compromised induction of p53 targets may contribute to a transformed phenotype of MCF10A cells expressing Bmi-1 and H-Ras.
The differential behavior of early and late passage H-Ras overexpressing MCF10A cells with respect to the transformed phenotype explains the different results that are reported in the literature (36-40). Our data suggest that in cases where H-Ras expressing MCF10A cells showed a transformed phenotype and gave rise to tumors in nude mice assays, late passage H-Ras expressing cells with defective p53 regulation may have been used. In other studies, where transformation of H-Ras expressing MCF10A cells was not reported, early passage H-Ras expressing MCF10A cells may have been used. Alternatively, the transforming potential of H-Ras cells could be correlated with the level of expression of H-Ras. In studies where H-Ras alone was reported to be transforming, the expression of H-Ras may be low, which permits proliferation and expression of other oncogenic functions of Ras. On the other hand, in cases where Ras was reported to be insufficient for transformation, the expression of Ras may be very high, which causes proliferation arrest and OIS. Neither of these possibilities is mutually exclusive and both possibilities are likely to contribute to transformation of HMECs by H-Ras. Recently it was shown that low levels of K-Ras induce proliferation and mammary epithelial cell hyperplasias, while high expression of K-Ras induces proliferation arrest and OIS in dox-inducible K-Ras transgenic mice (51). In this report, it was also shown that inactivation of p53 permits transformation of mammary epithelial cells and tumor formation by high expression of Ras (51). Our in vitro data are consistent with this report.
The results of histopathology, including special stains and immunohistochemistry, confirm that the MCF10A+H-Ras tumors are composed of multiple different populations of varying phenotypes (smooth muscle, hemangiomatous and mast cells), suggesting that these populations may be in part an in-vivo response to the xenografted tumor population, rather than original components of the neoplastic population that have undergone dedifferentiation and redifferentiation along multiple lines. The MCF10A-Bmi-1+H-Ras tumors on the other hand, represent a pure population of highly atypical, poorly-differentiated, and infiltrative spindle cells consistent with a mesenchymal phenotype. Although the α-SMA immunohistochemistry was negative in these tumors, the Masson's Trichrome stain along with positive immunohistochemistry for vimentin would suggest that these cells may represent a myoepithelial phenotype consistent with EMT.
The striking differences seen in tumor morphology both grossly and histologically are reflected in the survival data of these animals over time. For example, due to the hemorrhagic nature of their tumors, many animals injected with MCF10A+H-Ras cells were removed from study prematurely due to morbidity as a result of anemia, rather than tumor burden. On the other hand, animals receiving MCF10A-Bmi-1+H-ras cells generally survived until they became moribund due to tumor burden. These data would suggest that expression of Bmi-1 concurrently with H-Ras promotes a more poorly differentiated tumor phenotype that is directly responsible for clinical morbidity due to tumor burden in comparison to H-Ras alone, in which morbidity is primarily due to anemia.
Our preliminary data also suggest that tumors formed by MCF10A-Bmi-1+H-Ras (LP) cells are much more metastatic and invasive compared to tumors formed by MCF10A-H-Ras (LP) cells. Although, MCF10A-Bmi-1+H-Ras(LP) and MCF10A-H-Ras cells give rise to histologically distinct type of tumors, biochemically, these cells show only minor differences in regulation of growth regulatory pathways. The only significant difference between these two cell lines is that H-Ras (LP) cells expressed higher levels of BCL2 oncoprotein, which may contribute to the oncogenicity of these cells. The other significant difference between MCF10A-Bmi-1+H-Ras (LP) and MCF10A-H-Ras cells appears to be that MCF10A-Bmi-1+H-Ras (LP) cells can proliferate despite high Ras. The biochemical basis for proliferation of MCF10A-Bmi-1+H-Ras (LP) cells despite high Ras remains to be elucidated. In any case, we did not observe tumor formation by MCF10A-Bmi-1 (LP) cells suggesting the involvement of additional oncogenic events such as downregulation of p53, overexpression of CDK4 and cyclin D1, and upregulation of AKT and ERK activities in the transformation of HMECs and breast cancer progression. Our data also indicate that Bmi-1 may cooperate with Ras in transformation by simply allowing high Ras expressing cells to proliferate. The additional oncogenic events then may be largely contributed to by H-Ras in the experiments described here. It remains to be determined which of these oncogenic lesions, together with Bmi-1, are sufficient to transform HMECs and form tumors in vivo.
Acknowledgements
This work was supported by the grants from the National Cancer Institute (RO1CA 094150) and the US Department of Defense (DAMD17-02-1-0509) to GD.
Supplementary Material
References
- 1.Ringrose L, Paro R. Epigenetic regulation of cellular memory by the Polycomb and Trithorax group proteins. Annu Rev Genet. 2004;38:413–43. doi: 10.1146/annurev.genet.38.072902.091907. [DOI] [PubMed] [Google Scholar]
- 2.Raaphorst FM. Deregulated expression of Polycomb-group oncogenes in human malignant lymphomas and epithelial tumors. Hum Mol Genet. 2005;14(Spec No 1):R93–R100. doi: 10.1093/hmg/ddi111. [DOI] [PubMed] [Google Scholar]
- 3.Bea S, Tort F, Pinyol M, et al. BMI-1 gene amplification and overexpression in hematological malignancies occur mainly in mantle cell lymphomas. Cancer Res. 2001;61(6):2409–12. [PubMed] [Google Scholar]
- 4.van Kemenade FJ, Raaphorst FM, Blokzijl T, et al. Coexpression of BMI-1 and EZH2 polycomb-group proteins is associated with cycling cells and degree of malignancy in B-cell non-Hodgkin lymphoma. Blood. 2001;97(12):3896–901. doi: 10.1182/blood.v97.12.3896. [DOI] [PubMed] [Google Scholar]
- 5.Sawa M, Yamamoto K, Yokozawa T, et al. BMI-1 is highly expressed in M0-subtype acute myeloid leukemia. Int J Hematol. 2005;82(1):42–7. doi: 10.1532/IJH97.05013. [DOI] [PubMed] [Google Scholar]
- 6.Vonlanthen S, Heighway J, Altermatt HJ, et al. The bmi-1 oncoprotein is differentially expressed in non-small cell lung cancer and correlates with INK4A-ARF locus expression. Br J Cancer. 2001;84(10):1372–6. doi: 10.1054/bjoc.2001.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim JH, Yoon SY, Kim CN, et al. The Bmi-1 oncoprotein is overexpressed in human colorectal cancer and correlates with the reduced p16INK4a/p14ARF proteins. Cancer letters. 2004;203(2):217–24. doi: 10.1016/j.canlet.2003.07.009. [DOI] [PubMed] [Google Scholar]
- 8.Glinsky GV, Berezovska O, Glinskii AB. Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J Clin Invest. 2005;115(6):1503–21. doi: 10.1172/JCI23412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim JH, Yoon SY, Jeong SH, et al. Overexpression of Bmi-1 oncoprotein correlates with axillary lymph node metastases in invasive ductal breast cancer. Breast. 2004;13(5):383–8. doi: 10.1016/j.breast.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 10.Kang MK, Kim RH, Kim SJ, et al. Elevated Bmi-1 expression is associated with dysplastic cell transformation during oral carcinogenesis and is required for cancer cell replication and survival. Br J Cancer. 2007;96(1):126–33. doi: 10.1038/sj.bjc.6603529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song LB, Zeng MS, Liao WT, et al. Bmi-1 is a novel molecular marker of nasopharyngeal carcinoma progression and immortalizes primary human nasopharyngeal epithelial cells. Cancer Res. 2006;66(12):6225–32. doi: 10.1158/0008-5472.CAN-06-0094. [DOI] [PubMed] [Google Scholar]
- 12.Iwama A, Oguro H, Negishi M, et al. Enhanced self-renewal of hematopoietic stem cells mediated by the polycomb gene product Bmi-1. Immunity. 2004;21(6):843–51. doi: 10.1016/j.immuni.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 13.Lessard J, Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 2003;423(6937):255–60. doi: 10.1038/nature01572. [DOI] [PubMed] [Google Scholar]
- 14.Molofsky AV, He S, Bydon M, Morrison SJ, Pardal R. Bmi-1 promotes neural stem cell self-renewal and neural development but not mouse growth and survival by repressing the p16Ink4a and p19Arf senescence pathways. Genes & Dev. 2005;19(12):1432–7. doi: 10.1101/gad.1299505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Molofsky AV, Pardal R, Iwashita T, Park IK, Clarke MF, Morrison SJ. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature. 2003;425(6961):962–7. doi: 10.1038/nature02060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu S, Dontu G, Mantle ID, et al. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006;66(12):6063–71. doi: 10.1158/0008-5472.CAN-06-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu S, Dontu G, Wicha MS. Mammary stem cells, self-renewal pathways, and carcinogenesis. Breast Cancer Res. 2005;7(3):86–95. doi: 10.1186/bcr1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Itahana K, Zou Y, Itahana Y, et al. Control of the replicative life span of human fibroblasts by p16 and the polycomb protein Bmi-1. Mol Cell Biol. 2003;23(1):389–401. doi: 10.1128/MCB.23.1.389-401.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 1999;397(6715):164–8. doi: 10.1038/16476. [DOI] [PubMed] [Google Scholar]
- 20.Dimri GP. What has senescence got to do with cancer? Cancer cell. 2005;7(6):505–12. doi: 10.1016/j.ccr.2005.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dimri GP, Martinez JL, Jacobs JJ, et al. The Bmi-1 oncogene induces telomerase activity and immortalizes human mammary epithelial cells. Cancer Res. 2002;62(16):4736–45. [PubMed] [Google Scholar]
- 22.Kim RH, Kang MK, Shin KH, et al. Bmi-1 cooperates with human papillomavirus type 16 E6 to immortalize normal human oral keratinocytes. Exp Cell Res. 2007;313(3):462–72. doi: 10.1016/j.yexcr.2006.10.025. [DOI] [PubMed] [Google Scholar]
- 23.Guo WJ, Datta S, Band V, Dimri GP. Mel-18, a polycomb group protein, regulates cell proliferation and senescence via transcriptional repression of Bmi-1 and c-Myc oncoproteins. Mol Biol Cell. 2007;18(2):536–46. doi: 10.1091/mbc.E06-05-0447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee K, Adhikary G, Balasubramanian S, et al. Expression of Bmi-1 in Epidermis Enhances Cell Survival by Altering Cell Cycle Regulatory Protein Expression and Inhibiting Apoptosis. J Invest Dermatol. 2007 doi: 10.1038/sj.jid.5700949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo WJ, Zeng MS, Yadav A, et al. Mel-18 acts as a tumor suppressor by repressing Bmi-1 expression and down-regulating Akt activity in breast cancer cells. Cancer Res. 2007;67(11):5083–9. doi: 10.1158/0008-5472.CAN-06-4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berman H, Zhang J, Crawford YG, et al. Genetic and epigenetic changes in mammary epithelial cells identify a subpopulation of cells involved in early carcinogenesis. Cold Spring Harb Symp Quant Biol. 2005;70:317–27. doi: 10.1101/sqb.2005.70.051. [DOI] [PubMed] [Google Scholar]
- 27.Cowell JK, LaDuca J, Rossi MR, Burkhardt T, Nowak NJ, Matsui S. Molecular characterization of the t(3;9) associated with immortalization in the MCF10A cell line. Cancer Genet Cytogenet. 2005;163(1):23–9. doi: 10.1016/j.cancergencyto.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 28.Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30(3):256–68. doi: 10.1016/s1046-2023(03)00032-x. San Diego, Calif. [DOI] [PubMed] [Google Scholar]
- 29.Dimri GP, Lee X, Basile G, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92(20):9363–7. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Band V, Zajchowski D, Kulesa V, Sager R. Human papilloma virus DNAs immortalize normal human mammary epithelial cells and reduce their growth factor requirements. Proc Natl Acad Sci U S A. 1990;87(1):463–7. doi: 10.1073/pnas.87.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dimri M, Naramura M, Duan L, et al. Modeling breast cancer-associated c-Src and EGFR overexpression in human MECs: c-Src and EGFR cooperatively promote aberrant three-dimensional acinar structure and invasive behavior. Cancer Res. 2007;67(9):4164–72. doi: 10.1158/0008-5472.CAN-06-2580. [DOI] [PubMed] [Google Scholar]
- 32.Elenbaas B, Spirio L, Koerner F, et al. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes & Dev. 2001;15(1):50–65. doi: 10.1101/gad.828901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seeburg PH, Colby WW, Capon DJ, Goeddel DV, Levinson AD. Biological properties of human c-Ha-ras1 genes mutated at codon 12. Nature. 1984;312(5989):71–5. doi: 10.1038/312071a0. [DOI] [PubMed] [Google Scholar]
- 34.Landis MW, Pawlyk BS, Li T, Sicinski P, Hinds PW. Cyclin D1-dependent kinase activity in murine development and mammary tumorigenesis. Cancer Cell. 2006;9(1):13–22. doi: 10.1016/j.ccr.2005.12.019. [DOI] [PubMed] [Google Scholar]
- 35.Yu Q, Sicinska E, Geng Y, et al. Requirement for CDK4 kinase function in breast cancer. Cancer Cell. 2006;9(1):23–32. doi: 10.1016/j.ccr.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 36.Dawson PJ, Wolman SR, Tait L, Heppner GH, Miller FR. MCF10AT: a model for the evolution of cancer from proliferative breast disease. Amer J Path. 1996;148(1):313–9. [PMC free article] [PubMed] [Google Scholar]
- 37.Giunciuglio D, Culty M, Fassina G, et al. Invasive phenotype of MCF10A cells overexpressing c-Ha-ras and c-erbB-2 oncogenes. Int J Cancer. 1995;63(6):815–22. doi: 10.1002/ijc.2910630612. [DOI] [PubMed] [Google Scholar]
- 38.Moon A, Kim MS, Kim TG, et al. H-ras, but not N-ras, induces an invasive phenotype in human breast epithelial cells: a role for MMP-2 in the H-ras-induced invasive phenotype. Int J Cancer. 2000;85(2):176–81. doi: 10.1002/(sici)1097-0215(20000115)85:2<176::aid-ijc5>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 39.Santner SJ, Dawson PJ, Tait L, et al. Malignant MCF10CA1 cell lines derived from premalignant human breast epithelial MCF10AT cells. Breast Cancer Res Treat. 2001;65(2):101–10. doi: 10.1023/a:1006461422273. [DOI] [PubMed] [Google Scholar]
- 40.Shin I, Kim S, Song H, Kim HR, Moon A. H-Ras-specific activation of Rac-MKK3/6-p38 pathway: its critical role in invasion and migration of breast epithelial cells. J Biol Chem. 2005;280(15):14675–83. doi: 10.1074/jbc.M411625200. [DOI] [PubMed] [Google Scholar]
- 41.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88(5):593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 42.Sun P, Yoshizuka N, New L, et al. PRAK is essential for ras-induced senescence and tumor suppression. Cell. 2007;128(2):295–308. doi: 10.1016/j.cell.2006.11.050. [DOI] [PubMed] [Google Scholar]
- 43.Wang W, Chen JX, Liao R, et al. Sequential activation of the MEK-extracellular signal-regulated kinase and MKK3/6-p38 mitogen-activated protein kinase pathways mediates oncogenic ras-induced premature senescence. Mol Cell Biol. 2002;22(10):3389–403. doi: 10.1128/MCB.22.10.3389-3403.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen G, Hitomi M, Han J, Stacey DW. The p38 pathway provides negative feedback for Ras proliferative signaling. J Biol Chem. 2000;275(50):38973–80. doi: 10.1074/jbc.M002856200. [DOI] [PubMed] [Google Scholar]
- 45.el-Deiry WS. Regulation of p53 downstream genes. Sem Cancer Biol. 1998;8(5):345–57. doi: 10.1006/scbi.1998.0097. [DOI] [PubMed] [Google Scholar]
- 46.Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Molecular Cell. 2001;7(3):683–94. doi: 10.1016/s1097-2765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- 47.Flatt PM, Polyak K, Tang LJ, et al. p53-dependent expression of PIG3 during proliferation, genotoxic stress, and reversible growth arrest. Cancer Lett. 2000;156(1):63–72. doi: 10.1016/s0304-3835(00)00441-9. [DOI] [PubMed] [Google Scholar]
- 48.Boehm JS, Hahn WC. Understanding transformation: progress and gaps. Cur Opin Genet & Dev. 2005;15(1):13–7. doi: 10.1016/j.gde.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 49.Clark GJ, Der CJ. Aberrant function of the Ras signal transduction pathway in human breast cancer. Breast Cancer Res Treat. 1995;35(1):133–44. doi: 10.1007/BF00694753. [DOI] [PubMed] [Google Scholar]
- 50.von Lintig FC, Dreilinger AD, Varki NM, Wallace AM, Casteel DE, Boss GR. Ras activation in human breast cancer. Breast Cancer Res Treat. 2000;62(1):51–62. doi: 10.1023/a:1006491619920. [DOI] [PubMed] [Google Scholar]
- 51.Sarkisian CJ, Keister BA, Stairs DB, Boxer RB, Moody SE, Chodosh LA. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nature Cell Biol. 2007;9(5):493–505. doi: 10.1038/ncb1567. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.