

SUMMARY
Although TGFβ is a potent inhibitor of proliferation, epithelia lacking the essential receptor (TβRII) for TGFβ signaling display normal tissue homeostasis. By studying asymptomatic TβRII-deficient stratified epithelia, we show that tissue homeostasis is maintained by balancing hyperproliferation with elevated apoptosis. Moreover, rectal and genital epithelia, which are naturally proliferative, develop spontaneous squamous cell carcinomas with age when TβRII is absent. This progression is associated with a reduction in apoptosis and can be accelerated in phenotypically normal epidermis by oncogenic mutations in Ras. We show that TβRII deficiency leads to enhanced keratinocyte motility and integrin-FAK-Src signaling. Together, these mechanisms provide a molecular framework to account for many of the characteristics of TβRII-deficient invasive SQCCs.
INTRODUCTION
The development of cancers depends on the ability of tumor cells to enhance growth-promoting programs and restrict growth-inhibiting mechanisms and apoptotic cell death. This endows tumors with the advantage to overcome growth limitations, which they accomplish by acquiring multiple mutations in oncogenes and tumor suppressor genes. Featured prominently in cancers are oncogenic mutations that elevate Ras-MAPK signaling to enhance the growth potential of cells. Yet the activation of growth-promoting factors alone is not sufficient to sustain tumorigenesis, which requires additional mutations to abrogate growth-inhibiting factors. Retinoblastoma (Rb), which sequesters the E2F transcription factor to arrest cell cycle progression, is also frequently mutated in cancers. Together, Ras and Rb pathways represent the core program controlling tissue homeostasis and their activities are finely tuned by a variety of signaling mechanisms.
These nodes of interaction between peripheral signaling pathways and the core machinery enable a diverse set of oncogenes and tumor suppressor genes to hijack the tissue homeostasis program and endow the mutated cells with a growth advantage which eventually promotes tumorigenesis. Understanding these complex mechanisms involving cell autonomous as well as nonautonomous effects holds the promise of therapeutic approaches to counteract tumorigenesis.
Under normal circumstances, the transforming growth factor beta (TGFβ) signaling pathway restricts tumorigenesis, particularly in epithelial tissues (Bierie and Moses, 2006; Massague and Gomis, 2006). TGFβs bind to a bidimeric surface receptor complex composed of receptor types I (TβRI) and II (TβRII) to phosphorylate and activate receptor-bound Smad (Smad2/3) transcription factors enabling them to translocate into the nucleus and regulate TGFβ-responsive genes. In cultured epithelial cells in vitro, TGFβs act as potent inhibitors of proliferation. They act by causing an upregulation of cyclin D kinase inhibitors p15Ink4B and p21Cip to prevent Rb inactivation, resulting in a concomitant downregulation of c-Myc expression (Iavarone and Massague, 1997; Pietenpol et al., 1990; Reynisdottir et al., 1995). Thus, TGFβ signaling shows tumor suppressor characteristics, and TβRII is frequently mutated or transcriptionally suppressed, in human epithelial tumors.
Given the ascribed effects of TGFβ signaling on growth inhibition, it is surprising that many epithelia—including mammary gland, oral mucosa, esophagus, pancreas and intestine—still develop normally upon quantitative ablation of TGFβ signaling through conditional targeting of the TβRII gene (Biswas et al., 2004; Forrester et al., 2005; Ijichi et al., 2006; Lu et al., 2006; Munoz et al., 2006). That said, progression to cancers occurs rapidly when the TβRII null epithelial tissues are exposed to activated oncogenes (often oncogenic Ha-Ras) and/or loss of additional tumor suppressors, suggesting that some as yet unidentified feature of the homeostasis balancing mechanism must be compromised in the absence of TGFβ signaling. Another intriguing twist is the appearance of spontaneous invasive squamous cell carcinomas (SQCCs) of the forestomach epithelium that arises non-cell autonomously from ablation of TβRII in stromal fibroblasts (Bhowmick et al., 2004).
Analysis of the function of TGFβ signaling in surface epithelia has thus far been limited to dominant negative and overexpression strategies. Perhaps not surprisingly, results have often been conflicting, and both positive and negative effects on normal epidermal homeostasis and wound healing have been described (Amendt et al., 1998; 2002; Crowe et al., 2000; Ito et al., 2001; Wang et al., 1997b). In addition, despite TGFβ’s well-documented function as a suppressor of proliferation in cultured keratinocytes, skin tumorigenesis is paradoxically promoted when carcinogenesis protocols are applied to transgenic mice that either display superactivated TGFβ signaling (through overexpression of TGFβs) or suppressed TGFβ signaling (through overexpression of dominant negative TβRII) (Amendt et al., 1998; Cui et al., 1996; Cui et al., 1995; Go et al., 2000; Han et al., 2005; Wang et al., 1997b; Wang et al., 1999; Weeks et al., 2001).
Some of these disparate results are likely to arise from combinatorial extrinsic and intrinsic effects, since both dermal and epidermal cells respond to TGFβs. Additionally, some studies suggest that TGFβs act as growth suppressors early but act as metastasis promoters later in tumor progression (Bierie and Moses, 2006). To this end, both gain and loss of TGFβ signaling have been reported to promote invasive cell migration (Bhowmick et al., 2001; Ozdamar et al., 2005; Wang et al., 2005) as well as apoptosis (Amendt et al., 2002; Forrester et al., 2005), depending on cellular context and stage of tumorigenesis. Overall, these data suggest that the cell’s signaling profile may define the functional consequences of TGFβ signaling.
In the current study, we use a keratin 14 (K14) promoter, active in surface, oral, and anogenital stratified squamous epithelia as well as some glandular and ductal epithelia (Wang et al., 1997a) to conditionally target the loss of TβRII in mice. We show that mice lacking TGFβ receptor signaling in K14-positive cells develop spontaneous invasive SQCCs in their anal and genital epithelia, and that TGFβ signaling is diminished in human genital SQCCs. By contrast, TβRII null epidermis is phenotypically normal, and although wounds heal faster, oncogenic transformation with Ha-Ras is required to promote invasive SQCC and metastasis. Exploiting the ability to culture primary epidermal keratinocytes from our mice, we employ a combination of in vitro and in vivo strategies to explore the intrinsic and extrinsic mechanisms underlying how asymptomatic TβRII null stratified epithelia are able to maintain homeostasis, how they lose it, and why this happens at a higher frequency in anogenital epithelium. We also investigate how elevated Ras-MAPK signaling accelerates an imbalance in homeostasis and progression to SQCC when TGFβ signaling is defective. Finally, we address why loss of TβRII leads to enhanced cell motility and provide insights as to how this may promote invasive metastatic SQCCs and accelerated wound healing in epithelial tissues.
RESULTS
Conditional Targeting of the TGFβ Receptor II Gene in Mice Results in Spontaneous Anal and Genital SQCCs with Age
Mice harboring the TβRII floxed exon 4 (Leveen et al., 2002) were bred to mice expressing Cre recombinase under the control of the human K14 promoter, strongly active by embryonic day 15 (E15) in proliferative cells of most surface stratified squamous and glandular epithelia, as well as oral, anal, and genital stratified squamous epithelia (Vasioukhin et al., 1999) (Figure S1A in the Supplemental Data available with this article online). K14-Cre/TβRII (fl/fl) conditional knockout (cKO) mice were viable and appeared phenotypically normal through early adulthood. As judged by real-time PCR, in situ hybridization, and immunoblot analyses, intact TβRII mRNA and protein were absent by postnatal day 2 (P2) in skin epithelium and in cultured primary keratinocytes (MKs) derived from neonatal backskin epidermis (Figure 1A, and Figures S1A–S1D).
Figure 1. Adult TβRII cKO Mice Develop Spontaneous Anogenital SQCCs.
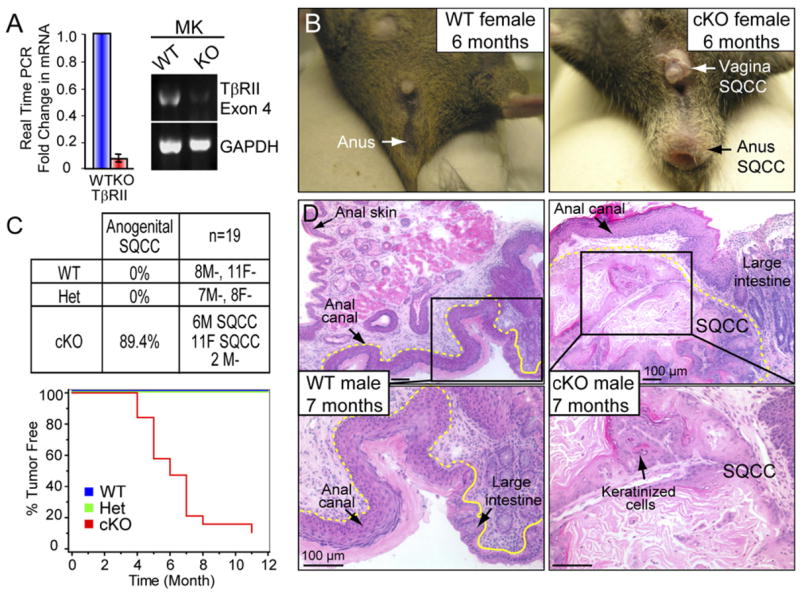
(A) Efficient targeting and loss of TβRII mRNA was assessed by real-time PCR using primer pairs corresponding to the floxed exon 4 of TβRII. MK, primary keratinocytes. Mean ± SD.
(B) Anal and vaginal tumors were visible in 6-month-old cKO, but not WT mice.
(C) Kaplan-Meier curves depicting the probability of tumor-free survival with age. M, male; F, female. Note that >89% of cKO mice developed anogenital tumors within 7 months, while WT and heterozygous (Het) animals were tumor-free.
(D) Hematoxylin- and eosinstained longitudinal sections of the distal colon, rectal, and anal/perianal tissues revealed classical SQCC pathology in 7-month-old cKO, but not in WT mice. SQCCs displayed anastomosing trabeculae and nests of keratinized squamous cells organized around central cavities, and were associated with moderate, multifocal, and chronic lymphoplasmacytic inflammation around the tumor masses.
Adult TβRII cKO mice developed SQCCs in their anal and genital regions, starting as early as 4 months of age (Figure 1B). By 7 months, 89% of cKO mice displayed visible signs of tumor formation (Figure 1C). Histological analyses revealed SQCCs arising within the anal canal in a transitional zone between stratified squamous epithelium of anal skin and mucosal epithelium of the large intestine. Typified by keratinized cells, the SQCCs infiltrated surrounding stroma and tunica muscularis of the rectum (Figure 1D). In males, genital SQCCs arose from preputial ductal epithelium and invaded surrounding stroma (Figure S2A). In females, SQCC tumors developed from mucosal stratified squamous epithelium of vagina and rectum and from adjacent follicular/adnexal epithelium (Figure S2B).
Immunostaining of anal and genital SQCCs confirmed the loss of TβRII protein and phosphorylated (active) p-Smad2 in these keratin 14 (K14) and keratin 5 (K5)-positive tumors (Figures S3 and S4). By contrast, the TβRII-positive stromal tissue surrounding the tumors exhibited p-Smad2, indicative of TGFβ signaling, and also infiltration of smooth muscle actin-positive myofibroblasts. Similar changes were observed in the stroma surrounding spontaneous genital SQCCs.
Human Genital SQCCs Exhibit Diminished TβRII Signaling
Given that TβRII function seemed to suppress development of spontaneous anogenital tumors in mice, we wondered whether TGFβ signaling might also be affected in human genital cancers. As judged by immunohistochemistry, TβRII was reduced or absent in 73% of the 80 male genital SQCC samples tested (Figures 2A and 2B). Concomitant with this reduction was a corresponding loss of activated Smad2 within the tumor tissue, despite the presence of TGFβ1 ligand (Figures 2A and 2B) Similarly, 76% (n = 41) of female genital SQCCs displayed reduced or absent TβRII protein which correlated with reduced or absent p-Smad2 (Figures 2C and 2D). Interestingly, these features were even observed in early-stage grade I genital tumors of both sexes, suggesting that loss of TβRII signaling may be an early event in tumor progression.
Figure 2. Diminished TGFβ Signaling in Human Genital SQCCs.
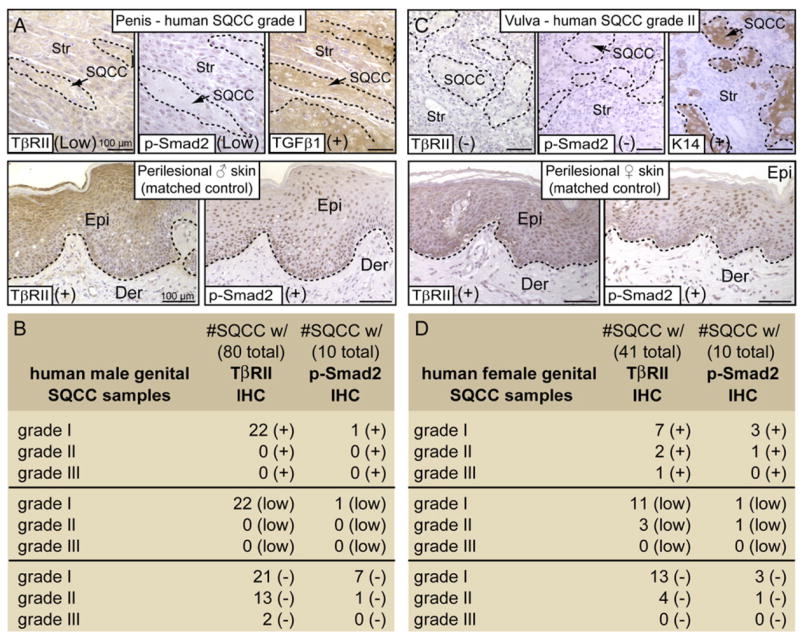
Male (B) and female (D) human genital SQCCs were scored according to their grade of severity (I-lowest to III-highest) and analyzed for TβRII, phosphorylated (activated) Smad2, TGFβ1 and/or K14 by immunohistochemistry (IHC). Examples of IHC staining from a male genital SQCC grade I (A) and from a female SQCC grade II (C) show reduced or no anti-TβRII, in contrast to perilesional, matched control skins. TβRII loss correlated with a lack of p-Smad2 staining in 10 out of 10 SQCC samples. TGFβ1 and K14 served as internal quality controls and were detected in TβRII-deficient human samples. Scoring: (+), positive staining; (−), negative staining; (low), reduced expression. Epi, epidermis; Der, dermis; Str, stroma. Lines encircle SQCCs in the top panels.
In K14-TβRII Null Animals, Spontaneous SQCCs Develop at Transitional Zones between Two Merging Epithelial Tissue Types
It was notable that the spontaneous SQCCs arising in our TβRII cKO mice frequently arose within transitional zones between two merging but distinct epithelial tissue types. Tumor susceptibility was especially prominent at the juncture of K5/K14-positive mucosal stratified squamous epithelium of the anal canal and K8/K18-positive simple epithelial tissue of the large intestine. Interestingly, irrespective of the status of TβRII gene targeting, anal canal epithelium at this juncture displayed many features typically associated with hyperproliferative epidermis (Schäfer and Werner, 2007). This included expression of both suprabasal markers, e.g., keratin 17 (K17) and keratin 6 (K6), and basal markers, e.g., β6-integrin (β6) and Tenascin C (TnC) (Figure 3A and Figure S5). Further reflective of an atypical hyperproliferative epidermal state was the presence of a large number of macrophages (Mac1) within the underlying anogenital stroma (Figures 3Ba and 3Bb).
Figure 3. Tissue Homeostasis Is Impaired in the Anal Canal of Older TβRII cKO Mice.

(A) The anal canal epithelium is a transitional epithelium bordering stratified squamous epithelium of anal skin and simple epithelium of large intestine. Immunofluorescence microscopy of frozen tissue sections revealed that this epithelium naturally expresses many markers of a hyperproliferative state, including K17, K6, β6-integrin (β6), and tenascin C (TnC). β4-integrin (β4) or K5 label the basal-like cells of the anal canal. Epithelial-stromal border is denoted by white lines. DAPI (blue) labels nuclear chromatin. Str, stroma.
(B) (Ba) Inflammatory Macrophages (Mac1) are prevalent in the stroma underlying the anal epithelium. (Bb) Mac1-positive cells are much more prevalent in the anal region (Anal) than in backskin epidermis (Epi) in both WT and cKO mice.
(C–F) TβRII null anal epithelium exhibits sustained hyperproliferation but maintains homeostasis through enhanced apoptosis, which is lost in spontaneous tumors.
(Ca–E) Proliferation was assayed by BrdU incorporation and correlates with pronounced Ras/p-MAPK staining. (Fa) Apoptosis was assayed by immunostaining for activated caspase 3 (Ac-casp3) (arrows). (Cb, Db, and Fb) Quantification of proliferation and apoptosis in WT and cKO anal canal and adjacent anal epidermis at 7 weeks when the cKO anal canal is histologically normal, and 7 months when it appears hyperplastic. Accelerated proliferation is balanced by increased apoptosis when homeostasis is maintained, while alleviated apoptosis correlated with tumor formation. Graphs represent the mean (±SD) of 3 different WT and cKO mice. *p < 0.05.
Overall, the natural state of anogenital transitional epithelium more closely resembled that of backskin epidermis when subjected to an imbalance in tissue homeostasis due to injury, microbial infection, inflammation, or precancerous lesions (Coulombe, 2003; Weiss et al., 1984).
Control of Epithelial Homeostasis in the Absence of TβRII Relies Upon the Ability to Balance Offsetting Increases in Proliferation and Apoptosis
Tissue homeostasis depends on a balance between proliferation, differentiation, and apoptosis, and aberrations in this equilibrium can result in the development of tumors. To test whether tissue homeostasis was affected in the anogenital tissue of asymptomatic cKO mice we pulsed these animals for 48 hours with BrdU and analyzed the level of proliferation. We detected a significant increase in the number of S phase cells within the epithelia of both anal canal and anal skin of 7 week cKO animals when compared to their wild-type (WT) littermates (Figure 3Cb). As mice aged, BrdU incorporation waned in anal epithelium of WT mice and in asymptomatic cKO epithelium, but it remained high in spontaneous tumors (Figure 3Cb).
Whether WT or cKO, wherever homeostasis was maintained, BrdU-positive epithelial cells were largely confined to a single epithelial layer adjacent to the basement membrane (Figure 3Ca). By contrast, proliferation often extended to suprabasal layers in the anal canal of 7-month-old cKO mice. Elevated proliferation was also noted in underlying stroma, further reflective of mesenchymal-epithelial interactions (Figures 3Ca and 3Cb). Consistent with the marked hyperproliferation, anal canal epithelium displayed signs of elevated Ras-MAPK signaling (Figures 3Da, 3Db, and 3E). Antibodies against p-MAPK (Erk1, Erk2) and pan-Ras both stained the tumor more strongly than adjacent or asymptomatic tissues. Thus, the increase in Ras-MAPK signaling correlated with tumorigenesis and a loss of homeostasis in TβRII-deficient anal epithelium.
Tissue homeostasis can be maintained in a hyperproliferative tissue either by increased cell death or accelerated differentiation. To test whether the hyperproliferation seen in asymptomatic anal epithelium of cKO mice might be counterbalanced by an increase in apoptosis, we conducted immunofluorescence using antibodies against activated caspase 3 (Ac-casp3). At 7 weeks of age, while apoptotic cells were rare in WT, they were frequent within the basal layer of cKO anal epithelium (Figures 3Fa and 3Fb). By 7 months of age, however, when anal tumorigenesis was common in cKO mice, the numbers of apoptotic cells were low in both WT and cKO anal epithelia.
Taken together, these findings suggest that when TβRII is deficient in anal epithelium of younger mice, homeostasis is still maintained by balancing enhanced proliferation with apoptosis; however, as animals age, the spontaneous transition to tumorigenesis is accompanied by a decline in apoptosis, tipping this equilibrium between proliferation and cell death.
Malignant Conversion of Ha-Ras-Induced Papillomas to SQCCs in Backskin Lacking TβRII
Given our findings with anal epithelia, we wondered whether the counterbalancing of elevated proliferation and apoptosis might also account for why loss of TβRII did not overtly seem to affect backskin epidermis and its appendages. We analyzed proliferation and apoptosis levels in two different stem cell compartments of skin epithelium: the slow cycling α6 integrin-positive, CD34-positive cells from the hair follicle bulge (Morris et al., 2004) and the more proliferative α6 integrin-positive, CD34-negative cells from the basal layer of interfollicular epidermis. Following a 48 hour pulse of BrdU administered to 7-week-old mice, fluorescence-activated cell sorting (FACS) was used to analyze the two populations from skin.
Despite signs of elevated TGFβ signaling normally displayed by bulge stem cells (Figure S7A) (Tumbar et al., 2004), the loss of TβRII did not appear to alter their quiescent state nor affect apoptosis (Figure 4A). By contrast, basal interfollicular epidermal cells exhibited an ~2× increase in BrdU incorporation within 48 hours, which was counterbalanced by elevated apoptosis (Figure 4A). Although perturbations in proliferation and apoptosis were lower in cKO backskin epidermis than in cKO anogenital epithelium, the outcome was similar, namely, a morphologically normal epithelium. In contrast to anogenital epithelium, however, epidermal homeostasis was maintained as animals aged.
Figure 4. TGFβ and Ras-MAPK Signaling Also Cooperate to Control Tissue Homeostasis in the Epidermis.
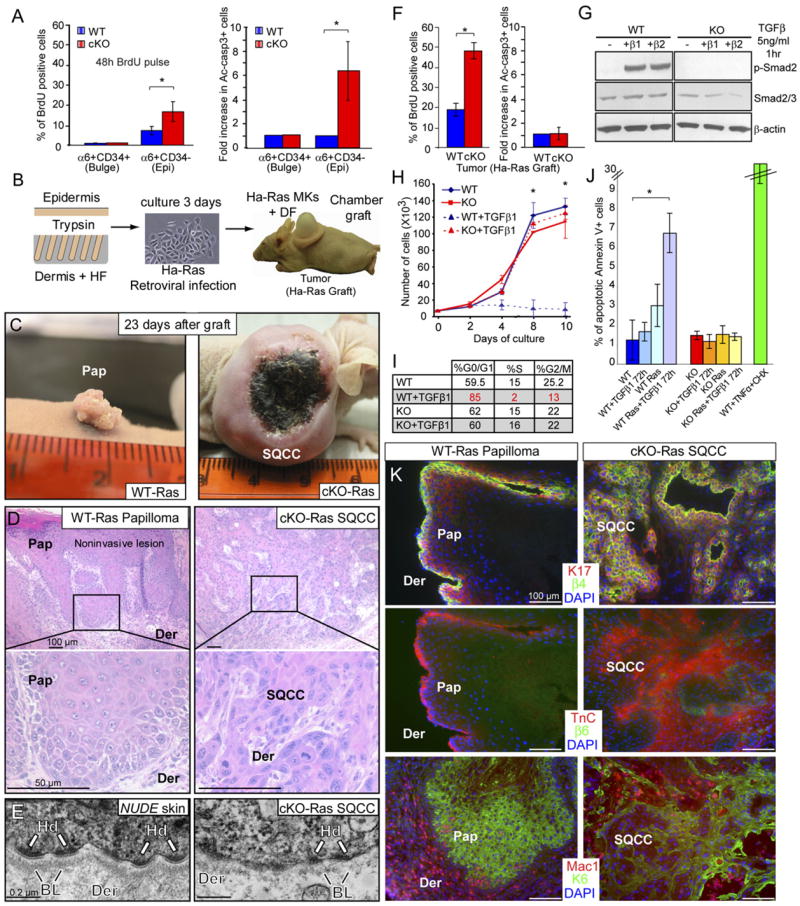
(A) Proliferation and apoptosis rates are increased in the stem cell compartment (basal layer) of TβRII null epidermis (Epi) (α6+CD34+), but not in the stem cell compartment (bulge) of the hair follicle (α6+CD34+). Analyses were performed on adult mice (7 wk). Graphs represent the mean (±SD) of 3 different WT and cKO mice. *p < 0.05.
(B) Schematic of tumor susceptibility assay where primary keratinocytes (MKs) from WT or cKO backskins were isolated, infected with a retrovirus expressing constitutively active (oncogenic) Ha-Ras, and grafted together with WT dermal fibroblasts (DF) onto the backs of Nude mice. HF, Hair follicle. (C) At 23 d, papillomas formed from Ha-Ras MKs, while invasive SQCCs formed from TβRII null/Ha-Ras MKs. Note necrosis in center of large tumor. (D) Hematoxylin- and eosin-stained tissue sections revealed papilloma (Pap) pathology in Ha-Ras graft and SQCC pathology in TβRII null/Ha-Ras graft.
(E) Ultrastructural analysis indicated a compromised basal lamina underlying Ha-Ras transformed, TβRII null SQCCs (right frame). Scarcity of hemidesmosomes (Hd) and perturbed basal lamina (BL) is typical of invasive SQCCs. The basal lamina appeared uncompromised at the boundary between Epi and dermis (Der) in adjacent nude skin (left frame).
(F) TβRII null/Ha-Ras-induced SQCCs exhibited sustained hyperproliferation without enhanced apoptosis. Graphs represent the mean (±SD) of 3 different WT and cKO mice. *p < 0.05.
(G) TβRII null MKs fail to activate Smad2 and remain resistant to a G1 growth arrest when stimulated with TGFβs as judged by growth curves and cell cycle profiles.
(H and I) MKs were cultured in low Ca2+ medium in the presence or absence of 5 ng/ml TGFβ1, added at day 2. Growth curves (H) indicate the average value of 3 independent experiments performed in triplicate (±SD). (Note: by this assay and Elisas, active TGFβs are low/absent in epidermal culture medium.) (I) Cell cycle analyses with propidium iodide staining.
(J) FACS analysis for the presence of the apoptotic marker annexin-V revealed that Ha-Ras and TGFβ signaling cooperate to induce apoptosis in WT MKs, while apoptosis rates remain low and unaltered in KO MKs under these conditions. TNFα (100 ng/ml) and cycloheximide (5 μg/ml) induced apoptosis in WT MKs served as a positive control. Bar graphs indicate mean value (±SD) on duplicate experiments.
(K) Keratin 17 (K17), Tenascin C (Tnc), and β6-integrin are expressed in both Ha-Ras papillomas and TβRII null/Ha-Ras SQCCs. β4-integrin marks the basal cell layer and Macrophages (Mac1) are recruited to the tumor stroma. Note similarities between the expression patterns in Ha-Ras papillomas (here) and WT anogenital epithelia (Figure 3).
Since Ras-MAPK activity was strongly enhanced in TβRII-deficient anogenital tumors, we wondered whether oncogenic mutations in this pathway might tilt tissue homeostasis in TβRII-deficient epidermis to promote SQCC formation. To test this hypothesis, we briefly cultured freshly isolated epidermal cells from WT and cKO mice, infected them with a retroviral vector expressing an oncogenic mutation in the GTPase domain of Ha-Ras (Ha-RasV12), and grafted these cells onto backs of athymic Nude mice (Figure 4B; Figures S6A and S6B). By 4 weeks, control grafts from Ha-Ras infected WT epidermal cells displayed visible and morphological signs of benign papilloma expected from Ha-Ras transformation of skin (Bailleul et al., 1990) (Figures 4C and 4D, and Figure S6C). Notably, TβRII expression was maintained in these papillomas, as judged by immunohistochemistry (Figure S6E).
In striking contrast, grafts from Ha-Ras-infected KO MKs displayed overt aberrations soon after grafting (Figure S6D). By 4 weeks, 100% of cKO-Ras grafts had developed large, aggressive tumors, which often developed signs of ulcerations and necroses in the tumor center (Figure 4C). Ethical concerns mandated sacrificing mice within 4 weeks, at which time the cKO-Ras engrafted skin displayed signs of poorly differentiated SQCCs, including cellular atypia, mitoses, cellular disorganization, and invasion (Figure 4D). At the ultrastructural level, an intact basal lamina demarcated the dermal-epidermal boundary in normal skin of the Nude mouse (Figure 4E, left frame), while in invasive, Ha-Ras transformed TβRII null SQCCs, it was discontinuous and often absent (Figure 4E, right frame).
Both Ha-Ras papillomas and TβRII null/Ha-Ras SQCCs displayed elevated BrdU incorporation relative to WT backskin (compare Figures 4F and 4A). While BrdU-positive cells were confined to the basal and first few supra-basal layers of papillomas, they were dispersed throughout the TβRII null/Ha-Ras SQCCs (Figure S6G). Outside necrotic regions, ~2.5× more BrdU-positive cells were found in the TβRII null/Ha-Ras SQCCs when compared to Ha-Ras papillomas (Figure 4F), while apoptoses were comparable and low compared to Ha-Ras papillomas (Figures S6H, Figure 4F).
To determine whether alterations in epithelial proliferation in TβRII null cells are cell-intrinsic or dependent upon their environment, we examined KO MKs in vitro. In contrast to their WT counterparts, these cells were refractory to TGFβ signaling and displayed no signs of activated phospho-Smad2 when exposed to recombinant active TGFβ1 or TGFβ2 (Figure 4G). KO MKs also failed to undergo TGFβ-mediated growth arrest (Figure 4H), and did not accumulate in the G0/G1 phase of the cell cycle at the expense of S phase cells (Figure 4I and Figure S7B). Similarly, only WT MKs responded to TGFβ1 by enhanced expression of cyclin-dependent kinase inhibitors p15INK4b and p27Kip1 (Figure S7C). Thus, although KO and WT MK were comparably hyperproliferative in the absence of TGFβ signaling, marked differences were noted when TGFβs were present. Notably, p-Smad2 and TGFβ1 were both upregulated in the stroma surrounding Ha-Ras cKO SQCCs (Figure S6F).
To test whether differences in apoptosis are intrinsic to the loss of TβRII function, we treated MKs with TGFβ1 (test) or the potent apoptosis-inducer TNFα (control), and used FACS to quantify expression of the apoptosis marker Annexin V. Even after treating with TGFβ1 for 72 hours, apoptosis levels remained low, and no significant differences were noted between WT and KO MKs (Figure 4J, blue and red bars, respectively). We next examined the ability of Ha-Ras transformation to impact apoptosis. Even without exogenously added TGFβ1, Ha-Ras MKs exhibited an ~2.5× increase in apoptosis, and when TGFβ1 was added, this level rose another 2×. In striking contrast, apoptosis levels remained low in the TβRII null/Ha-Ras MKs (Figure 4J). Thus, the loss of TβRII appeared to render Ha-Ras transformed MKs refractory to TGFβ1-mediated apoptosis.
The similarities in Ha-Ras transformed epidermis and WT anal epithelium to loss of TβRII prompted us to examine the status of the molecular markers that we previously found to distinguish the anal canal from backskin of WT mice. Ha-Ras-induced papillomas expressed K17, K6, and TnC, and caused a marked macrophage infiltration in underlying dermis (Figure 4K, left). These alterations were as or more pronounced in TβRII null/Ha-Ras-induced SQCCs, and in addition, β6-integrin was selectively found at invasive fronts (Figure 4K, right).
Taken together, these data suggest that loss of TβRII in epidermal keratinocytes cooperates with oncogenic mutations in Ha-Ras to promote hyperproliferation and maintain low levels of apoptosis, resulting in a gross imbalance in tissue homeostasis, which progresses to SQCC. Ha-Ras transformation of epidermis also induced features of WT anogenital epithelium, and although the initiating event remains to be uncovered, Ras-MAPK signaling was elevated in spontaneous anogenital tumors that formed in older TβRII cKO mice. These findings may account for differences in susceptibility of TβRII-deficient anogenital and backskin epithelium to homeostatic imbalancing and tumor progression.
Elevated Integrin and FAK Activity Is Intrinsic to the Loss of TβRII in Stratified Epithelia
The increased susceptibility of TβRII null epidermis to Ha-Ras induced SQCC formation was further tested by treating mice with the chemical mutagen 7,12-dimethyl-benz [a] anthracene (DMBA), which typically induces mutations in Ha-Ras (Balmain et al., 1984) but requires additional promoting agents to generate SQCCs (Yuspa et al., 1995). In contrast to WT mice, cKO mice developed visible backskin tumors within 5–6 weeks following DMBA treatments (Figure 5A). After 15–20 weeks, cKO mice had an average of 7 tumors each, and pathological analyses revealed signs of K14-positive SQCCs in the lungs, suggestive of metastases (Figure 5B).
Figure 5. Invasive Metastatic Skin Tumors Correlate with Increased Integrin, FAK, Src, and MAPK Activities.

(A) DMBA treatment of mice revealed a marked increase in DMBA-induced tumor formation in the absence of TβRII.
(B) Lung metastasis from TβRII-cKO mouse topically treated with DMBA to induce backskin SQCCs. Note typical keratinized SQCC histology as revealed by hematoxylin and eosin staining and K14 immunohistochemistry.
(C–F) Anti-FAK and p-FAK immunohistochemistry on sections from WT anal canal (C), cKO pretumor and tumor stage of anogenital skin (D and E), and cKO-Ras backskin SQCC (F). Str, stroma; Epi, epidermis; Der, dermis.
(G–I) Immunoblots of protein lysates from MKs grown in the presence or absence of serum, ±4 min serum stimulation where indicated. Note: Src activity was assessed by immunoprecipitation/immunoblot analysis using Src and pY417-Src antibodies.
(J–M) Immunofluorescence with antibodies against β1- and active β1-integrin (Ac-β1) performed on tissue sections as indicated.
(N) Quantitative FACS analysis of KO (red) versus WT (blue) cultured MKs with antibodies against the cell surface integrins indicated. Gray lines depict secondary antibody-only control.
Elevated focal adhesion kinase (FAK) has been detected in head and neck SQCCs (Canel et al., 2006), and mice lacking epidermal FAK are more resistant to DMBA/TPA-induced SQCCs (McLean et al., 2004). To determine whether FAK expression and/or activation might be involved in SQCCs formed in TβRII cKO animals, we first conducted immunohistochemistry. Whereas antibodies against FAK only weakly labeled WT tissue, staining was readily detected in cKO anal epithelium (Figures 5C and 5D). Signs of activated (pY397) FAK were even more pronounced in the SQCCs that formed (Figures 5E and 5F). Elevated FAK activity was observed in both basal and suprabasal layers, consistent with the atypical supra-basal expression of integrins known to occur in SQCCs (Owens and Watt, 2001).
To better assess the relationship between loss of TβRII and elevated FAK activity, we next plated MKs on fibronectin (FN)-coated dishes and cultured them in absence or presence of serum. Immunoblot analyses of protein extracts revealed that in vitro as in vivo, KO MKs displayed elevated FAK activity (Figure 5G). Using analogous phosphospecific antibodies, we found that Src and MAPK activities, but not the cell survival kinase AKT, were elevated in TβRII null MKs compared to WT MKs (Figures 5H and 5I and data not shown). FAK, Src, and MAPK activities were enhanced even in serum-free media (Figures 5G–5I), suggesting that signaling through integrins and cell matrix interactions might be responsible for these differences. We tested this hypothesis by analyzing the in vivo expression and activity of β1-integrin, a key activator of FAK. In adult cKO backskin, anti-β1-integrin labeled the basal epidermal layer as well as the dermis, while antibodies against the activated form of β1-integrin only showed strong labeling in the dermis (Figure 5J). By contrast, both antibodies labeled the cKO anal canal prominently, in stroma and in SQCC cells (Figures 5K and 5L). TβRII null/Ha-Ras SQCC cells also exhibited enhanced β1-integrin signaling (Figure 5M). The association of increased β1-integrin activation with TβRII-ablation was also observed in vitro, where we could use FACS analysis to quantify surface integrin levels (Figure 5N).
TβRII-Deficient Epidermal Keratinocytes Possess a Cell-Autonomous, Enhanced Ability to Migrate, Degrade Extracellular Matrix, and Invade: In Vivo and In Vitro Studies
The data presented in Figure 5 suggest that loss of TβRII might enhance keratinocyte migration through activation of integrin-FAK-Src signaling. To explore this possibility further, we next monitored the response of our mice to 4 mm punch biopsy wounds. Interestingly, cKO wounds healed faster (Figure 6A). FAK and MAPK were hyperactivated at the wound edge in both WT and cKO skins, although the wound edge was noticeably thicker in cKO skin (Figures 6B and 6C). BrdU labeling substantiated the hyperproliferative status of cKO epidermis in the wound area, and anti-Ac-casp 3 immunofluorescence suggested that apoptosis was selectively reduced at the wound site, thereby accounting for the corresponding thickened tissue (Figures 6D and 6E). When tissue explants were transferred from backskin to FN-coated culture dishes, epidermal outgrowth was also accelerated when TβRII was absent (frames in Figure 6F). These differences were striking when 5 ng/ml active TGFβ1 was added to the culture medium, a feature which abruptly inhibited outgrowth from WT explants while leaving cKO explants unaffected (graph in Figure 6F).
Figure 6. Wound Closure Is Accelerated in cKO versus WT Animals.
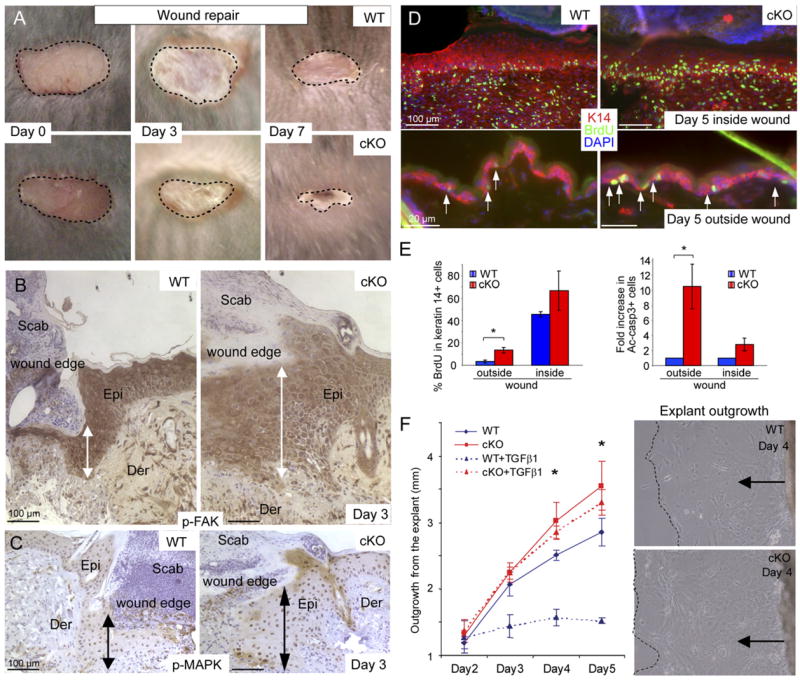
(A) Shown are representative examples at 0, 3, and 7 days after wounding.
(B and C) Immunohistochemistry reveals activated FAK and MAPK at the epidermal wound edge by day 3 after wounding. Note hyperthickening of wounded cKO skin. Epi, epidermis; Der, dermis.
(D and E) Quantification of proliferation and apoptosis inside and outside the wound area. Bar graphs depict mean ± SD. *p < 0.05.
(F) WT and cKO skin explant outgrowth in serum-containing media indicate accelerated epidermal outgrowth from cKO skin explants, a difference that becomes even more pronounced upon addition of active TGFβ (5 ng/ml) to the culture medium on day 2. Graph indicates the mean value of three independent experiments (±SD). *p < 0.03. Phase contrast images show extent of outgrowth at 4 days in the absence of active TGFβ .
A priori, the enhanced wound healing and explant outgrowth could be due solely to the imbalance in tissue homeostasis caused by loss of TβRII. Alternatively, it might reflect an additional migratory and/or invasive advantage. To distinguish between these possibilities, we conducted transwell migration assays in which primary MKs were placed in the upper compartment of a Boyden chamber, while fibroblast-conditioned medium was placed as a stimulus in the bottom chamber. In these assays, KO MKs exhibited a migratory advantage over WT cells with and without additional expression of Ha-Ras, but Ha-Ras enhanced the effects (Figure 7A). When the assay was repeated, this time after coating the chamber filter with Matrigel (extracellular matrix), KO MKs exhibited an increased ability to invade the matrix and traverse the filter (Figure 7B). Again, the presence of oncogenic Ha-Ras enhanced the invasive behavior both in KO and WT cells. Moreover, since the stimulus used in the bottom chamber was always the same, the observed differences in migration and invasion must initiate from keratinocyte-autonomous changes resulting from TβRII-deficiency and/or elevated Ha-Ras activity, which appeared to act synergistically.
Figure 7. Elevated Cell Motility in the Absence of TbRII.
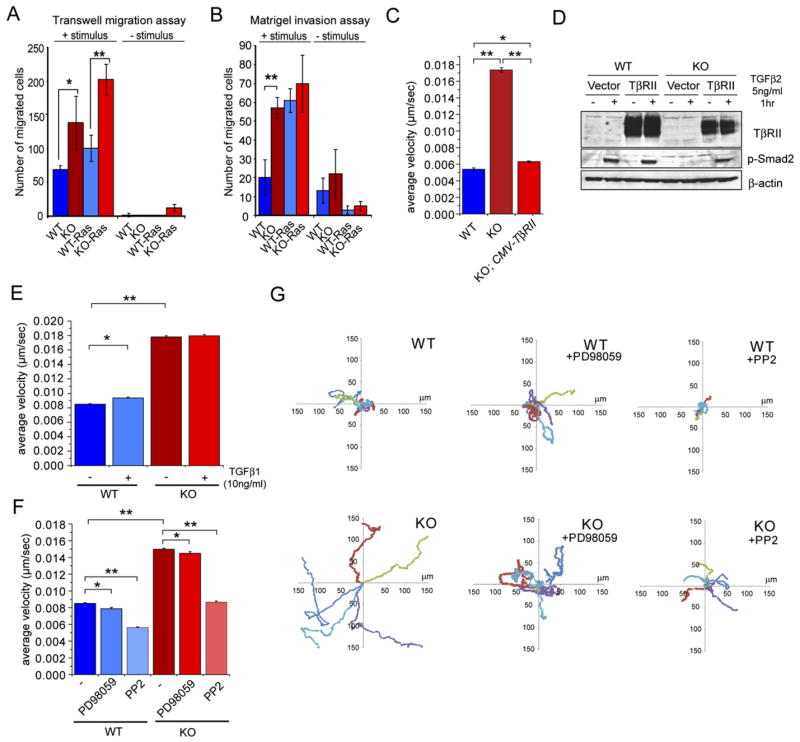
(A and B) Elevated cell motility in the absence of TβRII. Transwell migration assays were carried out on Boyden chambers ± coating with Matrigel ECM on the top chamber and in the absence and presence of fibroblast-conditioned medium, used as stimulus in the bottom chamber. Bar graphs depict means of 3 independent experiments ± SD. *p < 0.03. **p < 0.004. Note that KO MKs showed elevated migration and invasion over WT MKs, and Ha-Ras transformation enhanced these effects.
(C and D) Rescue experiment to show that the enhanced migratory ability of KO MKs is specifically attributable to TβRII loss. The CMV promoter was used to drive TβRII cDNA, which restored cell velocities to WT levels (C). Immunoblot shows that re-expression of TβRII in KO MKs restores TGFβ2 responsiveness (D).
(E) TGFβ1 treatment has only a slight effect on the velocity of WT MKs, which are still significantly less motile than KO MKs.
(F and G) Enhanced MAPK (Erk) and Src/FAK activities promote cell motility in KO MKs. (F) Cell velocities were measured ± 50 μM of the MEK1 inhibitor PD98059 and ± 5 μM of the Src inhibitor PP2, which also inhibits FAK activity. (C, E, and F) Thirty cells were tracked by phase-contrast microscopy with an acquisition rate of 1 frame/min over 12 hr. Bar graphs indicate average velocity ± SEM. *p < 0.05. **p < 0.0001. (G) Scatter plots depict the migration tracks of 5 representative cells for each condition.
We quantified cell velocities by imaging the migrations of MKs on FN-coated slides (n = 30 over 12 hr). By video-microscopy, the significantly faster velocities of KO MKs were readily visualized (Figure 7C). Moreover, the accelerated movement was directly due to the loss of TβRII, as re-expression of TβRII restored p-Smad2 activity and rescued the cell motility advantage (Figures 7C and 7D). Furthermore, although treatment of WT MKs with TGFβ1 slightly accelerated their motility, they did not reach the pace of KO cells, which were insensitive to TGFβ1 treatment (Figure 7E). Taken together, these findings underscore the intrinsic importance of TβRII-deficiency in the enhanced migratory behavior of MKs.
Finally, we treated MKs with the MEK1 inhibitor PD98059 and the Src/FAK inhibitor PP2 to test the degree to which downstream elevation of MAPK or FAK/Src activities might account for the differences in cell motility. Although inhibition of MEK1 only modestly reduced motilities, inhibition of Src/FAK had a more robust effect (Figures 7F and 7G). These findings suggest that the elevated Src/FAK activities contribute to the enhanced migratory behavior of TβRII cKO epithelia, which could be important in their increased susceptibility to homeostatic imbalance and tumorigenesis.
DISCUSSION
Balancing Hyperproliferation and Apoptosis to Maintain Homeostasis
The development of spontaneous anogenital SQCC in our TβRII cKO mice was unexpected. No significant pathological changes have been identified in internal stratified or simple epithelia lacking TβRII expression (Biswas et al., 2004; Ijichi et al., 2006; Lu et al., 2006), and while TβRII null mammary epithelia exhibited a transient hyperplasia in neonatal mice, this regressed during adulthood, and tissue homeostasis was maintained thereafter (Forrester et al., 2005). Similarly in our own studies, tissue homeostasis was maintained in the absence of TβRII, and both in backskin epidermis and in juvenile anogenital epithelium, enhanced hyperproliferation was counterbalanced by an increased apoptosis rate.
As cKO mice aged, apoptosis decreased significantly in the anogenital epithelium, while cells remained hyperproliferative and sustained elevated Ras-MAPK signaling. This age-related imbalance in TβRII-deficient tissue homeostasis correlated with the appearance of spontaneous anogenital SQCCs. Moreover, even though we did not detect spontaneous SQCCs in cKO backskin, invasive metastatic SQCCs were readily induced by oncogenic mutations in Ha-Ras or treatment with the mutagen DMBA.
Like the spontaneous anogenital tumors which formed, Ha-Ras transformed cKO backskin tumors exhibited significantly reduced apoptosis rates while simultaneously remaining hyperproliferative. Our data on cultured MKs revealed that TGFβ signaling in cells transformed by oncogenic Ha-Ras induces apoptosis in WT cells, and this effect is quantitatively lost when TβRII is mutated. Taken together, these findings suggest that loss of TβRII signaling stresses the homeostatic circuitry of stratified squamous epithelia by uncoupling the ability of keratinocytes to execute apoptosis in the face of oncogenic mutations that promote a sustained hyperproliferative state.
Hyperactivation of Regulatory Circuitries Involving Integrin-FAK-Src-MAPK Signaling and Cell Migration: Links between Loss of TβRII and Enhanced Wound Healing, SQCC Progression, and Metastasis
In exploring the underlying basis for why the junction between simple epithelium of the intestine and squamous epithelium of the anal canal might be particularly sensitive to SQCC progression, we began to realize that this transitional epithelium naturally possesses many of the same features that are displayed by the epidermis when it either responds to wounding in normal individuals or exists in patients with hyperproliferative skin disorders (Coulombe, 2003; Weiss et al., 1984). Thus, WT anogenital epithelium is not only substantially thicker and more proliferative than backskin epidermis, but in addition it expresses hyperproliferative-associated suprabasal keratins as well as migration-associated integrins and ECM ligands, and even displays a high number of inflammatory cells in the underlying stroma. These findings suggest that this zone of stratified epithelium may be naturally subjected to a chronic hyperproliferative state, a trait which renders the epithelium more susceptible to loss of tissue homeostasis when TGFβ signaling is compromised.
It is particularly intriguing that TnC and β6-integrin are expressed at this site as their expression is known to correlate with invasive growth during wound healing and tumorigenesis (Bates et al., 2005; Munger et al., 1999). Furthermore, αvβ6 can activate latent TGFβ1 and TGFβ3 leading to TGFβ stimulation in mice (Annes et al., 2004). In this way, the activation of αvβ6/TnC may provide a mechanism for hyperproliferative epithelial tissues to keep homeostasis in check by stimulating and responding to TGFβ signaling, a process interrupted by conditionally targeting TβRII.
Another link between loss of TβRII and altered integrin activation came from our studies in cultured MKs, which like anal epithelia, naturally display elevated αvβ6 signaling (Schober et al., 2007). In the absence of TβRII, however, cultured MKs additionally displayed higher β1-integrin, FAK, Src, and MAPK activities, and enhanced migration and invasion. This was the case even when cells were cultured in the absence of exogenous growth factors, suggesting that the mechanism is intrinsic to the keratinocyte, and is mediated through integrin signaling. It is also dependent upon TβRII deficiency, as reintroduction of TβRII expression into TβRII null cells reduced their migratory behavior to WT levels.
TGFβ signaling has been attributed to wound healing and cell motility, although a variety of Smad-dependent and independent mechanisms have been implicated, and both positive and negative effects have been described (Arany et al., 2006; Ashcroft et al., 1999; Crowe et al., 2000; Koch et al., 2000; Oft et al., 1998; Zhu et al., 1998). Our studies provide compelling evidence that loss of TGFβ signaling in keratinocytes promotes wound closure in vivo and leads to enhanced epidermal outgrowth from explants in vitro. The advantage of TβRII null epidermis in wound healing does not appear to be attributable solely to the alterations in proliferation and apoptosis, as the average velocities of migrating TβRII null MKs were significantly higher than WT MKs even in vitro and in the absence of active TGFβs, i.e., conditions where proliferative and apoptotic levels were comparable.
At first glance, our finding might seem paradoxical in light of those studies where TGFβ stimulates cell motility in MKs or in other cell types. That said, a biphasic concentration-dependent response is often observed in migrating cells where suboptimal cell velocities have resulted from either elevated or abrogated chemotactic signaling or elevated or decreased adhesiveness to the underlying substratum (Lauffenburger and Horwitz, 1996). In this regard, hyperactivation of β1-integrin-FAK-Src-MAPK signaling can profoundly influence cell motility and conversely pharmacological inhibition of FAK and Src activities strongly suppressed the migratory advantage of TβRII null MKs. These data argue for a role for FAK and Src downstream of TβRII in controlling cell motility in keratinocytes.
The increased activity of β1, FAK, Src, and MAPK activities coupled with enhanced migratory activity could also explain why cKO mice repaired their wounds faster than their WT counterparts. Not only is FAK hyperactive in normal wound repair, but conversely, conditional ablation of FAK in keratinocytes impairs epidermal outgrowth from skin explants in vivo and causes defects in hair follicle downgrowth in vivo (Mitra et al., 2006; Schober et al., 2007). Interestingly, FAK deficiency also renders stratified squamous epithelia resistant to Ha-Ras-induced SQCC formation (McLean et al., 2004), further strengthening the importance of the connection between loss of TβRII, elevated integrin-FAK signaling, and enhanced cell migration that we’ve uncovered here.
In closing, although future studies will be necessary to illuminate more of the details underlying the circuitry we’ve described, our data provide a mechanistic framework for understanding the role of TGFβ signaling in regulating homeostasis, injury response, and carcinogenesis in epithelial tissues, and offer a molecular explanation for the surface epithelial tumorigenesis that occurs when TGFβ signaling is compromised.
EXPERIMENTAL PROCEDURES
Generation of cKO Mice, Carcinogenesis Protocols, and Analysis of Tumors
TβRII floxed mice and K14-Cre transgenic mice were generated as described (Leveen et al., 2002; Vasioukhin et al., 1999). Genotyping was conducted by PCR of tail skin DNAs. Complete carcinogenesis protocols were performed on cohorts of 6 WT and 5 cKO mice at 6–8 weeks of age as described (Harper et al., 1987). Tumor pathologies were analyzed by at least two independent pathologists in the Laboratory of Comparative Pathology at Cornell University Medical College. All studies were approved by the IACUC committee and follow the NIH guidelines.
Immunofluorescence, Antibodies, Inhibitors, and In Situ Hybridization
Primary antibodies are described in the Supplemental Experimental Procedures. For detection of apoptosis Annexin V (Molecular Probes) was used. Chemical inhibitors used: PD98059 (MEK, Calbiochem, 50 μM for 6 hr), and PP2 (Src, Calbiochem, 5 μM).
Human Tumor Studies and Anatomic Pathology
Tissue microarray from 118 human skin SQCCs (SK241, SK242, SK801, SK802) and from 80 male and 41 female genital SQCCs (UV241, UV801, PE241, PE801) were obtained from US Biomax, Rockeville, MD. Paraffin slides were deparaffinized and stained with either TβRII or p-Smad2 antibodies, followed by the DAB substrate kit for peroxidase visualization of secondary antibodies (Vector Laboratories).
Wound, Explant, and Migration/Invasion Assays
In vivo wounds on 7-week-old animals were performed using a 4 mm dermal biopsy punch (Miltex). The size of the closing wound was monitored daily until day 8. Skin explants and migration assays were performed as described (Schober et al., 2007). Invasion assays were performed in precoated Matrigel invasion chamber (BD Biosciences). Individual MKs were imaged with an Olympus phase-contrast microscope (20X) for 12 hr at 1 frame/min and manually tracked in Metamorph (Universal Imaging).
Supplementary Material
The Supplemental Data include Supplemental Experimental Procedures and seven supplemental figures and can be found with this article online at http://www.cancercell.org/cgi/content/full/12/4/313/DC1/.
Acknowledgments
We extend a special thank you to those who provided us with mice, antibodies, and reagents, and we cite their gifts in the text. We are grateful to Stefan Karlsson (Institute of Laboratory Medicine, The Lund Strategic Research Center for Stem Cell Biology and Cell Therapy, Lund University) for floxed-TβRII mice; Stuart Yuspa (National Institutes of Health) for the oncogenic Haras viral stock; Christophe Cataisson for numerous scientific discussions; Krista Laperle and Suzanna Couzo (Tri-Institutional Laboratory of Comparative Pathology & Genetically Engineered Mouse Phenotyping Core, Memorial Sloan-Kettering Cancer Center); June de la Cruz, Maria Nikolova, and Nicole Stokes for technical assistance; Xuan Wang, Agnieska Kobielak, Jonathan Nowak, and Valerie Horsley for valuable discussions and advice. E.F. is an Investigator of the Howard Hughes Medical Institute, G.G. is the recipient of an HFSP fellowship, and M.S. is the recipient of a Jane Coffin Childs fellowship. This work was funded in part by a grant from the National Institutes of Health. None of the authors have financial interest related to this work.
Footnotes
SIGNIFICANCE: By generating a mouse model where an essential receptor (TβRII) for TGFβ signaling is lost in stratified epithelia, we’ve discovered an unexpected and specific link between spontaneous anogenital cancers and TβRII deficiency in mice and in humans. We show that the transition from phenotypically normal homeostasis to tumor progression is associated with a reduction in apoptosis, which unmasks hyperproliferative and migratory defects also caused by TβRII deficiency. Finally, progression to carcinogenesis in TβRII-deficient stratified epithelial tissues is accelerated upon oncogenic mutations or otherwise sustained signaling of the Ras/MAPK pathway. These findings provide insights into our understanding of squamous cell carcinomas, which are among the most prevalent and life-threatening cancers in the world.
References
- Amendt C, Schirmacher P, Weber H, Blessing M. Expression of a dominant negative type II TGF-β receptor in mouse skin results in an increase in carcinoma incidence and an acceleration of carcinoma development. Oncogene. 1998;17:25–34. doi: 10.1038/sj.onc.1202161. [DOI] [PubMed] [Google Scholar]
- Amendt C, Mann A, Schirmacher P, Blessing M. Resistance of keratinocytes to TGFβ-mediated growth restriction and apoptosis induction accelerates re-epithelialization in skin wounds. J Cell Sci. 2002;115:2189–2198. doi: 10.1242/jcs.115.10.2189. [DOI] [PubMed] [Google Scholar]
- Annes JP, Chen Y, Munger JS, Rifkin DB. Integrin αVβ6-mediated activation of latent TGF-β requires the latent TGF-β binding protein-1. J Cell Biol. 2004;165:723–734. doi: 10.1083/jcb.200312172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arany PR, Flanders KC, Kobayashi T, Kuo CK, Stuelten C, Desai KV, Tuan R, Rennard SI, Roberts AB. Smad3 deficiency alters key structural elements of the extracellular matrix and mechanotransduction of wound closure. Proc Natl Acad Sci USA. 2006;103:9250–9255. doi: 10.1073/pnas.0602473103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft GS, Yang X, Glick AB, Weinstein M, Letterio JL, Mizel DE, Anzano M, Greenwell-Wild T, Wahl SM, Deng C, et al. Mice lacking Smad3 show accelerated wound healing and an impaired local inflammatory response. Nat Cell Biol. 1999;1:260–266. doi: 10.1038/12971. [DOI] [PubMed] [Google Scholar]
- Bailleul B, Surani MA, White S, Barton SC, Brown K, Blessing M, Jorcano J, Balmain A. Skin hyperkeratosis and papilloma formation in transgenic mice expressing a ras oncogene from a suprabasal keratin promoter. Cell. 1990;62:697–708. doi: 10.1016/0092-8674(90)90115-u. [DOI] [PubMed] [Google Scholar]
- Balmain A, Ramsden M, Bowden GT, Smith J. Activation of the mouse cellular Harveyras gene in chemically induced benign skin papillomas. Nature. 1984;307:658–660. doi: 10.1038/307658a0. [DOI] [PubMed] [Google Scholar]
- Bates RC, Bellovin DI, Brown C, Maynard E, Wu B, Kawakatsu H, Sheppard D, Oettgen P, Mercurio AM. Transcriptional activation of integrin β6 during the epithelial-mesenchymal transition defines a novel prognostic indicator of aggressive colon carcinoma. J Clin Invest. 2005;115:339–347. doi: 10.1172/JCI23183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhowmick NA, Ghiassi M, Bakin A, Aakre M, Lundquist CA, Engel ME, Arteaga CL, Moses HL. Transforming growth factor-β1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol Biol Cell. 2001;12:27–36. doi: 10.1091/mbc.12.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S, Washington MK, Neilson EG, Moses HL. TGF-β signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 2004;303:848–851. doi: 10.1126/science.1090922. [DOI] [PubMed] [Google Scholar]
- Bierie B, Moses HL. Tumour microenvironment: TGFβ: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer. 2006;6:506–520. doi: 10.1038/nrc1926. [DOI] [PubMed] [Google Scholar]
- Biswas S, Chytil A, Washington K, Romero-Gallo J, Gorska AE, Wirth PS, Gautam S, Moses HL, Grady WM. Transforming growth factor β receptor type II inactivation promotes the establishment and progression of colon cancer. Cancer Res. 2004;64:4687–4692. doi: 10.1158/0008-5472.CAN-03-3255. [DOI] [PubMed] [Google Scholar]
- Canel M, Secades P, Rodrigo JP, Cabanillas R, Herrero A, Suarez C, Chiara MD. Overexpression of focal adhesion kinase in head and neck squamous cell carcinoma is independent of fak gene copy number. Clin Cancer Res. 2006;12:3272–3279. doi: 10.1158/1078-0432.CCR-05-1583. [DOI] [PubMed] [Google Scholar]
- Coulombe PA. Wound epithelialization: accelerating the pace of discovery. J Invest Dermatol. 2003;121:219–230. doi: 10.1046/j.1523-1747.2003.12387.x. [DOI] [PubMed] [Google Scholar]
- Crowe MJ, Doetschman T, Greenhalgh DG. Delayed wound healing in immunodeficient TGF-β 1 knockout mice. J Invest Dermatol. 2000;115:3–11. doi: 10.1046/j.1523-1747.2000.00010.x. [DOI] [PubMed] [Google Scholar]
- Cui W, Fowlis DJ, Cousins FM, Duffie E, Bryson S, Balmain A, Akhurst RJ. Concerted action of TGF-β 1 and its type II receptor in control of epidermal homeostasis in transgenic mice. Genes Dev. 1995;9:945–955. doi: 10.1101/gad.9.8.945. [DOI] [PubMed] [Google Scholar]
- Cui W, Fowlis DJ, Bryson S, Duffie E, Ireland H, Balmain A, Akhurst RJ. TGFβ1 inhibits the formation of benign skin tumors, but enhances progression to invasive spindle carcinomas in transgenic mice. Cell. 1996;86:531–542. doi: 10.1016/s0092-8674(00)80127-0. [DOI] [PubMed] [Google Scholar]
- Forrester E, Chytil A, Bierie B, Aakre M, Gorska AE, Sharif-Afshar AR, Muller WJ, Moses HL. Effect of conditional knockout of the type II TGF-β receptor gene in mammary epithelia on mammary gland development and polyomavirus middle T antigen induced tumor formation and metastasis. Cancer Res. 2005;65:2296–2302. doi: 10.1158/0008-5472.CAN-04-3272. [DOI] [PubMed] [Google Scholar]
- Go C, He W, Zhong L, Li P, Huang J, Brinkley BR, Wang XJ. Aberrant cell cycle progression contributes to the early-stage accelerated carcinogenesis in transgenic epidermis expressing the dominant negative TGFβRII. Oncogene. 2000;19:3623–3631. doi: 10.1038/sj.onc.1203701. [DOI] [PubMed] [Google Scholar]
- Han G, Lu SL, Li AG, He W, Corless CL, Kulesz-Martin M, Wang XJ. Distinct mechanisms of TGF-β1-mediated epithelial-to-mesenchymal transition and metastasis during skin carcinogenesis. J Clin Invest. 2005;115:1714–1723. doi: 10.1172/JCI24399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JR, Reynolds SH, Greenhalgh DA, Strickland JE, Lacal JC, Yuspa SH. Analysis of the rasH oncogene and its p21 product in chemically induced skin tumors and tumor-derived cell lines. Carcinogenesis. 1987;8:1821–1825. doi: 10.1093/carcin/8.12.1821. [DOI] [PubMed] [Google Scholar]
- Iavarone A, Massague J. Repression of the CDK activator Cdc25A and cell-cycle arrest by cytokine TGF-β in cells lacking the CDK inhibitor p15. Nature. 1997;387:417–422. doi: 10.1038/387417a0. [DOI] [PubMed] [Google Scholar]
- Ijichi H, Chytil A, Gorska AE, Aakre ME, Fujitani Y, Fujitani S, Wright CV, Moses HL. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-β signaling in cooperation with active Kras expression. Genes Dev. 2006;20:3147–3160. doi: 10.1101/gad.1475506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y, Sarkar P, Mi Q, Wu N, Bringas P, Jr, Liu Y, Reddy S, Maxson R, Deng C, Chai Y. Overexpression of Smad2 reveals its concerted action with Smad4 in regulating TGF-β-mediated epidermal homeostasis. Dev Biol. 2001;236:181–194. doi: 10.1006/dbio.2001.0332. [DOI] [PubMed] [Google Scholar]
- Koch RM, Roche NS, Parks WT, Ashcroft GS, Letterio JJ, Roberts AB. Incisional wound healing in transforming growth factor-β1 null mice. Wound Repair Regen. 2000;8:179–191. doi: 10.1046/j.1524-475x.2000.00179.x. [DOI] [PubMed] [Google Scholar]
- Lauffenburger DA, Horwitz AF. Cell migration: a physically integrated molecular process. Cell. 1996;84:359–369. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- Leveen P, Larsson J, Ehinger M, Cilio CM, Sundler M, Sjostrand LJ, Holmdahl R, Karlsson S. Induced disruption of the transforming growth factor β type II receptor gene in mice causes a lethal inflammatory disorder that is transplantable. Blood. 2002;100:560–568. doi: 10.1182/blood.v100.2.560. [DOI] [PubMed] [Google Scholar]
- Lu SL, Herrington H, Reh D, Weber S, Bornstein S, Wang D, Li AG, Tang CF, Siddiqui Y, Nord J, et al. Loss of transforming growth factor-β type II receptor promotes metastatic head-and-neck squamous cell carcinoma. Genes Dev. 2006;20:1331–1342. doi: 10.1101/gad.1413306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J, Gomis RR. The logic of TGFbeta signaling. FEBS Lett. 2006;580:2811–2820. doi: 10.1016/j.febslet.2006.04.033. [DOI] [PubMed] [Google Scholar]
- McLean GW, Komiyama NH, Serrels B, Asano H, Reynolds L, Conti F, Hodivala-Dilke K, Metzger D, Chambon P, Grant SG, et al. Specific deletion of focal adhesion kinase suppresses tumor formation and blocks malignant progression. Genes Dev. 2004;18:2998–3003. doi: 10.1101/gad.316304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra SK, Mikolon D, Molina JE, Hsia DA, Hanson DA, Chi A, Lim ST, Bernard-Trifilo JA, Ilic D, Stupack DG, et al. Intrinsic FAK activity and Y925 phosphorylation facilitate an angiogenic switch in tumors. Oncogene. 2006;25:5969–5984. doi: 10.1038/sj.onc.1209588. [DOI] [PubMed] [Google Scholar]
- Morris RJ, Liu Y, Marles L, Yang Z, Trempus C, Li S, Lin JS, Sawicki JA, Cotsarelis G. Capturing and profiling adult hair follicle stem cells. Nat Biotechnol. 2004;22:411–417. doi: 10.1038/nbt950. [DOI] [PubMed] [Google Scholar]
- Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, et al. The integrin α v β 6 binds and activates latent TGF β 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–328. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- Munoz NM, Upton M, Rojas A, Washington MK, Lin L, Chytil A, Sozmen EG, Madison BB, Pozzi A, Moon RT, et al. Transforming growth factor β receptor type II inactivation induces the malignant transformation of intestinal neoplasms initiated by Apc mutation. Cancer Res. 2006;66:9837–9844. doi: 10.1158/0008-5472.CAN-06-0890. [DOI] [PubMed] [Google Scholar]
- Oft M, Heider KH, Beug H. TGFβ signaling is necessary for carcinoma cell invasiveness and metastasis. Curr Biol. 1998;8:1243–1252. doi: 10.1016/s0960-9822(07)00533-7. [DOI] [PubMed] [Google Scholar]
- Owens DM, Watt FM. Influence of β1 integrins on epidermal squamous cell carcinoma formation in a transgenic mouse model: α3β1, but not α2β1, suppresses malignant conversion. Cancer Res. 2001;61:5248–5254. [PubMed] [Google Scholar]
- Ozdamar B, Bose R, Barrios-Rodiles M, Wang HR, Zhang Y, Wrana JL. Regulation of the polarity protein Par6 by TGFβ receptors controls epithelial cell plasticity. Science. 2005;307:1603–1609. doi: 10.1126/science.1105718. [DOI] [PubMed] [Google Scholar]
- Pietenpol JA, Stein RW, Moran E, Yaciuk P, Schlegel R, Lyons RM, Pittelkow MR, Munger K, Howley PM, Moses HL. TGF-β 1 inhibition of c-myc transcription and growth in kerati-nocytes is abrogated by viral transforming proteins with pRB binding domains. Cell. 1990;61:777–785. doi: 10.1016/0092-8674(90)90188-k. [DOI] [PubMed] [Google Scholar]
- Reynisdottir I, Polyak K, Iavarone A, Massague J. Kip/ Cip and Ink4 Cdk inhibitors cooperate to induce cell cycle arrest in response to TGF-β. Genes Dev. 1995;9:1831–1845. doi: 10.1101/gad.9.15.1831. [DOI] [PubMed] [Google Scholar]
- Schäfer M, Werner S. Transcriptional control of wound repair. Annu Rev Cell Dev Biol. 2007 doi: 10.1146/annurev.cellbio.23.090506.123609. in press. Published online May 2, 2007. [DOI] [PubMed] [Google Scholar]
- Schober M, Raghavan S, Nikolova M, Polak L, Pasolli HA, Beggs HE, Reichardt LF, Fuchs E. Focal adhesion kinase modulates tension signaling to control actin and focal adhesion dynamics. J Cell Biol. 2007;176:667–680. doi: 10.1083/jcb.200608010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumbar T, Guasch G, Greco V, Blanpain C, Lowry WE, Rendl M, Fuchs E. Defining the epithelial stem cell niche in skin. Science. 2004;303:359–363. doi: 10.1126/science.1092436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasioukhin V, Degenstein L, Wise B, Fuchs E. The magical touch: genome targeting in epidermal stem cells induced by tamoxifen application to mouse skin. Proc Natl Acad Sci USA. 1999;96:8551–8556. doi: 10.1073/pnas.96.15.8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SE, Wu FY, Shin I, Qu S, Arteaga CL. Transforming growth factor β (TGF-β)-Smad target gene protein tyrosine phosphatase receptor type κ is required for TGF-β function. Mol Cell Biol. 2005;25:4703–4715. doi: 10.1128/MCB.25.11.4703-4715.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zinkel S, Polonsky K, Fuchs E. Transgenic studies with a keratin promoter-driven growth hormone transgene: prospects for gene therapy. Proc Natl Acad Sci USA. 1997a;94:219–226. doi: 10.1073/pnas.94.1.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XJ, Greenhalgh DA, Bickenbach JR, Jiang A, Bundman DS, Krieg T, Derynck R, Roop DR. Expression of a dominant-negative type II transforming growth factor β(TGF-β) receptor in the epidermis of transgenic mice blocks TGF-β-mediated growth inhibition. Proc Natl Acad Sci USA. 1997b;94:2386–2391. doi: 10.1073/pnas.94.6.2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XJ, Liefer KM, Tsai S, O’Malley BW, Roop DR. Development of gene-switch transgenic mice that inducibly express transforming growth factor β1 in the epidermis. Proc Natl Acad Sci USA. 1999;96:8483–8488. doi: 10.1073/pnas.96.15.8483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks BH, He W, Olson KL, Wang XJ. Inducible expression of transforming growth factor β1 in papillomas causes rapid metastasis. Cancer Res. 2001;61:7435–7443. [PubMed] [Google Scholar]
- Weiss RA, Eichner R, Sun TT. Monoclonal antibody analysis of keratin expression in epidermal diseases: a 48- and 56-kdalton keratin as molecular markers for hyperproliferative keratino-cytes. J Cell Biol. 1984;98:1397–1406. doi: 10.1083/jcb.98.4.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuspa SH, Hennings H, Dlugosz A, Tennenbaum T, Glick A. The role of growth factors in mouse skin tumor promotion and premalignant progression. Prog Clin Biol Res. 1995;391:39–48. [PubMed] [Google Scholar]
- Zhu Y, Richardson JA, Parada LF, Graff JM. Smad3 mutant mice develop metastatic colorectal cancer. Cell. 1998;94:703–714. doi: 10.1016/s0092-8674(00)81730-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The Supplemental Data include Supplemental Experimental Procedures and seven supplemental figures and can be found with this article online at http://www.cancercell.org/cgi/content/full/12/4/313/DC1/.