

Abstract
Mouse cytomegalovirus (MCMV), a β-herpesvirus that establishes latent and persistent infections in mice, is a valuable model for studying complex virus-host interactions. MCMV encodes the m145 family of putative immunoevasins with predicted MHC-I structure. Functions attributed to some family members include downregulation of host MHC-I (m152) and NKG2D ligands (m145, m152, m155) and interaction with inhibitory or activating NK receptors (m157). We present the cellular, biochemical and structural characterization of m153, which is a heavily glycosylated homodimer, that does not require β2m or peptide, and is expressed at the surface of MCMV-infected cells. Its 2.4 Å crystal structure confirms that this compact molecule preserves an MHC-I-like fold and reveals a novel mode of dimerization, confirmed by site-directed mutagenesis, and a distinctive disulfide-stabilized extended amino terminus. The structure provides a useful framework for comparative analysis of the divergent members of the m145 family.
A major strategy for immune evasion, employed by the large DNA viruses like herpes- and poxviruses, is the expression of a variety of molecules that attenuate the host’s immune response. Some of these impair the cellular pathways of antigen processing and presentation, while others serve as decoys, mimicking host molecules crucial to natural killer (NK) or T cell recognition (1). The genome of MCMV, which serves as a model for the human virus (HCMV), encodes a set of proteins that are predicted to be structural analogs of class I major histocompatibility (MHC-I) glycoproteins (2–4). The detailed function of each of these viral MHC-I-like molecules (MHC-Iv) has not yet been determined, but several examples suggest that they either downregulate host MHC-I or MHC-I-like molecules or directly bind to NK cell inhibitory receptors. Thus, they can interfere with key components of T cell recognition or NK cell activation, thereby promoting virus survival. MHC-I molecules, transmembrane cell surface receptors consisting of a heavy chain, a β2-microglobulin (β2m) light chain, and bound peptides, are poised for recognition by clonotypic αβ receptors on CD8+ T cells or by inhibitory or activating receptors on NK cells. MHC-I are classical (Ia) or non-classical (Ib) molecules based on their amino acid sequence polymorphism, function, and tissue-specific expression. The heavy chain consists of an α1α2 domain unit containing two α-helices set atop an eight-stranded β-sheet platform and an Ig-like α3 domain (5). MHC-Ib molecules exhibit less polymorphism, most do not bind peptide, and they vary in their association with β2m (6). MHC-Ib proteins function both in immune recognition and in other physiological settings (7).
Only one MCMV protein, m144, like its HCMV counterpart UL18 (8), is clearly related to MHC-I based on amino acid sequence similarity (9). m144 allows virus-infected cells to evade the NK cell response, but its mechanism of action and the nature of any ligand remain unclear. Expression studies of m144 indicate that, unlike MHC-Ia molecules, it does not require peptide for cell surface expression (10,11). The crystal structure of m144 reveals an MHC-I-like protein, bound to β2m but not associated with peptide, confirming predictions based on sequence comparison. The molecule is characterized by a large angle between α1α2 and α3, a unique stabilizing disulfide bond in α1α2, and relatively loose association of the heavy chain with β2m (11).
In addition to m144, MCMV encodes the eight molecules of the m145 family, all of which are predicted to have an MHC-I protein fold (2–4), despite the lack of significant sequence similarity to m144 or to MHC-Ia molecules. Four of the family members (m145, m152, m155, and m157) function as immunoevasins. m145, m152 and m155 downregulate the cell surface expression of ligands of the activating receptor NKG2D (12–15). m152 also causes the retention of MHC-I molecules in the endoplasmic reticulum-Golgi intermediate compartment (16). m157 functions by binding the NK inhibitory receptor Ly49I129 in MCMV sensitive mouse strains such as 129. Alternatively it can interact with the NK activating receptor Ly49H in strains (e.g. C57BL/6) that are resistant to MCMV infection (2,17). m157 expression does not require either β2m or peptide (2). Its crystal structure revealed several deviations from the MHC-Ia fold: an extended, helical N-terminus, designated α0, that precedes the canonical α1 helix and imparts a three-helix structure to the amino terminal α1α2 domain; a long α2 helix that extends into the α3 domain; short interhelical distances and close interactions of the α1α2 domain with α3 resulting in a molecule significantly more compact than classical MHC-I; and an α3 domain whose Ig fold is less twisted than typical Ig folds (18).
The unique functions of m145, m152, m155, and m157 suggest that each member of the m145 family is important in modulating the response of the host to MCMV infection. The structural features of m144 and m157 imply that other members of the m145 family also may have evolved significant structural deviations from typical MHC-I to solve novel difficulties in their encounter with the immune system. An understanding of the evolution and function of the MHC-I-like molecules of MCMV demands examination of the cellular expression, immunological function, and molecular structure of each of these molecules. To gain further insight into another member of the m145 family, we have examined m153 in detail. We first demonstrate that m153 is a functional gene that directs the expression of its encoded protein to the surface of MCMV-infected cells. Surface expression is independent of the transporter associated with antigen processing (TAP) or β2m. In surprising contrast to MHC-I molecules, recombinant soluble m153 is homodimeric. The crystal structure of m153 described here reveals the mode of dimerization in addition to other distinguishing features. Thus, although prediction algorithms group members of the m145 family together within the broad MHC-I structural family, the details of the individual structures show wide variation from MHC-I and demonstrate the robustness and adaptability of the MHC-I fold in the service of diverse biological functions.
EXPERIMENTAL PROCEDURES
Cell lines
NIH3T3, RMA and RMA-S were grown in DMEM containing 10% fetal bovine serum, 5 × 10−5 M 2-mercaptoethanol, 1x non-essential amino acids (Biowhittaker) and 50 μg/ml gentamicin at 37°C in 5% CO2. R1.1 and R1E were cultured in RPMI containing the same supplements as above. Drosophila S2 cells were maintained in Insect Express medium (BioWhittaker) supplemented with 50 μg/ml gentamicin except where mentioned otherwise.
Antibodies
Rabbit antiserum was produced against m153 expressed as inclusion bodies in E. coli. Reduced, denatured SDS-gel purified m153 was emulsified in complete Freund adjuvant prior to immunization. The mouse monoclonal antibody mAb153.16 was generated with the extracellular domain of mature m153 (amino acids 1-314) expressed in S2 cells and stains m153 transfected cells and immunoprecipitates m153 from transfected or virus-infected cells.
Vector constructs – Insect cell expression
cDNA encoding the extracellular domain of the mature form of m153 (amino acids 1-314) was PCR-amplified from MCMV-infected cell culture supernatants (Smith strain – American Type Culture Collection, catalog no. VR-1399) using primers 5′GGATCCGAGGTCGTGCGGCCCGAAGTTAAC and 5′GGTACCTCAATGGTGATGGTGATGATGGCTGCCGCGCGGCACCAGGGTCAGT CTCGAATCGTTGATCGTC. A BamHI restriction site was incorporated in the forward primer and a thrombin cleavage site (LVPRG), a 6xHis tag, a termination codon, and a KpnI restriction site were incorporated in the reverse primer. The m153 PCR product was directly cloned into the pCR4-Topo-TA vector (Invitrogen), the sequence verified, and then the excised BamHI/KpnI m153 fragment was inserted in the BglII and KpnI sites of pMT-Bip-V5-His (Invitrogen).
Mammalian cell expression
The full-length m153 gene (amino acids -22-382, including the signal peptide sequence) was amplified from MCMV infected cell culture supernatants and an N-terminal FLAG-tag introduced by sequential PCR. The sequential forward primers used were A-5′TCAGCGGAGGTCGACTACAAGGATGACGATGACAAGGTGCGGCCCGAAGTTAACAGG B-5′TTGGTTCTCATCTCAGGAGGGTTCTGGGCGTGCGGCTCAGCGGAGGTCGACTACAAG C-5′GCTACGGGATCCATGATTCCCCTTCTCCTTCTGCCGTTGGTTCTCATCTCAGGAGGG, and the reverse primer was 5′TACGCTGAATTCTTACACCACATTCTCCTCCGTATCCGAGCA. 5′ BamHI and a 3′ EcoRI restriction sites were incorporated in the forward and reverse primers respectively for subsequent cloning into the pIRES-hr-GFP-II vector (Stratagene).
BiFC vectors
Mammalian expression vectors encoding the N-terminal 1-154 residues (N-YFP) or the C-terminal 155-238 residues (C-YFP) of YFP were a kind gift from Dr. K. Ozato, NICHD, NIH. m153 and m144 were amplified from existing full-length constructs using primers m153_5YFP 5′ACGGGATCCATGATTCCCCTTCTCCTTCTGCCG and m153_3YFP 5′ACGGAATTCGCCACCACATTCTCCTCCGTATCCG and m144 was amplified using primers m144_5YFP 5′ACGAGATCTATGAGGGCTCTGGCGCTG and m144_3YFP 5′ACGGAATTCGCAATGCTGGGATCCGGGACCGTG. The PCR products were digested and inserted in the BglII and EcoRI sites of N-YFP and C-YFP and the sequences verified. The resulting constructs encode fusion proteins of m153 and m144 linked at their C-termini to either the N- or C-terminal fragments of YFP. A 12–13 amino acid spacer separates the viral proteins from the YFP fragments.
β2m and TAP-dependence
The Amaxa nucleofector (Amaxa Biosystems) was used to introduce the pIRES-hr-GFP II vector (Stratagene) encoding either FLAG-m153 or H-2Dd into R1.1 and R1E cells (solution V, program A30) or into RMA and RMA-S cells (solution T, program A30) according to the manufacturer’s instructions. 18–24 hours post transfection the cells were stained for surface expression of FLAG-tagged m153 with the M2 monoclonal antibody (Sigma) and a PE labeled anti-mouse IgG1 antibody (Southern Biotech). H-2Dd was detected using the PE-labeled 34-5-8S antibody (BD Biosciences). Cells were analyzed by flow cytometry (FACSCalibur, BD Biosciences).
MCMV infection, cell surface staining and FACS
70% confluent NIH3T3 cells were infected with MCMV-GFP (a virus containing GFP at an innocuous site (19)) at an m.o.i. of two. Twenty-four hours post infection (p.i.) the cells were detached with a non-enzymatic cell dissociation solution (Cellstripper, Cellgro) and resuspended in PBS containing 5% FCS and 0.1% NaN3. F(ab′)2 fragments were generated from monoclonal antibody mAb153.16 with the Immunopure IgG1 F(ab′)2 kit (Pierce). Infected and uninfected cells were incubated with the F(ab′)2 followed by anti-mouse IgG-APC. Dead cells were excluded by propidium iodide gating. Cells were analyzed with a FACSCalibur and virus-infected cells were gated on GFP expression.
Bimolecular fluorescence complementation (BiFC)
NIH3T3 cells were transfected with 153-N-YFP and 153-C-YFP or 144-N-YFP and 144-C-YFP using the Amaxa nucleofector (solution R, program U30). Cells were then seeded on chambered coverslides and incubated for 48 hours at 37 °C. A Leica TCS AOBS SP2 confocal microscope (Leica Microsystems) equipped with a 63 × 1.4 NA PL APO objective was used to examine live cells at room temperature for YFP fluorescence complementation. Leica TCS v2.1547 software was used to capture the images. After analysis the cells were detached with trypsin and the YFP positive cells were quantified by flow cytometry.
Immunoprecipitation and western blots
Transiently transfected and virus-infected cell lines were processed for immunoprecipitation and western blotting as detailed in the supplementary Experimental Procedures.
Expression and purification of m153 in S2 cells
The pMT-Bip-m153-His construct together with puromycin and blasticidin resistance- encoding plasmids (10:1:1 ratio by weight) were transfected into S2 cells using Fugene6 (Roche) according to the manufacturer’s protocol. 48 hours after transfection the cells were transferred to selective medium containing 5 μg/ml puromycin (Sigma) and 5 μg/ml blasticidin (Calbiochem). Resistant cells were maintained in selective medium in shaker cultures (28 °C, 100 rpm). Expression of m153 was induced with 1 mM CuSO4 at a cell density of 2 × 107/ml. m153 was purified on a Ni-NTA column (Qiagen) and then subjected to SEC (Shodex KW802.5 or Pharmacia Superdex 75). The His-tag was removed by thrombin cleavage at 37 °C for 2h, and the protein was purified by anion exchange chromatography (Mono Q, Pharmacia). For preparation of a selenomethionine (SeMet) derivative of m153, the m153/S2 cells were grown in Orbigen insect cell medium to 2 × 107 cells/ml. The cells were then washed in methionine-free medium (Orbigen) and starved in this medium for 6 hours under normal growth conditions. Protein expression was induced by simultaneous addition of 1 mM CuSO4 and 60 μg/ml SeMet (Sigma). The SeMet labeled protein was purified as described for the native protein.
Crystallization, structure determination and refinement
Crystals of the SeMet derivative of m153 were grown in hanging drops at 18°C over 25% PEG 2000 MME, 0.1 M TRIS-HCl pH8 (Solution 24, The PEGs Suite, Qiagen). Addition of 0.5% β-D-maltoside to the drops yielded crystals that diffracted to 2.3 Å. Diffraction data for SAD phasing were collected on a single crystal on beamline X29A at the NSLS, Brookhaven, at a wavelength of 0.9791 Å and processed with HKL2000 (20). The automated SOLVE/RESOLVE scripts as incorporated in the AutoSol module of the PHENIX suite (21) were used with data truncated at 2.8 Å to obtain SAD phases followed by automated model building and docking to the m153 sequence. Ambiguously placed residues were rebuilt manually into the SAD-phased map in Coot as alanines (22). To extend the resolution to 2.4 Å a molecular replacement search was carried out in Phaser (23) with the partially built model yielding Z-scores of 11.0 and 23.7 for the rotation and translation functions, respectively, for one dimer in the asymmetric unit. Rigid body refinement specifying α1α2 and α3 as separate domains yielded a model with initial R and Rfree of 36.9% and 38.6% respectively. Refinement at 2.4 Å was continued in Refmac5 (24) with tight NCS restraints between chains A and B with further model building guided by 2Fo-Fc and Fo-Fc maps generated in Coot. Although data were collected to 2.3 Å, data in the 2.38–2.3 shell were of insufficient quality and completeness. Therefore only data to 2.4 Å were used in refinement. Residues in the 231–237 loop which had little or no electron density were omitted from the NCS restraints. Optimal domains for TLS refinement were identified and input files for TLS refinement were generated using the TLSMD web server (25). TLS refinement using 10 TLS groups per chain decreased Rfree from 29.9% to 28%. Finally, waters were added with CNS (26) and a final round of refinement was performed without NCS restraints. The final model has Rcryst=23% and Rfree=27.9%, includes residues 2-230 and 238-277 of chain A and residues 5-277 of chain B and has 57 waters. The atomic coordinates and structure factors have been deposited in the Protein Data Bank (27) under accession number 2O5N. Buried surface area calculations were performed in CNS. Figures were generated using PyMOL (28).
Hinge angles were calculated using HINGE, a program written by Peter Sun. HINGE calculates an ellipsoid (defined by axes a, b and c) for each indicated domain, and reports the angle between the long axes of the adjacent domains as the hinge angle. HINGE is available at: http://sis.niaid.nih.gov/programs/hinge.html.
Transient expression of wild type and mutant m153 in SF9 cells
Alanine mutations were introduced simultaneously at four residues (T128, S131, R225 and S241) in the m153-N-YFP construct using the QuickChange Multi mutagenesis kit (Stratagene). Wild type and mutated m153 (residues -22-314) genes were PCR-amplified from the m153-N-YFP and m153-N-YFP-mutant constructs using primers 5′CGTCACCCATGGCAATTCCCCTTCTCCTTCTG CCGTTGGTTCTC and 5′ACTTCGCTCGAGGGTGAGTCTCGAATCGTTGATCGTCCTCTG. The PCR products were digested with NcoI and XhoI and inserted into the corresponding sites of the pIEx-4 SF9 expression vector (Novagen). 200 μg of m153-pIEx-4 or quadruple mutant-m153-pIEx-4 were transfected into 1 × 108 exponentially growing SF9 cells in 100 ml with 1ml of Insect GeneJuice transfection reagent (Novagen). 72h after transfection the cell supernatants were collected. Wild type and quadruple mutant m153 were then purified on Ni-NTA and SEC columns as described for S2 insect cell expression.
Analytical ultracentrifugation
Sedimentation velocity experiments were conducted with a ProteomeLab XLI ultracentrifuge (Beckman Coulter). 400 μl samples were loaded at concentrations of 0.3 mg/ml for m153 wild type and 0.5 and 0.1 mg/ml for the m153 mutant and centrifuged at 50,000 rpm at 20°C. Sedimentation data were acquired with the Rayleigh interference optical system at 1 min intervals, and data were analyzed using SEDFIT software (29). Additional details on experimental and data analysis protocols can be found at http://www.analyticalultracentrifugation.com.
RESULTS
m153 is expressed on the surface of MCMV-infected fibroblasts
The open reading frame of the m153 gene predicts a type I transmembrane glycoprotein with a core molecular weight of 45 kDa. As a first step in the functional characterization of m153, we examined its expression in MCMV-infected fibroblasts. NIH3T3 cells, infected with MCMV at an m.o.i. of 2, expressed readily detectable levels of m153 at the cell surface 24h p.i. as detected by the monoclonal antibody mAb153.16, specific for the extracellular domain of m153 (Fig. 1A). Uninfected cells did not stain with this antibody. By immunoprecipitation m153 was detected in lysates of infected cells as early as 3 and up to 48 hours after infection with protein levels peaking at 24 hours p.i. (data not shown). Immunoprecipitation of cell lysates followed by SDS-PAGE and western blot analysis of m153 revealed an unexpectedly high apparent molecular weight of 80 kDa, which is decreased to 45 kDa upon deglycosylation with PNGaseF (Fig. 1B). Such heavy glycosylation is consistent with six predicted sites for asparaginyl-carbohydrate addition.
Fig. 1.
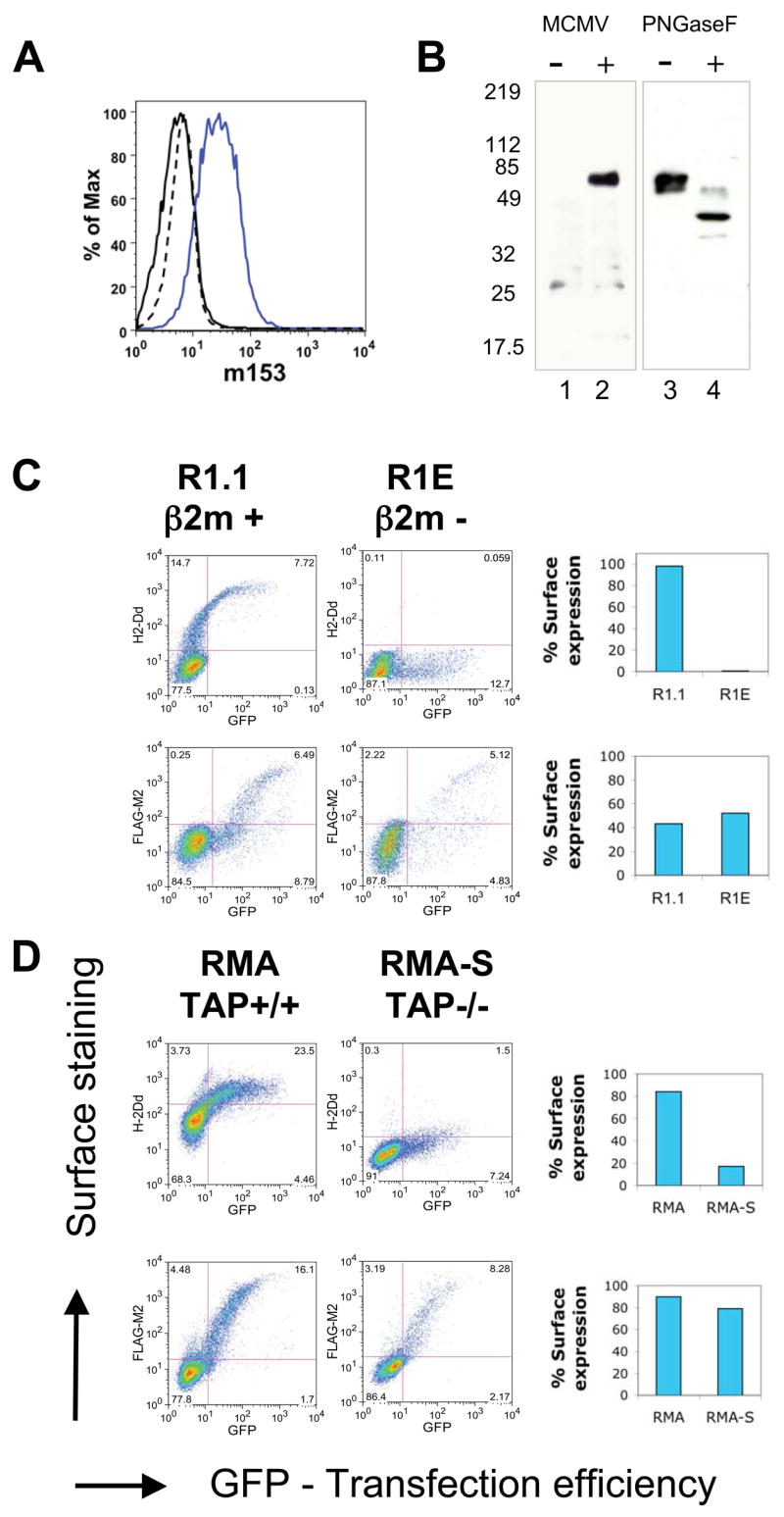
m153 expression at the surface of MCMV-infected cells, β2m- and TAP-deficient cells. A. MCMV infected NIH3T3 cells were surface stained for m153 expression at 24 hours post infection (p.i.). Blue-infected cells + mAb153.16, Black-infected cells + control Ab, Dashed-uninfected cells + mAb153.16. B. Immunoprecipitation (IP) and western blot (WB) analysis of m153 from uninfected (Lane 1) and MCMV- infected (Lane 2) NIH3T3 cell lysates at 24 hours p.i. IP of m153 from transfected NIH3T3 cells (IP-mAb153.16, WB-anti-m153 rabbit serum). Samples were mock treated (Lane 3) or treated with PNGase F (Lane 4) before SDS-PAGE and western blot. C. R1.1 (β2m+) and R1E (β2m−) or D. RMA (TAP+) and RMA-S (TAP−) cells were transfected with FLAG-m153-pIRES-GFP or H-2Dd-pIRES-GFP as a control. 18 hours post transfection surface levels of m153 and H-2Dd were determined as described in Experimental Procedures. The number of GFP positive cells indicates transfection efficiency. The percentage surface expression is shown in the bar graphs on the right (percentage of GFP+ cells that stain for either FLAG-M2 or H-2Dd).
Cell surface expression of m153 is independent of β2m or TAP expression
Since the MHC-I fold was predicted for all members of the MCMV m145 family (4), we next determined whether m153 shares the requirements of classical MHC-I molecules for β2m and peptide for stable expression. To investigate its requirement for β2m we transfected N-terminally FLAG-tagged m153 (FLAG-m153) into a cell line deficient in β2m (R1E (30)) as well as its β2m sufficient counterpart (R1.1). m153 surface expression was equivalent in both cell types indicating that m153 does not require β2m for stable surface expression (Fig. 1C).
To evaluate the requirement for bound peptide we examined surface expression of m153 in RMA-S, a TAP deficient cell line that fails to transfer peptides from the cytosol to the ER (31). MHC-Ia molecules expressed in such cells are unstable at the cell surface. We transfected RMA-S and RMA (TAP+) cells with FLAG-m153 and evaluated surface expression of the viral protein in comparison to transfected H-2Dd, an MHC-Ia molecule (Fig. 1D). TAP deficiency had no effect on m153 surface expression, whereas expression of H-2Dd was clearly reduced. We conclude that m153 does not require β2m or bound peptide for stable surface expression.
The MHC-I fold of m153
To elucidate the structure of m153 a SeMet derivative was prepared by biosynthetic labeling of the protein expressed in Drosophila S2 cells. Size exclusion chromatography indicated that soluble m153 was a dimer (Fig. S1). Diffraction data from a single crystal were collected, the structure was solved by SAD phasing, and the model was refined at 2.4 Å resolution to final Rcryst and Rfree of 23 and 27.9 respectively (Table 1). There is a single dimer in the asymmetric unit with chains designated A and B. We observed continuous electron density for residues 2-230 and 238-277 of chain A and residues 5-277 of chain B. No electron density was observed for the C-terminal residues 278-314 of either chain and these were omitted from the model. The average B-factor was 42 Å2 for chain A and 32 Å2 for chain B. We selected chain B to describe the structural features of the monomer as they relate to the MHC-I fold because it is more complete and has lower B-factors.
Table 1.
Data collection, refinement and validation statistics
| Data Collection | |
| Space Group | P21 |
| Unit cell dimensions | |
| a, b, c | 61.7, 78.2, 62,7 |
| α, β, γ | 90, 90.4, 90.0 |
| Molecules per asymmetric unit | 2 |
| Resolution (Å) | 2.3 |
| Total observations | 335, 101 |
| Unique reflections | 26, 191 |
| Completeness (%)a | 97.5 (94.2) |
| I/σI a | 9.7 (2.0) |
| Rsym a/b | 0.081 (0.44) |
| Refinement | |
| Resolution range | 62.75 – 2.4 |
| Reflections | |
| Working set | 23443 |
| Test set | 1183 |
| Rcryst (%)c | 23.0 |
| Rfree (%)c | 27.9 |
| Number of non-H protein atoms | 4393 |
| Number of water molecules | 57 |
| Carbohydrate chains | 3 |
| r.m.s deviations from ideality | |
| Bond lengths (Å) | 0.01 |
| Bond angles (deg.) | 1.318 |
| Average B values (Å2) | A, B |
| Main-chain | 42.2, 32.2 |
| Side-chain | 42.5, 32.7 |
| Whole chain | 42.4, 32.4 |
| Overall | 37.6 |
| Model Validation | |
| Ramachandran plot statistics (%) | |
| Procheck | |
| Most favored | 87.8 |
| Allowed | 12.2 |
| Generous | 0 |
| Disallowed | 0 |
Values in parentheses are statistics for the highest resolution shell used in refinement (2.48–2.38 Å).
Rsym(I) = σ|Ij − <I>|/σIj, where Ij is the intensity of the jth observation of a reflection and <I> is the mean intensity from multiple measurements of that reflection.
Rcryst = σ||Fo| − |Fc||/σ|Fo|, where Fo and Fc are the observed and calculated structure factor amplitudes, respectively. Rfree is as for Rcryst but calculated for a randomly selected 5.0% of reflections not included in the refinement. Procheck was used for model validation (49).
The structure of m153 (Fig. 2) reveals basic structural elements of the MHC-I fold. The m153 α1α2 domain consists of a unit in which the α1 helix and a discontinuous α2 helix rest atop a platform of seven β-strands. Three of the strands derive from the α1 domain and four from α2. The fourth β–strand of the α1 domain, commonly found in MHC-Ia and Ib molecules, is not observed in m153. Unlike either MHC-Ia or Ib molecules the C-terminal end of the α2-helix extends downwards into a third helix (designated H2b) that connects the α1α2 domain with the Ig-like α3 domain (Fig. 2A).
Fig. 2.
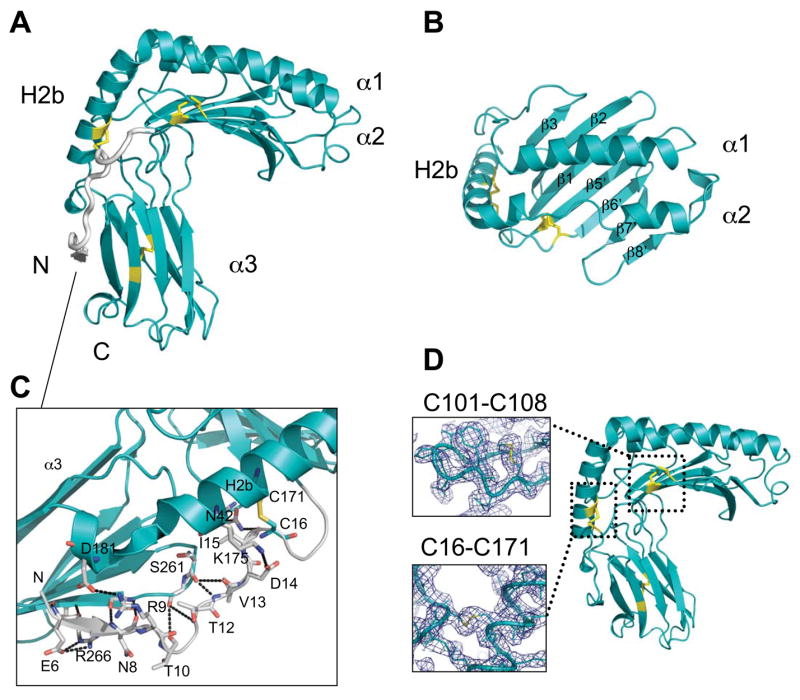
Conserved and unique features of the m153 monomer. A. Side view of m153 chain B. The α1, α2 and H2b helices, the α3 domain, N- and C-termini are labeled and the disulfide bonds are in yellow. N-terminal residues 2-20 are colored gray. B. Top view of the 7 β-strand platform and α1α2 helixes of m153. C. Extended N-terminus of m153 showing extensive interactions between the N-terminus (gray) and H2b helix, loops in the α1α2 domain and the G strand of the α3 domain (cyan). Hydrogen bonds are in black. D. Side view of m153 monomer. Two unique disulfide bonds link the N-terminal fragment to the H2b helix (C16-C171) and the β5′ platform strand to the loop connecting the β5′ and β6′ strands (C101-C108). Electron density around the unique disulfides is contoured at 1.25σ. The conserved disulfide bond (C203-255) is located in the Ig-like α3 domain.
Several structural features of the m153 monomer differ substantially from classical MHC-I molecules. Most striking is the N-terminus, extended by 20 residues, that starts adjacent to the G strand of the α3 domain, forming a short β strand. From there it extends upwards alongside the H2b helix and continues as the first strand of the platform beginning at residue 21 (Fig. 2A). This feature is absent in all MHC-I molecules described thus far and differs substantially from m157, whose extended N-terminus forms instead a third, short helix adjacent to the α2 helix (Fig. 3A & 3C). A unique disulfide between C16 and C171 anchors the extended N-terminus of m153 to the H2b helix (Fig. 2D). In addition several hydrogen bonds link the extended N-terminal strand (residues 6-15) and the loop between platform strands β2 and β3. Other hydrogen bonds link the extended N-terminus to the H2b helix and the α3 F-G loop and serve to further stabilize the structure (Fig. 2C). The α1 and α2 helices are shorter than those observed in MHC-Ia proteins and are closely juxtaposed above the β sheet floor. Close interactions between these helices preclude the binding of peptide or other ligand (Fig. 2B), in agreement with cellular expression studies indicating that m153 does not depend on bound peptide for stability (Fig. 1D). The platform is stabilized by a second unusual disulfide (C101-C108) which connects strands β5′ and β6′ of the floor (Fig. 2D). A similar bond is seen in m157 (C103-C112) but MHC-Ia molecules preserve a disulfide bond in the same region which links β-strand 5 of the platform to the α2 helix (C101-C164) (32). The presence of the unusual disulfide bonds in m153 was confirmed by mass spectrometry (Figs. S3 & S4).
Fig. 3.

Comparison of m153 with m157, H-2Kb, Rae-1β and rat neonatal FcR. A. Ribbon diagrams of the platform domains of m153, m157, Rae1β, FcRn and H-2Kb. The width of the groove of each molecule is indicated in Å and was measured at both ends and in the middle at comparable residues of the α-helices. B. Surface representation of the peptide-binding domain of H-2Kb (groove shown without any peptide) and the corresponding region of m153. Black rectangles indicate the groove. C. Comparison of m153 chain B with m157 and H-2Kb. The α1α2 and H2b-helices, α3-domain, and N- and C-termini are labeled. PDB accession codes: 2NYK, 2VAA, 1JFM and 3FRU.
The α3 domain of m153 has a C2 type Ig-fold, which is characterized by a more compact domain and shorter intercysteine spacing than the C1 or V type fold (33). The A, B and E strands make up one sheet of the domain and the C, C′, F and G strands make up the opposite sheet (Figure 2A). The canonical Ig-domain disulfide bond (C203-C255) connects strands B and F. Searches against the DALI database (34) identified m144, also with a C2 type Ig-fold, and CD1d as proteins with α3 domains that are structurally most closely related to m153. The difference between the strand dispositions in the α3 domains of m153 and m157 is indicated by the relatively high rmsd of 2.3 Å for the superposition of 67 Cα atoms. The α3 domain of m153 has longer β-strands that exhibit the characteristic β-sandwich twist, whereas the m157 α3 domain has strands that are shorter and form flatter sheets (Fig. 3C). The terminal residues that were modeled in both structures adopt different conformations, in m153 they form a short α-helix, whereas in m157 the C-terminal strand forms a circular loop that contacts both sheets of the α3 domain (18).
In Figure 3 we compare the α1α2 domain of m153 chain B to that of m157, H-2Kb (35), rat FcRn (36), and the NKG2D ligand Rae-1β (37) The α1 and H2b helices of m153 and m157 are in equivalent positions, whereas their α2 helices differ. The N-terminal end of the m157 α2 helix was not modeled due to lack of electron density (18). The m153 α2 helix starts with two full turns (residues 140-146), followed by an extended region that stretches along the platform ending in two turns of helix before bending sharply downward to continue as the H2b helix.
The maximal distance between the α1 and α2 helices of m153 is 8.1 Å compared to 16.8 Å in H-2Kb (Fig. 3A). Surface representations of the α1α2 domains of m153 and H-2Kb (Fig. 3B) illustrate the lack of a peptide-binding groove in m153. The groove region of m153 is comparable to that of Rae-1β and FcRn, two molecules that also do not bind peptide. The maximal distance between Cα atoms of the α1 and α2 helices of Rae-1β and FcRn are 8.2 Å and 9.2 Å respectively. The α1 helices of m153, Rae-1β, and FcRn have a similar arrangement on the platform but the m153 helix is shorter. The m153 platform has seven strands compared to the eight found in classical MHC-I proteins (Fig. 3A).
The hinge angle between α1α2 and α3 of m153 chain B is 76°, which is within the range of the MHC-I molecules H-2Kb, CD1d and T22 (71–74°), but is smaller than that of m157 (81°), MICA (116°) and m144 (99°) (Table S1). The m153 α3 domain makes extensive contacts with the H2b helix, with the loop connecting strands β2 and β3 of the platform as well as with the loop connecting β3 and α1. The buried surface area between α1α2 and α3 of m153 is 1777 Å2, which is considerably larger than the analogous interfaces in m157 (1265 Å2) and H-2Kb (455 Å2).
The m153 dimer
SEC of the insect cell expressed extracellular portion of m153 indicated that the molecule formed a stable non-covalent homodimer (Fig. S1), an observation confirmed by equilibrium and velocity sedimentation (data not shown). The crystal structure reveals the details of the mode of dimerization. Two MHC-I-like chains of m153 are organized in a head-to-tail arrangement forming a compact dimer (Fig. 4A). The two chains are almost identical (rmsd of 0.624 Å for superposition over 268 Cα atoms). The interaction of the platform domain of one subunit with the α3 domain of the other imparts a coronal shape forming a central cavity with a volume of about 9500 Å3. The conical cavity measures 46 Å × 35 Å at the top narrowing down to 22 Å × 18 Å. It is closed off at the bottom where the K115 sidechains of each subunit are only 4 Å apart. We propose an orientation of the m153 dimer with respect to the plasma membrane shown in Figure 4B. The subunits lie with their long axes parallel to the membrane and with the α1 helices closest to the membrane. In this orientation the C-termini of both chains point towards the membrane (Fig. 4B – lower). The 37 C-terminal residues for which no electron density was observed in either chain may form a flexible stalk connecting the ectodomain to the transmembrane region. m153 conserves only two of the 31 β2m-contacting residues present in H-2Kb. It is evident from the dimer orientation of m153 that association with β2m would be sterically hindered, an observation consistent with the β2m independence of m153 expression (Fig. 1C).
Fig. 4.

Overall structure of the m153 dimer. A. Ribbon diagram of the m153 dimer as viewed from the top. Chain A - salmon, Chain B - cyan. The α1α2 helices, the α3 Ig-like domains and N- and C-termini are labeled. Dashed line in chain A indicates a disordered loop. Blue sticks - carbohydrates observed in the crystal structure, Yellow sticks - N-linked glycosylation sites confirmed by mass spectrometry. B. Two 90° rotations reveal different side views of the m153 dimer, and its proposed orientation on the cell membrane. C. Top and bottom views of the electrostatic surface representation of the m153 dimer (red – acidic; blue – basic).
Three sugar moieties were modeled in the m153 structure (at N107 in chains A and B and N252 in chain B). These all point upward from the dimer away from the cell surface. Mass spectrometry indicated glycosylation at five of the six predicted N-linked carbohydrate addition sites (data not shown). These sites occur in both α1α2 and α3, and are distributed around the periphery of the m153 dimer, with none in the central cavity or at the proposed membrane proximal face of the molecule (Fig. 4A). The electrostatic surface of the m153 dimer reveals four nearly symmetrical basic patches inside the central cavity, and two highly acidic regions on the bottom surface that are formed by the loop that connects the A and B strands of the α3 domain and the C-terminal α-helical fragment (Fig. 4C).
The m153 dimer interface is formed by parallel β-strand interactions between the platform (β8′ strand) of one subunit and the α3 domain (C′ strand) of the second subunit (Fig. 5A), resulting in two extended, twisted β-sheets of 12 strands each that form the core of the dimer. The two interfaces are not perfectly symmetrical as 21 residues of chain A and 22 residues of chain B are involved in dimerization. There are 16 hydrogen bonds at the interface, most of which represent backbone interactions (Fig. 5B). In addition there are 114 non-bonded contacts. The area buried at the dimer interface is 1088 Å2 which is larger than that observed in HLA-G (676 Å2), which forms a disulfide-linked MHC-Ib dimer (Fig. 5C). The shape complementarity (Sc) index of the dimer was calculated as 0.72, which is higher than that of most antibody-antigen interfaces (0.62–0.68), consistent with the stability of the homodimer (Sc=1 represents a perfect fit, (38)). To confirm the observed interface we tested the effects of mutating four interface residues T128, S131, R225, and S241, all of which form side-chain/side-chain hydrogen bonds, to alanine (Fig. 5C). Wild type m153 and a mutant containing these four alanine mutations were expressed and analyzed by ultracentrifugation and SEC. The mutant protein had a smaller sedimentation coefficient indicating disruption of the dimer (Fig. 5D). SEC confirmed the smaller size of mutant m153 (Fig. S5). Thus the four mutations destabilized the m153 dimer and confirm the mode of dimerization visualized in the structure.
Fig. 5.
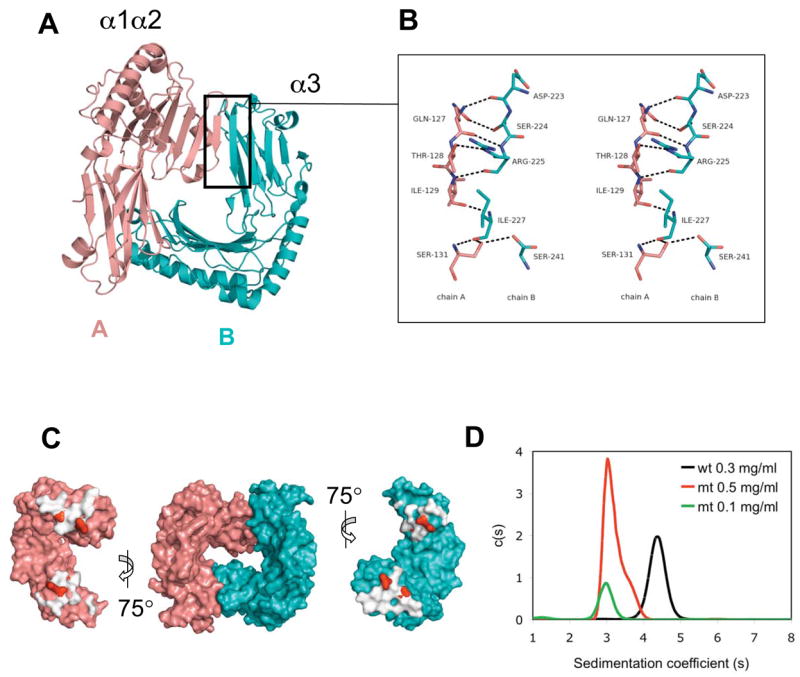
Dimer interface of m153. A. Chain A-platform/Chain B-α3 interface indicated by boxed region. B. Stereo view of the parallel β strand interactions between the β8′ strand of chain A (salmon) and the G strand of chain B (cyan). The nine hydrogen bonds are shown as black dashes. O-red, N-blue. C. Molecular surface representation of m153 dimer (center). At the left and right the two chains were rotated 75° out of the plane of the paper to display the buried interaction surfaces (white). The positions of the interface residues that were mutated to alanine (T128, S131, R225, S241) are shown in red. D. Mutations at the interface destabilize the m153 dimer. Sedimentation coefficients of wild type m153 (black − 0.3 mg/ml) and quadruple mutant m153 (T128A, S131A, R225A, S241A, red − 0.5 mg/ml, green 0.1 mg/ml) as determined by sedimentation velocity ultracentrifugation.
Full-length m153 is dimeric in mammalian cells
To exclude the possibility that dimerization is a result of the engineering of soluble m153, we used two approaches to examine m153 in mammalian cells. In BiFC (39) experiments we fused full-length m153 to either N- or C-terminal segments of YFP. While transfectants of either construct alone did not show fluorescence, co-transfection of m153-N-YFP and m153-C-YFP resulted in the complementation of YFP fluorescence in transfected cells (Fig. 6A and Fig. S2), indicating that m153 does form dimers. Cells transfected with YFP fusion constructs of m144, a monomeric MHC-Iv protein, showed fewer than 1% YFP positive cells. In a second approach we co-transfected N-terminal FLAG- and HA-tagged m153 in NIH3T3 cells. Successful immunoprecipitation of m153 with anti-FLAG antibodies and western blot detection with anti-HA antibodies confirmed that m153 dimerizes in transfected fibroblasts (Fig. 6B). Together these results provide strong evidence that m153 is a dimer when expressed as the full-length protein in mammalian cells.
Fig. 6.
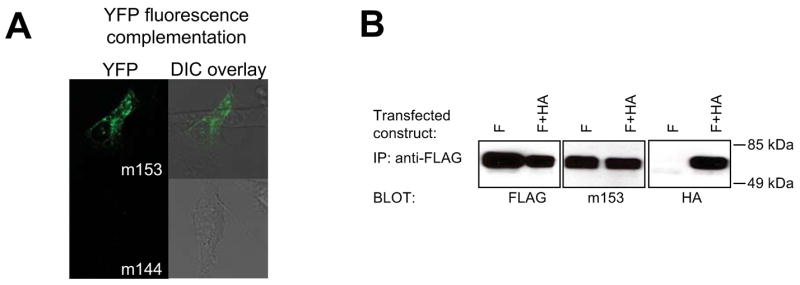
m153 forms a homodimer in mammalian cells. A. BiFC analysis. m153 constructs, fused to the N-terminal or C-terminal portions of YFP, were cotransfected in NIH3T3 cells and the live cells were examined by confocal microscopy for fluorescence complementation after 48 hours. As a negative control YFP-fusion constructs of MCMV m144 were cotransfected. Left - YFP channel, Right - overlay of the YFP on the differential interference contrast (DIC) channel. Quantification of the number of YFP positive cells is shown in Figure S2. B. Co-immunoprecipitation of differentially tagged m153. FLAG-m153 (F) or F + HA-m153 (F+HA) constructs were transfected in NIH3T3 cells. 24 hours post transfection post-nuclear cell lysates were immunoprecipitated with anti-FLAG antibodies and detected on a western blot with anti-FLAG (left), anti-m153 rabbit antibodies (middle) or HA-specific antibodies (right).
Comparison of m153 to the m145 family
m153 is related by sequence to proteins of the mouse and rat CMV 145 families but has no direct homolog in the rat virus (3,40). The m153 protein is most closely related to m152 (28% identity), a known regulator of MHC-I and NKG2D ligands in MCMV-infected cells (14,16). Figure 7 shows a sequence alignment of the m145 family highlighting shared structural features. Based on comparison of m153 and m157 we suggest that the core of the viral MHC-I-like fold of the m145 family is comprised of the α1α2 domain, the extended H2b helix and an Ig-like α3 domain. The number of platform strands, the length of α2 on top of the platform and the position of some disulfide bonds may vary. A crucial stabilizing interaction for this MHC-I-like fold is that between E70 (α1 helix), R166 and W167 (H2b helix). Apart from a cysteine in the α3 domain, these three residues are the only amino acids that are conserved throughout the mouse and rat 145 families, emphasizing the importance of this salt-bridge. Within the m145 family stretches of similarities are found in α helix and β sheet regions, whereas loop regions and the α3 domain show less conservation. The m153 residues involved in dimerization are not conserved in the rest of the family consistent with our observation that m151 and m152 do not dimerize in solution (unpublished). Thus dimerization may be unique to m153. Five family members contain cysteines that align with those of the unusual m153 C16-C171 disulfide. The C101-C108 and C203-C255 disulfides are also conserved among some of the family members. The limited sequence similarity of the N- and C-terminal regions of the extracellular domains of the m145 family members likely indicate a structural divergence in these parts of these molecules.
Fig. 7.

Amino acid sequence alignment of the m145 family. The predicted extracellular domains of all m145 family members (Refseq: NC_004065 (3)) were aligned using ClustalW (47) and optimized based on m153 and m157 structure. Secondary structure elements of m153 are shown above and those of m157 shown below the alignment. Cysteines involved in disulfide bonds are indicated by red (m153) and blue (m157) squares, and the respective pairs are indicated. Cysteines that are not conserved throughout the family, but are predicted to form disulfide bonds are boxed in red. Residues involved in a salt bridge (E70 and R166) are also indicated. N-linked glycosylation sites are indicated by green stars. The residues involved in m153 dimer interface contacts are indicated by blue ovals. The figure was generated with ESPRIPT (48).
DISCUSSION
MHC-I molecules play a crucial role in the protection of the host against virus infection. Not only are MHC-I molecules essential for the detection of infection and priming of the cytotoxic CD8+ T cell response, but their presence on the cell surface protects healthy cells from attack by natural killer cells. Viral molecules with an MHC-I-like structure are likely to be excellent decoys for inhibitory natural killer receptors and may be able to interact with numerous molecules involved in the immune response. It is therefore not surprising that herpesviruses have incorporated MHC-I-like molecules in their arsenal of immunoevasins. Human, mouse and rat CMV encode MHC-I-like molecules that exhibit different levels of similarity to MHC-Ia molecules (3,40). HCMV encodes the MHC-I homolog UL18 which binds both β2m and peptide and engages the inhibitory receptor LIR-1 (41). It also encodes UL142, which functions to inhibit NK cell lysis, has less sequence similarity to MHC-I and is predicted to consist of only the MHC-I α1α2 domain (42). MCMV encodes proteins like m144 that can associate with β2m, and others like m153 and m157, members of the m145 family, that are expressed without β2m. It is likely that several host genes have been captured by viruses during the course of coevolution (43). In the case of the MHC-I viral homologs it is probable that the closely related MHC-I genes like m144 and r144 were originally acquired from the host and that the distantly related m145 and r145 families arose via gene duplication and expansion of the 144 gene shaped by the selective pressures of the host immune response.
The crystal structure of MCMV m153 provides an example of the versatility of the MHC-I fold and is the first report of an obligate non-covalent dimer composed of two MHC-I-like chains. Together with the recently reported m157 structure, it highlights both the conserved and divergent features of the m145 family. The m153 monomer retains the core MHC-I characteristics, a β-sheet platform that supports two α-helices and an Ig-like α3 domain, but it exploits several structural adaptations to yield a stable molecule in the absence of peptide and β2m. Three main features combine to stabilize the m153 monomer: 1) the closely spaced α1 and α2 helices on the platform do not form a groove and alleviate the need for peptide binding; 2) the extended H2b helix bridges the α1α2 domain to the α3 domain; and 3) numerous interdomain contacts and two unique disulfide bonds (C16-C171 and C101-C108) stabilize the structure. Some of these features are shared among the other members of the m145 family. Whereas m157 is linked to the cell membrane via a glycophosphatidylinositol moiety and functions as a monomer (2), m153 has a bona fide transmembrane region and a large (47 amino acid) intracellular domain. We have shown that m153 is a stable dimer when expressed in mammalian cells, and the crystal structure of the soluble ectodomain reveals the mode of dimerization.
Dimerization is unusual for MHC-I molecules. Although dimers of some classical MHC-I molecules have been reported i.e., HLA-B27 (44) and HLA-G (45), these are disulfide-linked and their physiological relevance remains unclear. A non-disulfide linked dimer has been observed in crystals of the rat FcRn. Unlike m153, the two MHC-I molecules of the FcRn interact via their α3 and β2m domains, utilizing extensive protein-protein and protein carbohydrate interactions at the interface (36). In the m157 crystal, a dimer of the same orientation as m153 is formed with one of the symmetry related molecules. In m157 this dimer interface is formed by anti-parallel beta strand interactions between the β6 strand of the platform and the D strand of the α3 domain. Thus the same chain orientation but an opposite sheet of the α3 domain (ABED) is utilized in the observed crystal packing.
To gain insight into possible ligand binding sites on m153 we have examined regions corresponding to known interaction sites on murine MHC-I like molecules. These include the CD8αα binding site (α3 domain residues 220-228 and residues of the α1α2 domain platform), the Ly49A NK receptor binding site (“site 2” including residues from the α3 domain, the α1α2 domain platform, and key residues of β2m), and the region of the FcRn-Fc interaction site (N-terminal portions of the α2 domain and of β2m) (7). The compact nature of the m153 dimer, its lack of β2m association, and the regions that are masked by dimerization make it unlikely that any of the above mentioned sites are available for ligand binding.
Since m153 is clearly related to other immunoevasins by both amino acid sequence and structure, it may be expected also to function to subvert the host immune response. Several features strongly indicate that m153 has a significant role in the viral life cycle. 1) Several regions of the m153 sequence are conserved among related molecules with known immunoregulatory function. m152 and m155 conserve the C16-C171 disulfide, and m152 and m145, as well as m157, conserve the β5′-β6′ C101-C108 disulfide. 2) This is a readily expressed viral glycoprotein easily detected both intracellularly and at the cell surface in MCMV-infected fibroblasts. 3) Heteroduplex mobility assay and DNA sequence analysis have been used to examine the levels of sequence variation of a number of MCMV genes of isolates from wild mice and laboratory strains. The MHC-Iv immunoevasins m144, m155 and m157 have significant variation, while m152 is highly conserved (17,46). m153, like m152, is highly conserved among many strains showing little variation by both heteroduplex mobility assay and sequencing8. Such conservation implies not only a role in viral survival, but suggests that m153, like m152, may interact with an invariant site on a host molecule (46). Further experiments will explore the identification of cellular and molecular ligands, and test the contribution of m153 to acute and chronic stages of viral infection.
The abbreviations used are
- MHC-I
major histocompatibility class I
- NK
natural killer
- MCMV
mouse cytomegalovirus
- PE
phycoerythrin
- APC
allophycocyanin
- TAP
transporter associated with antigen processing
- SeMet
selenomethionine
- SAD
single anomalous dispersion
- rmsd
root mean square deviation
- m.o.i
multiplicity of infection
- p.i
post infection
- SEC
size exclusion chromatography
- BiFC
bimolecular fluorescence complementation
- YFP
yellow fluorescent protein
JM was supported by a predoctoral fellowship from the NIH and the Polio Research Foundation of South Africa. DE and SH (while at NIDDK) performed mass spectrometry experiments. Support for beamline X29 of the National Synchrotron Light Source comes principally from the Offices of Biological and Environmental Research and of Basic Energy Sciences of the US DOE, and from the NCRR of the NIH. Research was supported by the Intramural Research Program of the NIH, NIAID and NIDDK. We thank Barbara Newman and Rose Mage for production of rabbit antiserum, members of the MBS, LI, NIAID for their help and Sam Xiao and Jack Bennink for comments on the manuscript.
The atomic coordinates for the crystal structure of m153 were deposited in the Research Collaboratory for Structural Bioinformatics Protein Databank = PDB # 2O5N
Personal communication: Lee Smith and Alec Redwood. University of Western Australia
References
- 1.Tortorella D, Gewurz BE, Furman MH, Schust DJ, Ploegh HL. Annu Rev Immunol. 2000;18:861–926. doi: 10.1146/annurev.immunol.18.1.861. [DOI] [PubMed] [Google Scholar]
- 2.Arase H, Mocarski ES, Campbell AE, Hill AB, Lanier LL. Science. 2002;296(5571):1323–1326. doi: 10.1126/science.1070884. [DOI] [PubMed] [Google Scholar]
- 3.Rawlinson WD, Farrell HE, Barrell BG. J Virol. 1996;70(12):8833–8849. doi: 10.1128/jvi.70.12.8833-8849.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith HR, Heusel JW, Mehta IK, Kim S, Dorner BG, Naidenko OV, Iizuka K, Furukawa H, Beckman DL, Pingel JT, Scalzo AA, Fremont DH, Yokoyama WM. Proc Natl Acad Sci U S A. 2002;99(13):8826–8831. doi: 10.1073/pnas.092258599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bjorkman PJ, Parham P. Annu Rev Biochem. 1990;59:253–288. doi: 10.1146/annurev.bi.59.070190.001345. [DOI] [PubMed] [Google Scholar]
- 6.Natarajan K, Li H, Mariuzza RA, Margulies DH. Rev Immunogenet. 1999;1(1):32–46. [PubMed] [Google Scholar]
- 7.Margulies DH, Natarajan K, Rossjohn J, McCluskey J. Major Histocompatibility Complex (MHC) Molecules: Structure, Function, and Genetics. In: Paul WE, editor. Fundamental Immunology. Lippincott, Williams, and Wilkins; Philadelphia: 2007. [Google Scholar]
- 8.Beck S, Barrell BG. Nature. 1988;331(6153):269–272. doi: 10.1038/331269a0. [DOI] [PubMed] [Google Scholar]
- 9.Farrell HE, Vally H, Lynch DM, Fleming P, Shellam GR, Scalzo AA, Davis-Poynter NJ. Nature. 1997;386(6624):510–514. doi: 10.1038/386510a0. [DOI] [PubMed] [Google Scholar]
- 10.Chapman TL, Bjorkman PJ. J Virol. 1998;72(1):460–466. doi: 10.1128/jvi.72.1.460-466.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Natarajan K, Hicks A, Mans J, Robinson H, Guan R, Mariuzza RA, Margulies DH. J Mol Biol. 2006;358(1):157–171. doi: 10.1016/j.jmb.2006.01.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hasan M, Krmpotic A, Ruzsics Z, Bubic I, Lenac T, Halenius A, Loewendorf A, Messerle M, Hengel H, Jonjic S, Koszinowski UH. J Virol. 2005;79(5):2920–2930. doi: 10.1128/JVI.79.5.2920-2930.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krmpotic A, Hasan M, Loewendorf A, Saulig T, Halenius A, Lenac T, Polic B, Bubic I, Kriegeskorte A, Pernjak-Pugel E, Messerle M, Hengel H, Busch DH, Koszinowski UH, Jonjic S. J Exp Med. 2005;201(2):211–220. doi: 10.1084/jem.20041617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lodoen M, Ogasawara K, Hamerman JA, Arase H, Houchins JP, Mocarski ES, Lanier LL. J Exp Med. 2003;197(10):1245–1253. doi: 10.1084/jem.20021973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lodoen MB, Abenes G, Umamoto S, Houchins JP, Liu F, Lanier LL. J Exp Med. 2004;200(8):1075–1081. doi: 10.1084/jem.20040583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ziegler H, Thale R, Lucin P, Muranyi W, Flohr T, Hengel H, Farrell H, Rawlinson W, Koszinowski UH. Immunity. 1997;6(1):57–66. doi: 10.1016/s1074-7613(00)80242-3. [DOI] [PubMed] [Google Scholar]
- 17.Voigt V, Forbes CA, Tonkin JN, Degli-Esposti MA, Smith HR, Yokoyama WM, Scalzo AA. Proc Natl Acad Sci U S A. 2003;100(23):13483–13488. doi: 10.1073/pnas.2233572100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adams EJ, Juo ZS, Venook RT, Boulanger MJ, Arase H, Lanier LL, Garcia KC. Proc Natl Acad Sci U S A. 2007;104(24):10128–10133. doi: 10.1073/pnas.0703735104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mathys S, Schroeder T, Ellwart J, Koszinowski UH, Messerle M, Just U. The Journal of infectious diseases. 2003;187(6):988–999. doi: 10.1086/368094. [DOI] [PubMed] [Google Scholar]
- 20.Otwinowski Z, Minor W. Methods Enzymol. 1997;276(part A):307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 21.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. Acta Crystallogr D Biol Crystallogr. 2002;58(Pt 11):1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 22.Emsley P, Cowtan K. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 23.Read RJ. Acta Crystallogr D Biol Crystallogr. 2001;57(Pt 10):1373–1382. doi: 10.1107/s0907444901012471. [DOI] [PubMed] [Google Scholar]
- 24.Murshudov GN, Vagin AA, Dodson EJ. Acta Crystallogr D Biol Crystallogr. 1997;53 (Pt 3):240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 25.Painter J, Merritt EA. Acta Crystallogr D Biol Crystallogr. 2005;61(Pt 4):465–471. doi: 10.1107/S0907444905001897. [DOI] [PubMed] [Google Scholar]
- 26.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Crystallogr D Biol Crystallogr. 1998;54(Pt 5):905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 27.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DeLano WL, editor. The PyMOL User’s Manual. DeLano Scientific; Palo Alto, CA, USA: 2002. [Google Scholar]
- 29.Schuck P. Biophys J. 2000;78(3):1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Allen H, Fraser J, Flyer D, Calvin S, Flavell R. Proc Natl Acad Sci U S A. 1986;83 (19):7447–7451. doi: 10.1073/pnas.83.19.7447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Townsend A, Ohlen C, Foster L, Bastin J, Ljunggren HG, Karre K. Cold Spring Harb Symp Quant Biol. 1989;54 Pt 1:299–308. doi: 10.1101/sqb.1989.054.01.038. [DOI] [PubMed] [Google Scholar]
- 32.Bjorkman PJ, Saper MA, Samraoui B, Bennett WS, Strominger JL, Wiley DC. Nature. 1987;329(6139):506–512. doi: 10.1038/329506a0. [DOI] [PubMed] [Google Scholar]
- 33.Halaby DM, Mornon JP. J Mol Evol. 1998;46(4):389–400. doi: 10.1007/pl00006318. [DOI] [PubMed] [Google Scholar]
- 34.Holm L, Sander C. Science. 1996;273(5275):595–603. doi: 10.1126/science.273.5275.595. [DOI] [PubMed] [Google Scholar]
- 35.Fremont DH, Matsumura M, Stura EA, Peterson PA, Wilson IA. Science. 1992;257(5072):919–927. doi: 10.1126/science.1323877. [DOI] [PubMed] [Google Scholar]
- 36.Burmeister WP, Gastinel LN, Simister NE, Blum ML, Bjorkman PJ. Nature. 1994;372(6504):336–343. doi: 10.1038/372336a0. [DOI] [PubMed] [Google Scholar]
- 37.Li P, McDermott G, Strong RK. Immunity. 2002;16(1):77–86. doi: 10.1016/s1074-7613(02)00258-3. [DOI] [PubMed] [Google Scholar]
- 38.Lawrence MC, Colman PM. J Mol Biol. 1993;234(4):946–950. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- 39.Hu CD, Chinenov Y, Kerppola TK. Mol Cell. 2002;9(4):789–798. doi: 10.1016/s1097-2765(02)00496-3. [DOI] [PubMed] [Google Scholar]
- 40.Vink C, Beuken E, Bruggeman CA. J Virol. 2000;74(16):7656–7665. doi: 10.1128/jvi.74.16.7656-7665.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cosman D, Fanger N, Borges L, Kubin M, Chin W, Peterson L, Hsu ML. Immunity. 1997;7(2):273–282. doi: 10.1016/s1074-7613(00)80529-4. [DOI] [PubMed] [Google Scholar]
- 42.Wills MR, Ashiru O, Reeves MB, Okecha G, Trowsdale J, Tomasec P, Wilkinson GW, Sinclair J, Sissons JG. J Immunol. 2005;175(11):7457–7465. doi: 10.4049/jimmunol.175.11.7457. [DOI] [PubMed] [Google Scholar]
- 43.Alcami A, Koszinowski UH. Trends Microbiol. 2000;8(9):410–418. doi: 10.1016/S0966-842X(00)01830-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bowness P. Rheumatology (Oxford) 2002;41(8):857–868. doi: 10.1093/rheumatology/41.8.857. [DOI] [PubMed] [Google Scholar]
- 45.Shiroishi M, Kuroki K, Ose T, Rasubala L, Shiratori I, Arase H, Tsumoto K, Kumagai I, Kohda D, Maenaka K. J Biol Chem. 2006;281(15):10439–10447. doi: 10.1074/jbc.M512305200. [DOI] [PubMed] [Google Scholar]
- 46.Smith LM, Shellam GR, Redwood AJ. Virology. 2006;352(2):450–465. doi: 10.1016/j.virol.2006.04.031. [DOI] [PubMed] [Google Scholar]
- 47.Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD. Nucleic Acids Res. 2003;31(12):3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gouet P, Courcelle E, Stuart DI, Metoz F. Bioinformatics. 1999;15(4):305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- 49.Laskowski RA, Moss DS, Thornton JM. J Mol Biol. 1993;231(4):1049–1067. doi: 10.1006/jmbi.1993.1351. [DOI] [PubMed] [Google Scholar]