

Abstract
A convenient optically active synthesis of (3aS,5R,6aR)-5-hydroxy-hexahydrocyclopenta[b]furan, a high-affinity nonpeptidyl ligand for HIV protease inhibitor 2, is described. The synthesis utilizes commercially available (1R,5S)-(+)-2-oxabicyclo[3.3.0]oct-6-en-3-one as the starting material and oxymercuration or bromohydrin reaction as the key step. Enantiopure ligand was converted to protease inhibitor 2.
Effective treatment of HIV/AIDS continues to be one of the most critical problems facing the medical community in the 21st century.1 The advent of highly active antiretroviral therapy (HAART) with HIV protease inhibitors (PIs) in combination with reverse transcriptase (RT) inhibitors significantly improved HIV management and the quality of life for HIV/AIDS patients. Despite these advances, there are major problems with HAART treatment regimens, particularly alarming is the adverse side effects including toxicity, complexity and the occurrence of various cancers due to survival elongation.2 Perhaps, most concerning is that a growing number of patients are developing drug-resistant HIV-1 variants and there are reports that these strains can be transmitted.3,4 Therefore, the development of new classes of antiretroviral drugs with minimal adverse effects and potent activity against mutant strains resistant to currently approved PIs, remains an important treatment objective.
Our recent structure-based design strategies specifically targeting the HIV-1 protease backbone led to a number of exceedingly potent PIs with impressive drug-resistant profiles.5 One of these PIs is darunavir (1, TMC-114, Figure 1) and it has been approved by the United States Food and Drug Administration for the treatment of HIV/AIDS patients harboring multidrug-resistant HIV-1 variants.6,7 Darunavir contains a structure-based designed and privileged bis-THF ligand which makes extensive interactions with the HIV-1 protease backbone. Subsequently, based upon darunavir-bound protein-ligand X-ray structures, we have designed a stereochemically defined cyclopentyltetrahydrofuran (Cp-THF)-derived urethane as the P2-ligand.9 The inhibitor 2, containing this ligand has shown remarkable potency against both wild-type and drug-resistant viruses (2, Ki = 4.5 pM and ID50 = 1.8 nM). Our initial synthesis of optically active Cp-THF ligand was based upon an enzymatic asymmetrization as the key step.9 Recently, Shibasaki and co-workers have reported an optically active synthesis of Cp-THF ligand through Al-Li-bis(binaphthoxide) catalyst-controlled Michael addition of dimethyl malonate to racemic 4-O-protected cyclopentenone.10 In our continuing studies aimed at broadening the scope and utility of the Cp-THF-derived PIs, we have carried out a convenient enantioselective synthesis of Cp-THF ligand utilizing commercially available (1R, 5S)-(+)-2-oxabicyclo [3.3.0] oct-6-en-3-one. The route is amenable to quantities of Cp-THF ligand in high optical purity.
Figure 1.
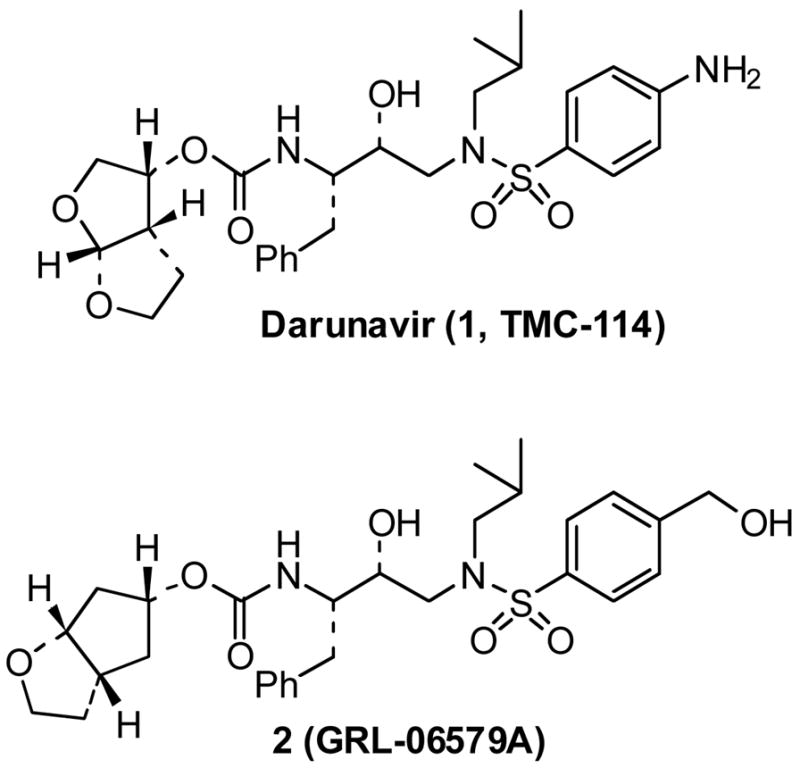
Structure of inhibitor 1 and 2
As shown in Scheme 1, commercially available (1R,5S)-(+)-2-oxabicyclo[3.3.0]oct-6-en-3-one 3 was utilized as a starting material. Stereoselective installation of the C5-hydroxyl group was accomplished by an oxymercuration-demercuration protocol in a one-pot operation as described by Olivo and co-workers.11 As shown, exposure of 3 to 2 equiv of mercuric acetate in a mixture of THF and water at 0 ºC to 23 ºC for 2 h provided the corresponding organomercurial derivative. Aqueous sodium hydroxide followed by NaBH4 were added to the mixture at 0 ºC to afford the endo-alcohol 4 in near quantitative yield as a single diastereomer by 1H-NMR analysis. Protection of the hydroxyl group with TIPSOTf in the presence of 2,6-lutidine in CH2Cl2 at −50 ºC for 3 h gave the corresponding silylether. Reduction of the resulting silyloxylactone with lithium aluminum hydride in THF at −20 ºC furnished diol 5 (89% yield for two steps). The diol was converted to a tetrahydrofuranyl derivative by selective mesylation of the primary alcohol with mesyl chloride and triethylamine followed by a reaction of the resulting mono-mesylate with sodium hydride in THF at 23 ºC for 2 h. This provided cyclopenta[b]furan 6 in a 70% yield for two steps. Removal of TIPS-group with TBAF in THF at 23 ºC for 2 h afforded optically active hexahydrocyclopentafuran-5-ol 7 in 97% yield.12
Scheme 1.
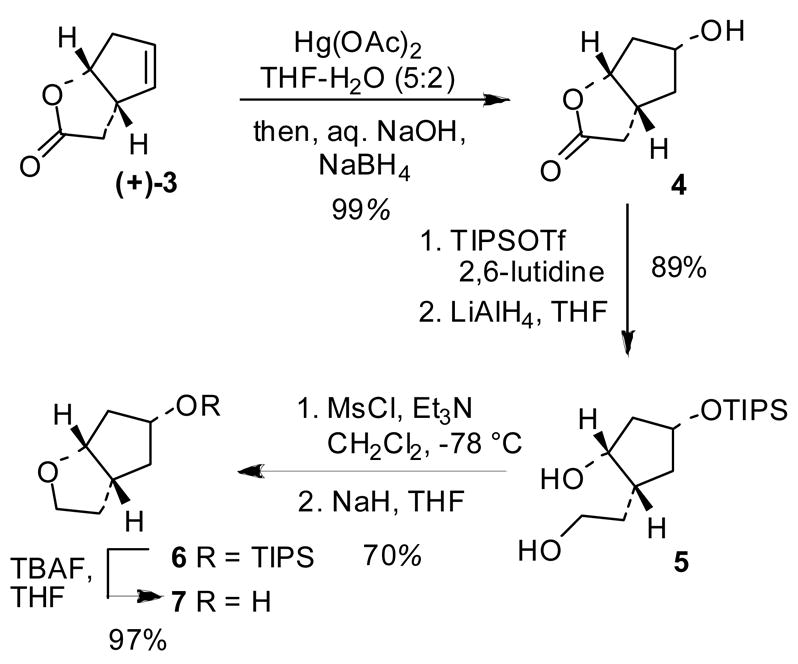
Synthesis of hexahydrocyclopentafuran-5-ol
As shown above, hydroxylation of alkene 3 by an oxymercuration-demercuration protocol provided excellent yield and diastereoselectivity. However, the overall procedure may be less suitable for scale up of Cp-THF ligand as it requires multi-gram quantities of mercuric acetate and a tedious work up with mercury byproducts. In this context, we have investigated more practical halohydrin reactions using NBS and NIS. As shown in Scheme 2, reaction of 3 with 1.1 equiv. of NBS in a mixture (10:1) of DME and water at 23 °C for 16 h provided bromohydrin 8 in 47% yield. This reaction was investigated under various reaction conditions as shown in Table 1. The choice of solvent and amount of water has a significant effect on the yield of the desired bromohydrin 8. Alternative bromohydrin 9 is formed as a significant byproduct. Among various solvents examined (entries 1–5), aqueous DME showed the most reproducible results. The amount of water in DME was found to be critical (entries 5–9). A DME-water mixture ratio of 10:1 provided the best result (entry 9) with 47% yield of bromohydrin 8. Reaction of NIS in DME also showed similar trends(entries 10, 11). Reduction of bromohydrin 8 using n-Bu3SnH in the presence of AlBN in refluxing toluene for 12 h afforded hydroxyl lactone 4 in near quantitative yield. We have also investigated catalytic hydrogenation conditions for reduction of bromohydrin 8.
Scheme 2.
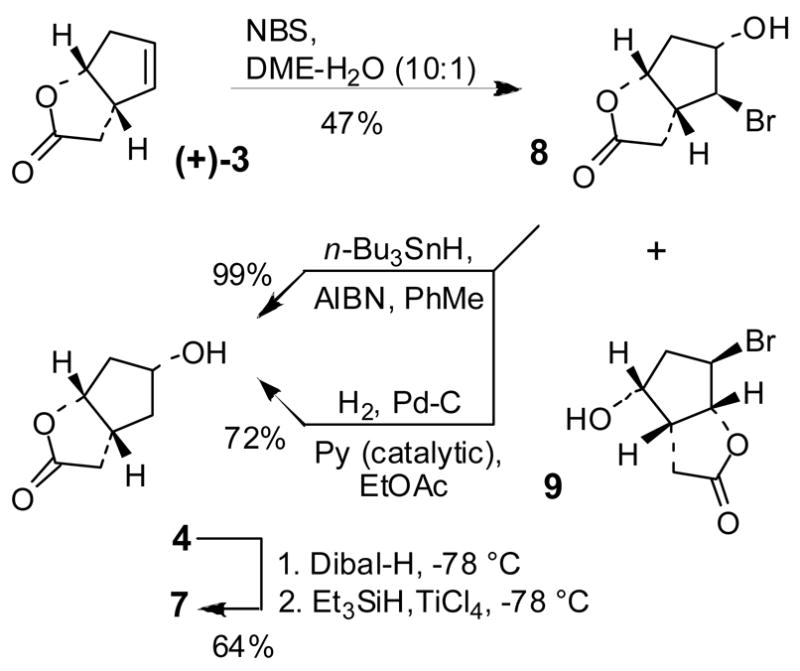
Synthesis of Cp-THF via bromohydrin reaction
Table 1.
Halohydrin reactions of lactone 3
| Entry | Reagenta | Solvent | (8 : 9)b | Yield (%)c |
|---|---|---|---|---|
| 1 | NBS | CH3CN/H2O(1:5) | 27:73 | 29 |
| 2 | NBS | THF/H2O(1:5) | 20:80 | 18 |
| 3 | NBS | acetone/H2O(1:1) | 50:50 | 34 |
| 4 | NBS | CH2Cl2/H2O(5:1) | --- | NR |
| 5 | NBS | DME/H2O(1:1) | 36:64 | 31 |
| 6 | NBS | DME/H2O(2:1) | 50:50 | 36 |
| 7 | NBS | DME/H2O(3:1) | 52:48 | 42 |
| 8 | NBS | DME/H2O(4:1) | 53:47 | 43 |
| 9 | NBS | DME/H2O(10:1) | 57:43 | 47 |
| 10 | NIS | DME/H2O(1:1) | 40:60 | 33 |
| 11 | NIS | DME/H2O(1:4) | 40:60 | 42 |
All reactions were carried out with 1 equiv of reagent.
Ratios determined after separation by silica gel chromatography.
Isolated yield of 8 after chromatography.
This reduction proceeded in good yield (72%) in the presence of 10% Pd-C and a catalytic amount of pyridine in ethyl acetate, providing the lactone 4.13 When ethanol was used as the solvent, the reaction yield was reduced to 54%. The choice of solvent and the presence of a catalytic amount of pyridine is critical to this reduction process. Lactone 4 was converted to Cp-THF 7 by Dibal-H reduction in CH2Cl2 at −78 °C for 2 h to provide the corresponding lactol. Further reduction of this lactol with 2 equiv of Et3SiH in the presence of 1 equiv. of TiCl4 in CH2Cl2 at −78 °C afforded optically active Cp-THF 7 in 64% yield over two steps.
To circumvent the formation of isomeric lactone 9, we then investigated the bromohydrin reaction with the corresponding 3-derived tetrahydro-2H-cyclopenta[b]furan derivative. As shown in Scheme 3, Dibal-H reduction followed by reduction of the resulting lactol with Et3SiH in the presence of TiCl4 provided 10 in 76% yield. Bromohydrin reaction of 10 with 1.1 equiv of NBS in a mixture (10:1) of acetone and water at −20 °C for 16 h provided a mixture (3.5:1) of diastereomeric bromohydrins 11 and 12 in 60% and 17% yields respectively after separation by silica gel chromatography (25% ethyl acetate in hexanes as the eluent).14 Reduction of bromohydrin 11 by catalytic hydrogenation over 10% Pd-C in the presence of a catalytic amount of pyridine in ethyl acetate afforded optically active Cp-THF 7 in 69% yield. The minor isomer 12 was reduced to epimeric alcohol 13 in 63% yield as above. Alcohol 13 was converted to desired Cp-THF 7 by an oxidation/reduction sequence. Thus, TPAP oxidation15 of 13 in CH2Cl2 at 23 °C for 2 h provided the corresponding ketone which was reduced with NaBH4 in methanol at −20 °C to furnish optically active Cp-THF alcohol 7 in 63% yield for two steps. It was converted to mixed carbonate 14 by treatment with N,N′-disuccinimidyl carbonate in the presence of Et3N in CH2Cl2 at 23 °C for 12 h.16 Mixed succinimidyl carbonate 14 was isolated in 68% yield. Reaction of this carbonate with previously described amine 15 in CH2Cl2 in the presence of diisopropylethylamine furnished inhibitor 2 in 78% yield.9
Scheme 3.
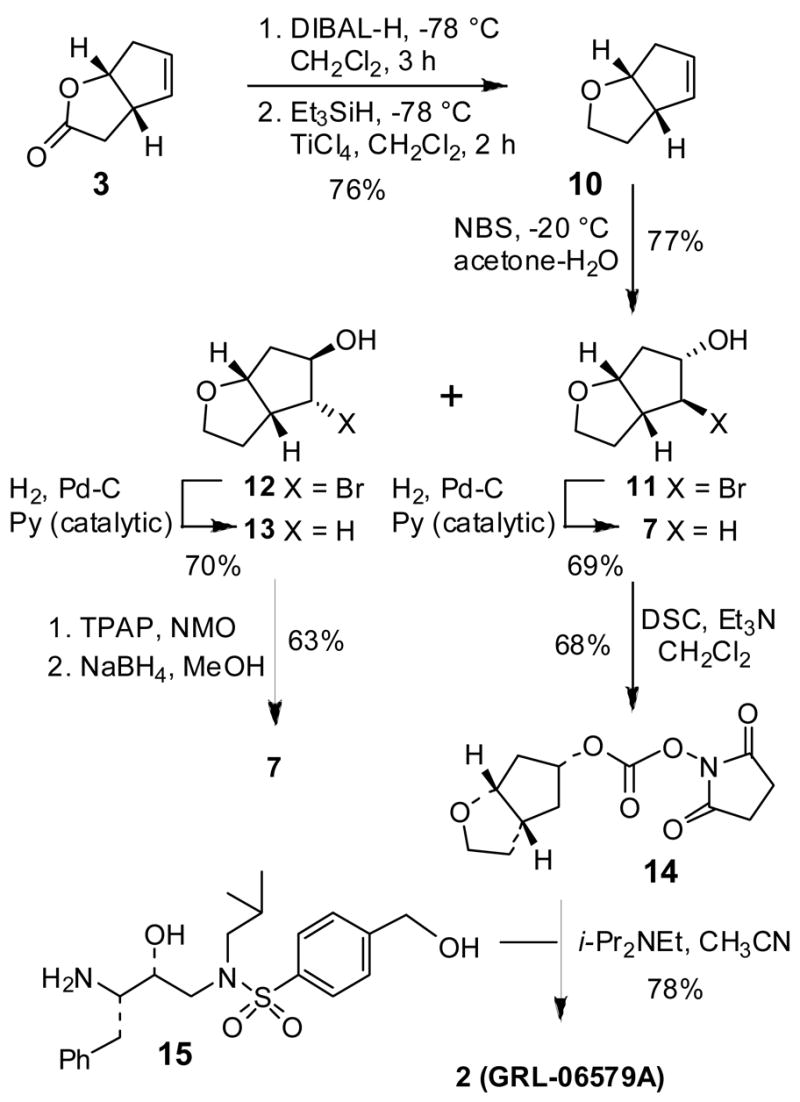
Synthesis of Cp-THF and inhibitor 2
In summary, we carried out a short synthesis of optically active (3aS, 5R, 6aR)-5-hydroxyhexahydrocyclopenta-[b]furan ligand for HIV protease inhibitors using commercially available (1R,5S)-(+)-2-oxabicyclo[3.3.0]oct-6-en-3-one. The current synthetic routes would provide rapid access to this important nonpeptide high affinity ligand for a variety of HIV protease inhibitors.
Acknowledgments
Financial support by the National Institutes of Health (GM53386) is gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.United Nations. Report on the global HIV/AIDS Epidemic: 4th global report. UN; New York: 2004. [Google Scholar]
- 2.Sepkowitz KA. N Engl J Med. 2001;344:1764. doi: 10.1056/NEJM200106073442306. [DOI] [PubMed] [Google Scholar]
- 3.Staszewski S, Morales-Ramirez J, Tashima KT, Rachlis A, Skiest D, Standord J, Stryker R, Johnson P, Labriola DF, Farina D, Manion DJ, Ruiz NM. N Engl J Med. 1999;341:1865. doi: 10.1056/NEJM199912163412501. [DOI] [PubMed] [Google Scholar]
- 4.Wainberg MA, Friedland G. J Am Med Assoc. 1998;279:1977. doi: 10.1001/jama.279.24.1977. [DOI] [PubMed] [Google Scholar]
- 5.Ghosh AK, Chapsal BD, Weber IT, Mitsuya H. Acc Chem Res. 2008;41:78. doi: 10.1021/ar7001232. [DOI] [PubMed] [Google Scholar]
- 6.On June 23, 2006, FDA approved new HIV treatment for patients who do not respond to existing drugs. Please see http://www.fda.gov/bbs/topics/NEWS/2006/NEW01395.htm.
- 7.Ghosh AK, Dawson ZL, Mitsuya H. Bioorg Med Chem. 2007;15:7576. doi: 10.1016/j.bmc.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghosh AK, Ramu Sridhar P, Kumaragurubaran N, Koh Y, Weber IT, Mitsuya H. ChemMedChem. 2006;1:939. doi: 10.1002/cmdc.200600103. [DOI] [PubMed] [Google Scholar]
- 9.Ghosh AK, Sridhar PR, Leshchenko S, Hussain AK, Li J, Kovalevsky AY, Walters DE, Wedekind JE, Grum-Tokars V, Das D, Koh Y, Maeda K, Gatanaga H, Weber IT, Mitsuya H. J Med Chem. 2006;49:5252. doi: 10.1021/jm060561m. [DOI] [PubMed] [Google Scholar]
- 10.Mihara H, Sohtome Y, Matsunaga S, Shibasaki M. Chem Asian J. 2008;3:359. doi: 10.1002/asia.200700330. [DOI] [PubMed] [Google Scholar]
- 11.Ríos M, Velázquez F, Olivo HF. Tetrahedron. 2003;59:6531. [Google Scholar]
- 12.Selected data for 7: [α]D23 -13 (c 1, CHCl3); 1H NMR (CDCl3, 300 MHz): δ4.37 (t, 1H, J = 5.7 Hz), 4.23-4.20 (m, 1H), 4.01 (dt, 1H, J = 4.2, 8.7 Hz), 3.59 (dd, 1H, J = 8.6, 15 Hz), 2.71-2.59 (m, 1H), 2.53 (br s, 1H), 2.25-2.14 (m, 1H), 2.06-1.96(m, 2H), 1.87-1.76 (m, 2H), 1.67-1.59 (m, 1H). 13C NMR (CDCl3, 75 MHz): 85.7, 74.8, 68.2, 42.5, 41.4, 41.4, 35.1. MS (CI): m/z 129.1 [M + H]+.
- 13.Cravero RM, Signorella SR, Gonzalez-Sierra M, Ruveda EA. Synth Comm. 1987;17:1287. [Google Scholar]
- 14.All new compounds gave satisfactory spectroscopic and analytical results. Compound 8: [α]D23 +14.7 (c 1, CHCl3). 1H NMR (CDCl3) δ 2.20 (d, 1H, J = 15.1 Hz), 2.56 (ddd, 1H, J = 4.8, 6.7 and 11.6 Hz), 2.63 (dd, 1H, J = 3.6 and 18.6 Hz), 2.84 (dd, 1H, J = 11.6 and 18.6 Hz), 3.31–3.37 (m, 1H), 4.09 (dd, 1H, J = 2.4 and 4.1 Hz), 4.45–4.48 (m, 1H), 5.16 (t, 1H, J = 7.0 Hz). 13C NMR (CDCl3): δ 35.6, 38.4, 48.0, 58.1, 79.4, 84.0, 176.8. FT-IR (NaCl): 1075, 1198, 1750, 3444 cm−1. MS (CI): m/z 220.98 [M + H]+.Compound 9: [α]D23 +15.2 (c 1, CHCl3). 1H NMR (CDCl3): δ 2.01–2.12 (m, 1H), 2.34–2.45 (m, 1H), 2.74–2.86 (m, 2H), 3.27–3.37 (m, 1H), 4.14–4.22 (m, 1H), 4.40 (dd, 1H, J = 5.1 and 11.1 Hz), 5.04 (dt, 1H, J = 3.0 and 6.9 Hz). 13C NMR (CDCl3): δ 29.5, 33.5, 38.2, 40.8, 58.3, 82.3, 176.5. FT-IR (NaCl): 1180, 1768, 3429 cm−1. MS (CI): m/z 220.98 [M + H]+.Compound 11: [α]D23 -15.5 (c 1, CHCl3). 1H NMR (CDCl3): δ 1.86–1.95 (m, 2H), 2.19–2.27 (m, 1H), 2.44 (dt, 1H, J = 6.1 and 14.5 Hz), 2.82 (bs, 1H), 2.99–3.05 (m, 1H), 3.65 (dd, 1H, J = 7.5 and 16.0 Hz), 3.91–3.99 (m, 2H), 4.25–4.27 (bm, 1H), 4.45 (dt, 1H, J = 2.4 and 6.5 Hz). 13C NMR (CDCl3): δ 32.8, 37.5, 52.4, 60.2, 67.8, 80.4, 83.1. MS (CI): m/z 207.1 [M + H]+.Compound 12: [α]D23 -11.6 (c 1, CHCl3). 1H NMR (CDCl3): δ 1.60–1.67 (m, 1H), 1.95–2.06 (m, 2H), 2.18 (dd, 1H, J = 6.7 and 14.0 Hz), 2.93–3.01 (m, 1H), 3.54 (dt, 1H, J = 5.7 and 8.9 Hz), 3.82–3.90 (m, 2H), 4.21–4.28 (m, 1H), 4.37 (t, 1H, J = 6.7 Hz). 13C NMR (CDCl3): δ 32.5, 38.0, 45.3, 59.9, 68.1, 76.4, 79.9. MS (CI): m/z 207.2 [M + H]+.
- 15.(a) Griffith WP, Ley SV. Aldrichim Acta. 1990;23:13. [Google Scholar]; (b) Griffith WP, Ley SV. Synthesis. 1994:639. [Google Scholar]
- 16.Ghosh AK, Duong TT, McKee SP, Thompson WJ. Tetrahedron Lett. 1992;33:2781. doi: 10.1016/S0040-4039(00)78856-3. [DOI] [PMC free article] [PubMed] [Google Scholar]