

Abstract
A systematic study into the effect of C16 and C17 substitution on the stereochemical outcome of bis-spirocyclization to form the ABC ring system of azaspiracid is disclosed. Successful construction of the natural 10R,13R bis-spirocyclic stereochemistry has been accomplished on the C16 benzyloxy-containing precursor.
The azaspiracids are an intriguing class of recently isolated natural products that possess a complex structural framework as well as considerable biological activity.1-3 As discussed in the previous paper,4 the D ring appears to exert considerable influence on the bis-spirocyclization. Based on these results, our efforts shifted toward the construction of selected substrates containing substitution at C16 or C17 (Scheme 1). The C17 series appeared more attractive as inspection of the potential chair conformations of bis-spiroketals 2 and 34 revealed that the transoidal and the cisoidal structures were both capable of placing the C17 allyl substituent equatorial on the basis of the proposed conformation for bis-spiroketals 5 and 6.
Scheme 1.
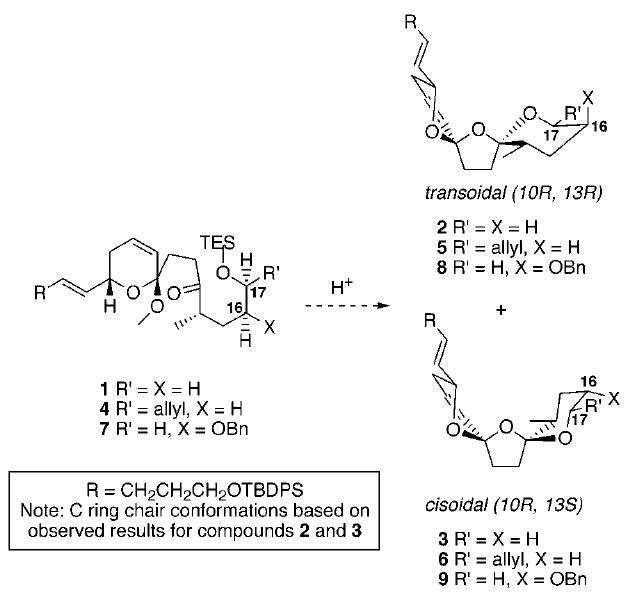
Potential Combinations for Substitution Patterns at C16 and C17
C17 Substitution
Spirocyclization of previously described keto sulfone 104 using our preferred conditions for keto sulfone substrates (CSA, MeCN)5 led to a mixture of stereoisomers (Scheme 2). Treatment of the spirocycle 11 with n-BuLi induced β-elimination to yield the elaborate enol ether 12 in 70% yield, along with 10% of the presumed C10 epimer. The strategy allowed for the protection of the C13 carbonyl function while selectively revealing the C17 hydroxyl group. Oxidation at C17 followed by Brown allylation6 yielded the homoallylic alcohol 13 in greater than 20:1 d.s. Removal of the sulfone functionality revealed the highly labile enol ether 14, which rapidly underwent spirocyclization under the standard conditions (0.04 M CSA, t-BuOH/PhMe, 14–18 h) to give the bis-spiroketal 6 as a single diastereomer. Unfortunately, NOESY and COSY NMR experiments (CDCl3) confirmed the cisoidal 10R,13S relationship of the bis-spiroketal. While it is important to point out that the enol ether spirocyclization precursor 14 is structurally different than ketone 4, previous work in our laboratory has shown that the C10 ketal is readily ionized under acidic conditions,5 thereby ensuring formation of comparable spirocyclization intermediates from both precursors. It is interesting to note, however, that the sulfone moiety once again provided added stability,7 as 13 was indefinitely stable in the freezer. Also, our ketalization/β-elimination/desulfonylation strategy represents, to the best of our knowledge, a unique strategy for the effective construction of highly labile enol ethers (e.g., 14).
Scheme 2.
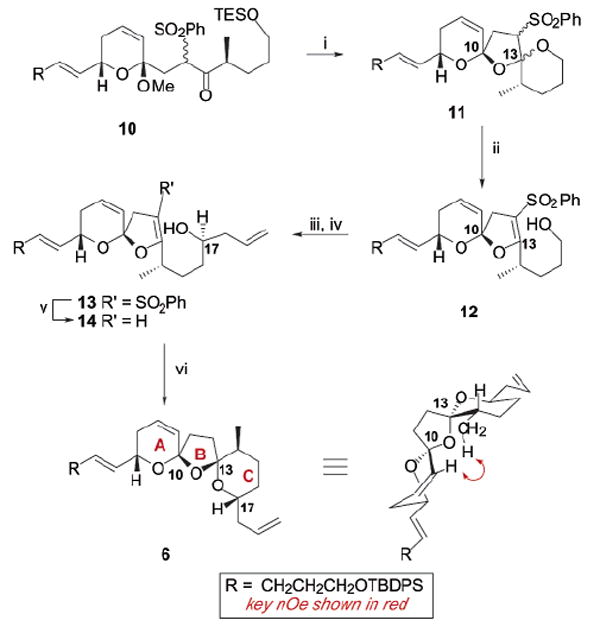
Bis-spirocyclization of C17 Allyl Substratea
a Key: (i) CSA, MeCN, 90%; (ii) n-BuLi, THF, −78 °C, 70%; (iii) TPAP, NMO, CH2Cl2, mol. sieves, 96%; (iv) (+)-Ipc2Ballyl, Et2O, pentane, 70%, >20:1 d.s.; (v) 5% Na/Hg, Na2HPO4, MeOH, THF, −10 °C; (vi) CSA, t-BuOH, PhMe, 76% (over two steps).
C16 Substitution
The synthesis of the required benzyloxy aldehyde 21 began with the known Evans alkylation product 158 (Scheme 3). Conversion to its benzyl ester 16 followed by Sharpless asymmetric dihydroxylation,9 in situ lactonization, and TIPS protection yielded the lactone 17, in overall 66% yield from 16, as a separable 3:1 ratio at C16 favoring the desired stereochemistry.10 While the diastereomeric selectivity in the dihydroxylation is less than optimum (3:1 d.s.), direct dihydroxylation of the chiral oxazolidinone-containing alkene 15 provided inferior results (1.5:1 d.s.). This observation is consistent with our previous findings with other oxazolidinone-containing alkenes.11 Subsequent reduction using LiBH4 provided the diol 18. Next, sequential protection at C13 and C16 produced the fragment 19, along with a small amount of impurities derived from migration of the pivaloate and silyl protecting groups. Removal of the TIPS ether under standard conditions allowed for easy purification. The C17 hydroxyl was reprotected as its TES ether 20. Cleavage of the C13 pivaloate protecting group did prove problematic with DIBAL-H;12 however, the use of LiBH4, in the presence of a small amount of saturated aqueous NH4Cl, cleanly provided the desired alcohol in 99% yield. Finally, TPAP oxidation yielded the target aldehyde 21.
Scheme 3.
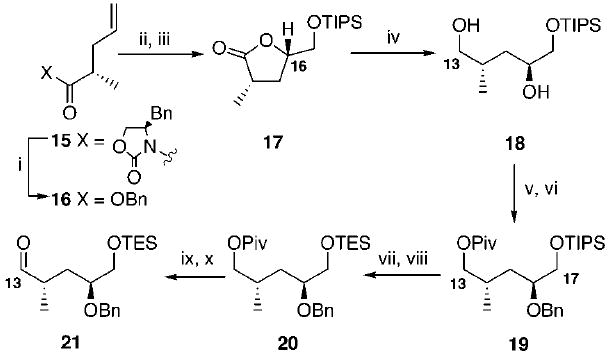
Synthesis of the C16 Benzyloxy-Substituted Aldehydea
a Key: (i) BnOLi, PMBOH, THF, 99%; (ii) AD mix α, NaHCO3, t-BuOH, H2O; (iii) TIPSOTf, 2,6-lutidine, CH2Cl2, −78 °C, 66% overall yield from 16 (3:1 d.s.); (iv) LiBH4, MeOH, THF, 0 °C, 99%; (v) PivCl, Et3N, DMAP, CH2Cl2, 66%; (vi) NaH, BnBr, DMF, −50 °C to −10 °C; (vii) TBAF, THF, 79% (over two steps); (viii) TESCl, DMAP, Et3N, CH2Cl2, 95%; (ix) LiBH4, saturated aqueous NH4Cl, THF, 0 °C to rt, 99%; (x) TPAP, NMO, CH2Cl2, molecular sieves, 87%.
Lithiation of sulfone A with LDA followed by addition of the aldehyde 21 provided the hydroxy sulfone adduct as a labile mixture13 of all four diastereomers (Scheme 4). Immediate oxidation to the ketone sulfone 22 was accomplished with TPAP in a 60% yield over two steps. Desulfurization under standard conditions followed by bis-spirocyclization (0.04 M CSA, t-BuOH/PhMe, 14–18 h) gave two spiroketals in a 3:5 ratio (8/9) in an overall 80% yield. After careful separation of the two isomers, the stereochemistry of the more polar isomer was determined to be the cisoidal ketal 9 (2D NMR, CDCl3). We were gratified to find, however, that the less polar spirocycle 8 possessed the natural transoidal stereochemistry at C10 and C13 as established by ROESY and COSY correlations (C6D5N). While the NOE between the C9 alkene to C17 hydrogen was previously observed in the transoidal product 2 from the D ring truncated series,4 the NOE between the C6 hydrogen and the methyl at C14 was not present. This lack of signal can be explained by the placement of the C13 furan oxygen and the C14 methyl substituents in axial orientations within the pyran C ring. Further NMR evidence supports this hypothesis: (1) a strong NOE is observed between the C14 methyl and the C16 hydrogen and (2) the coupling constants between the C16 and C17 hydrogens match the predicted data for the proposed C ring, chair conformation using the Karplus correlation.14 Furthermore, the alternate chair conformer (with the C13 furan oxygen and the C14 methyl substituent in equatorial orientations) is significantly higher in energy (3.4 kcal/mol) using the B3LYP density functional and a 6-31G(d) basis set. It should also be noted that the observed NOE and coupling constant data would not be expected for the alternate non-natural transoidal bis-spirocycle.5 Finally, the proposed conformation of transoidal spirocycle 8 is in contrast to the previously discussed transoidal spirocycle 2, which appeared to place the C13 furan oxygen and the C14 methyl substituents in equatorial orientations.
Scheme 4.
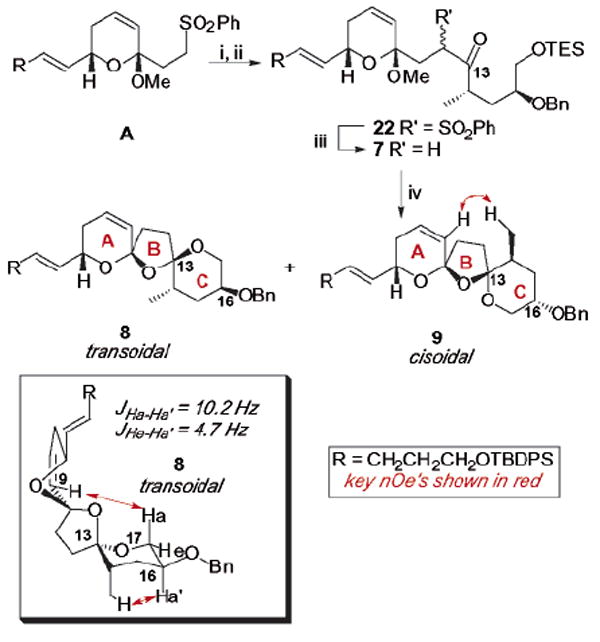
Bis-spirocyclization of C16 Benzyloxy Substratea
a Key: (i) LDA, THF, −78 °C then 21; (ii) TPAP, NMO, CH2Cl2, molecular sieves 60% (over two steps); (iii) 5% Na/Hg, Na2HPO4, MeOH, THF, −10 °C; (iv) CSA, PhMe/t-BuOH, 80%, 3:5 ratio (8/9).
To further study the nature of the bis-spiroketalization, the reaction with ketone 7 was performed at lower temperatures (−10 to +4 °C, 21 h) and reduced molarity of the acid catalyst (0.003 M) under otherwise identical reaction conditions (1:1 t-BuOH/PhMe). We were intrigued to discover that the predominate product was the cisoidal bis-spirocycle 9. Gratifyingly, further warming of the reaction to room temperature for an additional 48 h resulted in the previously observed (3:5 ratio of 8:9) equilibrium mixture.15 It would appear from these observations that the cisoidal bis-spirocycle 9 is the result of kinetic control while the transoidal bis-spirocycle 8 can be accessed under thermodynamic conditions. With one equilibration cycle, a 50% overall yield of the desired transoidal bis-spirocycle 8 can be obtained. Finally, use of Nicolaou and co-workers’ conditions16 for equilibration of their cisoidal bis-spirocycle to the natural transoidal species (3 equiv of TFA, CH2Cl2) provided inferior results for the conversion of 9 to 8 (approximately 1:3 ratio for 8:9).17
The first systematic study into the effect of substituents on the bis-spirocyclization of a series of precursors has been presented. The C16 oxygen substitution facilitated formation of a nearly equal mixture of the cisoidal and transoidal bis-spirocycles while C17 allyl substitution provided sole access to the cisoidal species. Our continuing progress toward the total synthesis will be reported in due course.
Supplemental Materials
Acknowledgments
We thank the National Institutes of Health (GM63723) and the University of Mississippi for partial support of this work. In addition, we thank Dr. Jeff Morré and Professor Max Dienzer (Oregon State University) for mass spectral data. Finally, we thank Dr. Roger Hanselmann (Rib-X Pharmaceuticals) for his helpful discussions.
Footnotes
This work was performed at the University of Mississippi.
Supporting Information Available: Experimental procedures and spectral characterization are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.MacMahon T, Silke J. Harmful Algae News. 1996;14:2. [Google Scholar]
- 2.Ofuji K, Satake M, McMahon T, Silke J, James KJ, Naoki H, Oshima Y, Yasumoto T. Nat Toxins. 1999;7:99. doi: 10.1002/(sici)1522-7189(199905/06)7:3<99::aid-nt46>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 3.Ofuji K, Satake M, McMahon T, James KJ, Naoki H, Oshima Y, Yasumoto T. Biosci Biotechnol Biochem. 2001;65:740. doi: 10.1271/bbb.65.740. [DOI] [PubMed] [Google Scholar]
- 4.Carter RG, Bourland TC, Graves DE. Org Lett. 2002;4:2177. doi: 10.1021/ol026033w. ol026033w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carter RG, Graves DE. Tetrahedron Lett. 2001;42:6035. [Google Scholar]
- 6.Racherla US, Brown HC. J Org Chem. 1991;56:401. [Google Scholar]
- 7.We have consistently observed an increased stabilization in substrates containing the sulfone function versus their corresponding desulfonylated counterparts, which are prone to elimination at C10,11 to the corresponding enol ether.
- 8.Evans DA, Ennis MD, Mathre DJ. J Am Chem Soc. 1982;104:1737. [Google Scholar]
- 9.Kolb HC, VanNieuwenhze MS, Sharpless KB. Chem ReV. 1994;94:2483. [Google Scholar]
- 10.A small, inseparable bi-product was present in 17. Fleming and coworkers recently reported the presence of an n-propyl silyl impurity in selected TIPS protection protocols. As silylation as alternate silyl protecting groups (such as TBS or TES) did not lead to a similar impurity, it would appear that the n-propyl silyl species is a reasonable explanation. Barden DJ, Fleming I. Chem Commun. 2001:2366.
- 11.Carter RG, Weldon DJ. Org Lett. 2000;2:3913. doi: 10.1021/ol006674w. [DOI] [PubMed] [Google Scholar]
- 12.It is unclear at this juncture as to the exact nature of the problem; however, the experimental data is consistent with silyl migration of the C17 TES protecting group.
- 13.The Julia adduct appeared to be prone to rapid spirocyclization of the C13 hydroxyl moiety onto a corresponding C10 oxonium ion. This problem could be easily circumvented by the immediate oxidation of the hydroxy sulfone intermediate (without purification or storage) to the corresponding keto sulfone 22.
- 14.Karplus M. J Phys Chem. 1959;30:11. [Google Scholar]
- 15.The same ratio (3:5, 92% overall yield) can also be observed by resubmission of the cisoidal bis-spirocycle 9 to the standard conditions (0.04 M CSA, t-BuOH/PhMe,14–18 h).
- 16.Nicolaou KC, Qian W, Bernal F, Uesaka N, Pihko PM, Hinrichs J. Angew Chem Int Ed. 2001;40:4068. doi: 10.1002/1521-3773(20011105)40:21<4068::AID-ANIE4068>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 17.While this protocol appeared to reach equilibrium in a similar time frame to Nicolaou and co-workers (4 h), the TFA/CH2Cl2 conditions led to a significantly more complex crude reaction mixture, presumably due to decomposition.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.