

Abstract
Interleukin-3 (IL-3) mediates hematopoietic cell survival and proliferation via several signaling pathways such as the Janus kinase/signal transducer and activator of transcription pathway, mitogen-activated protein kinase (MAPK) pathway, and phosphoinositide-3 kinase (PI-3K) pathway. Mammalian target of rapamycin (mTOR) is one of the downstream targets of the PI-3K pathway, and it plays an important role in hematopoiesis and immune cell function. To better elucidate how mTOR mediates proliferation signals from IL-3, we assessed the role of S6 kinase 2 (S6K2), one of the downstream targets of mTOR, in IL-3 signaling. We show that S6K2 is activated by IL-3 in the IL-3-dependent Ba/F3 cell line and that this is mediated by mTOR and its upstream activator PI-3K but not by the MAPK kinase/extracellular signal-regulated kinase pathway. S6K2 is also activated in primary mouse bone marrow-derived mast cells upon IL-3 stimulation. Expression of a rapamycin-resistant form of S6K2, T388E, in Ba/F3 cells provides a proliferation advantage in the absence or presence of rapamycin, indicating that S6K2 can potentiate IL-3-mediated mitogenic signals. In cells expressing T388E, rapamycin still reduces proliferation at all doses of rapamycin, showing that mTOR targets other than S6K2 play an important role in IL-3-dependent proliferation. Cell-cycle analysis shows that T388E-expressing Ba/F3 cells enter S phase earlier than the control cells, indicating that the proliferation advantage may be mediated by a shortened G1 phase. This is the first indication that S6K2 plays a role in IL-3-dependent cell proliferation.
Keywords: S6K2, mTOR, PI-3K, IL-3
INTRODUCTION
Hematopoietic cell development, survival, proliferation, and function are regulated by a wide range of cytokines. Interleukin (IL)-3 is one of the cytokines shown to play a role in survival and proliferation of hematopoietic progenitor cells [1]. The receptor for IL-3 (IL-3R) is composed of α and β subunits, and the β subunit is shared by the IL-5R and granulocyte macrophage-colony stimulating factor receptor and therefore called the βcommon chain (βc) [1]. Upon binding of IL-3, the cytoplasmic region of βc becomes phosphorylated on multiple sites, and this recruits and activates proteins in downstream signaling pathways such as the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway, mitogen-activated protein kinase (MAPK) pathway, and phosphoinositide-3 kinase (PI-3K) pathway [1]. Much research has focused on the mechanism of these downstream signaling pathways because of their possible therapeutic applications in a range of biomedical treatments using stem cells such as transplantation [2].
The PI-3K pathway plays a role in multiple cellular processes such as cell size regulation, proliferation, apoptosis, and cytoskeletal regulation in many different cell types [3, 4]. When PI-3K is activated by mitogenic stimuli, it phosphorylates phosphatidylinositol-4,5-bisphosphate at the D3 position to yield phosphatidylinositol-3,4,5-trisphosphate (PIP3), and proteins with pleckstrin homology domains dock at PIP3 and become activated [3, 4]. The PI-3K pathway plays a crucial upstream role in regulation of the mammalian target of rapamycin (mTOR) signaling pathway [4–6]. The significance of mTOR was first appreciated when an immunosuppressant rapamycin was shown to inhibit the function of mTOR [5]. Rapamycin blocks proliferation of cells in response to many mitogens and cytokines including IL-3, and it also inhibits hematopoiesis after myelodepression, suggesting that the mTOR pathway may play an important role in cytokine signaling [5, 7]. When PI-3K is active and generates PIP3, it leads to docking and activation of Akt, which phosphorylates tuberous sclerosis complex 2 (TSC2) [4, 6]. The TSC1/TSC2 heterodimer blocks the function of Rheb, a GTPase that activates mTOR. Phosphorylation of TSC2 by Akt lifts this inhibition of Rheb, thereby activating mTOR [4, 6], which activates several downstream signaling molecules involved in cell size regulation or translation, such as S6 kinase 1 (S6K1), S6K2, and eIF4E-binding protein 1 (4E-BP1) [5].
Although much has been elucidated about how mTOR is activated by PI-3K, the mechanism by which mTOR contributes to IL-3-mediated cell proliferation is less clear. To better understand the downstream events of the mTOR pathway in this system, we evaluated the role of S6K2, one of the downstream targets of mTOR, in IL-3-mediated cell function. S6K2 was first identified via its sequence homology to S6K1 [8–12]. S6K2 is regulated by proteins in the PI-3K, MAPK kinase (MEK), and mTOR pathways in fibroblasts [8–18], and S6K2 has been shown to phosphorylate S6 in vivo [19, 20]. However, the cellular function of S6K2 is largely unknown at this time. It is likely that S6K2 has a set of substrates in addition to S6 which plays a variety of roles in cell function. Indeed S6K1 has recently been shown to have a substrate that is unique and cannot be phosphorylated by S6K2 [21], indicating that S6K1 and S6K2 are likely to have different subsets of substrates in cells and, by extension, play different roles in cells. In addition, given the divergent noncatalytic region sequences and distinct subcellular locations of S6K1 and S6K2 [8, 13–15, 22, 23], it is likely that the two kinases have different roles in cells.
As S6K2 is a downstream signaling protein of mTOR and as mTOR plays a role in IL-3-mediated signaling, we assessed the role S6K2 may play in mTOR signaling in IL-3 function. We report that S6K2 is activated by IL-3, and this activation is mediated by mTOR and PI-3K. The MEK pathway does not seem to play a major role in S6K2 activation in Ba/F3 cells, unlike in fibroblast cell types. We show that when S6K2 is overexpressed in Ba/F3 cells, the cells have a proliferative advantage by entering S phase earlier, suggesting that the proliferation signal from the IL-3R is partially mediated by S6K2.
MATERIALS AND METHODS
Cell culture and treatment
Ba/F3 cells were cultured at 37°C in RPMI 1640 supplemented with 10% fetal calf serum (FCS), 10% WEHI-3 culture supernatant, penicillin (250 units/ml), streptomycin (250 μg/ml), and L-glutamine (292 μg/ml). The cells were IL-3-starved for 12 h prior to treatments by incubating them in RPMI supplemented with all of the above except the WEHI supernatant. Cells were then treated with or without murine IL-3 at indicated concentrations for designated times, and for experiments that required inhibitors, cells were incubated with rapamycin (20 ng/ml), LY294002 (10 μM), or U0126 (5 μM) for 20 min prior to IL-3 activation. For bone marrow-derived mast cell, mouse bone marrow cells were collected from femurs of Balb/c mice and cultured for 28 days in RPMI media containing 10% FCS, 20 ng/ml IL-3, 2 mM L-glutamine, 0.1 mM nonessential amino acids, 50 μg/ml gentamycin sulfate, and 50 μM β2-mercaptoethanol. Cells were cultured in IL-3-free media for 4 h and then incubated with or without rapamycin (20 ng/ml) for 20 min followed by activation with IL-3 (5 ng/ml) for 20 min. Mice were housed and killed according to procedures approved by the Institutional Animal Care and Use Committee at the University of California (UC) Irvine.
Retroviral expression of T388E and fluorescein-activated cell sorter (FACS)
The retroviral construct for CD4 [murine stem cell virus-internal ribosome entry site-human CD4 (MSCV-IRES-hCD4)] and CD4/T388E (MSCV-T388E-IRES-hCD4) was a gift from Dr. Craig Walsh (UC Irvine). The retroviral vector MSCV-IRES-hCD4 uses the MSCV long-terminal repeat to drive expression of the gene of interest and a tail-less human CD4 on a bicistronic message. The coding region of T388E was generated via polymerase chain reaction and inserted into MSCV-IRES-hCD4 to produce MSCV-T388E-IRES-hCD4. Retroviral stocks were prepared by transient transfection of 293T cells as described previously [24]. At 24 h post-infection, Ba/F3 cells were positively selected using human CD4 magnetic cell sorter microbeads (Miltenyi Biotec, Auburn, CA) per the manufacturer’s instruction. Expression level of CD4 for Ba/F3 cells post-sorting was assessed via staining of the cells with human CD4-allophycocyanin antibody (Caltag Laboratories, South San Francisco, CA) and analyzing via FACS (BD Biosciences, San Jose, CA).
Immunoprecipitation and immune complex kinase assay
Cells were lysed in cell lysis buffer as described previously [20]. S6K2 was immunoprecipitated from the cell lysates by incubating the lysates with sc9381 anti-S6K2 antibody (Santa Cruz Biotechnology, CA) or 07-173 anti-S6K2 antibody (Upstate Biotechnology, Lake Placid, NY), followed by protein-G plus-agarose beads. Immune complex kinase assays were carried out by incubating immunoprecipitated S6K2 with γ32P-adenosine 5′-triphosphate (ATP) and glutathione S-transferase (GST)-S6 (as exogenous substrate) at 30°C as described previously [20]. The samples were separated via sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and the gel was Coomassie-stained, dried, and exposed to autoradiographic film to visualize the degree of S6 phosphorylation, as described previously [20]. The level of S6 phosphorylation was quantified using Phosphorimager (BioRad, Hercules, CA) or Typhoon 8600 (Amersham Biosciences, Piscataway, NY).
Immunoblotting
Cells were lysed, and the lysates were separated via SDS-PAGE and immunoblotted as described previously [20]. The membrane was immunoblotted with one of the following antibodies: AP8009 anti-S6K2 antibody (Abgent, San Diego, CA), antiphospho S6 (pS235/236) antibody, antiextracellular signal-regulated kinase (Erk) antibody, antiphospho Erk antibody, antiphospho STAT5 (pT694) antibody, or anti-STAT5 antibody (all from Cell Signaling Technology, Beverly, MA). These primary antibodies were incubated with an anti-rabbit immunoglobulin secondary antibody conjugated with horseradish peroxidase. Immunoblots were visualized via enhanced chemiluminescence.
Cell proliferation assay
Cells were plated in 96-well plates at 1.2 × 104 cells/well in triplicates in the presence of varying concentrations of IL-3 (see Fig. 5A) or varying concentration of rapamycin in the presence of 2.5 ng/ml IL-3 (see Fig. 5B). After 24 h of incubation at 37°C, the wells were pulsed with 3H-thymidine at 1 μCi/well for 5 h. The cells were harvested onto filters (MachII-96, TomTec, Hamden, CT) and counted (BetaPlate, Wallac, Gaithersburg, MD).
Fig. 5.
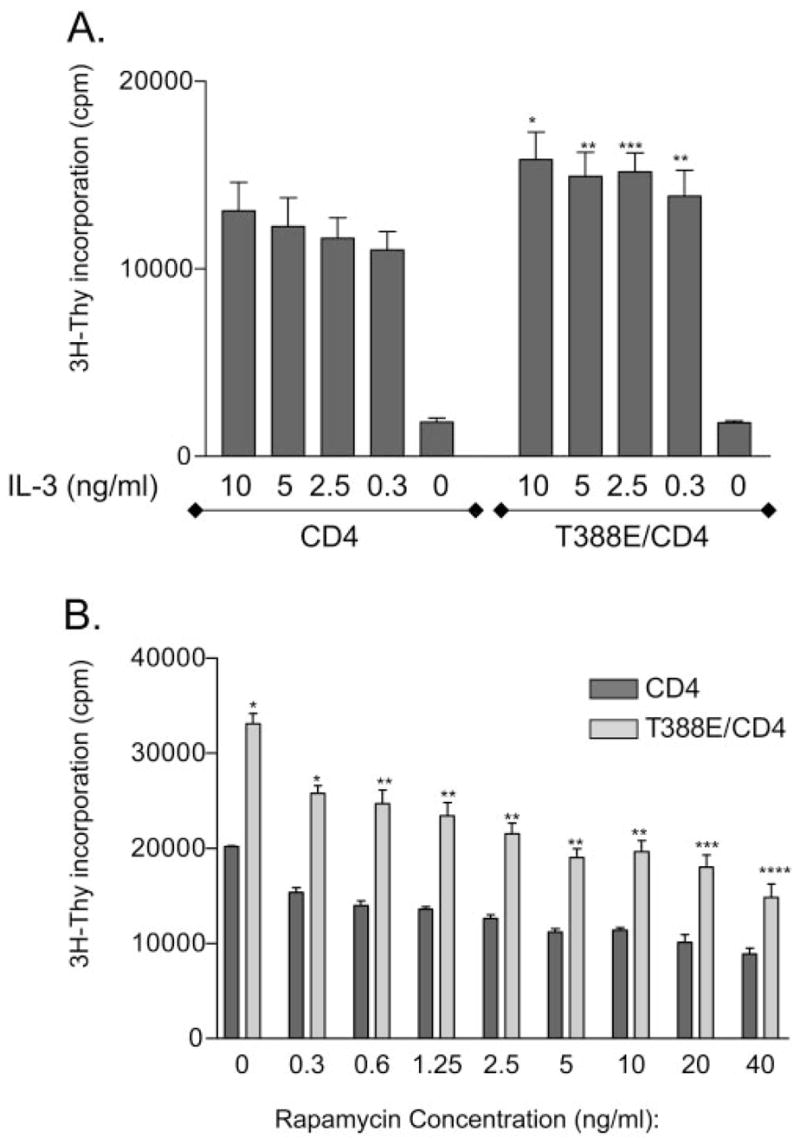
T388E S6K2 confers a proliferative advantage to Ba/F3 cells. (A) Cells expressing T388E and CD4 or CD4 only were cultured in media with varying concentrations of IL-3, and after 24 h, cell proliferation was measured by 3H-thymidine incorporation (n=6). The graph of mean counts per minute (cpm) ± SEM is shown (*, P<0.001; **, P<0.005; ***, P<0.01, compared with the equivalent IL-3 dose of CD4-only cells). (B) Ba/F3 cells expressing T388E and CD4 or CD4 only were cultured in media without IL-3 (2.5 ng/ml) in the presence of varying concentrations of rapamycin as shown. After 24 h of culture, cell proliferation was measured by 3H-thymidine incorporation (n=3). The graph of mean cpm ± SEM is shown (*, P<0.001; **, P<0.005; ***, P<0.01; ****, P<0.05, compared with the equivalent rapamycin dose of CD4-only cells).
Statistics
Data are presented as the means ± SE. Two-tailed homoscedastic Student’s t-tests were used to analyze the results.
Cell-cycle analysis
Cells were starved for IL-3 for 12 h, treated with IL-3 (5 ng/ml), and collected at indicated times. Cells were resuspended in 250 μl 1× phosphate-buffered saline (PBS)/5 mM EDTA and fixed by adding an equal volume of cold 100% ethanol. After 30 min, cells were centrifuged and resuspended in 250 μl 1× PBS/5 mM EDTA. Cells were then incubated for 30 min with RNase A. PBS (1×)/5 mM EDTA (250 μl) containing 0.1 mg/ml propidium iodide was added. Cells were analyzed via FACS, and calculation of percentage of cells in each cell-cycle phase was carried out using FlowJo software (Dean/Jett/Fox cell-cycle analysis, TreeStar, Ashland, OR).
RESULTS
S6K2 is activated by IL-3
To assess whether S6K2 plays a role in IL-3 signaling, we activated Ba/F3 cells with IL-3 and assessed the catalytic activity of endogenous S6K2. Cells that were cultured in media lacking IL-3 were stimulated with IL-3 for 20 min and lysed, and S6K2 was immunoprecipitated using two different antibodies specific to S6K2. Immune complex kinase assay was carried out by incubating immunoprecipitated S6K2 with γ32P-ATP and exogenous substrate, GST-S6. This is a widely accepted method of assessing S6K2 activity [20]. Figure 1A shows that the catalytic activity of S6K2 was increased when Ba/F3 cells were activated with IL-3 and the kinase activity assay was carried out on immunoprecipitated S6K2, and this was reproducible when two different S6K2-specific antibodies were used (Fig. 1A, i and iii). Immunoblotting for S6K2 shows that equal amounts of S6K2 were immunoprecipitated for the activity assays (Fig. 1A, ii and iv). To show that these Ba/F3 cells have a normal IL-3 signaling pathway, we immunoblotted the cell lysates with antibodies specific to STAT5 or phospho-STAT5. Our data show that STAT5 became phosphorylated upon IL-3 signaling and that the cell lysate contained the same amount of STAT5 (Fig. 1B). We next assessed the maximal activation time for S6K2 in Ba/F3 cells by stimulating the cells with IL-3 for up to 4 h and carrying out S6K2 immune complex kinase assays. Figure 1C shows that activation of S6K2 was maximal between 15 min and 2 h post-IL-3 treatment, and peak activity was at 60 min.
Fig. 1.

IL-3 activates S6K2. (A) IL-3-starved Ba/F3 cells were activated with IL-3 (5 ng/ml) for 20 min. S6K2 from the cell lysates was immunoprecipitated (IP) using sc9381 anti-S6K2 antibody (Ab) or 07-173 anti-S6K2 antibody, and immune complex kinase assays were performed by incubating S6K2 with γ32P-ATP and GST-S6 as exogenous substrate. The reaction mixture was separated via SDS-PAGE, and the degree of phosphorylation of GST-S6 was visualized via autoradiogram (i and iii). The immunoprecipitates were also separated via SDS-PAGE and immunoblotted with AP8009 anti-S6K2 antibody (Abgent) to verify that equal amounts of S6K2 were precipitated (ii and iv). The data are representative of four experiments. (B) Cell lysates from IL-3-activated Ba/F3 cells were immunoblotted with antibodies specific to phospho-STAT5 (pStat5) or STAT5. (C) IL-3-starved Ba/F3 cells were stimulated with IL-3 for the specified times, and cells were lysed. Immune complex kinase assay for S6K2 was carried out as above. S6 phosphorylation was quantified using a phosphorimager, and the mean fold activation (over quiescent S6K2 activity) ±SEM from three experiments is shown as a graph. The data are statistically significant (*, P<0.05; **, P<0.01; ***, P<0.005; ****, P<0.001, compared with 0 activation time-point). (i) The level of phosphorylation on the substrate S6 in a representative assay; (ii) Coomassie staining of S6 as a loading control.
Activation of S6K2 also occurs in primary cells and is mediated by the mTOR and PI-3K pathways but not by the MEK pathway
We assessed if activation of S6K2 by IL-3 is mediated via the mTOR pathway. Ba/F3 cells were treated with the mTOR inhibitor rapamycin prior to IL-3 stimulation, and S6K2 immune complex kinase assay was carried out as described above. Figure 2A shows that the catalytic activity of S6K2 upon IL-3 stimulation was inhibited when cells were treated with rapamycin. Immunoblotting the immunoprecipitates with an anti-S6K2 antibody showed that equal amounts of S6K2 were immunoprecipitated for the activity assay (data not shown). Our data indicate that mTOR mediates the activating signal from the IL-3R to S6K2, as in other mitogenic systems. To show that S6K2 plays a role in IL-3-mediated signaling in primary, IL-3-dependent cells, we generated primary mouse bone marrow-derived mast cells. The cells were starved from IL-3 for 4 h and then treated with IL-3 in the presence or absence of rapamycin, and immune complex kinase assay on S6K2 was carried out. Figure 2B shows that S6K2 was activated in primary bone marrow mast cells and that this activation was inhibited by rapamycin, indicating that S6K2 plays a role in IL-3-mediated mTOR signaling in primary bone marrow mast cells as well as in Ba/F3 cells. Next, we assessed whether PI-3K, an upstream activator of mTOR, is also involved in S6K2 activation by IL-3. We treated Ba/F3 cells with a PI-3K inhibitor, LY294002, prior to IL-3 stimulation and carried out an immune complex kinase assay on S6K2. Figure 3A shows that LY294002 inhibited activation of S6K2, indicating that PI-3K is required for activation of S6K2 by IL-3 in Ba/F3 cells. Immunoblotting the immunoprecipitates with an anti-S6K2 antibody showed that equal amounts of S6K2 were immunoprecipitated (data not shown).
Fig. 2.
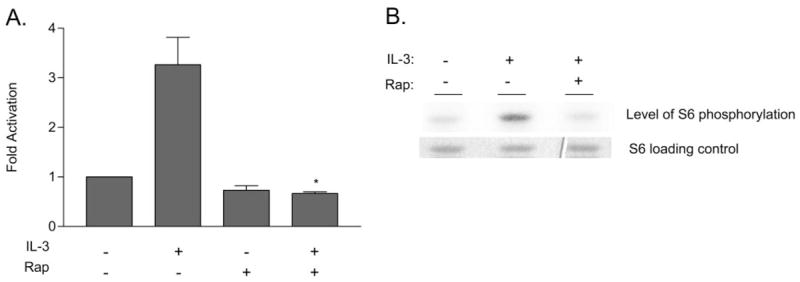
Effect of rapamycin on S6K2 activity. (A) IL-3-starved Ba/F3 cells were incubated for 20 min with rapamycin (Rap; 20 ng/ml) prior to stimulation with IL-3 (5 ng/ml) for 20 min. Cells were lysed, and immune complex kinase assays were carried out on immunoprecipitated S6K2 as described in Figure 1. The degree of phosphorylation on S6 substrate was quantified using a phosphorimager, and the fold activation over quiescent S6K2 activity from three experiments was used to generate the graph of mean activation ±SEM (*, P<0.01, compared with the activity in the presence of IL-3 alone). (B) Primary bone marrow-derived mast cells were starved for IL-3 and activated with IL-3 (5 ng/ml) for 20 min in the presence or absence of rapamycin (20 ng/ml). Cells were lysed, and immune complex kinase assay on S6K2 was carried out as described in Figure 1. The upper panel shows the degree of phosphorylation of exogenous substrate S6, and the lower panel shows Coomassie staining of S6 as a loading control.
Fig. 3.

Effect of LY294002 and U0126 on S6K2 activity. (A) IL-3-starved Ba/F3 cells were incubated with 10 μM LY294002 for 20 min prior to IL-3 stimulation (5 ng/ml, 20 min). Cells were lysed, and immune complex kinase assays were carried out on immunoprecipitated S6K2 as described in Figure 1. The degree of phosphorylation on the S6 substrate was quantified using a phosphorimager, and the fold activation over quiescent S6K2 activity from three experiments was used to generate the graph of mean activation ± SEM. The data are statistically significant (*, P<0.05, compared with the activity in the presence of IL-3 alone). (B) IL-3-starved Ba/F3 cells were incubated with 5 μM U0126 for 20 min prior to IL-3 stimulation (5 ng/ml, 20 min). Cells were lysed, and immune complex kinase assays were carried out on immunoprecipitated S6K2 as described in Figure 1. Fold activation over quiescent S6K2 activity from three experiments was used to generate the graph of mean activation ± SEM. The data are statistically significant (*, P=0.641 compared with the activity in the presence of IL-3 alone). (C) Cell lysates from B were immunoblotted with antibodies specific to ERK or phospho-Erk (pErk).
It has been shown that the MEK pathway plays a role in activating S6K2 via epidermal growth factor (EGF), fibroblast growth factor-2 (FGF-2), and serum in fibroblasts [16, 18, 20]. The MEK pathway is activated by IL-3R signals [1]. We therefore assessed whether MEK also plays a role in activation of S6K2. Ba/F3 cells were treated with the MEK inhibitor U0126 prior to IL-3 stimulation, and the catalytic activity of S6K2 was measured via immune complex kinase assays. Figure 3B shows that U0126 did not have inhibitory effects on S6K2 activation by IL-3 in Ba/F3 cells. Immunoblotting the immunoprecipitates with an anti-S6K2 antibody showed that equal amounts of S6K2 were immunoprecipitated (data not shown). When the cell lysates from the experiment were immunoblotted with antibodies specific to Erk and phospho-Erk, we observed that the MEK pathway was indeed activated in these Ba/F3 cells upon IL-3 stimulation and that U0126 inhibited phosphorylation of Erk-1 and -2 (Fig. 3B). This indicates that the MEK pathway, although activated by IL-3, does not have a significant role in activation of S6K2 by IL-3 stimulation in Ba/F3 cells.
T388E S6K2 expression increases cellular sensitivity to IL-3
The data above show that IL-3 activates S6K2 in a manner dependent on the PI-3K and mTOR pathways but not on the MEK signaling pathway in Ba/F3 cells. Next, we assessed the biological significance of S6K2 activation in IL-3 signaling. Blockade of mTOR by rapamycin normally inhibits catalytic activity of wild-type S6K2. The phosphomimetic point mutant at threonine 388 of S6K2, T388E, has full catalytic activity when activated in the presence of rapamycin in fibroblasts [14, 20]. Using retroviral transduction, we expressed T388E S6K2 in Ba/F3 cells to assess its effects on Ba/F3 cell function. Human CD4 that lacks the cytoplasmic domain was coexpressed on a bicistronic mRNA with T388E S6K2 and used as a cell-surface expression marker, and following retroviral infection, only the cells that expressed high levels of CD4 were sorted and used for experiments. When the sorted cells were stained for the CD4 surface marker and analyzed via FACS, high expresser cells were indeed obtained (Fig. 4A). We used Ba/F3 cells expressing CD4 only without T388E as control cells for all experiments below. To verify that T388E/CD4 cells overexpress T388E S6K2, we immunoblotted the cell lysates with an anti-S6K2 antibody. Figure 4B, i, shows that the T388E/CD4 cells indeed expressed higher levels of T388E over the endogenous S6K2 level seen in CD4-only cells. The antibody does not distinguish between the wild-type and the T388E S6K2 point mutant.
Fig. 4.

Retroviral expression of T388E. (A) Ba/F3 cells were infected with MSCV-IRES-hCD4 or MSCV-T388E-IRES-hCD4 retrovirus so that they would express a tail-less CD4 cell-surface marker or CD4 marker along with the S6K2 mutant T388E. Cells were sorted for high-expressing populations using magnetic sorting techniques as described in Materials and Methods. Sorted cells were stained with anti-CD4 antibody, and FACS analysis of CD4 surface expression after sorting is shown. (B) Ba/F3 cells expressing T388E and CD4 or CD4 surface marker only were activated with IL-3 (5 ng/ml) in the presence or absence of rapamycin (20 ng/ml). Cell lysates were separated via SDS-PAGE and immunoblotted with antibodies specific to S6K2 (top panel), phosphorylated S6 (pS6; middle panel), or STAT5α/ β isoforms (bottom panel).
We next set out to verify that expressing T388E confers higher S6K2 activity to the cells and that T388E behaves in a rapamycin-resistant manner in Ba/F3 cells, as was seen previously in human embryonic kidney (HEK)293 cells [20]. To this end, we assessed the phosphorylation state of the endogenous ribosomal protein S6 to see whether cellular S6 is phosphorylated at a greater degree and also to see whether this phosphorylation is rapamycin-resistant in T388E-expressing Ba/F3 cells. T388E-expressing Ba/F3 cells or control cells, expressing CD4 only, were activated with IL-3 in the presence or absence of rapamycin, and the total cell lysates were immunoblotted with an antibody for the phosphorylated form of the S6 ribosomal protein. Figure 4B, ii, left, shows that the lysates from the control Ba/F3 cells showed phosphorylation of S6 upon IL-3 stimulation, which was abrogated when rapamycin was added to cells. Cells that express T388E showed enhanced phosphorylation of S6 upon IL-3 stimulation (Fig. 4B, ii, right). Partial S6 phosphorylation was still observed when rapamycin was added to cells, indicating that T388E is active in the presence of rapamycin, although all other endogenous S6 kinase activity is inhibited by rapamycin in cells, resulting in diminished but still present S6 phosphorylation. This shows that T388E behaves as a rapamycin-resistant mutant in Ba/F3 cells, as it was shown to be in fibroblasts [20]. The enhanced S6K2 catalytic activity seen in T388E-expressing cells in the presence of IL-3 is consistent with the over-expression of T388E S6K2, as seen in Figure 4B, top panels. Immunoblotting with an antibody specific to STAT5α and -β isoforms showed that the loading levels of lysates in all lanes were equivalent (Fig. 4B, iii).
Having established that T388E behaves as a rapamycin-resistant S6K2 and that expressing T388E confers higher activity of S6K2 in cells, we next tested if expressing T388E has effects on Ba/F3 cell proliferation. We cultured Ba/F3 cells with or without T388E in decreasing doses of IL-3 for 24 h and carried out 3H-thymidine incorporation assays to measure cell proliferation (Fig. 5A). Ba/F3 cells that express T388E, exhibited greater cell proliferation at all doses of IL-3. At 0.3 ng/ml IL-3, T388E-expressing cells proliferated as well as control cells cultured in 10 ng/ml IL-3. Neither cell type proliferated appreciably when cultured in the absence of IL-3, indicating that expression of T388E is not sufficient to overcome the lack of IL-3 by itself. This suggests that S6K2 is not the sole mediator of proliferation in IL-3 signaling. Cell proliferation assay carried out by cell counts showed similar results (data not shown).
We next assessed whether the proliferative advantage that occurs when cells express T388E is inhibited when cells are cultured with rapamycin. As T388E is insensitive to the inhibitory effects of rapamycin, we hypothesized that T388E-expressing cells would still show proliferative advantage over CD4-expressing control cells in the presence of rapamycin. When the cells, which do or do not express T388E, were cultured in IL-3 with varying concentrations of rapamycin, we observed that T388E-expressing cells still had a proliferative advantage over CD4-only cells in the presence of rapamycin at all doses (Fig. 5B). Cell proliferation was nevertheless reduced by rapamycin in a dose-dependent manner in T388E cells, indicating that other molecule(s) play a role in inhibition of IL-3-mediated proliferation by rapamycin in these cells and that S6K2 is not the sole mediator of IL-3 signaling through mTOR.
Ba/F3 cells with T388E S6K2 enter S phase earlier
To elucidate the mechanism of T388E-mediated proliferation advantage, we assessed the cell-cycle profile of Ba/F3 cells that express this mutant. Ba/F3 cells, that express T388E and CD4 or CD4 marker only were starved for IL-3, and upon addition of IL-3, they were collected at different time-points and stained with propidium iodide, and their cell-cycle profiles were analyzed via FACS. Figure 6 shows that Ba/F3 cells expressing T388E entered S phase sooner, with 65% of cells in S phase at 10 h after addition of IL-3, whereas only 45% of the control cells were in S phase at 10 h. This suggests that the proliferative advantage seen in Ba/F3 cells expressing T388E is mediated in part by an earlier entry into S phase.
Fig. 6.
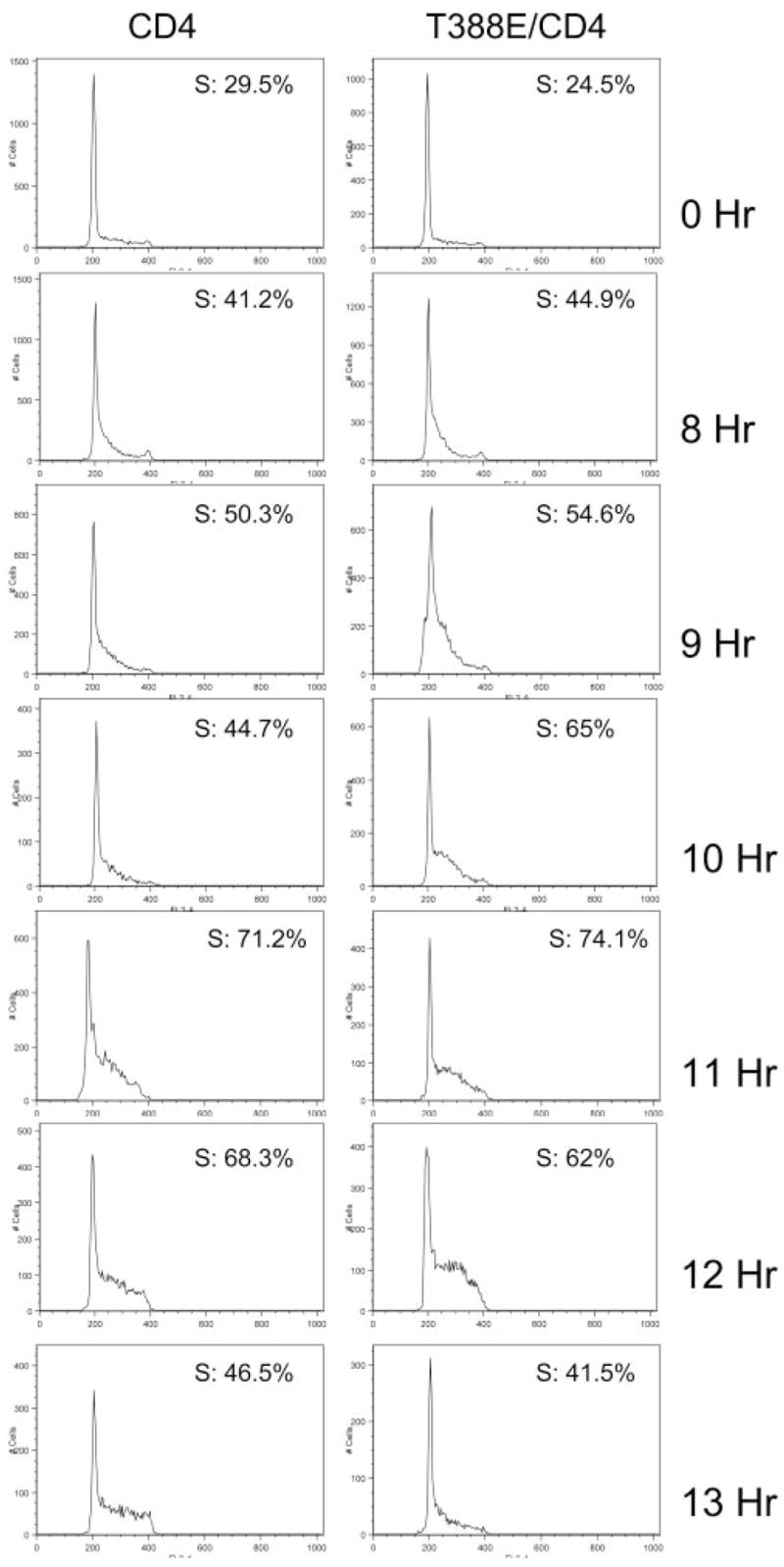
Ba/F3 cells expressing T388E S6K2 enter S phase earlier. Ba/F3 cells expressing T388E and CD4 or CD4 only were starved for IL-3, and upon addition of IL-3 (5 ng/ml), cells were collected at indicated times and stained with propidium iodide for cell-cycle analysis via FACS. The experiment was repeated with similar results.
DISCUSSION
In this report, we provide evidence that S6K2 plays a role in IL-3-mediated cell signaling. S6K2 is activated when Ba/F3 cells or primary mouse bone marrow-derived mast cells are stimulated with IL-3, and this signaling is mediated via the PI-3K and mTOR pathway but not by the MEK pathway in Ba/F3 cells. In fibroblasts, it has been shown that the MEK pathway plays a role in activating S6K2; S6K2 activity, which is induced by serum, EGF, or FGF-2, is sensitive to treatment with U0126 [16, 18, 20]. It is thought that serines 410, 417, and 423 in the C terminus of S6K2 play a role in autoinhibition by acting as a pseudosubstrate in quiescence, and phosphorylation of these sites upon mitogenic stimulation relieves autoinhibitory effects and initiates activation of S6K2 [16, 20]. When these serines are mutated to aspartic acid, S6K2 becomes insensitive to the inhibitory effects of U0126 when stimulated with EGF or serum [16, 20]. We observe in Ba/F3 cells that U0126 does not have inhibitory effects on S6K2 activity, which suggests that a mitogenic signal different from the MEK pathway contributes to phosphorylation and lift in autoinhibition of S6K2. Indeed, Martin et al. [16] have shown that U0126 inhibits activation of S6K2 by EGF but not insulin in the same cell line, HEK293, indicating that different growth factor receptors use different mechanisms even within the same cell.
It is interesting to note that T388E S6K2 has been shown to be constitutively active in HEK293 cells in the absence of serum [14, 20]. However, we did not observe constitutive catalytic activity of T388E when it was expressed in Ba/F3 cells in the absence of IL-3, although the IL-3-stimulated catalytic activity was still rapamycin-resistant. This suggests that different regulation of S6K2 catalytic activity occurs in the two cell types. One possible mechanism is that there exists in HEK293 cells a constitutively active signal to S6K2 that renders it partly ready to be activated, and with only addition of the T388E mutation, S6K2 can become fully active in the absence of serum. In Ba/F3 cells, this putative signal may not be constitutive but instead active only in the presence of IL-3.
Ba/F3 cells expressing T388E proliferate better at all doses of IL-3; yet, in the absence of IL-3, the cells fail to proliferate. This suggests that expressing T388E in itself is not sufficient to drive proliferation and/or survival of Ba/F3. This is consistent with the fact that IL-3 activates other signaling pathways known to contribute to proliferation and survival, including the JAK/STAT pathway, which are divergent from the mTOR pathway and need to be activated along with mTOR. Proliferation of T388E-expressing Ba/F3 cells was inhibited by rapamycin in a dose-dependent manner, although these cells still proliferate better than the control cells at all doses of rapamycin. The observation that expressing T388E in Ba/F3 cells is not sufficient to overcome inhibition of proliferation by rapamycin indicates that other molecule(s), which are downstream of mTOR, play an important role in IL-3-mediated cell proliferation and that T388E cannot substitute for their activity. We envision these molecule(s) to be parallel to S6K2 in the mTOR pathway. When rapamycin is added to cells, although S6K2 activity is overexpressed and may give some proliferative advantage, these other molecule(s) would still be inhibited by rapamycin, resulting in inhibition of proliferation.
How does T388E mediate a proliferative advantage in the presence of IL-3? There are two possibilities. T388E may influence IL-3-mediated signaling by partly activating one of the signaling pathways necessary for cell-cycle progression, thereby allowing easier activation and proliferation when IL-3 is added. In other words, T388E may influence the IL-3 signaling pathway in a quantitative manner. Alternatively, T388E may affect the IL-3 signaling pathway in a qualitative manner, by changing the way cells behave in response to proliferation signals. If T388E affects the proliferative response by partly activating one of the signaling pathways (i.e., in a quantitative manner), it could be predicted that at a maximal dose of IL-3, higher cell numbers in T388E-expressing cells would not necessarily be observed, as all signaling pathways would be activated maximally at that dose. However, T388E-expressing cells proliferate better at all IL-3 doses, including at a maximal dose, which suggests that T388E changes qualitatively the way Ba/F3 cells proliferate. One possibility is that T388E-expressing cells go through the cell cycle at a faster rate, resulting in greater cell numbers. Indeed, when we assessed the cell-cycle profile of cells expressing T388E, we observed that these cells enter S phase earlier. These findings suggest that S6K2 may play a role in G1-phase length regulation and/or S-phase entry. Rapamycin blocks or delays cell-cycle progression from G1 to S phase, and it has recently been shown that U2OS cells expressing a rapamycin-resistant form of mTOR can bypass a rapamycin-induced S-phase entry block when stimulated with serum [25]. This was partly mediated through S6K1 and 4E-BP1, as overexpressing eIF4E or a rapamycin-resistant form of S6K1 exhibited modest but significant acceleration in S-phase entry [25]. The study did not address whether S6K2 also plays a role in mTOR-mediated S-phase entry. Here, we present evidence that S6K2 can also play a role in S-phase entry in Ba/F3 cells proliferating with IL-3 stimulation. We do not rule out the possibility that there may exist other mechanisms by which IL-3-mediated cell proliferation is aided by expression of T388E. IL-3 in hematopoietic cells promotes not only cell proliferation but also cell survival by regulating apoptotic-signaling pathways, most notably by up-regulating expression of Bcl-x and Bcl-2 and by inhibiting BAD through phosphorylation [1]. It has also been reported that an S6K2-related kinase, S6K1, can phosphorylate BAD and may play a role in apoptosis [26]. Substrates for S6K2, other than S6, have not been found to date. Further examination about whether S6K2 plays a role in apoptosis may yield better insights into the mechanism of S6K2-mediated proliferation regulation.
It is not clear whether the T388E-mediated proliferative advantage is the result of increased phosphorylation of S6 or other substrates. S6K2 was first identified via its homology to S6K1, an in vivo kinase for S6. A S6K1/S6K2 double-knockout mouse study shows that cells derived from the double null mice have virtually no S6 phosphorylation, whereas cells that lack one or the other kinase have only partially reduced S6 phosphorylation, indicating that S6 is likely to be an in vivo substrate of S6K2 [19]. Our data show that S6 is phosphorylated in a rapamycin-resistant manner in cells expressing T388E, establishing that T388E behaves as an in vivo S6 kinase in this system as well. However, S6K2 might have other substrates relevant to cell-cycle progression. Indeed, recently an S6K1-specific substrate, SKAR, was identified, indicating that S6K1 and S6K2 may have nonoverlapping subsets of substrates as well as the shared substrate S6 [21]. Studies are currently under way for identifying the protein(s) whose phosphorylation is affected when S6K2 is diminished in or near S phase, with the purpose of elucidating the mechanism of S-phase entry regulation by S6K2.
In summary, we have shown that IL-3-mediated cell proliferation is potentiated by S6K2 and that S6K2 may play a role in regulating G1-phase duration and/or S-phase entry. Finding a full spectrum of S6K2 substrates in G1 phase may yield better insights to the mechanism of S6K2-mediated G1/S-phase regulation.
Acknowledgments
This work was supported by National Institutes of Health, 1 S06 GM63119-01 (MBRS-SCORE), to K.K.L-F., AI50831 to D.A.F., and T32-CA9054 to M.G.K.R.C. and L.H. contributed equally to this work. We thank Dr. Craig Walsh for the retroviral constructs and Travis Moore and Dr. Jonathan Deane for technical help.
References
- 1.Martinez-Moczygemba M, Huston DP. Biology of common β receptor-signaling cytokines: IL-3, IL-5, and GM-CSF. J Allergy Clin Immunol. 2003;112:653–665. doi: 10.1016/S0091. [DOI] [PubMed] [Google Scholar]
- 2.Ivanovic Z. Interleukin-3 and ex vivo maintenance of hematopoietic stem cells: facts and controversies. Eur Cytokine Netw. 2004;15:6–13. [PubMed] [Google Scholar]
- 3.Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC, Woscholski R, Parker PJ, Waterfield MD. Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem. 2001;70:535–602. doi: 10.1146/annurev.biochem.70.1.535. [DOI] [PubMed] [Google Scholar]
- 4.Richardson CJ, Schalm SS, Blenis J. PI3-kinase and TOR: PIKTORing cell growth. Semin Cell Dev Biol. 2004;15:147–159. doi: 10.1016/j.semcdb.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 5.Fingar DC, Blenis J. Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene. 2004;23:3151–3171. doi: 10.1038/sj.onc.1207542. [DOI] [PubMed] [Google Scholar]
- 6.Krymskaya VP. Tumor suppressors hamartin and tuberin: intra-cellular signaling. Cell Signal. 2003;15:729–739. doi: 10.1016/s0898-6568(03)00040-8. [DOI] [PubMed] [Google Scholar]
- 7.Quesniaux VF, Wehrli S, Steiner C, Joergensen J, Schuurman HJ, Herrman P, Schreier MH, Schuler W. The immunosuppressant rapamycin blocks in vitro responses to hematopoietic cytokines and inhibits recovering but not steady-state hematopoiesis in vivo. Blood. 1994;84:1543–1552. [PubMed] [Google Scholar]
- 8.Koh H, Jee K, Lee B, Kim J, Kim D, Yun YH, Kim JW, Choi HS, Chung J. Cloning and characterization of a nuclear S6 kinase, S6 kinase-related kinase (SRK); a novel nuclear target of Akt. Oncogene. 1999;18:5115–5119. doi: 10.1038/sj.onc.1202895. [DOI] [PubMed] [Google Scholar]
- 9.Lee-Fruman KK, Kuo CJ, Lippincott J, Terada N, Blenis J. Characterization of S6K2, a novel kinase homologous to S6K1. Oncogene. 1999;18:5108–5114. doi: 10.1038/sj.onc.1202894. [DOI] [PubMed] [Google Scholar]
- 10.Saitoh M, ten Dijke P, Miyazono K, Ichijo H. Cloning and characterization of p70(S6Kβ) defines a novel family of p70 S6 kinases. Biochem Biophys Res Commun. 1998;253:470–476. doi: 10.1006/bbrc.1998.9784. [DOI] [PubMed] [Google Scholar]
- 11.Shima H, Pende M, Chen Y, Fumagalli S, Thomas G, Kozma SC. Disruption of the p70(s6k)/p85(s6k) gene reveals a small mouse phenotype and a new functional S6 kinase. EMBO J. 1998;17:6649–6659. doi: 10.1093/emboj/17.22.6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gout I, Minami T, Hara K, Tsujishita Y, Filonenko V, Waterfield MD, Yonezawa K. Molecular cloning and characterization of a novel p70 S6 kinase, p70 S6 kinase β containing a proline-rich region. J Biol Chem. 1998;273:30061–30064. doi: 10.1074/jbc.273.46.30061. [DOI] [PubMed] [Google Scholar]
- 13.Park IH, Bachmann R, Shirazi H, Chen J. Regulation of ribosomal S6 kinase 2 by mammalian target of rapamycin. J Biol Chem. 2002;277:31423–31429. doi: 10.1074/jbc.M204080200. [DOI] [PubMed] [Google Scholar]
- 14.Minami T, Hara K, Oshiro N, Ueoku S, Yoshino K, Tokunaga C, Shirai Y, Saito N, Gout I, Yonezawa K. Distinct regulatory mechanism for p70 S6 kinase β from that for p70 S6 kinaseα. Genes Cells. 2001;6:1003–1015. doi: 10.1046/j.1365-2443.2001.00479.x. [DOI] [PubMed] [Google Scholar]
- 15.Valovka T, Verdier F, Cramer R, Zhyvoloup A, Fenton T, Rebholz H, Wang ML, Gzhegotsky M, Lutsyk A, Matsuka G, Filonenko V, Wang L, Proud CG, Parker PJ, Gout IT. Protein kinase C phosphorylates ribosomal protein S6 kinase βII and regulates its subcellular localization. Mol Cell Biol. 2003;23:852–863. doi: 10.1128/MCB.23.3.852-863.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martin KA, Schalm SS, Romanelli A, Keon KL, Blenis J. Ribosomal S6 kinase 2 inhibition by a potent C-terminal repressor domain is relieved by mitogen-activated protein-extracellular signal-regulated kinase kinase-regulated phosphorylation. J Biol Chem. 2001;276:7892–7898. doi: 10.1074/jbc.M009972200. [DOI] [PubMed] [Google Scholar]
- 17.Martin KA, Schalm SS, Richardson C, Romanelli A, Keon KL, Blenis J. Regulation of ribosomal S6 kinase 2 by effectors of the phosphoinositide 3-kinase pathway. J Biol Chem. 2001;276:7884–7891. doi: 10.1074/jbc.M006969200. [DOI] [PubMed] [Google Scholar]
- 18.Pardo OE, Arcaro A, Salerno G, Tetley TD, Valovka T, Gout I, Seckl MJ. Novel cross talk between MEK and S6K2 in FGF-2 induced proliferation of SCLC cells. Oncogene. 2001;20:7658–7667. doi: 10.1038/sj.onc.1204994. [DOI] [PubMed] [Google Scholar]
- 19.Pende M, Um SH, Mieulet V, Sticker M, Goss VL, Mestan J, Mueller M, Fumagalli S, Kozma SC, Thomas G. S6K1(−/−)/S6K2(−/−) mice exhibit perinatal lethality and rapamycin-sensitive 5′-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol Cell Biol. 2004;24:3112–3124. doi: 10.1128/MCB.24.8.3112-3124.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Phin S, Kupferwasser D, Lam J, Lee-Fruman KK. Mutational analysis of ribosomal S6 kinase 2 shows differential regulation of its kinase activity from that of ribosomal S6 kinase 1. Biochem J. 2003;373:583–591. doi: 10.1042/BJ20021794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Richardson CJ, Broenstrup M, Fingar DC, Julich K, Ballif BA, Gygi S, Blenis J. SKAR is a specific target of S6 kinase 1 in cell growth control. Curr Biol. 2004;14:1540–1549. doi: 10.1016/j.cub.2004.08.061. [DOI] [PubMed] [Google Scholar]
- 22.Reinhard C, Fernandez A, Lamb NJ, Thomas G. Nuclear localization of p85s6k: functional requirement for entry into S phase. EMBO J. 1994;13:1557–1565. doi: 10.1002/j.1460-2075.1994.tb06418.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coffer PJ, Woodgett JR. Differential subcellular localization of two isoforms of p70 S6 protein kinase. Biochem Biophys Res Commun. 1994;198:780–786. doi: 10.1006/bbrc.1994.1112. [DOI] [PubMed] [Google Scholar]
- 24.Kharas MG, Deane JA, Wong S, O’Bosky KR, Rosenberg N, Witte ON, Fruman DA. Phosphoinositide 3-kinase signaling is essential for ABL oncogene-mediated transformation of B-lineage cells. Blood. 2004;103:4268–4275. doi: 10.1182/blood-2003-07-2193. [DOI] [PubMed] [Google Scholar]
- 25.Fingar DC, Richardson CJ, Tee AR, Cheatham L, Tsou C, Blenis J. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol Cell Biol. 2004;24:200–216. doi: 10.1128/MCB.24.1.200-216.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harada H, Andersen JS, Mann M, Terada N, Korsmeyer SJ. p70S6 kinase signals cell survival as well as growth, inactivating the pro-apoptotic molecule BAD. Proc Natl Acad Sci USA. 2001;98:9666–9670. doi: 10.1073/pnas.171301998. [DOI] [PMC free article] [PubMed] [Google Scholar]