

Abstract
The therapeutic, antibiotic potential of antimicrobial peptides can be prohibitively diminished because of the cytotoxicity and hemolytic profiles they exhibit. Quantifying and predicting antimicrobial peptide toxicity against host cells is thus an important goal of AMP related research. In this work, we present quantitative structure activity relationships for toxicity of protegrin-like antimicrobial peptides against human cells (epithelial and red blood cells) based on physicochemical properties, such as interaction energies and radius of gyration, calculated from molecular dynamics simulations of the peptides in aqueous solvent. The hypothesis is that physicochemical properties of peptides, as manifest by their structure and interactions in a solvent and as captured by atomistic simulations, are responsible for their toxicity against human cells. Protegrins are β-hairpin peptides with high activity against a wide variety of microbial species, but in their native state are toxic to human cells. Sixty peptides with experimentally determined toxicities were used to develop the models. We test the resulting relationships to determine their ability to predict the toxicity of several protegrin-like peptides. The developed QSARs provide insight into the mechanism of cytotoxic action of antimicrobial peptides. In a subsequent blind test, the QSAR correctly ranked four of five protegrin analogues newly synthesized and tested for toxicity.
Keywords: Antimicrobial peptides, Toxicity prediction, Protegrin, Molecular dynamics simulation, Quantitative structure activity relationship (QSAR)
1. Introduction
In recent years there has been an alarming increase in the number of cases of antibiotic resistant bacterial infections, in particular the emergence of multi-resistant tuberculosis and vancomycin-resistant Enterococcus strains [46]. For over 30 years at the end of the last century no new antibiotics were introduced, and in the last decade there have only been four structurally novel classes of antibiotics entering the clinic [17]. Since their discovery nearly twenty years ago, there have been great efforts put forth to develop new antibiotics based on antimicrobial peptides (AMPs). AMPs are found in nature as part of the innate immune response of most organisms and can kill Gram-negative and Gram-positive bacteria, fungi, and enveloped viruses [48]. In general, they are cationic due to the presence of 3 or more arginine or lysine residues; they are small, generally less than 50 residues; and they tend to contain approximately fifty percent hydrophobic amino acid content that is generally segregated from the positive amino acids in the folded structure [17]. Some of the more well-studied peptides include magainin [48], human LL-37 [13], and human β-defensin-3 [11], among many others. Unfortunately AMPs are often toxic to healthy host cells at the concentrations needed to fight infections. The goal of research over the past decade has been on designing new peptides through rational modifications of existing peptides to reduce host cell toxicity and also to improve selectivity for the desired microbe [8,10,22,44].
There are currently two main methods for designing new AMPs. One is primary sequence modification of existing peptides. This method has produced few successful mutants [17], mainly due to the difficulty in predicting the response of a peptide to single mutations. For example, one peptide may have increased activity with a mutation to increase its charge, while another peptide may exhibit decreased activity for the same increase in charge. The other method is random combinatorial peptide libraries or mutagenesis of DNA sequences encoding peptides [15,20,21]. These techniques suffer from the technical and financial constraints of synthesizing an extremely large number of peptides and testing them for activity and toxicity. The number of possible 18-residue peptides is on the order of 1023. Though this number can be reduced by placing constraints on certain positions, it is still time and resource consuming to design peptides using this method.
Over the past decade, efforts have been made to develop computational simulation methods to design new AMPs. We have used molecular dynamics simulations to observe the molecular level interactions between AMPs and membrane mimics [4,25–30,33–35] and have been able to relate these interactions to the known toxicity of the peptides studied. We hypothesize that the activity of any drug is a function of its physicochemical properties and that we can predict the toxicity of new peptides by measuring these properties from simulations. In earlier work we developed a QSAR for the activity of protegrin molecules based on peptide properties calculated in vacuo [24,40]. In this work we have developed a QSAR for toxicity based on atomistic simulations of protegrins in an aqueous solvent. We calculate physicochemical properties of the peptide such as solvent exposed surface area, electrostatic interactions between peptide and solvent and radius of gyration. In principle simulations of peptides with lipid bilayers mimicking the mammalian cell membranes should be conducted and the free energy of the interaction would be used for predicting the toxicity of the peptide. This approach is computationally prohibitive for many peptides and thus not amenable to high throughput screening. Simulations in water require significantly less computing time than AMPs in lipid membrane mimic environments, yet, as shown in this work, provide meaningful results.
Because most antimicrobial peptides are active at some level, difficulties in determining how mutations affect toxicity hinder the design of new sequences. What we will present in the following is evidence that molecular dynamics simulations can be used to predict the toxicity of peptides. We believe that if they are used as a part of a sequence screening process, will reduce the number of peptides that must be synthesized and screened.
In what follows we focus on protegrin-like peptides. Protegrins are a family of five potent cationic antimicrobial peptides originally purified from porcine leukocytes [31,42]. Protegrin-1 (PG-1) [H3N-R G G R L C Y C R R R F C V C V G R-CONH2] has a β-hairpin structure that is stabilized by disulfide bonds linking Cys-6 and Cys-15, and Cys-8 and Cys-13. PG-1 has remarkable activity against Gram-positive and Gram-negative bacteria and certain fungi [12,31]; however, PG-1 also damages human cells at concentrations necessary for therapeutic uses [47]. We used sixty protegrin analogues for which toxicity profiles have been measured in developing the model. To test the predictive ability of our methods, a set of experiments was conducted recently with five new analogues. Though toxicity data against epithelial and lymphocyte cells are available for PG-5 [6], we have still included it in the rankings as a benchmark.
2. Methods
2.1. Simulation methods
Our previous work on protegrins has involved simulations of a set of 60 protegrin analogue sequences with known activity and toxicity data (Table 1). Hemolysis was measured as the percent of red blood cells killed by an 80 μg/mL concentration of the AMP. This was converted to a scale of 0–5 as shown in Table 2. Cytotoxicity was measured as the concentration of the peptide (in μg/mL) required to kill fifty percent of a sample of cervical epithelial cells. This was also converted to a scale from 0 to 5 (Table 2).
Table 1.
Protegrin sequences used in building the models
| Name | Sequence | Hemolysis | Cytotoxicity |
|---|---|---|---|
| PC1 | RGGRLCYCRRRFCVCVGR | 5 | 4 |
| PC3 | RGGGLCYCRRRFCVCVGR | 5 | 4 |
| PC4 | RGGRLCYCRGWICFCVGR | 5 | |
| PC5 | RGGRLCYCRPRFCVCVGR | 5 | 3 |
| PC9 | RGGRLAYCRRRFCVAVGR | 0 | 2 |
| PC10 | RGGRLCYARRRFAVCVGR | 0 | 4 |
| PC11 | LCYCRRRFCVCVGR | 5 | 5 |
| PC12 | RCYCRRRFCVCVGR | 3 | 4 |
| PC13 | RGGRLCYCRRRFCVCV | 5 | 5 |
| PC14 | RGGRLCYCRRRFCICV | 5 | 4 |
| PC15 | RGGRLCYCRRRFCVCR | 1 | 3 |
| PC16 | RCYCRRRFCVCR | 0 | 2 |
| PC17 | LCYCRRRFCVCV | 5 | 3 |
| PC18 | LCYARRRFAVCV | 1 | 2 |
| PC19 | RCYARRRFAVCR | 0 | 0 |
| PC20 | LAYCRRRFCVAV | 0 | 1 |
| PC21 | RAYCRRRFCVAR | 0 | 0.5 |
| PC37 | CYCRRRFCVCVGR | 1 | 3 |
| PC45 | RGGRLCYCRRRFCVC | 1 | 3 |
| PC64 | LCYTRRRFTVCV | 1 | 3 |
| PC64a | LTYCRRRFCVTV | 0 | 2 |
| PC65 | LCYTRPRFTVCV | 0 | 0.5 |
| PC69 | LCYTFRPRFVCV | 0 | 0.5 |
| PC70 | LCYTFRGRFVCV | 0 | 1 |
| PC71 | CYCRRRFCVCV | 0 | 3 |
| PC72 | LCYCRRRFCVC | 2 | 3 |
| PC73 | CYCRRRFCVC | 0 | 1 |
| PC74 | CYCFRRFCVC | 0 | 2 |
| PC77 | LCYCRRRRCVCV | 1 | 3 |
| PC78 | LCYCFRRRCVCV | 3 | 0.5 |
| PC79 | LCYCRFRRCVCV | 1 | 0.5 |
| PC80 | LCYCRRFRCVCV | 3 | 0.5 |
| PC91 | YCYCRRRFCVCVGR | 4 | 4 |
| PC92 | TCYCRRRFCVCVGR | 3 | 3 |
| PC93 | ACYCRRRFCVCVGR | 4 | 4 |
| PC94 | VCYCRRRFCVCVGR | 5 | 4 |
| PC95 | ICYCRRRFCVCVGR | 5 | 4 |
| PC96 | FCYCRRRFCVCVGR | 5 | 4 |
| PC97 | WCYCRRRFCVCVGR | 5 | 4 |
| PC98 | ECYCRRRFCVCVGR | 1 | 3 |
| PC100 | RGGRLCYCRRRFCVCY | 5 | 4 |
| PC101 | RGGRLCYCRRRFCVCT | 3 | 2 |
| PC102 | RGGRLCYCRRRFCVCA | 4 | 3 |
| PC103 | RGGRLCYCRRRFCVCL | 5 | 4 |
| PC104 | RGGRLCYCRRRFCVCI | 5 | 4 |
| PC105 | RGGRLCYCRRRFCVCF | 5 | 3 |
| PC106 | RGGRLCYCRRRFCVCW | 5 | 3 |
| PC107 | RGGRLCYCRRRFCVCE | 0 | 2 |
| PC109 | RLCYTRGRFTVCV | 3 | |
| PC110 | LCYTRGRFTVCVR | 0 | |
| PC111 | RLCYTRGRFTVCVR | 3 | |
| PC112 | LCYCHHHFCVCV | 2 | 2 |
| PC113 | LCYTHHHFTVCV | 0 | 1 |
| PC146 | LCYCRRRFCYCV | 2 | 2 |
| PC147 | LCYCRRRFCFCV | 5 | 2 |
| PC148 | LCYCRRRFCTCV | 0 | 2 |
| PC149 | LCYCRRRFCGCV | 0 | 2 |
| PC150 | LCYCRRRFCWCV | 4 | 2 |
These sequences were published along with with experimental MIC values against a variety of microbial species in ref. [40]. For a few of the sequences only hemolysis or cytotoxicity data was available.
Table 2.
Scale values for cytotoxicity and hemolysis
| Scale | Cytotoxicity | Hemolysis |
|---|---|---|
| EC50 (μg/mL) | %RBC killed | |
| 0 | >400 | 0–3 |
| 1 | 200–400 | 3–6 |
| 2 | 100–200 | 6–12 |
| 3 | 50–100 | 12–25 |
| 4 | 25–50 | 25–50 |
| 5 | <25 | >50 |
Using the structures from Ostberg and Kaznessis [40], CHARMM molecular dynamics simulations were carried out for a single peptide in a rhombic dodecahedron (RHDO) crystal with side length of 56.1 Å filled with 3869 TIP3P water molecules and a NaCl concentration of 150 mM. Appropriate counterions were added to each system to neutralize the simulation box. The system was minimized with the peptide and bulk water initially kept under weak harmonic constraints with spring constants of 15 and 5 kcal/mol Å, respectively. The constraints on the bulk water were gradually reduced during 20,000 steps of steepest descent minimization. The entire system was then minimized for 20,000 additional steps, with 15 kcal/mol Å constraints on the peptide. After the minimizations, the system was gradually heated to 303.15 K. This temperature was chosen based on previous simulations run at the same temperature, though the peptides were tested for toxicity at body temperature. The 7° difference is not expected to be substantial enough to influence the results. After 500 ps of equilibration, the entire assembly was subjected to NPT dynamics. Over the first 0.5 ns, the harmonic constraints on the peptide were gradually removed. The constant pressure–temperature module of CHARMM was used for the simulations with a leap-frog integrator (2 fs time step). A Hoover temperature control was used to set the temperature at 303.15 K [19]. All components of the piston mass array were set to 500 amu for the extended system pressure algorithm [2,45]. Particle mesh Ewald (PME) summation [9] without truncation and a real space Gaussian width of 0.25 Å−1, a β-spline order of 4, and a FFT grid of about one point per Angstrom was employed. All simulations were carried out using CHARMM version c30b2 with the param22 parameter set [5,36]. A total of 2 ns of simulation after equilibration was conducted for each peptide. This is adequate time since the structure of the peptides is not expected to change significantly because of the double cysteine bonds that limit the flexibility of the β-hairpin. A snapshot of the system is shown in Fig. 1.
Fig. 1.
Sample snapshot of PG-5 from the simulation. The peptide is shown in blue ribbons, with sodium and chloride ions in red and yellow, respectively, and water in cyan. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
Though the simulations were short, the systems were at steady state, as evidenced by the total energy of the system (Fig. 2A) and the RMSD of the backbones of the peptides (Fig. 2B). The RMS increases at 0.5 ns after the constraints are removed from the peptides.
Fig. 2.
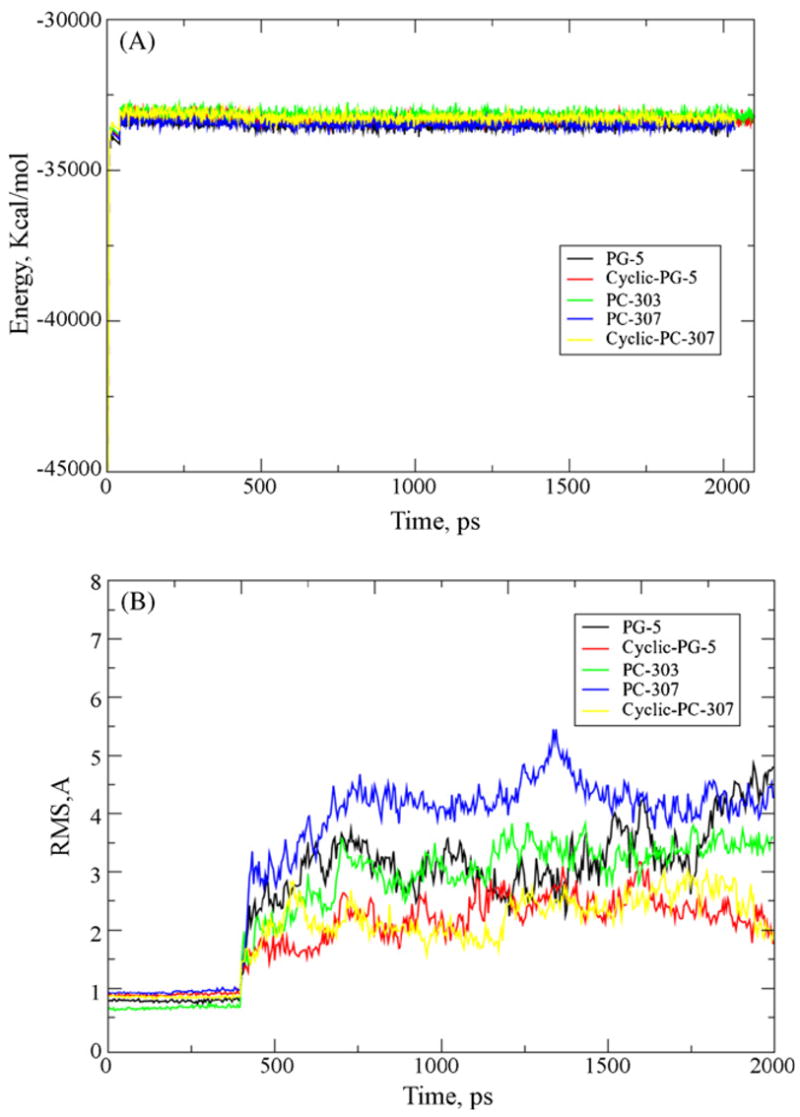
(A) Total energy of the system as a function of time for each simulation of the test peptides. (B) RMS of the backbone of each peptide. The increase in RMSD for each peptide at 500 ps is due to the removal of constraints on the peptides at this time. The RMSD for each peptide does level off as the simulation progresses.
The sequences used in the blind test of the model are given in Table 3. Simulations for these peptides were performed in the same manner as described above.
Table 3.
Sequences of the five test peptides
| Peptide | Sequence |
|---|---|
| PG-5 | RGGRLCYCRPRFCVCVGR-CONH2 |
| Cyclic-PG-5 | RGGRLCYCRPRFCVCVGR |
| PC-303 | RWRLCYCRPRFCVCV-CONH2 |
| PC-307 | RGWRACYCRPRFCACVGR-CONH2 |
| Cyclic-PC-307 | RGWRACYCRPRFCACVGR |
PG-5, PC-303, and PC-307 are β-hairpin peptides with amidated C-terminus and are not cyclic. Cyclic-PG-5 and cyclic-PC-307 are cyclic peptides joined at residues Arg-1 and Arg-18, and therefore do not have an amidated C-terminus. The structures for these sequences were determined through homology modeling.
2.2. QSAR model building
In the development of these QSAR models, a total of 29 properties were initially considered, some of which were determined prior to the simulation, such as sequence length. The properties and their descriptions are listed in Table 4 (Fig. 3).
Table 4.
Properties used in QSAR development
| Property | Description |
|---|---|
| BRIDGES | Number of disulfide bridges |
| EXAREA | Exposed area of the peptide |
| CHARGE | Net charge on peptide |
| DIPOLE | Dipole moment |
| ECCEN | Eccentricity |
| ELE-PEP | Internal electrostatic energy of the peptide |
| ELECE-PEP-WAT | Electrostatic energy between the water and the peptide |
| HB-PEP-PEP-ENERGY | Energy of H-bonds within the peptide |
| HB-PEP-PEP | Number of H-bonds within the peptide |
| HB-PEP-WATER-ENERGY | Energy of H-bonds between the peptide and water |
| HB-PEP-WATER | Number of H-bonds between the peptide and water |
| HYDROPHOBICTY | Total hydrophobicity of the peptide from individual amino acid hydrophobicities |
| LEN | Average length of folded peptide |
| MOM-INERTIA | Moment of inertia |
| NEG-SASA | Negatively charged solvent exposed surface area |
| NUM-ACCEPTORS | Number of peptide’s H-bond acceptors |
| NUM-DONORS | Number of peptide’s H-bond donors |
| PEX | Percent of peptide exposed to aqueous phase |
| PLANE | Deviation of the folded peptide from a planar conformation |
| POS-SASA | Positively charged solvent exposed surface area |
| RGYR | Radius of gyration |
| RMS | RMS deviation of peptide’s structure during the simulation |
| SASA | Total solvent exposed surface area |
| SEQ-LEN | Number of amino acids |
| TOT-AREA | Total peptide area |
| TOT-E-PEP | Total internal energy of the peptide |
| TOT-E-PEP-WAT | Total energy due to peptide–water interactions |
| VDWE-PEP | Van der Waals interaction energy within the peptide |
| VDWE-PEP-WAT | Van der Waals interaction energy between peptide and water |
Properties were calculated based on simulations of the peptides in water.
Fig. 3.

Surface view of PG-5 (A), PC-72 (B) and PC-100 (C). Positively charged residues are shown in blue, polar in yellow and non-polar in white. These surface areas were included in the QSAR model building. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
Statistical analyses were performed using the JMP [23] and SAS [1] software packages. Preliminary analysis revealed significant multicollinearity in the data set, which was expected because many of the physical properties are related to each other. A correlation matrix revealed a number of pairwise correlations with coefficients greater than 0.9. Several variables, such as sasa-all, which was correlated with nearly half of the other properties, with coefficients greater than 0.85, were removed from the data set. To further reduce the multicollinearity, several of the variables were redefined (Table 5).
Table 5.
Combinations of properties that were found to be useful in building the QSAR
| Name | Property combination |
|---|---|
| EPF | ele-pep/tot-e-pep |
| EPWF | ele-pep-wat/tot-e-pep-wat |
| VPF | vdwe-pep/tot-e-pep |
| VPWF | vdwe-pep-wat/tot-e-pep-wat |
| MHYD | hydrophob/seq-length |
| MCHG | charge/seq-len |
| MNA | num-acceptors/seq-len |
| MND | num-donors/seq-len |
| NSASA | neg-sasa/exarea |
| PSASA | pos-sasa/exarea |
Variable selection was performed using the all-possible-models method in SAS. RSQUARE all-possible-models method efficiently performs all possible subset regressions and displays the models in decreasing order of R2 magnitude within each subset size. The all-possible models method is preferred to stepwise methods, as stepwise methods are not guaranteed to find the models with the highest R2 values. We focused on the best regression equations having five or fewer variables. After taking the top models (based on high R2 values) predicted by SAS, we entered the models in JMP, and determined outlier values using studentized residuals and eliminated these data points from the model. Final statistical analysis was performed using JMP on the reduced set of data.
The best model for hemolysis is (the terms in these models are described in Tables 4 and 5):
n = 50, R2 = 0.8222, F = 50.8949.
The p-values corresponding to the t-ratios for each property were all less than 0.0001. R2 is the squared correlation coefficient, and F is the ratio of the model square divided by the error mean square.
Removing a single term from this model still provides a good fit with the data:
n = 48, R2 = 0.7299, F = 40.5354.
The p-values corresponding to the t-ratios for each property were all less than 0.0001.
In general, hemolysis was more easily described by the properties in the data set. For cytotoxicity, the best model required five properties:
n = 49, R2 = 0.7322, F = 22.9717.
The p-values corresponding to the t-ratios for each property were all 0.0001 or less.
Plots of experimental versus predicted values for each model are shown in Fig. 4.
Fig. 4.
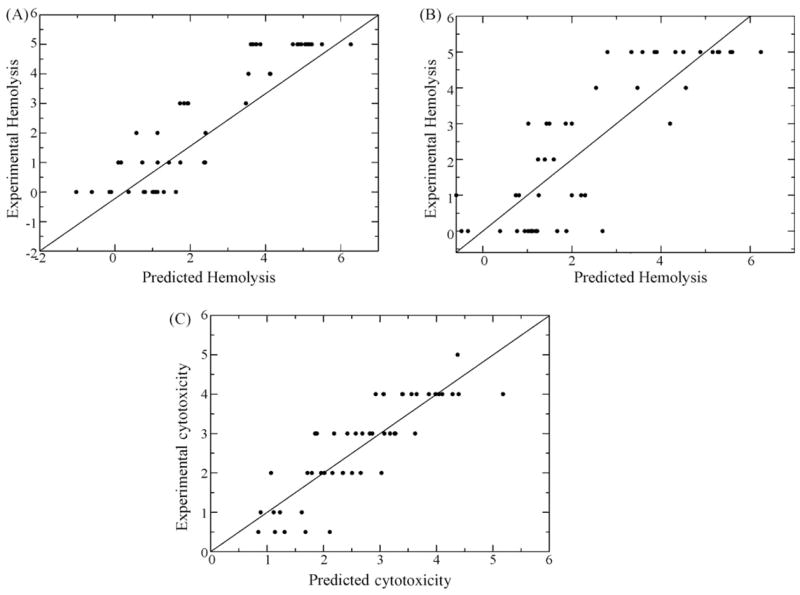
Plots of experimental values for Hemolysis 1 (A), Hemolysis 2 (B) and Cytotoxicity (C) for vs. predicted values obtained by using the QSAR models.
2.3. Peptide synthesis
PG-1 analogs were synthesized on an ABI 431A synthesizer using Fastmoc chemistry [14] with an ABI 431A peptide synthesizer, oxidized and purified by reverse phase high performance chromatography described previously [35]. Cyclic protegrin analogs were assembled from linear peptides using a disulfide oxidation strategy followed by coupling the N-terminal and C-terminal ends to form an amide linkage similar to that used for cyclic retrocyclin peptides [7].
2.4. Experimental testing of toxicity
Hemolysis and cytotoxicity data for the set of 60 peptides was previously published in ref. [40]. Each of the test peptides was tested with 8 different concentrations (5, 12.5, 25, 50, 100, 150, 200, 250 μg/mL) in triplicates. Experiments were carried out at 37°C. Results show the concentration, in μg/mL, that caused 50% cytotoxicity in 24 h. The lower the IC50, the more cytotoxic the peptide. For H9 human lymphocyte cells, RPMI-1640 media + 10% FCS + antibiotic was used. In each sample well (96-well plate), 100 μl of 105 cells per milliliter was incubated overnight in the incubator before the addition of peptide. For ME180 human cervical epithelial cells, 5A McCoy’s media + 10% FCS + antibiotic was used. In each sample well (96-well plate), 100 μl of 5 × 104 cells per milliliter was incubated overnight in the incubator before the addition of peptide. H9 and ME180 cell experiments were done in media that contained 10% serum, so peptides that bind extensively to serum components appear less cytotoxic than they do in media with less serum. The values obtained from these experiments were used to compare the ranking of the peptides in order of toxicity determined by the QSAR models.
3. Results
3.1. QSAR ranking
To test the predictive ability of the models, the five test peptides were simulated in water and properties were calculated. These were then applied to the models to generate values for hemolysis and cytotoxicity. Results are given in Table 6.
Table 6.
Properties for the five test peptides used in the QSAR models and the results for hemolysis and toxicity
| Peptide | PG-5 | Cyclic-PG-5 | PC-303 | PC-307 | Cyclic-PC-307 |
|---|---|---|---|---|---|
| EPWF | 0.811 | 0.887 | 0.907 | 0.912 | 0.873 |
| VPWF | 0.081 | 0.105 | 0.084 | 0.088 | 0.100 |
| MNA | 1.056 | 1.056 | 1.067 | 1.056 | 1.056 |
| LEN | 39.786 | 28.230 | 30.528 | 27.977 | 31.031 |
| RGYR | 10.487 | 9.148 | 8.267 | 10.114 | 9.711 |
| PEX | 69.058 | 74.316 | 74.575 | 70.481 | 70.820 |
| PSASA | 0.345 | 0.350 | 0.371 | 0.397 | 0.356 |
| HBPW | −9.424 | −8.599 | −9.424 | −11.057 | −9.314 |
| Hemolysis | 7.9 | 5.2 | 3.5 | 3.0 | 5.7 |
| Hemolysis 2 | 7.4 | 4.8 | 3.8 | 3.1 | 5.4 |
| Cytotoxicity | 8.7 | 8.0 | 5.8 | 7.1 | 7.4 |
Some of the peptides have calculated properties that are higher than the ones from which the model was built (i.e., hemolysis/cytotoxicity above 5), but the QSARs still provide values that we can use to rank the peptides relative to each other. We see that in order of decreasing hemolytic activity, the peptides are ranked: PC-5, cyclic-PC-307, cyclic-PC-5, PC-303, and PC-307. In terms of decreasing cytotoxicity the peptides are ranked: PC-5, cyclic-PC-5, cyclic-PC-307, PC-307, and PC-303. The results from the QSAR suggest that we can group the peptides as follows: PG-5 is the most toxic; cyclic-PG-5 and cyclic-PC-307 are moderately toxic; and PC-307 and PC-303 have low toxicity.
3.2. Comparison to experimental data
The experimental results are given in Table 7. Each peptide was tested as described in Section 2.3.
Table 7.
Experimental values of cytotoxicity for the five peptides against human epithelial cervical cells (ME180) and human lymphocyte cells (H9), in order of decreasing toxicity
| Peptide | IC50 (μg/mL)
|
|
|---|---|---|
| H9 | ME180 | |
| PG-5 | 38.9 | 74.9 |
| PG-307 | 48.7 | 122 |
| PG-307cy | 89.9 | 140.9 |
| PG-5cy | 79.2 | 166.3 |
| PG-303 | 154.8 | 472.1 |
4. Discussion
In this study we developed empirical relations based on properties from molecular dynamics simulations to rank antimicrobial peptides of unknown toxicity in order of toxicity. In Tables 6 and 7 we present the final ranking of the blind test in order of toxicity as predicted from the simulations and the experimentally determined toxicity. We see that the simulation methods correctly determine the most and least toxic peptides, PG-5 and PC-303, respectively. The toxicities of cyclic-PC-307 and cyclic-PG-5 were also correctly determined to be in the same range as each other and moderately toxic.
All of the peptides are β-hairpin, as they contain at least one cysteine-cysteine disulfide bond. Protegrins in which cysteine residues have been replaced with alanine do not appear to form β-hairpins and are both non-active and non-toxic [18]. Protegrin mutants in which the cysteines have been replaced with residues with a propensity to form β-hairpins or non-typical residues which maintain the original structure and charge distribution are active, but toxicity of these peptides was not reported [32]. Similar results have been found for tachyplesin and polyphemusins [41,43].
The high R2 values for the QSAR models for cytotoxicity and hemolytic activity suggested that there was significant correlation between the measured properties and the toxicity of the peptides, and we have seen here that the QSAR formalism can be useful as a ranking tool.
The models for hemolysis both contain length and mean number of acceptors indicating that these are statistically significant properties. All models for both toxicity and cytotoxicity contain an energy term, either VPWF or EPWF. The two models for hemolysis differ only by the exclusion of the fraction of electrostatic energy between the peptide and the solvent. At least for our test peptides, we can get as good a ranking from the smaller number of properties than the four properties model, implying that the term EPWF is less statistically significant than the other terms in the models. Using the same data set as used to build the three property hemolysis model, we see that length alone fits the hemolysis data with an R2 value of 0.649, implying a high statistical significance for this property. Recent evidence suggests that protegrin-1 does not form pores in zwitterionic mammalian membrane mimics but rather disrupts from the surface [37]. The meaning of this relationship, then, between length and hemolysis is not as it would be if the peptides needed to transverse the membrane to form a pore. However, due to the similarity of the protegrin sequences in our data set, length does have certain implications about the number of positively charged residues and hydrophobic residues present in peptide, which can be important for membrane disruption. Length may also be affecting the stability of protegrin multimers on the surface of mammalian lipid bilayers, which in turn may be important for membrane disruption.
It is possible that AMPs form aggregates before inserting or otherwise interacting with the cell membrane, as seen for magainin [3,16,38,39]. There is currently no information available that explains how protegrin-1 goes from being in solution to interacting with membranes, but as mentioned previously, it is know that protegrin-1 does not form pores in mammalian cell membrane mimics [37]. The possibility of the peptides aggregating in solution before interacting with the lipid bilayer is important to keep in mind with the current study, in which the properties are based on monomer properties in solution. The fact that the models based on monomers correctly predicted the order in terms of toxicity of four out of five peptides suggests that this information is being captured, at least indirectly.
The best five-property model for cytotoxicity still only fits the data as well as the three term model for hemolysis, and the fit is not as good, as evidenced by the lower F value for this model. This could be because the data is not spread out in such a way that models of higher significance can be built. In studying the data, we see that the peptides are clustered more with cytotoxicities between 2 and 4. Perhaps the small, discrete-number scale used for cytotoxicity measurements is hindering the model building.
There is one failure in our predictions, for PC-307, which was predicted to be non-toxic when in fact it is highly toxic. Though it is possible that the structure of PC-307 we obtained through homology modeling is different from its actual structure influencing the results, this is highly unlikely, due to the constraints on the structure (same length as PG-1, small size, two cysteine-cysteine bonds). It is interesting to note the shape that PC-307 takes on during the simulation, however. The C-terminal Arg-18 interacts with residues on the same strand of the peptide resulting in a significant bend in the C-terminal strand. This is most likely a conformation particular to the aqueous environment, not necessarily the conformation that we would expect PC307 to take on when interacting with other peptides to disrupt the membrane. It would have been possible for PG-5 to exhibit similar behavior, as it has an unrestricted C-terminal strand, but in our simulation we did not see such behavior. The three remaining peptides are unable to distort their structure in such a way, as PC-303 has a shortened C-terminus and the other two are cyclic.
We conclude that the results are encouraging with respect to predicting toxicity of AMPs through simulations of single peptides, though we caution that care should be taken when interpreting results of simulations in which the C-terminus of the peptide is having strong intrapeptide interactions. Although, the predictive accuracy of an empirical approach like the one presenting may be surprising at first, it reaffirms the belief that the biological function of antimicrobial peptides is the result of the physicochemical principles. Certainly, antimicrobial activity and toxicity are manifestations of a multitude of interaction phenomena, from peptide–membrane binding, to peptide–peptide association, to disruption of the membrane integrity and the movement of ions through it, to, finally, the collapse of the transmembrane potential and the lysis of the cell. As such, simple peptide–solvent simulations cannot point to the actual mechanism underlying biological function. And although this paper makes a major step in the linkage of simulation and prediction components involved in molecular design of antimicrobials, it is probable that there are parameters such as peptide–peptide association strength that are critical for biological function and simply not taken into account by the presented models. Nonetheless, the success of the method we have presented here can be viewed as an important step that will allow us to begin testing of new peptide sequences that have not been synthesized previously. Using these methods we may be able to screen out potential mutations to the sequences before costly synthesis and experimental testing. In the future it will be possible to refine this set of tools to include other parameters such as possible activity-related peptide self association strength, with the hope to increase the predictive accuracy further, not only for protegrins, but for other AMPs. The same methods can easily be used to build predictive models like those developed in this study for any set of related peptides for which structure and toxicity data is available.
Acknowledgments
Benjamin Schuster was supported by a University of Minnesota Bioinformatics Summer Institute internship, NSF award EEC-0234112. This work was supported by a grant from NIH (GM 070989). Computational support from the Minnesota Supercomputing Institute (MSI) is gratefully acknowledged. This work was also partially supported by National Computational Science Alliance under MCA04T033 and utilized the marvel cluster at the Pittsburgh Supercomputing Center.
References
- 1.SAS, 9.1.3 ed. Cary, NC: SAS Institute Inc.; 2004. [Google Scholar]
- 2.Andersen HC. Molecular dynamics simulations at constant pressure and/or temperature. J Chem Phys. 1980;72:2384–93. [Google Scholar]
- 3.Andreu D, Rivas L. Animal antimicrobial peptides: an overview. Biopolymers. 1998;47:415–33. doi: 10.1002/(SICI)1097-0282(1998)47:6<415::AID-BIP2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 4.Bolintineanu D, Langham A, Davis HT, Kaznessis Y. Molecular dynamics simulations of three protegrin-type anti-microbial peptides: interplay between charges at the termini, β-sheet structure and amphiphilic interactions. Mol Simul. 2007;33(9–10):809–19. doi: 10.1080/08927020701393481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: a program for macromolecular energy, minimization, and dynamics simulations. J Comp Chem. 1983;4:187–217. [Google Scholar]
- 6.Cho Y, Turner JS, Dinh NN, Lehrer RI. Activity of protegrins against yeast-phase Candida albicans. Infect Immun. 1998;66:2486–93. doi: 10.1128/iai.66.6.2486-2493.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cole AM, Hong T, Boo LM, Nguyen T, Zhao C, Bristol G, et al. Retrocyclin: a primate peptide that protects cells from infection by T- and M-tropic strains of HIV-1. Proc Natl Acad Sci USA. 2002;99:1813–8. doi: 10.1073/pnas.052706399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Conlon JM, Al-Ghaferi N, Abraham B, Leprince J. Strategies for transformation of naturally occurring amphibian antimicrobial peptides into therapeutically valuable anti-infective agents. Methods. 2007;42:349–57. doi: 10.1016/j.ymeth.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 9.Darden T, York D, Pedersen LG. Particle mess Ewald: an Nxlog(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–92. [Google Scholar]
- 10.Dathe M, Nikolenko H, Meyer J, Beyermann M, Bienert M. Optimization of the antimicrobial activity of magainin peptides by modification of charge. FEBS Lett. 2001;501:146–50. doi: 10.1016/s0014-5793(01)02648-5. [DOI] [PubMed] [Google Scholar]
- 11.Dhople V, Krukemeyer A, Ramamoorthy A. The human beta-defensin-3, an antibacterial peptide with multiple biological functions. Biochim Biophys Acta. 2006;1758:1499–512. doi: 10.1016/j.bbamem.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 12.Drin G, Temsamani J. Translocation of protegrin I through phospholipid membranes: role of peptide folding. Biochim Biophys Acta. 2002;1559:160–70. doi: 10.1016/s0005-2736(01)00447-3. [DOI] [PubMed] [Google Scholar]
- 13.Durr UH, Sudheendra US, Ramamoorthy A. LL-37, the only human member of the cathelicidin family of antimicrobial peptides. Biochim Biophys Acta. 2006;1758:1408–25. doi: 10.1016/j.bbamem.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 14.Fields CG, Lloyd DH, Macdonald RL, Otteson KM, Noble RL. HBTU activation for automated Fmoc solid-phase peptide synthesis. Pept Res. 1991;4:95–101. [PubMed] [Google Scholar]
- 15.Goodson B, Ehrhardt A, Ng S, Nuss J, Johnson K, Giedlin M, et al. Characterization of novel antimicrobial peptoids. Antimicrob Agents Chemother. 1999;43:1429–34. doi: 10.1128/aac.43.6.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hallock KJ, Lee DK, Ramamoorthy A. MSI-78, an analogue of the magainin antimicrobial peptides, disrupts lipid bilayer structure via positive curvature strain. Biophys J. 2003;84:3052–60. doi: 10.1016/S0006-3495(03)70031-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hancock RE, Patrzykat A. Clinical development of cationic antimicrobial peptides: from natural to novel antibiotics. Curr Drug Targets Infect Disord. 2002;2:79–83. doi: 10.2174/1568005024605855. [DOI] [PubMed] [Google Scholar]
- 18.Harwig SS, Waring A, Yang HJ, Cho Y, Tan L, Lehrer RI. Intramolecular disulfide bonds enhance the antimicrobial and lytic activities of protegrins at physiological sodium chloride concentrations. Eur J Biochem. 1996;240:352–7. doi: 10.1111/j.1432-1033.1996.0352h.x. [DOI] [PubMed] [Google Scholar]
- 19.Hoover WH. Canonical dynamics: equilibirum phase-space distributions. Phys Rev A. 1985;31:1695–7. doi: 10.1103/physreva.31.1695. [DOI] [PubMed] [Google Scholar]
- 20.Houghten RA. The broad utility of soluble peptide libraries for drug discovery. Gene. 1993;137:7–11. doi: 10.1016/0378-1119(93)90244-w. [DOI] [PubMed] [Google Scholar]
- 21.Houghten RA, Pinilla C, Blondelle SE, Appel JR, Dooley CT, Cuervo JH. Generation and use of synthetic peptide combinatorial libraries for basic research and drug discovery. Nature. 1991;354:84–6. doi: 10.1038/354084a0. [DOI] [PubMed] [Google Scholar]
- 22.Imura Y, Nishida M, Ogawa Y, Takakura Y, Matsuzaki K. Action mechanism of tachyplesin I and effects of PEGylation. Biochim Biophys Acta. 2007;1768:1160–9. doi: 10.1016/j.bbamem.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 23.JMP. JMP: the statistical discovery software. Cary, NC: SAS Institute Inc.; 1999. [Google Scholar]
- 24.Kaznessis YN, Snow ME, Blankley CJ. Prediction of blood–brain partitioning using Monte Carlo simulations of molecules in water. J Comput Aided Mol Des. 2001;15:697–708. doi: 10.1023/a:1012240703377. [DOI] [PubMed] [Google Scholar]
- 25.Khandelia H, Kaznessis Y. Cation-p interactions stabilize the structure of the antimicrobial peptide indolicidin near membranes: molecular dynamics simulations. J Phys Chem B. 2007;111:242–50. doi: 10.1021/jp064776j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khandelia H, Kaznessis YN. Molecular dynamics investigation of the influence of anionic and zwitterionic interfaces on antimicrobial peptides’ structure: implications for peptide toxicity and activity. Peptides. 2006;27:1192–200. doi: 10.1016/j.peptides.2005.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khandelia H, Kaznessis YN. Molecular dynamics simulations of helical antimicrobial peptides in SDS micelles: what do point mutations achieve? Peptides. 2005;26:2037–49. doi: 10.1016/j.peptides.2005.03.058. [DOI] [PubMed] [Google Scholar]
- 28.Khandelia H, Kaznessis YN. Molecular dynamics simulations of the helical antimicrobial peptide ovispirin-1 in a zwitterionic dodecylphosphocholine micelle: insights into host-cell toxicity. J Phys Chem B. 2005;109:12990–6. doi: 10.1021/jp050162n. [DOI] [PubMed] [Google Scholar]
- 29.Khandelia H, Kaznessis YN. Structure of the antimicrobial beta-hairpin peptide protegrin-1 in a DLPC lipid bilayer investigated by molecular dynamics simulation. Biochim Biophys Acta. 2007;1768:509–20. doi: 10.1016/j.bbamem.2006.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khandelia H, Langham AA, Kaznessis YN. Driving engineering of novel antimicrobial peptides from simulations of peptide–micelle interactions. Biochim Biophys Acta. 2006;1758:1224–34. doi: 10.1016/j.bbamem.2006.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kokryakov VN, Harwig SS, Panyutich EA, Shevchenko AA, Aleshina GM, Shamova OV, et al. Protegrins: leukocyte antimicrobial peptides that combine features of corticostatic defensins and tachyplesins. FEBS Lett. 1993;327:231–6. doi: 10.1016/0014-5793(93)80175-t. [DOI] [PubMed] [Google Scholar]
- 32.Lai JR, Huck BR, Weisblum B, Gellman SH. Design of non-cysteine-containing antimicrobial beta-hairpins: structure–activity relationship studies with linear protegrin-1 analogues. Biochemistry. 2002;41:12835–42. doi: 10.1021/bi026127d. [DOI] [PubMed] [Google Scholar]
- 33.Langham A, Kaznessis Y. Effects of mutations on the C-terminus of protegrin-1: a molecular dynamics simulation study. Mol Simul. 2006;32:193–201. [Google Scholar]
- 34.Langham AA, Khandelia H, Kaznessis YN. How can a beta-sheet peptide be both a potent antimicrobial and harmfully toxic? Molecular dynamics simulations of protegrin-1 in micelles Biopolymers. 2006;84:219–31. doi: 10.1002/bip.20397. [DOI] [PubMed] [Google Scholar]
- 35.Langham AA, Waring AJ, Kaznessis YN. Comparison of interactions between β-hairpin decapeptides and SDS/DPC micelles from experimental and simulation data. BMC Biochem. 2007;8:11. doi: 10.1186/1471-2091-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.MacKerell ADJ, Brooks B, Brooks CL, III, Nilsson L, Roux B, Won Y, et al. The encyclopedia of computational chemistry. 1. Chichester: John Wiley & Sons; 1998. [Google Scholar]
- 37.Mani R, Tang M, Wu X, Buffy JJ, Waring AJ, Sherman MA, et al. Membrane-bound dimer structure of a beta-hairpin antimicrobial peptide from rotational-echo double-resonance solid-state NMR. Biochemistry. 2006;45:8341–9. doi: 10.1021/bi060305b. [DOI] [PubMed] [Google Scholar]
- 38.Matsuzaki K. Magainins as paradigm for the mode of action of pore forming polypeptides. Biochim Biophys Acta. 1998;1376:391–400. doi: 10.1016/s0304-4157(98)00014-8. [DOI] [PubMed] [Google Scholar]
- 39.Oren Z, Shai Y. Mode of action of linear amphipathic alpha-helical antimicrobial peptides. Biopolymers. 1998;47:451–63. doi: 10.1002/(SICI)1097-0282(1998)47:6<451::AID-BIP4>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 40.Ostberg N, Kaznessis Y. Protegrin structure–activity relationships: using homology models of synthetic sequences to determine structural characteristics important for activity. Peptides. 2005;26:297–306. doi: 10.1016/j.peptides.2004.09.020. [DOI] [PubMed] [Google Scholar]
- 41.Powers JP, Tan A, Ramamoorthy A, Hancock RE. Solution structure and interaction of the antimicrobial polyphemusins with lipid membranes. Biochemistry. 2005;44:15504–13. doi: 10.1021/bi051302m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qu XD, Harwig SS, Shafer WM, Lehrer RI. Protegrin structure and activity against Neisseria gonorrhoeae. Infect Immun. 1997;65:636–9. doi: 10.1128/iai.65.2.636-639.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ramamoorthy A, Thennarasu S, Tan A, Gottipati K, Sreekumar S, Heyl DL, et al. Deletion of all cysteines in tachyplesin I abolishes hemolytic activity and retains antimicrobial activity and lipopolysaccharide selective binding. Biochemistry. 2006;45:6529–40. doi: 10.1021/bi052629q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reddy KV, Yedery RD, Aranha C. Antimicrobial peptides: premises and promises. Int J Antimicrob Agents. 2004;24:536–47. doi: 10.1016/j.ijantimicag.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 45.Ryckaert JP, Ciccotti G, Berendesen HJC. Numerical integration of the cartesian equations of motion for a system with constraints: molecule dynamics of n-alkanes. J Comp Phys. 1977;23:327–41. [Google Scholar]
- 46.Travis J. Reviving the antibiotic miracle? Science. 1994;264:360–2. doi: 10.1126/science.8153615. [DOI] [PubMed] [Google Scholar]
- 47.Yasin B, Harwig SS, Lehrer RI, Wagar EA. Susceptibility of Chlamydia trachomatis to protegrins and defensins. Infect Immun. 1996;64:709–13. doi: 10.1128/iai.64.3.709-713.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415:389–95. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]