

Abstract
OBJECTIVE
Ischemia-reperfusion (I/R) injury, often encountered clinically, results in myocardial apoptosis and necrosis. Hydrogen sulfide (H2S) is produced endogenously in response to ischemia and thought to be cardioprotective, although its mechanism of action is not fully known. This study investigates cardioprotection provided by exogenous H2S, generated as sodium sulfide on apoptosis following myocardial I/R injury.
METHODS
The mid-LAD coronary artery in Yorkshire swine (n=12) was occluded for 60 minutes, followed by reperfusion for 120 minutes. Controls (n=6) received placebo, and treatment animals (n=6) received sulfide 10 minutes prior to and throughout reperfusion. Hemodynamic, global, and regional functional measurements were obtained. Evans blue/TTC staining identified the area-at-risk (AAR) and infarction. Serum CK-MB, troponin I, and FABP were assayed. Tissue expression of bcl-2, bad, apoptosis-inducing-factor (AIF), total & cleaved caspase-3, and total & cleaved PARP were assessed. PAR and TUNEL staining were performed to assess apoptotic cell counts, and poly-ADP ribosylation, respectively.
RESULTS
Pre-I/R hemodynamics were similar between groups. Post-I/R, mean arterial pressure (mmHg) was reduced by 30.2±4.3 in controls vs. 8.2±6.9 in treatment animals (p=0.01). +LV dP/dt (mmHg/sec) was reduced by 1308±435 in controls vs. 403±283 in treatment animals (p=0.001). Infarct size (% of AAR) in controls was 47.4±6.2% vs. 20.1±3.3% in the treated group (p=0.003). In treated animals, CK-MB and FABP were lower by 47.0% (p=0.10) and 45.1% (p=0.01), respectively. AIF, caspase-3, and PARP expression was similar between groups, whereas cleaved caspase-3 and cleaved PARP was lower in treated animals (p=0.04). PAR staining was significantly reduced in sulfide treated groups (p=0.04). TUNEL staining demonstrated significantly fewer apoptotic cells in sulfide treated animals (p=0.02).
CONCLUSIONS
Sodium sulfide is efficacious in reducing apoptosis in response to I/R injury. Along with its known effects on reducing necrosis, sulfide’s effects on apoptosis may partially contribute to providing myocardial protection. Exogenous sulfide may have therapeutic utility in clinical settings in which I/R injury is encountered.
Keywords: Ischemia, Reperfusion, Apoptosis, Cardiac, Sulfide, Protection
Introduction
Coronary artery disease remains the leading cause of mortality in the industrialized world. Despite lifestyle modification and advances in pharmacotherapy, chronic and acute myocardial ischemia require intervention to salvage viable myocardium at risk. Presently, three approaches are generally employed – thrombolysis, percutaneous transluminal coronary angioplasty (with and without coronary stenting) (PTCA), and coronary artery bypass grafting (CABG). While essential for reestablishment of perfusion, these modalities can induce iatrogenic injury, commonly referred to as ischemia – reperfusion (I/R) injury. In addition to causing myocardial necrosis, I/R injury can result in additional damage to the heart independent of the ischemic insult manifesting as cardiomyocyte death through the process of apoptosis[1]. While reducing the number of viable cardiomyocytes, apoptosis is also thought to contribute to myocardial stunning[2], and in the cardiac surgical setting, apoptotic markers have been correlated to short and long term mortality[3].
Investigation in to the pathogenesis of myocardial I/R injury has identified an endogenous inflammatory response and the release of damaging free radicals and oxidants associated with this response to be key in inducing myocardial damage[4, 5]. Recently, research in to the gaseous signaling molecule hydrogen sulfide (H2S) has demonstrated it may in fact serve as an endogenous mediator to limit inflammation and free radical damage[6]. Specifically, H2S has been shown to limit neutrophil adhesion and activation In response to inflammatory stimuli, as well as suppress the release of the pro-inflammatory mediator tumor necrosis factor-alpha (TNF-α)[7, 8]. In addition to its effects on neutrophils, H2S, which is a strong reducing agent, is able to react with multiple oxidant stressors including superoxide radical anion[9], hydrogen peroxide [10], peroxynitrite [6], and hypochlorite[7]. H2S, which is synthesized endogenously from L-cysteine via cystathionine-γ-lyase (CSE) in the heart and vasculature, has also become the subject of recent investigation in the context of myocardial protection. These studies, which have all utilized rodent models of myocardial injury, have demonstrated sulfide is able to limit myocardial infarction size and oxidative stress in response to I/R injury[10–12]. Given the ability of H2S to mitigate the production of stimuli known to induce apoptosis – free radical stressors[13] and inflammatory cytokines such as TNF α[14] – we hypothesized administration of exogenous therapeutic H2S prior to the onset of reperfusion after acute myocardial ischemia would reduce the apoptotic response to I/R injury. Utilizing a pre-clinical large animal model, this study investigates the effects of H2S on apoptotic signaling in response to acute myocardial ischemia followed by reperfusion.
Material & Methods
Animals
Animals were housed individually and provided with laboratory chow and water ad libitum. All experiments were approved by the Beth Israel Deaconess Medical Center animal care and use committee and the Harvard Medical Area standing committee on animals and conformed to the US National Institutes of Health guidelines regulating the care and use of laboratory animals (NIH publication 5377-3, 1996).
Experimental Design
Yorkshire pigs of either sex (35 to 40 kg) were divided randomly into control (n=6) and sulfide treatment (n=6) groups. Animals were subjected to regional left ventricular (LV) ischemia by left anterior descending (LAD) arterial occlusion distal to the second diagonal branch for 60 minutes. The treatment group received sodium sulfide (100μg/kg bolus + 1mg/kg/hr infusion) 10 minutes prior to the onset of reperfusion, whereas the control group received a placebo carrier solution of equal volume. Sodium sulfide was produced and formulated to pH neutrality and iso-osmolarity by Ikaria Inc. (Seattle, WA)using H2S gas (Mattheson, Newark, CA) as the starting material. The myocardium was reperfused for 120 minutes following ischemia. Arterial blood gas (ABG), arterial blood pressure, hematocrit (Hct), LV pressure, heart rate (HR), EKG, O2 saturation, core temperature, and intravenous fluid requirements were measured and recorded. Myocardial segmental shortening in the long axis (parallel to the LAD) and short-axis (perpendicular to the LAD) were recorded at baseline prior to the onset of ischemia, and prior to harvest after 120 minutes of reperfusion. At the completion of the protocol, the heart was excised, and tissue samples from the ischemic-reperfused, distal LAD territory were collected for molecular analyses as described below.
Surgical Protocol
Swine were sedated with ketamine hydrochloride (20 mg/kg, intramuscularly, Abbott Laboratories, North Chicago, IL), and anesthetized with a bolus infusion of thiopental sodium (Baxter Healthcare Corporation, Inc, Deerfield, IL; 5.0 to 7.0 mg/kg intravenously), followed by endotracheal intubation. Ventilation was begun with a volume-cycled ventilator (model Narkomed II-A; North American Drager, Telford, PA; oxygen, 40%; tidal volume, 600cc; ventilation rate, 12 breaths/min; positive end-expiratory pressure, 3 cm H2O; inspiratory to expiratory time, 1:2). General endotracheal anesthesia was established with 3.0% sevoflurane (Ultane; Abbott Laboratories) at the beginning of the surgical preparation, and then maintained with 1.0% throughout the experiment. One liter of Lactated Ringer’s intravenous (IV) fluid was administered after induction of anesthesia and continued thereafter throughout the surgical protocol at 150cc/hour. A right groin dissection was performed and the femoral vein and common femoral artery were isolated and cannulated utilizing 8F sheaths (Cordis Corporation, Miami, FL). The femoral vein was cannulated for intravenous access, drug / placebo delivery, and the right common femoral artery was cannulated for arterial blood sampling and continuous intra-arterial blood pressure monitoring (Millar Instruments, Houston, TX). A median sternotomy was performed exposing the pericardial sac, which was then opened to form a pericardial cradle. A catheter-tipped manometer (Millar Instruments, Houston, TX) was introduced through the apex of the left ventricle to record LV pressure. Segmental shortening in the area-at-risk was assessed utilizing a sonometric digital ultrasonic crystal measurement system (Sonometrics Corp, London, ON, Canada) using four 2-mm digital ultrasonic probes implanted in the subepicardial layer approximately 10 mm apart within the ischemic LV area. Cardiosoft software (Sonometrics Corp, London, ON, Canada) was used for data recording (LV dP/dt, segmental shortening, arterial blood pressure, heart rate) and subsequent data analysis to determine myocardial function. Baseline hemodynamic, functional measurement (global: +LV dP/dt, regional: segmental shortening), arterial blood gas analysis, and hematocrit were obtained. ABG analysis was continued every 15 minutes throughout the protocol and hematocrit was measured every 20 minutes. All animals received 75mg of lidocaine and 20 mEq of potassium chloride as prophylaxis against ventricular dysrhythmia, as well as 60 units /kg of intravenous heparin bolus prior to occlusion of the LAD. The LAD coronary artery was occluded 3mm distal to the origin of the second diagonal branch utilizing a Rommel tourniquet. Myocardial ischemia was confirmed visually by regional cyanosis of the myocardial surface. Fifty minutes after the initiation of regional ischemia (10 minutes prior to the onset of reperfusion), control pigs received a placebo carrier solution infusion intravenously, and treatment animals received exogenous sulfide, generated as sodium sulfide (NaH2S), (100μg/kg bolus + 1mg/kg/hr infusion) until the end of the experimental protocol. The Rommel tourniquet was released 60 minutes after the onset of acute ischemia and the myocardium was reperfused for 120 minutes. At the end of the reperfusion period, hemodynamic and functional measurements were recorded as described above, followed by re-ligation of the LAD and injection of monastryl blue pigment (Engelhard Corp, Louisville, KY) at a 1:150 dilution in PBS into the aortic root after placement of an aortic crossclamp distal to the coronary arterial ostia to demarcate the area-at-risk. The heart was rapidly excised and the entire left ventricle, including the septum, was dissected free. The LV was cut in to 1cm thick slices perpendicular to the axis of the LAD. The area-at-risk was clearly identified by lack of blue pigment staining. Tissue from the area-at-risk of the slice 1cm proximal to the LV apex was isolated and divided for use in molecular and microvascular studies. The remaining slices were weighed utilized for infarct size calculation as described below. Ventricular dysrhythmia (ventricular fibrillation or pulseless ventricular tachycardia) events were recorded and treated with immediate electrical cardioversion (50 J, internal paddles).
Measurement of Global and Regional Myocardial Function
Global myocardial function was assessed by calculating the maximum positive first derivative of LV pressure over time (+dP/dt). Regional myocardial function was determined by using subepicardial 2-mm ultrasonic probes to calculate the percentage segment shortening (%SS), which was normalized to the baseline. Measurements were taken at baseline prior to the onset of ischemia and at the end of reperfusion. The ventilator was stopped during data acquisition to eliminate the effects of respiration. Measurements were made during at least three cardiac cycles in normal sinus rhythm and then averaged. Digital data were inspected for the correct identification of end-diastole and end-systole. End-diastolic segment length (EDL) was measured at the onset of the positive dP/dt, and the end-systolic segment length (ESL) at the peak negative dP/dt.
Quantification of Myocardial Infarct Size
The left ventricle was isolated and cut in to 1cm slices perpendicular to the axis of the LAD. Briefly, slices were immediately immersed in 1% triphenyl tetrazolium chloride (TTC, Sigma Chemical Co, St Louis, MO) in phosphate buffer (pH 7.4) at 38°C for 30 minutes. The infarct area (characterized by absence of staining), the non-infarcted area-at-risk (characterized by red tissue staining), and the non-ischemic portion of the LV (characterized by purple tissue staining) were sharply dissected from one another and weighed. The percentage area-at-risk was defined as: (Infarct mass + non-infarct area-at-risk mass) / Total LV mass x 100. Infarct size was calculated as a percentage of area at risk to normalize for any variation in AAR size using the following equation: (Infarct mass / total mass AAR) x 100.
Western Blotting
Whole-cell lysates were isolated from the homogenized myocardial samples with a RIPA buffer (Boston Bioproducts, Worcester, MA) and centrifuged at 12,000g for 10 min at 4 °C to separate soluble from insoluble fractions. Protein concentration was measured spectrophotometrically at a 595-nm wavelength with a DC protein assay kit (BioRAD, Hercules, CA). Forty to eighty micrograms of total protein were fractionated by 4-20% gradient, SDS polyacrylamide gel electrophoresis (Invitrogen, San Diego, CA) and transferred to PVDF membranes (Millipore, Bedford, MA). Each membrane was incubated with specific antibodies (Cell Signaling Technology, Beverly, MA) as follows: anti-Apoptosis Inducing Factor (AIF) (1:1000 dilution), anti-Bcl-2 (1:1000 dilution), anti-Bad (1:1000 dilution), anti-phospho Bad (Serine 136) (1:500 dilution), anti-phospho Bad (Serine 112) (1:2000 dilution), anti-caspase-3 (1:1000 dilution), anti-cleaved caspase-3 (1:1000 dilution), anti-poly (ADP) ribose polymerase (PARP) (1:1000 dilution), anti-cleaved PARP (1:1000 dilution). The membranes were subsequently incubated for 1 hour in diluted appropriate secondary antibody (Jackson Immunolab, West Grove, PA). Immune complexes were visualized with the enhanced chemiluminescence detection system (Amersham, Piscataway, NJ). Bands were quantified by densitometry of radioautograph films. Ponceau S staining was performed to confirm equivalent protein loading.
Serum Creatine Kinase-MB, Troponin I, and Fatty Acid Binding Protein Quantification
Serum collected prior to sacrifice was utilized for quantification of Creatine Kinase-MB (CK-MB), Troponin I, and Fatty Acid Binding Protein (FABP) utilizing a protein microarray (Allied Biotech Inc., Ijamsville, MD) in triplicate as described previously[15]. Serum levels of markers were calculated based on standards provided by the manufacturer.
Immunohistochemical Staining
Myocardial tissue from the ischemic territory was place in 10% buffered formalin for 24 hours, followed by paraffin mounting and sectioning in to 4μm slices. Poly (ADP) Ribosylation Staining - For the immunohistochemical detection of poly(ADP-ribose) polymerase activity, mouse monoclonal anti-poly(ADP-ribose) (PAR) antibody (Calbiochem, San Diego, CA) (1:1000, overnight, 4°C) was used. Secondary labeling was achieved by using biotinylated horse anti-mouse antibody (Vector Laboratories, Burlingame, CA) (30min room temperature). Horseradish peroxidase-conjugated avidin (30min, room temperature) and brown colored diaminobenzidine (6min, room temperature) was used to visualize the labeling (Vector Laboratories, Burlingame, CA). The sections were counterstained with hematoxylin (blue color). The intensity of specific staining of individual sections was determined by a blinded experimenter. The semiquantitative PAR-positivity score is the following : 1: no specific staining, 2: light cytoplasmic staining, 3: few positive nuclei, 4: light nuclear staining in approximately 10% of cells, 5: light nuclear staining in approximately 25% of cells, 6: light nuclear staining in approximately 50% of cells, 7: strong nuclear staining in approximately 50% of cells, 8: approximately 75% of the nuclei are positive, 9: approximately 90% of the nuclei are positive, 10: few negative cells). TUNEL Staining - The apoptotic cells were identified by dUTP nick-end labeling (TUNEL) using an apoptosis detection kit according to the manufacturer’s protocol (Chemicon Inc, Temecula, CA). Five photographs (magnification 20x) of each tissue section were taken. The nuclei were viewed and manually counted by an observer blinded to the experimental conditions. The number of TUNEL-positive cardiomyocytes, indicating apoptosis, was expressed in mean number per / 100 cells / microscopic field.
Data Analysis
Data are reported as means ± SD. Immunoblots are expressed as a ratio of protein to loading band density and were analyzed after digitization and quantification of x-ray films with ImageJ 1.33 (National Institutes of Health, USA). Blots were analyzed using an unpaired t-test. Bonferroni corrections were applied to multiple tests and probability values of less than 0.05 were considered statistically significant.
Results
Arterial Blood Gas, Hematocrit, and Core Temperature
No significant differences were observed between arterial pH, pCO2, pO2, Hct, or core temperature at baseline, or at the end of reperfusion.
Hemodynamic Parameters
Heart rate (HR) and mean arterial blood pressure (MAP) were similar between groups at baseline (HR Placebo 74.0 ± 7.2 vs. Sulfide 68.2 ± 20.4 beats /minute, p=0.53), MAP Placebo 62.5 ± 7.2 vs. Sulfide 56.4 ± 9.6 mmHg, p=0.24) (Figure 1). The HR remained similar between groups at the end of reperfusion (Placebo 82.0 ± 9.0 vs. Sulfide 78.00 ± 10.0 beats/min, p=0.49), but MAP was significantly lower in the placebo treated group (Placebo 32.3 ± 7.7 vs. Sulfide 49.6 ± 11.6 mmHg, p=0.01) (Figure 1).
Figure 1.
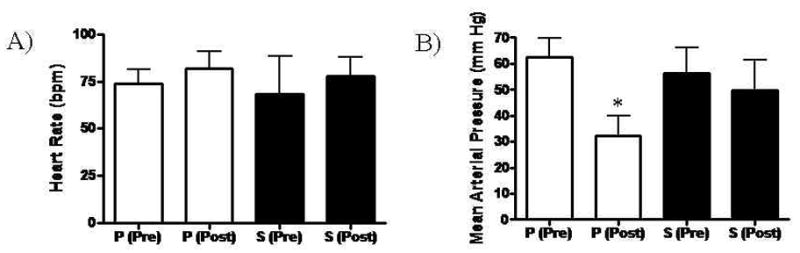
(A) Heart rate remained similar between both Placebo and Sulfide treated groups Pre and Post ischemia – reperfusion. (B) Mean arterial pressure was similar prior to ischemia – reperfusion in both groups, but was significantly lower in the Placebo treated group after reperfusion. P = Placebo, S = Sulfide, Pre = pre ischemia – reperfusion, Post = post ischemia – reperfusion
Global and Regional Myocardial Function
Global systolic LV function as determined from LV +dP/dt was similar between groups prior to the onset of ischemia (Placebo 2436 ± 1051 mmHg/sec vs. Sulfide 2430 ± 525 mmHg/sec, p=0.99). At the end of the reperfusion period, LV dP/dt was significantly lowered in the Placebo group (relative to pre-I/R LV dP/dt) compared to the Sulfide treated group (Placebo 1128 ± 178 mmHg/sec vs. Sulfide 2026 ± 452 mm Hg/sec, p=0.001). This reflected a 53.7% reduction in LV dP/dt in the Placebo group and a 16.8% reduction in LV dP/dt in the Sulfide group after I/R injury (Figure 2). Regional myocardial function in the area at risk was similar between groups in both longitudinal (Placebo 14.3 ± 6.8 vs. Sulfide 14.4 ± 3.1 % segmental shortening(SS), p=0.97) and horizontal axes (Placebo 17.6 ± 5.4 vs. Sulfide 16.8 ± 4.8 %SS, p=0.78) prior to the onset of ischemia. At the end of reperfusion, segmental shortening in the longitudinal axis was impaired significantly in both groups, without a significant difference between groups (Placebo 4.8 ± 1.8 vs. Sulfide 7.8 ± 3.7 % SS, p=0.11) (Figure 3). Horizontal segmental shortening was also significantly impaired in both groups at the end of reperfusion and no significant differences were seen between groups post I/R (Placebo 7.5 ± 3.0 vs. Sulfide 9.3 ± 3.0 % SS, p=0.35) (Figure 3).
Figure 2.
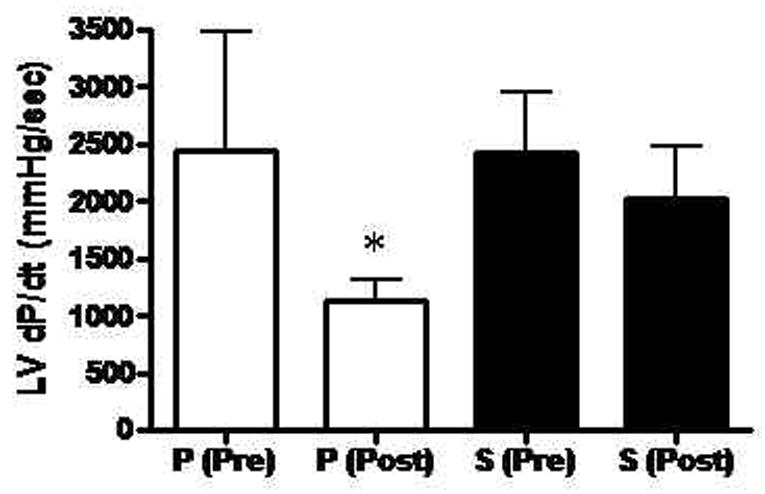
Left ventricular systolic function, as determined by +LV dP/dt was significantly lower in placebo treated animals after reperfusion. P = Placebo, S = Sulfide, Pre = pre ischemia – reperfusion, Post = post ischemia - reperfusion
Figure 3.
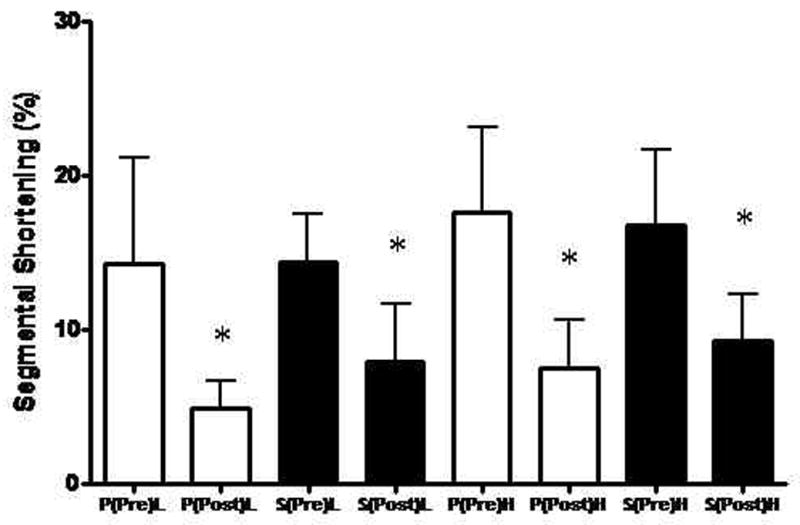
Segmental shortening in both longitudinal (L) and horizontal (H) axes was similar between groups prior to ischemia. Both groups demonstrated significant impairments in both longitudinal and horizontal segmental shortening after ischemia – reperfusion, without significant differences between groups. P = Placebo, S = Sulfide, Pre = pre ischemia – reperfusion, Post = post ischemia – reperfusion
Incidence of VF/VT
The incidence of ventricular fibrillation or pulseless ventricular tachycardia was similar between groups during the period of ischemia (Placebo 0.33 ± 0.2 vs. Sulfide 0.50 ± 0.22 episodes / animal, p=0.60), and during the period of reperfusion (Placebo 1.50 ± 0.50 vs. Sulfide 0.83 ± 0.31 episodes /animal, p=0.28). All dysrrythmias were successfully terminated with electrical cardioversion.
Myocardial Infarct Size
The ischemic area at risk, as a percentage of total LV mass was similar between groups (Placebo 31.1 ± 5.1 vs. Sulfide 33.5 ± 4.2 % of LV mass, p=0.48). Myocardial infarct size was significantly reduced by 2.36-fold in Sulfide treated groups relative to placebo (Placebo 47.4 ± 15.2 vs. Sulfide 20.1 ± 7.9 % of area at risk, p=0.003) (Figure 4).
Figure 4.
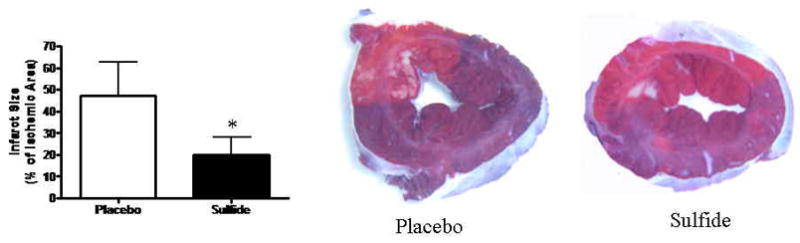
Myocardial infarct size was significantly smaller in Sulfide treated animals relative to Placebo.
Serum Markers of Myocardial Injury
Serum levels of CK-MB trended towards reduction in the sulfide treated group relative to placebo, but did not reach statistical significance (Placebo 4181 ± 2546 pg/mL vs. Sulfide 1965 ± 1643 pg/mL, p=0.10) (Figure 5A). Serum levels of FABP, an early marker of myocardial injury, were 2.2-fold higher in the Placebo group relative to the Sulfide treated group (Placebo 97.4 ± 39.3 pg/mL vs. Sulfide 43.9 ± 19.9 pg/mL, p=0.01) (Figure 5B). Serum Troponin I levels were not detectable in either group (detection threshold of 16 pg/mL) consistent with the early time point of serum harvest.
Figure 5.
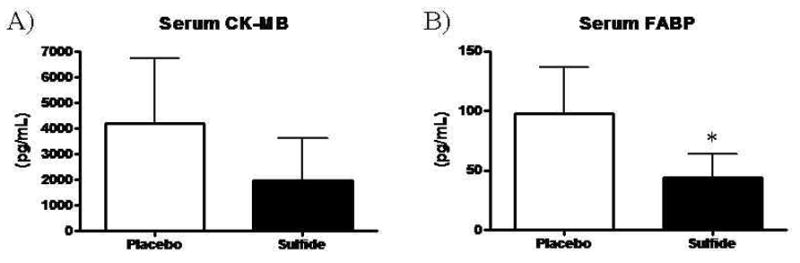
(A) Serum levels of CK-MB trended towards reduction in Sulfide animals. (B) Serum levels of FABP were significantly reduced in Sulfide treated animals.
Apoptotic Signaling
Expression of the anti-apoptotic Bcl-2 demonstrated no significant differences between groups (Placebo 30.8 ± 9.8 vs. Sulfide 42.2 ± 14.4 DU, p=0.21). Myocardial levels of total Bad, phospho-Bad (Serine 112), and phosphoBad (Serine 136) also demonstrated no significant differences between groups (total Bad: Placebo 40.7 ± 8.3 vs. Sulfide 37.8 ± 16.5 DU, p=0.76, pBad (Ser112): Placebo 48.8 ± 16.9 vs. Sulfide 47.5 ± 22.5 DU, p=0.93, pBad (Ser136): Placebo 60.0 ± 4.4 vs. Sulfide 46.4 ± 13.1 DU, p=0.12). Western blotting for AIF was similar between groups (Placebo 56.03 ± 3.6 vs. Sulfide 52.2 ± 5.2 densitometry units (DU), p=0.17). While total caspase-3 levels were similar between groups (Placebo 46.4 ± 2.6 vs. Sulfide 44.9 ± 8.2 DU, p=0.67), myocardial levels of cleaved caspase-3 were increased by 2.89-fold in Placebo treated animals relative to the sulfide group (Placebo 17.8 ± 12.2 vs. Sulfide 6.1 ± 3.3 DU, p=0.04) (Figure 6A and 6B). Similar findings occurred for PARP, with total PARP levels similar between groups (Placebo 33.1 ± 5.9 vs. Sulfide 33.7 ± 11.2 DU, p=0.92), but cleaved PARP 1.72-fold higher in Placebo treated animals (Placebo 24.1 ± 7.3 vs. Sulfide 13.4 ± 8.7 DU, p=0.04) (Figure 6C and 6D).
Figure 6.
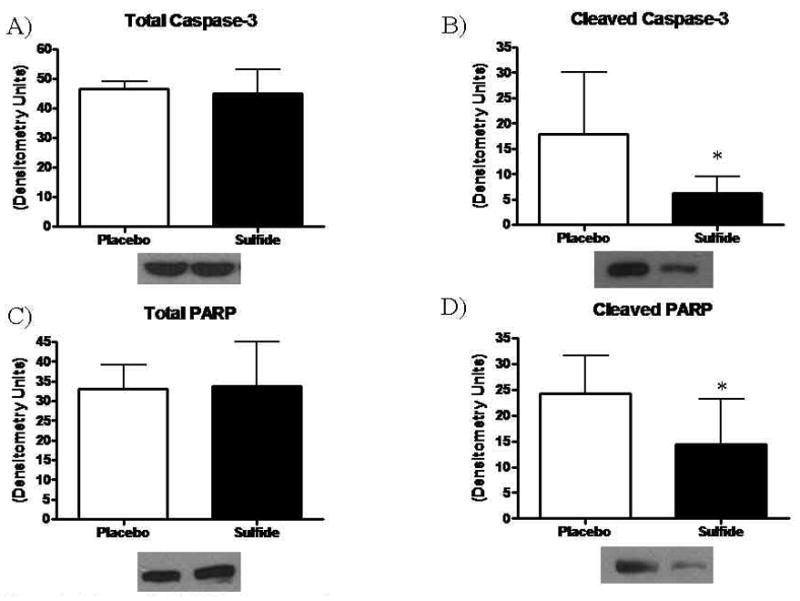
Myocardial (A) Total caspase-3 remained similar between groups, whereas (B) cleaved caspase-3 levels were significantly lower in Sulfide treated animals. (C) Total PARP remained similar between groups, and (D) cleaved PARP levels were significantly lower in Sulfide treated animals.
Poly ADP Ribosylation Staining
The degree of staining for poly-ADP ribosylated proteins was significantly greater in placebo treated animals, with sulfide treated animals exhibiting a mean PAR staining score of 4.3 ± 1.8 vs. 7.0 ± 2.0 in the placebo treated groups (p=0.04) (Figure 7A).
Figure 7.
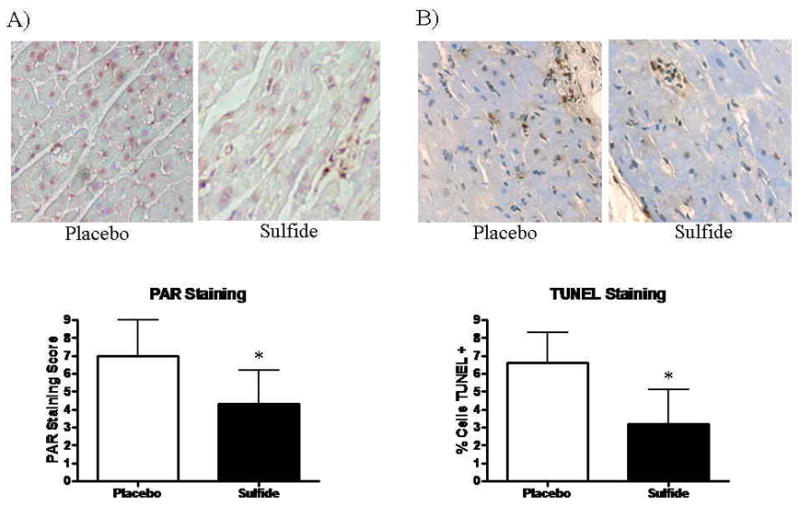
Immunostaining for (A) PAR demonstrated significantly lower PAR-staining scores in Sulfide treated animals. (B) TUNEL Staining demonstrated significantly fewer apoptotic cells in Sulfide treated animals. Upper panels display representative histologic images.
TUNEL Staining for Apoptosis
Apoptotic cell counts were significantly higher in the Placebo treated group as identified by TUNEL staining. The Placebo groups demonstrated 6.6 ± 1.6 % TUNEL+ cells/hpf vs. 3.2 ± 1.9 % TUNEL + cells/hpf in the Sulfide treated group (p=0.02) (Figure 7B).
Discussion
The current study demonstrates that parenteral therapeutic administration of sodium sulfide during the end of ischemia and throughout the reperfusion period, provides significant myocardial protection in response to acute I/R injury, limiting myocardial necrosis and infarct size and improving global systolic LV function, after I/R injury in a large animal model of myocardial infarction. The primary novel findings of this study demonstrate administration of exogenous sulfide reduces apoptosis in response to myocardial I/R injury, specifically decreasing levels of cleaved caspase-3 and PARP at 2 hours after the onset of reperfusion.
Previous work in murine models has demonstrated exogenous administration of sulfide can reduce myocardial infarct size in response to acute I/R injury[10–12]. This study, which utilizes a more clinically relevant porcine model found results consistent with these prior reports. Additionally, we evaluated clinically utilized serum biomarkers of myocardial injury – CK-MB, Troponin I, and FABP. Our study found a trend towards reduction in CK-MB levels (not reaching significance), and a significant reduction in serum levels of FABP, whereas Troponin I levels were not detectable. Our time point for serum collection (3 hours after the onset of ischemia) likely failed to capture a rise in Troponin I, which generally occurs several hours after this timepoint. While a trend towards differences in serum CK-MB was observed, the single timepoint collection also may have limited discerning the true differences in this marker of myocardial injury. Serum FABP, a protein released rapidly from cardiomyocytes in response to ischemic injury[16] and detectable as early as 90–120 minutes after injury[16], was significantly lower in sulfide treated animals. This finding may add clinical relevance to the therapeutic benefits of sulfide administration as increased levels of FABP after acute coronary syndrome are prognostic for an increased risk of death, recurrent MI, and congestive heart failure in patients[17].
While myocardial necrosis likely produces the greatest degree of injury after acute I/R injury, myocardial apoptosis in response to I/R injury continues to be a relatively unavoidable consequence during restoration of perfusion. While essential to maximize salvage of viable tissue, the resulting initiation of the apoptotic cascade may have important clinical consequences relating to stunning[2] and survival[3]. The apoptotic signaling cascade can be initiated by free radicals and TNFα[1, 18], both of which are produced in myocardial I/R injury[13, 14]. H2S can act as both an effective anti-oxidant[6], and limit the generation of TNFα[8], and in this study has been shown to reduce expression of two key downstream effectors of cell death – cleaved caspase-3 and cleaved PARP, with a resulting decrease in the number of apoptotic cardiomyocytes.
Early in apoptotic signaling the anti-apoptotic Bcl-2, and pro-apoptotic Bad are in opposition. A shift in favor of the pro-apoptotic proteins subsequently results in increased mitochondrial pore permeability, releasing cytochrome C and AIF, which can activate caspase-3, and facilitate DNA fragmentation, respectively[1]. Upon cleavage of terminal caspases, such as caspase-3, cell death is generally thought to be inevitable. In this study we found no significant differences in early mediators of apoptotic signaling, such as Bcl-2, and Bad, but did observe significant reduction in expression of downstream effectors including caspase-3 and PARP. The absence of significant differences in expression of the phosphorylated forms of Bad may be due to tissue collection at a single time point – 3 hours after the onset of ischemia, which may not capture transient changes in the activation status of these proteins. This time point was able to capture a significant reduction in the expression of cleaved caspase-3, a terminal effector of apoptosis which is responsible for intracellular degradation of structural proteins, regulatory proteins, and DNA repair enzymes. The reduction in I/R injury seen in our study is consistent with prior investigations which have demonstrated caspase inhibition reduces myocardial apoptosis and improves function in response to I/R injury[19]. Poly (ADP) ribose polymerase (PARP) is a nuclear enzyme activated by DNA damage which catalyzes the synthesis of poly (ADP) ribose (PAR) from nicotine adenine dinucleotide (NAD). Over-activation of PARP results in the depletion of NAD and subsequent ATP formation leading to cell death[20]. PARP inhibition has been shown to ameliorate myocardial damage in response to I/R injury[21]. The findings of this study indicate sulfide treated animals display reduced expression of the active form of PARP – cleaved PARP, and reduced poly ADP ribosylation of proteins, providing mechanistic insight in to sulfide’s ability to provide myocardial protection. Taken in sum, the ability of sulfide to limit production of key stimuli for initiation of apoptosis may allow for it limit the generation of multiple pro-apoptotic signaling molecules which have previously been individual targets for myocardial protection agents.
Limitations
While providing functional and molecular data in to the effects of sulfide therapy in myocardial I/R injury, this study has several limitations. Our time course for tissue harvest (3 hours after the onset of ischemia) is not able to account for long-term effects of sulfide on myocardial function and infarct extension, and conversely may miss rapid changes in the activation / phosphorylation status of certain apoptotic signaling proteins such as Bad. Additionally, the use of TUNEL staining may not capture cells in the initial stages of the apoptotic process and thus may underestimate the total number of apoptotic cells.
Conclusions
Therapeutic administration of H2S, generated as sodium sulfide, prior to the onset of reperfusion markedly attenuates myocardial ischemia-reperfusion injury. In addition to sulfide’s effects on limiting myocardial necrosis, this study demonstrates sulfide also effects pro-apoptotic signaling, limiting apoptosis. The anti-apoptotic properties of sulfide may provide myocardial protection in addition to sulfide’s ability to limit myocardial necrosis. H2S may have a valuable therapeutic role in the clinical setting when administered concomitantly with the coronary revascularization process.
Acknowledgments
We thank the staff of the Animal Research Facility at the Beth Israel Deaconess Medical Center for their efforts. The sodium sulfide solution used in the current studies was produced and formulated by Paul Hill (Ikaria Inc., Seattle, WA).
Supported by NHLBI grant HL46716. Drs. Sodha and Clements are supported in part by NIH grant T-32HL076130-02 and the Irving Bard Memorial Fellowship
Partial funding for this project was provided by Ikaria, Inc., Seattle, Washington
Footnotes
Presented at the 21st European Association for Cardio-Thoracic Surgery Annual Meeting, Geneva, Switzerland, September 18th, 2007.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Baines CP, Molkentin JD. STRESS signaling pathways that modulate cardiac myocyte apoptosis. J Mol Cell Cardiol. 2005;38(1):47–62. doi: 10.1016/j.yjmcc.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 2.Anselmi A, Abbate A, Girola F, Nasso G, Biondi-Zoccai GG, Possati G, Gaudino M. Myocardial ischemia, stunning, inflammation, and apoptosis during cardiac surgery: a review of evidence. Eur J Cardiothorac Surg. 2004;25(3):304–311. doi: 10.1016/j.ejcts.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 3.Zorc M, Vraspir-Porenta O, Zorc-Pleskovic R, Radovanovic N, Petrovic D. Apoptosis of myocytes and proliferation markers as prognostic factors in end-stage dilated cardiomyopathy. Cardiovasc Pathol. 2003;12(1):36–39. doi: 10.1016/s1054-8807(02)00134-5. [DOI] [PubMed] [Google Scholar]
- 4.Roberts MJ, Young IS, Trouton TG, Trimble ER, Khan MM, Webb SW, Wilson CM, Patterson GC, Adgey AA. Transient release of lipid peroxides after coronary artery balloon angioplasty. Lancet. 1990;336(8708):143–145. doi: 10.1016/0140-6736(90)91661-s. [DOI] [PubMed] [Google Scholar]
- 5.Kim KB, Chung HH, Kim MS, Rho JR. Changes in the antioxidative defensive system during open heart operations in humans. Ann Thorac Surg. 1994;58(1):170–175. doi: 10.1016/0003-4975(94)91094-4. [DOI] [PubMed] [Google Scholar]
- 6.Whiteman M, Armstrong JS, Chu SH, Jia-Ling S, Wong BS, Cheung NS, Halliwell B, Moore PK. The novel neuromodulator hydrogen sulfide: an endogenous peroxynitrite’scavenger’? J Neurochem. 2004;90(3):765–768. doi: 10.1111/j.1471-4159.2004.02617.x. [DOI] [PubMed] [Google Scholar]
- 7.Whiteman M, Cheung NS, Zhu YZ, Chu SH, Siau JL, Wong BS, Armstrong JS, Moore PK. Hydrogen sulphide: a novel inhibitor of hypochlorous acid-mediated oxidative damage in the brain? Biochem Biophys Res Commun. 2005;326(4):794– 798. doi: 10.1016/j.bbrc.2004.11.110. [DOI] [PubMed] [Google Scholar]
- 8.Fiorucci S, Antonelli E, Distrutti E, Rizzo G, Mencarelli A, Orlandi S, Zanardo R, Renga B, Di Sante M, Morelli A, Cirino G, Wallace JL. Inhibition of hydrogen sulfide generation contributes to gastric injury caused by anti-inflammatory nonsteroidal drugs. Gastroenterology. 2005;129(4):1210–1224. doi: 10.1053/j.gastro.2005.07.060. [DOI] [PubMed] [Google Scholar]
- 9.Mitsuhashi H, Yamashita S, Ikeuchi H, Kuroiwa T, Kaneko Y, Hiromura K, Ueki K, Nojima Y. Oxidative stress–dependent conversion of hydrogen sulfide to sulfite by activated neutrophils. Shock. 2005;24(6):529– 534. doi: 10.1097/01.shk.0000183393.83272.de. [DOI] [PubMed] [Google Scholar]
- 10.Geng B, Chang L, Pan C, Qi Y, Zhao J, Pang Y, Du J, Tang C. Endogenous hydrogen sulfide regulation of myocardial injury induced by isoproterenol. Biochem Biophys Res Commun. 2004;318(3):756–763. doi: 10.1016/j.bbrc.2004.04.094. [DOI] [PubMed] [Google Scholar]
- 11.Johansen D, Ytrehus K, Baxter GF. Exogenous hydrogen sulfide (H2S) protects against regional myocardial ischemia-reperfusion injury--Evidence for a role of K ATP channels. Basic Res Cardiol. 2006;101(1):53–60. doi: 10.1007/s00395-005-0569-9. [DOI] [PubMed] [Google Scholar]
- 12.Zhu YZ, Wang ZJ, Ho P, Loke YY, Zhu YC, Huang SH, Tan CS, Whiteman M, Lu J, Moore PK. Hydrogen sulfide and its possible roles in myocardial ischemia in experimental rats. J Appl Physiol. 2007;102(1):261– 268. doi: 10.1152/japplphysiol.00096.2006. [DOI] [PubMed] [Google Scholar]
- 13.Maulik N, Yoshida T, Das DK. Oxidative stress developed during the reperfusion of ischemic myocardium induces apoptosis. Free Radic Biol Med. 1998;24(5):875. doi: 10.1016/s0891-5849(97)00388-2. [DOI] [PubMed] [Google Scholar]
- 14.Krown KA, Page MT, Nguyen C, Zechner D, Gutierrez V, Comstock KL, Glembotski CC, Quintana PJ, Sabbadini RA. Tumor necrosis factor alpha-induced apoptosis in cardiac myocytes. Involvement of the sphingolipid signaling cascade in cardiac cell death . J Clin Invest. 1996;98(12):2854–2865. doi: 10.1172/JCI119114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, Rudensky AY. Foxp3-dependent programme of regulatory T-cell differentiation. Nature. 2007;445(7129):771–775. doi: 10.1038/nature05543. [DOI] [PubMed] [Google Scholar]
- 16.Kleine AH, Glatz JF, Van Nieuwenhoven FA, Van der Vusse GJ. Release of heart fatty acid-binding protein into plasma after acute myocardial infarction in man. Mol Cell Biochem. 1992;116(1–2):155–162. doi: 10.1007/BF01270583. [DOI] [PubMed] [Google Scholar]
- 17.O’Donoghue M, de Lemos JA, Morrow DA, Murphy SA, Buros JL, Cannon CP, Sabatine MS. Prognostic utility of heart-type fatty acid binding protein in patients with acute coronary syndromes. Circulation. 2006;114(6):550– 557. doi: 10.1161/CIRCULATIONAHA.106.641936. [DOI] [PubMed] [Google Scholar]
- 18.Wang S, El-Deiry WS. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene. 2003;22(53):8628–8633. doi: 10.1038/sj.onc.1207232. [DOI] [PubMed] [Google Scholar]
- 19.Yaoita H, Ogawa K, Maehara K, Maruyama Y. Attenuation of ischemia/reperfusion injury in rats by a caspase inhibitor. Circulation. 1998;97(3):276–281. doi: 10.1161/01.cir.97.3.276. [DOI] [PubMed] [Google Scholar]
- 20.Szabo C, Zingarelli B, O'Connor M, Salzman AL. DNA strand breakage, activation of poly (ADP-ribose) synthetase, and cellular energy depletion are involved in the cytotoxicity of macrophages and smooth muscle cells exposed to peroxynitrite. Proc Natl Acad Sci U S A. 1996;93(5):1753–1758. doi: 10.1073/pnas.93.5.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khan TA, Ruel M, Bianchi C, Voisine P, Komjati K, Szabo C, Sellke FW. Poly(ADP-ribose) polymerase inhibition improves postischemic myocardial function after cardioplegia-cardiopulmonary bypass. J Am Coll Surg. 2003;197(2):270–277. doi: 10.1016/S1072-7515(03)00538-6. [DOI] [PubMed] [Google Scholar]