

Abstract
The striatum, which processes cortical information for behavioral output, is a key target of Huntington's disease (HD), an autosomal dominant condition characterized by cognitive decline and progressive loss of motor control. Increasing evidence implicates deficient glutamate uptake caused by a down-regulation of GLT1, the primary astroglial glutamate transporter. To test this hypothesis, we administered ceftriaxone, a β-lactam antibiotic known to elevate GLT1 expression (200 mg/kg, ip, for 5 days), to symptomatic R6/2 mice, a widely studied transgenic model of HD. Relative to vehicle, ceftriaxone attenuated several HD behavioral signs: paw clasping and twitching were reduced, while motor flexibility, as measured in a plus maze, and open-field climbing were increased. Assessment of GLT1 expression in striatum confirmed a ceftriaxone-induced increase relative to vehicle. To determine if the change in behavior and GLT1 expression represented a change in striatal glutamate handling, separate groups of behaving mice were evaluated with no-net-flux microdialysis. Vehicle treatment revealed a glutamate uptake deficit in R6/2 mice relative to wild-type controls that was reversed by ceftriaxone. Vehicle-treated animals, however, did not differ in GLT1 expression, suggesting that the glutamate uptake deficit in R6/2 mice reflects dysfunctional rather than missing GLT1. Our results indicate that impaired glutamate uptake is a major factor underlying HD pathophysiology and symptomology. The glutamate uptake deficit, moreover, is present in symptomatic HD mice and reversal of this deficit by up-regulating the functional expression of GLT1 with ceftriaxone attenuates the HD phenotype.
Keywords: Huntington's disease, ceftriaxone, R6/2, striatum, microdialysis, glutamate
Huntington's disease (HD) is an autosomal dominant condition caused by an expansion of a polyglutamine (CAG) repeat in the coding region of the huntingtin gene (Huntington's Disease Collaborative Research Group, 1993). Symptoms, including adventitious movements (e.g. chorea, dystonia, tremor), cognitive impairment, and emotional disturbances, typically begin in middle age and progressively worsen until death (Harper, 1991). Although much work has aimed to characterize the mechanisms underlying HD, the function of the huntingtin protein and the pathogenesis of HD are far from understood.
The hallmark pathology is preferential deterioration of GABAergic medium-sized spiny neurons in striatum, which integrates cortical information for behavioral output (Graybiel, 1995). The onset and progression of the behavioral phenotype, however, is likely caused by deficits in corticostriatal information processing that precede cell death (Cepeda et al., 2003, 2007). In fact, several lines of transgenic mice that model HD show behavioral symptoms long before significant cell loss (Carter et al., 1999, Menalled et al., 2003). It is likely, therefore, that striatal dysfunction rather than death underlies the HD behavioral phenotype.
This view is consistent with evidence that in behaving, symptomatic R6/2 mice striatal neurons discharge at a significantly faster rate than in wild-type (WT) animals (Rebec et al., 2006). Glutamate, an excitatory amino acid, is likely to play a key role. When studied in vitro, for example, striatal neurons in HD mice show exaggerated glutamate-dependent responses, more depolarized membrane potentials, and increased intracellular Ca2+ levels after N-methyl-D-aspartate receptor activation compared to WT (Cepeda et al., 2001; Klapstein et al., 2001; Laforet et al., 2001; Zeron et al., 2002; Li et al., 2004). Although the nature of the glutamate problem in HD is unclear, increasing evidence implicates GLT1, the Na+-dependent glial transporter of glutamate. GLT1 is responsible for the removal of most extracellular glutamate (Robinson, 1998) and there is also mounting evidence that GLT1 actively participates in the regulation of synaptic transmission (see, Tzingounis and Wadiche, 2007). In HD mouse models, GLT1 is down-regulated and appears responsible for decreased striatal glutamate uptake (Lievens et al., 2001; Behrens et al., 2002; Shin et al., 2005). Decreased GLT1 mRNA (Arzberger et al., 1997) and deficient glutamate uptake (Hassel et al., 2007) also has been reported in postmortem brain tissue taken from HD patients. Thus, reduction of GLT1 is likely a key component of HD pathophysiology and symptomology.
Ceftriaxone, a β-lactam antibiotic, has recently been shown to increase the functional expression of GLT1 (Rothstein et al., 2005). Thus, we tested the hypothesis that up-regulation of GLT1 by ceftriaxone improves HD signs in symptomatic R6/2 mice. Western blot and immunohistochemistry were used to confirm a ceftriaxone-induced increase in GLT1 expression, and no-net-flux microdialysis coupled online to capillary electrophoresis with laser-induced fluorescence (CE-LIF) was used to monitor functional changes in extracellular glutamate.
Experimental Procedures
Animals
Male transgenic R6/2 mice (B6CBA-TgN[HDexon1]62Gpb) and WT controls were obtained from The Jackson Laboratories (Bar Harbor, ME). The R6/2 line is characterized by a rapidly progressive HD phenotype that leads to death in ∼13-14 weeks (Mangiarini et al., 1996). Mice were housed individually in the departmental animal colony under standard conditions (12 hr light/dark cycle with lights on at 07:30) with access to food and water ad libitum. Both the housing and experimental use of animals followed the National Institutes of Health guide-lines and were approved by the Institutional Animal Care and Use Committee.
Genotype and CAG Repeat Length
Genomic DNA was extracted from tail tissue samples in 25 μL cell lysis buffer (50 mM Tris, pH 8.0; 25 mM EDTA; 100 mM NaCl; 0.5% IGEPAL CA-630; 0.5% Tween 20) and proteinase K (10 mg/mL; 60 μg/reaction) at 55°C for 2 hours with gentle mixing after the first hour. DNA was diluted with 300 μL filter-sterilized HPLC water, heated to 100°C for 10 min, centrifuged for 2 min at 17,000× g, and stored at 4°C. PCR and agarose gel electrophoresis were used to determine CAG repeat length. Primers were 31329 (5′-ATGAAGGCCTTCGAGTCCCTCAAGTCCTTC-3′) and 33934 (5′-GGCGGCTGAGGAAGCTGAGGA-3′) (Mangiarini et al., 1996). Each reaction consisted of 2.0 μL DNA template (40 to 100 ng/μL), 0.4 μl each primer (20 μM), 7.2 μL filter-sterilized HPLC water, and 10.0 μL 2× Biomix™ Red (Bioline USA Inc., Taunton, MA) for 20 μL total volume. Cycling conditions were 94°C for 90 s followed by 30 cycles of 94°C for 30 s, 62°C for 45 s, 72°C for 90 s with a final elongation at 72°C for 10 min. Electrophoresis of samples was performed in 1.7% agarose with 0.1 μg/mL ethidium bromide at 5V/cm for 90 min using ΦX174/HaeIII digest as DNA standard.
Gels were evaluated with Kodak Image Station 4000R and Kodak Molecular Imaging software (Carestream Molecular Imaging, New Haven, CT) to confirm genotype and determine CAG repeat length. Using Clone Manager software (Sci-Ed Software, Cary, NC) primers were aligned to the huntingtin gene sequence acquired from the National Center for Biotechnology Information (www.ncbi.nlm.nih.gov). Alignment of primers to template showed that primer 31329 is immediately upstream of the CAG repeat region, and primer 33934 overlaps the downstream region of CCG (proline) repeats by one codon plus one nucleotide. Consequently, the DNA fragment amplified by PCR is 86 bp longer than the CAG repeat region. Computer analysis of fragment migration distance against ΦX174/HaeIII digest standard showed that our experimental R6/2 mice had 121 ± 1.8 (mean ± sem) repeated CAG codons.
Treatment Protocol
R6/2 and WT mice were treated with intraperitoneal injections of 200 mg/kg ceftriaxone (Sigma, St. Louis, MO), or equivolume saline vehicle once daily for 5 consecutive days between 09:00 and 10:00. It has been well established that this ceftriaxone dosing regimen increases the functional expression of GLT1 in in vivo preparations (Chu et al., 2006, Lipski et al., 2007, Mineur et al., 2007, Ouyang et al., 2007, Rawls et al., 2007, Rothstein et al., 2005). In addition, ceftriaxone is very tolerable at this dose and is non-toxic to the central nervous system. At high doses, diarrhea, nausea, vomiting, and candidiasis can occur (Lamb et al., 2002). Mice were monitored daily for these effects, and they never appeared. Thus, our treatment protocol elicited no overt side effects.
Treatment Groups and Experimental Timeline
Animals were 6 weeks of age when treatment began, which corresponds to a time of early symptom development in R6/2 mice (Carter et al., 1999). Paw-clasping was assessed daily before injection to confirm symptom expression. Twenty-four hours after the final injection when the animals were 7 weeks of age (post-treatment day 1), paw-clasping was re-assessed, along with other behavioral responses. Separate groups of similarly aged mice received the same treatment protocol but were assessed for glutamate or were sacrificed for Western blot or immunohistochemistry on post-treatment day 1. To test whether ceftriaxone has enduring effects on behavior, all behavioral assessments were repeated one week after the final treatment (post-treatment day 7).
Behavioral assessments
All behavioral tests were videotaped and coded by independent observers who were blind to both genotype and treatment.
Body Weight
There was no difference in body weight between genotype and treatment across all test days (p > 0.05). This is consistent with previous work showing no difference in body weight until 12 weeks of age, at which time R6/2 mice lose weight compared to WT (Carter et. al., 1999).
Paw-Clasping
Mice were suspended from the tail for 1 min to elicit the clasping phenotype, which was scored on a scale from 0 to 3, where 0 represented no clasping, 1 = forepaws clasping, 2 = forepaws and one hindpaw clasping, and 3 = all paws clasping.
Plus-maze
Mice were placed in a start box in one arm of an enclosed plus-maze (each clear Plexiglas® arm was 30 cm long, 11 cm wide, 11 cm high) and allowed to explore freely. The total number of turns into a perpendicular arm in each session was calculated. A criterion was established so that only mice that made 20 arm choices in 10 min were included in the analysis; this criterion excluded three R6/2 mice treated with saline and two R6/2 mice treated with ceftriaxone.
Open-field
Mice were placed individually in an open-field arena (45 X 26 cm) having clear Plexiglas® walls (20 cm) with standard bedding. A wire-mesh climbing box (11.5 X 9 cm) was placed in the center of the arena. Animals explored freely for 10 min. Climbing and twitching behaviors were analyzed using BEST Analysis software (Educational Consulting Inc., Las Vegas, NV). Inter-rater reliability was confirmed with Cohen's Kappa values (∼0.8-1.0). Each rater practiced coding behaviors until reliability was achieved. Climbing was defined as having all four feet on the side of the climbing box. Twitching, a manifestation of the shuddering phenotype (Mangiarini et al., 1996), occurred primarily in R6/2 mice but because it occurred in WT mice, albeit rarely, all WT mice were included in the analysis.
Western Blot
Mice were sacrificed by spinal translocation, brains were removed, and the striatum from both hemispheres was dissected and frozen for immunoblotting. Brain tissue was homogenized in 500 μl of lysis buffer (50 mM TrisHCl, 150 mM NaCl, 1 mM EDTA, 0.5% NP-40, 1% Triton, 0.1% SDS) supplemented with protease inhibitor cocktail. The tissue mixture was centrifuged at 14,000 rpm for 15 min at 4 C. Protein in the tissue extracts was measured by the Bio-Rad protein assay (Bio-Rad Laboratories, Hercules, CA). Samples, in Laemmli buffer, were heated for 4 min at 95 C and loaded onto 4%-20% glycine gel. Equal amounts of cell extracts from each sample were run in the same gel with protein standards of known molecular weight (Sigma) under identical conditions. Samples were separated in 4%-20% glycine gel (Invitrogen, Carlsbad, CA) in a Mini-Cell apparatus for 1h at 200 V at room temperature. Proteins were then transferred onto a nitrocellulose membrane electrophoretically at 30 V for 1h. The transferred proteins were visualized by staining the membrane with 0.1% Ponceau S solution in 5% acetic acid (Sigma). Unbound sites on the membrane were blocked by incubating the nitrocellulose membrane with 3% milk in TBST (0.5M Tris HCl; 1.5M NaCl, pH7.4; 10ml of 10% Tween20) for 30 min. The guinea pig anti-GLT1 antibody (Chemicon), diluted 1:5000 in 3% milk in TBST, was incubated overnight at 4 C. Membranes were washed with TBST five times, each for 5 min, and then incubated for 2 h with horseradish peroxidase secondary antibody at 1:10000 dilution with 3% milk in TBST. Membranes were washed five times in TBST for 5 min. Equivalent protein loading was assessed by β-actin immunoblotting of the nitrocellulose membrane to confirm quality and relative loading. After incubation with the horseradish peroxidase kit (SuperSignal West Pico., Pierce Inc., Rockford, IL) for 1 min, the membranes were exposed to Kodak BioMax MR film (Fisher Inc., Pittsburgh, PA), and were developed on a SRX-101A machine (Pacific Northwest X-Ray Inc., Gresham, OR). Immunoblots were digitized using a Kodak Image Station 4000R (Kodak, New Haven, CT) to obtain the optical density of the bands.
Immunohistochemistry
We used immunohistochemical techniques described by Wood et al. (2005). Mice were deeply anesthetized with chloropent and transcardially perfused with cold phosphate-buffered saline (PBS, 0.1 M, pH 7.4) followed by 4% paraformaldehyde. Brains were removed, post-fixed by immersion in 4% paraformaldehyde for 1 h, and cryoprotected in 10% sucrose in 0.1 M PBS. Cryosections were cut coronally at 30μm on a sliding microtome. Serial sections through the striatum were processed free-floating for GLT1. Sections were rinsed in immunophosphate buffer (0.1 M PBS, 1% bovine serum albumin and 0.1% Triton-X100) followed by incubation in a blocking solution (4% normal goat serum in immunophosphate buffer). Sections were then incubated overnight at 4 C in guinea pig anti-GLT1 polyclonal Ab (1:5000; Chemicon International, Temecula, CA). On the following day, sections were rinsed in immunophosphate buffer and incubated in secondary antibody (1:200, biotinylated goat anti-guinea pig IgG; Vector Laboratories, Burlingame, CA). After rinsing in 0.1 M PBS, sections were incubated in PBS with ABC Complex using the Vector Standard Elite kit (Vector Laboratories). PBS rinses were followed by rinses in 1.0 M acetate buffer (pH 7.2), 0.2 M imidazole buffer (pH 9.2) and acetate-imidazole buffer. Sections were developed in a nickel-intensified diaminobenzidine chromagen reaction. After a final rinse in acetate-imidazole buffer, sections were transferred to PBS, and placed on pig-gelatin treated slides. The sections were counterstained with neutral red. The sections were then dehydrated, cleared, and coverslipped. Control sections processed without primary antibody demonstrated virtually no immunoreactivity. Optical density of immunolabeling in striatum (sample area = 5,000 μm2) was measured (Stereo Investigator, MicroBrightField, Inc., Williston, VT) as average luminosity per pixel (in 256 gray levels, where black = 0 and white = 256) at a final magnification of 1940X under brightfield illumination. Measurements were randomly sampled from anterior 0.38 to 0.98 mm, lateral 1.4 to 2.4 mm, and ventral 2.8 to 3.5 mm from bregma. Three random sections per animal (12 sections for each group) were used in the analysis. To control for differences in immunoreactivity across sections and animals, optical density measures within each section were expressed relative to immunostaining in striatal tissue devoid of GLT1 label.
In Vivo Glutamate Measurements
Mice were anesthetized with intraperitoneal injections of ketamine (65 mg/kg) and Domitor (0.5 mg/kg) for stereotaxic surgery to place guide cannula into striatum (anterior +0.5 mm, lateral +2.0 mm, and ventral -2.0 mm from bregma). Animals recovered for 7 days after cannula implantation. Microdialysis probes (2 mm long and 220 μm diameter) were inserted as described previously (Shou et al., 2006). No-net-flux measurements began 3 hours after insertion, which is sufficient to record active glutamate release (Lada et al., 1998).
Glutamate was measured at 30 s intervals in the dialysate by CE-LIF coupled on-line to the microdialysis probe (Shou et al., 2004). CE-LIF recordings began immediately after probe insertion and were monitored until glutamate levels stabilized. After 150-180 min of perfusion, no-net-flux measurements of glutamate began (Smith and Justice, 1994; Miele et al., 1996; Melendez et al., 2005). Glutamate dissolved in the balanced salt solution was infused into the probes at 4 or 5 concentrations between 0.5 and 30 μM to bracket the initial concentration recorded in dialysate. The dialysate glutamate concentration was determined by averaging the glutamate peak area from 15-20 electropherograms and comparing to a calibration curve. The difference between the infused glutamate and the measured glutamate was calculated for the y-axis and the infused concentration was used for the x-axis; thus, a linear regression was constructed for the no-net-flux plot. Probe recovery, which reflects the degree of glutamate uptake by striatal tissue, was obtained from the slope (m) of the linear regression. Extracellular glutamate concentration (zero point on the y-axis) was obtained as –b/m from solving y = mx + b for x when y = 0, where b is the y intercept and m is the slope of the regression. In a separate group of WT mice, L-trans-2,4-pyrrolidine dicarboxylate (PDC; Sigma) was infused into the microdialysis probes to confirm that decreased recovery corresponded to decreased uptake in striatum.
Statistics
Group data were calculated as mean ± SEM. Two-way analyses of variance (ANOVA) were used to analyze basal glutamate levels (y = 0), glutamate uptake (slope), optical density of immunoblots, and relative luminosity of immunohistochemistry between genotype and treatment. A one-way repeated-measures ANOVOA (RMANOVA) was used to evaluate clasping. Two-way RMANOVAs were used to analyze number of turns in the plus-maze, percent time climbing, and percent time twitching between genotype and treatment. Group means were compared with Bonferroni post-tests. Statistically significant data were reported if p < 0.05.
Results
Ceftriaxone attenuates multiple manifestations of the HD behavioral phenotype
Unlike WT, R6/2 mice engage in a paw-clasping response that mimics dystonic-like movements in HD patients and worsens with disease progression. Over the course of ceftriaxone treatment, R6/2 mice showed a reduction in paw-clasping compared to saline-treated R6/2s (Fig. 1). In fact, after only two days of treatment, paw-clasping was significantly attenuated on test days 3, 4, and 5 (P < 0.05) and on post-treatment day 1 (P < 0.01). This effect, however, was not long lasting as revealed by only a trend for decreased clasping in ceftriaxone-treated R6/2 mice on post-treatment day 7 (P = 0.066).
Figure 1.

Ceftriaxone reduced the clasping phenotype in R6/2 mice after only 2 days of treatment, and this effect persisted through post-treatment day 1 (i.e. test day 1 after the break) (n = 7 for saline, n = 8 for ceftriaxone). WT mice were not included in this analysis as they showed no clasping phenotype. *p < 0.05, **p < 0.01. Error bars represent SEM.
Another behavioral symptom in R6/2 mice is motor inflexibility, as revealed by fewer perpendicular arm choices (turns) in a plus-maze (Rebec et al., 2003). In this test, saline-treated R6/2s made fewer turns than saline-treated WT mice on post-treatment days 1 and 7 (Fig. 2A). In contrast, ceftriaxone treatment increased R6/2 turning above saline and matched the WT level on pos-treatment day 1. Moreover, although there was not a difference between the number of turns in the plus-maze between R6/2 mice treated with saline and those treated with ceftriaxone on post-treatment day 7, the ceftriaxone-treated R6/2 were also not significantly different than WT (P > 0.05).
Figure 2.

Ceftriaxone attenuates the R6/2 behavioral phenotype as measured in the plus maze and open-field. A, R6/2 mice treated with ceftriaxone made significantly more turns in the plus-maze than saline-treated R6/2 mice on post-treatment day 1, and did not differ from WT mice on both post-treatment days (n = 9 for WT saline and ceftriaxone, n = 5 for R6/2 saline, n = 7 for R6/2 ceftriaxone). B, R6/2 mice treated with ceftriaxone engaged in climbing more than saline treated R6/2 mice on post-treatment day 1, and did not differ from WT mice on both post-treatment days. Saline treated R6/2 mice climbed less than WT mice on both post-treatment days. C, Treatment with ceftriaxone attenuated expression of the twitching phenotype in R6/2 mice on post-treatment day 7 (n= 9 for WT saline, WT ceftriaxone, and R6/2 ceftriaxone, n = 8 for R6/2 saline). *p < 0.05, **p < 0.01, ***p < 0.001. Error bars represent SEM.
Our final test focused on behavioral activity in an open-field arena. On post-treatment days 1 and 7, saline-treated R6/2s spent less time climbing than WTs (P < 0.001), but with ceftriaxone treatment, climbing time was comparable in R6/2 and WT mice on both post-treatment days and significantly more than saline-treated R6/2 mice on post-treatment day 1 (P < 0.05) (Fig. 2B). Furthermore, R6/2 mice treated with saline twitch more than R6/2 mice treated with ceftriaxone on post-treatment day 7 (P < 0.01), and there was no difference in twitching between WT and ceftriaxone-treated R6/2 mice on the same day (Fig. 2C). There was, however, no difference in twitching between saline and ceftriaxone-treated R6/2 mice on post-treatment day 1.
Ceftriaxone increases GLT1 expression in striatum
To confirm that ceftriaxone is able to increase GLT1 expression in HD mice, we quantified striatal GLT1 expression in separate groups of similarly treated WT and R6/2 mice using Western blot. Relative to saline, ceftriaxone increased striatal GLT1 expression in both WT and R6/2 mice (P < 0.05) (Fig. 3A, B), providing additional support for the ceftriaxone-induced increase in glutamate uptake. Interestingly, at 7 weeks of age, GLT1 expression was comparable in WT and R6/2 mice treated with saline. Since previous findings have shown reduced GLT1 expression in older R6/2 mice compared to WTs (Lievens et al., 2001; Behrens et al., 2002; Shin et al., 2005), we quantitatively confirmed our Western blot results with immunohistochemistry (Fig. 3C, D).
Figure 3.
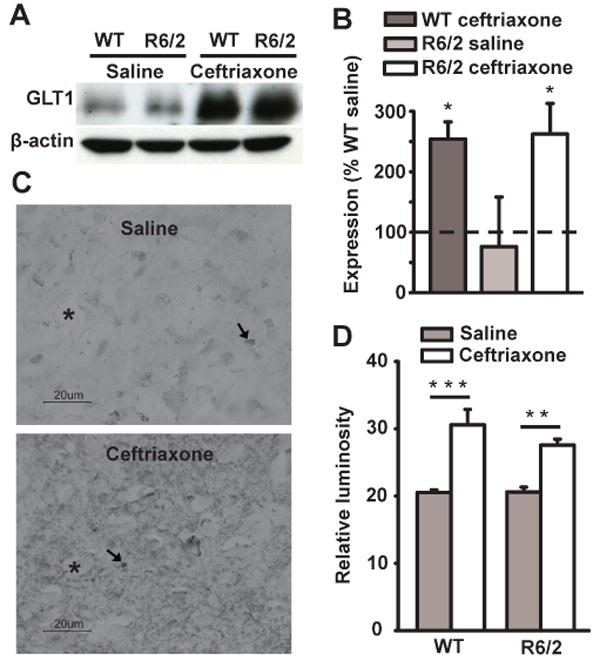
Ceftriaxone increases GLT1 expression in striatum. A, Digitized print of Western blot for GLT1 and β-actin protein levels. Anti-β-actin was used to control for differences in protein loading and showed no change across genotype and treatment. B, Quantitative analysis of Western blots. The difference in mean optical density is expressed as percentage of GLT1 levels in WT saline. Ceftriaxone increased GLT1 expression in striatum of ceftriaxone-treated WT and R6/2 animals compared to saline-treated groups. C, Digital light micrographs of GLT1 immunohistochemistry. Each image is a coronal section of striatal tissue from an R6/2 mouse treated with either saline or ceftriaxone and shown at the magnification at which data were collected. Examples of punctate GLT1-immunopositive glia are denoted by arrows. Neuronal perikarya are denoted by asterisks. Note that the density of GLT1 label is greater in ceftriaxone-treated animals revealing more prominent non-labeled (ghost-like) perikarya compared to the saline groups. D, Mean relative luminosity of GLT1-positive immunoreactivity in striatum of R6/2 and WT mice. For B and D, *p < 0.05, **p < 0.01, ***p < 0.001, n = 4 animals in each group. Error bars represent s.e.m.
Ceftriaxone restores glutamate uptake in striatum
Extracellular glutamate was assessed in striatum of freely behaving R6/2 and WT mice by combining no-net-flux microdialysis with CE-LIF. This approach permitted direct quantification of basal glutamate levels and glutamate uptake under naturally occurring behavioral conditions. No-net flux curves represent in vivo recovery of the probes, which is strongly dependent on cellular uptake such that increases in uptake increase recovery and the no-net-flux slope (Melendez et al., 2005). To confirm that recovery indicated cellular uptake, we infused the selective competitive glutamate uptake inhibitor PDC (200 μM) through the microdialysis probes in saline-treated WT mice (n = 4) and found a significant decrease in recovery (cellular uptake, indicated by flattening of the slope) from 57% to 16% (P < 0.001) and an increase (480%) in basal glutamate (P < 0.001) (Fig.4A, inset).
Figure 4.

Ceftriaxone decreases the basal level of extracellular glutamate and increases its uptake as measured by no-net-flux microdialysis with CE-LIF for glutamate. A, Averaged linear regressions of the no-net-flux plots. Positive numbers on the y-axis indicate diffusion to and negative numbers indicate diffusion from the probe based on the concentration of applied exogenous glutamate. The point of no-net-flux (y = 0 μM; horizontal dotted line) corresponds to glutamate equilibrium between tissue and microdialysis probe revealing the level of extracellular glutamate. The slope of each line for each group corresponds to probe recovery, reflecting the degree of glutamate uptake (i.e., increases in slope indicate increases in uptake). Note that PDC markedly decreased the slope of the no-net-flux regression (inset). B, Mean basal glutamate levels. Ceftriaxone decreased basal glutamate in WT and R6/2 mice. C, Mean glutamate uptake. R6/2 mice exhibited reduced glutamate uptake compared to WT mice. Ceftriaxone increased glutamate uptake in both WT and R6/2 mice. Note that ceftriaxone treatment restored glutamate uptake in R6/2 mice (mean slope = 0.558) to that of WT saline (mean slope = 0.544). Although glutamate uptake in ceftriaxone-treated WT mice was greater than in R6/2 mice treated with ceftriaxone, the magnitude of the ceftriaxone-mediated increase in glutamate uptake was equivalent. For A, B and C, n = 10 for WT saline, n = 8 for WT ceftriaxone and R6/2 ceftriaxone, n = 9 for R6/2 saline. *p < 0.05, **p < 0.01, ***p < 0.001. Error bars represent SEM.
Basal levels of glutamate were similar in WT (8.2 ± 1.4 μM) and R/62 (8.6 ± 1.6 μM) mice treated with saline, but uptake was decreased in R6/2 relative to WT (P < 0.05) (Fig. 4A,B,C). Treatment with ceftriaxone significantly increased glutamate uptake in WT (24%) (P < 0.001) and R6/2 (22%) (P < 0.05) mice (Fig. 4C), which was concomitant with a reduction in basal glutamate (WT = -59% (P < 0.001), R6/2 = -53% (P < 0.01); Fig. 4B). In fact, R6/2 mice treated with ceftriaxone had the same degree of glutamate uptake as saline-treated WT mice.
Discussion
Our results show that increasing striatal GLT1 expression attenuates the neurological signs of HD in R6/2 mice. The GLT1 increase, moreover, enhances glutamate uptake, suggesting that a dysregulation of striatal glutamate transmission plays a key role in HD. It also is interesting that, although glutamate uptake is attenuated in symptomatic R6/2 mice relative to wild-type, there is no difference in the expression level of GLT1. Thus, the GLT1 deficit appears to represent a deficiency in function rather than level of protein. The increase in GLT1 expression after ceftriaxone treatment, however, is accompanied by an increase in uptake, suggesting that the newly expressed protein is functional. Our results, therefore, pave the way for assessment of the potential therapeutic benefits of ceftriaxone and other compounds that may increase GLT1 expression in HD.
We assessed several behavioral signs of HD in R6/2 mice, and in each case ceftriaxone treatment led to a significant improvement relative to vehicle. Clasping or dyskinesia of the limbs when R6/2 mice are suspended by the tail is one of the first noticeable signs of the HD phenotype (Mangiarini et al., 1996). A ceftriaxone-induced improvement was evident by the third treatment day and persisted for each treatment day thereafter as well as the first post-treatment test day. Although the loss of this effect one week after treatment argues against a long-term change by ceftriaxone, our results in the plus-maze and open-field tests suggest a more complex picture. For example, on post-treatment day 1, R6/2 mice treated with ceftriaxone matched WT performance in both the plus maze and climbing in the open-field. Their performance, moreover, was not different than WT or saline-treated R6/2 mice on post-treatment day 7. In addition, R6/2 open-field twitching, which did not improve on the first post-treatment day in ceftriaxone-treated animals, showed a significant improvement over saline one week later. In fact, twitching had declined so dramatically by post-treatment day 7 that ceftriaxone-treated R6/2 mice were not significantly different from WT. Conceivably, different neural circuits changing at different time courses may explain these results, a conclusion supported by evidence of multiple behavioral pathways within striatal circuitry (Alexander et al., 1986). Other interpretations, however, are equally likely, including a complex interaction among the behaviors themselves such that a change in one could, over time, force a change in another. Thus, although it may be difficult to link a specific mechanism to a specific behavioral change, our results reveal significant improvement in the HD phenotype with ceftriaxone.
Another interesting observation is that ceftriaxone failed to alter the behavioral response of WT mice. None of our behavioral tests revealed a difference between WT animals treated with saline or ceftriaxone. Even open-field activity, which includes multiple movements and motor sequences (Dorner et al., 2007), was unaffected in WT mice. Although we cannot rule out some subtle behavioral effects of ceftriaxone not detected by our measurements, it appears that the drug does not alter common WT behavioral responses.
The behavioral effects of ceftriaxone in R6/2 mice cannot be explained by an antibiotic action since there is no indication of sepsis at this stage of HD. In fact, we focused on animals at 7-8 weeks of age because of the clear HD behavioral signs without the complications (e.g., diabetes, weight loss) that emerge at later ages. It also is unlikely that ceftriaxone exerted a general sedative or muscle-relaxing effect on behavior since the improvements with ceftriaxone include increases in turning and climbing behavior. We also saw none of the side effects characteristic of high ceftriaxone doses (e.g., diarrhea and nausea), ruling out a non-selective dose effect on behavior. It is also relevant that 3.5 μmol/L ceftriaxone, the EC50 required to increase GLT1 expression (Rothstein et al., 2005), is comparable to levels of ceftriaxone found in the central nervous system of patients undergoing therapy for meningitis (0.3 to 6 μmol/L) (Nau et al., 1993). A related issue is that the polyglutamine repeat length in R6/2 mice may vary among litters between 110-150 repeats. Although repeat length influences onset and progression of the HD phenotype (Brandt et al., 1996), all our R6/2 mice had a quantitatively reproducible phenotype with little variation between animals. This result is consistent with our repeat-length analysis, which also showed little variation (see Experimental Procedures).
We chose our ceftriaxone dose and treatment schedule because of our interest in GLT1. At 200 mg/kg for 5 consecutive days, ceftriaxone is known to elicit a maximal increase in GLT1 expression without causing neurotoxic effects (Rothstein et al., 2005; Chu et al., 2007). Our immunohistochemistry and Western blot data not only confirm these results, but also indicate that HD itself is not an impediment to increasing GLT1 expression. Thus, additional compounds believed to increase glutamate transport (e.g., the synthetic neuroimmunophilin GPI-1046) (Ganel et al., 2006) also may effectively reverse HD signs in transgenic models. Moreover, further testing of ceftriaxone (e.g., lower doses, longer treatment duration) in models showing a relatively long pre-symptomatic period can be used to address the possibility that increasing GLT1 expression even before symptom onset could significantly delay and perhaps minimize HD progression.
Our no-net-flux microdialysis data reveal for the first time that striatal glutamate uptake in symptomatic R6/2 mice is, in fact, impaired. This finding in saline-treated animals not only confirms the importance of a glutamate dysregulation in HD, but also that the uptake problem occurs when animals are fully awake and behaving. Interestingly, however, a decline in glutamate uptake was not reflected in an increase in extracellular glutamate. Thus, glutamate transmission in these mice may adapt to the loss of uptake with a compensatory decrease in glutamate release. Although this hypothesis is difficult to confirm in that most extracellular glutamate sampled by microdialysis appears to originate from a non-synaptic source (Timmerman and Westerink, 1997), previous data based on microdialysis done in conjunction with CE-LIF technology show that basal glutamate levels in striatum are sensitive to both electrical stimulation of corticostriatal neurons and application of tetrodotoxin, suggesting that the source of glutamate is, at least in part, synaptic (Lada et al., 1998, Rebec et al., 2005). Furthermore, it has been well established that the no-net-flux curves represent in vivo recovery of the probes, such that increases in cellular uptake increase recovery as well as the slope of the nonet-flux linear regression (Bungay et al., 1990; Parsons and Justice, 1992; Smith and Justice, 1994; Melendez et al., 2005). Finally, to confirm that recovery represents cellular uptake, we applied PDC, a glutamate uptake inhibitor, through the microdialysis probe and showed a marked increase in basal glutamate concomitant with decreased recovery of the probe.
Treatment with ceftriaxone restored glutamate uptake in R6/2 mice to saline-treated WT levels. Immunohistochemistry and Western blot confirmed that this effect occurred in conjunction with up-regulation of GLT1 expression. Unlike our behavioral results, however, the increase in GLT1 appeared in both WT and R6/2 mice. In WT animals, therefore, an increase in GLT1 and a concomitant reduction in glutamate have little, if any, behavioral impact, perhaps owing to the non-synaptic nature of glutamate in extracellular fluid (see above). Even if this is the case, however, it is clear that the change in uptake and extracellular glutamate induced by ceftriaxone has a significant effect on HD mice. This effect may be due to other HD-related changes in glutamate transmission such as a change in the sensitivity of glutamate receptors (see Zeron et al., 2002). A related point is that we found no difference in GLT1 expression between WT and R6/2 mice regardless of treatment. Thus, the uptake deficit in R6/2 mice appears to be due to dysfunctional rather than missing GLT1. If so, then ceftriaxone not only increases the amount of GLT1 but the increase is also functional. Interestingly, Kalivas and colleagues have also reported deficits in glutamate uptake that were not accompanied by decreased glutamate transporter expression in rats that were exposed to repeated ethanol injections (Melendez et al., 2005). It is also relevant that deficient glutamate uptake was observed in postmortem brain tissue taken from HD patients, while no changes were found in glutamate transporter levels (Hassel et al., 2007).
Previous studies using cultured striatal tissue from relatively old (12-14 weeks of age) and presumably severely symptomatic R6/2 mice, have reported decreased GLT1 expression along with deficient glutamate uptake (Lievens et al., 2001; Behrens et al., 2002; Shin et al., 2005). The inconsistency with our data may be related to the preparation since our results are based on intact, behaving animals, and extracellular glutamate is sensitive to the level of behavioral activation (Sandstrom and Rebec, 2007), most likely due to changes in cortical input (Lada et al, 1998). It is also relevant that R6/2 mice >12 weeks of age are often severely symptomatic (Carter et al., 1999). Our evidence of a glutamate uptake deficit at a relatively early stage of symptom development is important because such a deficit could be precursory to other pathogenic mechanisms in HD.
The ability of ceftriaxone to reverse the glutamate uptake deficit indicates that the increase in GLT1 expression can overcome dysfunctional GLT1 in HD. Although we cannot rule out other central mechanisms of action of ceftriaxone in our HD animals, it is unlikely that other glutamate transporters (e.g., GLAST and EAAC1) can account for the change in uptake since ceftriaxone acts selectively on GLT1 (Rothstein et al., 2005). In addition, GLAST and EAAC1 are not differentially expressed in HD mouse models compared to WT (Lievens et al., 2001; Behrens et al., 2002).
Several mechanisms, acting alone or in combination, may contribute to GLT1 dysfunction in HD. One comes from evidence that operation of GLT1 is sensitive to oxidative damage (Trotti et al., 1998), which is common in HD pathology (Browne and Beal, 2006). That R6/2 mice have low striatal extracellular ascorbate (Rebec et al., 2002), an antioxidant vitamin linked to glutamate uptake (Rebec and Pierce, 1994), is consistent with this view. Inhibition of glutamate transport, moreover, creates sufficient oxidative stress to overwhelm antioxidant protection (Nagatomo et al., 2007). It also is noteworthy that treatment of R6/2 mice with ascorbate, which restores striatal ascorbate to WT levels, attenuates the abnormally high firing rate of R6/2 striatal neurons (Rebec et al., 2006) and attenuates the HD phenotype (Rebec et al., 2003), both of which could be explained by improved GLT1 function. Another mechanism relates to mutant huntingtin, which accumulates in astrocytes as early as ∼4 weeks of age (Shin et al., 2005). The resulting disruption of intracellular protein trafficking (DiFiglia et al., 1995; Gunawarden and Goldstein, 2005) may cause improper localization or insertion of GLT1 into the membrane. Finally, medium spiny neurons in R6/2 mice exhibit abnormal dendritic morphology (Klapstein et al., 2001), which may cause altered coupling of astrocytic processes to the synapse, leading to inefficient glutamate uptake and spillover into the extracellular space. Our results suggest a need to investigate the mechanisms by which GLT1 becomes dysfunctional in HD.
Conclusion
Our results support the emerging hypothesis that dysregulated glutamate signaling is a major factor underlying HD pathology. By increasing the functional expression of GLT1, ceftriaxone reverses the glutamate uptake deficit in R6/2 mice. The functional significance of this effect is evident in the attenuated HD behavioral phenotype. Although the molecular mechanisms for the ceftriaxone-mediated increase in GLT1 expression are not known, ceftriaxone is thought to work through activation of the GLT1 promoter (Rothstein et al., 2005). It appears that behavioral improvement in ceftriaxone-treated R6/2 mice can be explained by normalization of glutamate handling, which may allow for improved information flow in HD striatum.
Acknowledgments
We thank Anne Prieto for assistance with Western blots, Rafael White for help with immunohistochemistry, Tyler Brock and Emma Klein for coding behavioral tests, and Faye Caylor for editorial and administrative support. This work was supported by grants from the US National Institute of Neurological Disorders and Stroke (R01 NS35663), National Science Foundation Graduate Research Fellowship Program, the Indiana METACyt Initiative of Indiana University, which is funded, in part, by a grant from the Lilly Endowment, Inc., and the Indiana University Center for the Integrative Study of Animal Behavior.
Abbreviations
- ANOVA
analysis of variance
- CE-LIF
capillary electrophoresis with laser-induced fluorescence
- HD
Huntington's disease
- PDC
pyrrolidine dicarboxylate
- WT
wild-type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Section Editor: Dr. Miles Herkenham
References
- Alexander GE, DeLong MR, Strick PL. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu Rev Neurosci. 1986;9:357–381. doi: 10.1146/annurev.ne.09.030186.002041. [DOI] [PubMed] [Google Scholar]
- Arzberger T, Krampfl K, Leimgruber S, Weindl A. Changes of NMDA receptor subunit (NR1, NR2B) and glutamate transporter (GLT1) mRNA expression in Huntington's disease--an in situ hybridization study. J Neuropathol Exp Neurol. 1997;56:440–454. doi: 10.1097/00005072-199704000-00013. [DOI] [PubMed] [Google Scholar]
- Behrens PF, Franz P, Woodman B, Lindenberg KS, Landwehrmeyer GB. Impaired glutamate transport and glutamate-glutamine cycling: downstream effects of the Huntington mutation. Brain. 2002;125:1908–1922. doi: 10.1093/brain/awf180. [DOI] [PubMed] [Google Scholar]
- Brandt J, Bylsma FW, Gross R, Stine OC, Ranen N, Ross CA. Trinucleotide repeat length and clinical progression in Huntington's disease. Neurology. 1996;46:527–531. doi: 10.1212/wnl.46.2.527. [DOI] [PubMed] [Google Scholar]
- Browne SE, Beal MF. Oxidative damage in Huntington's disease pathogenesis. Antioxid Redox Signal. 2006;8:2061–2073. doi: 10.1089/ars.2006.8.2061. [DOI] [PubMed] [Google Scholar]
- Bungay PM, Morrison PF, Dedrick RL. Steady-state theory for quantitative microdialysis of solutes and water in vivo and in vitro. Life Sci. 1990;46:105–119. doi: 10.1016/0024-3205(90)90043-q. [DOI] [PubMed] [Google Scholar]
- Carter RJ, Lione LA, Humby T, Mangiarini L, Mahal A, Bates GP, Dunnett SB, Morton AJ. Characterization of progressive motor deficits in mice transgenic for the human Huntington's disease mutation. J Neurosci. 1999;19:3248–3257. doi: 10.1523/JNEUROSCI.19-08-03248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, Ariano MA, Calvert CR, Flores-Hernandez J, Chandler SH, Leavitt BR, Hayden MR, Levine MS. NMDA receptor function in mouse models of Huntington disease. J Neurosci Res. 2001;66:525–539. doi: 10.1002/jnr.1244. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Hurst RS, Calvert CR, Hernandez-Echeagaray E, Nguyen OK, Jocoy E, Christian LJ, Ariano MA, Levine MS. Transient and progressive electrophysiological alterations in the corticostriatal pathway in a mouse model of Huntington's disease. J Neurosci. 2003;23:961–969. doi: 10.1523/JNEUROSCI.23-03-00961.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, Wu N, Andre VM, Cummings DM, Levine MS. The corticostriatal pathway in Huntington's disease. Prog Neurobiol. 2007;81:253–271. doi: 10.1016/j.pneurobio.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu K, Lee ST, Sinn DI, Ko SY, Kim EH, Kim JM, Kim SJ, Park DK, Jung KH, Song EC, Lee SK, Kim M, Roh JK. Pharmacological Induction of Ischemic Tolerance by Glutamate Transporter-1 (EAAT2) Upregulation. Stroke. 2007;38:177–182. doi: 10.1161/01.STR.0000252091.36912.65. [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase K, Schwarz C, Meloni A, Young C, Martin E, Vonsattel JP, Carraway R, Reeves SA. Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron. 1995;14:1075–1081. doi: 10.1016/0896-6273(95)90346-1. [DOI] [PubMed] [Google Scholar]
- Dorner JL, Miller BR, Barton SJ, Brock TJ, Rebec GV. Sex differences in behavior and striatal ascorbate release in the 140 CAG knock-in mouse model of Huntington's disease. Behav Brain Res. 2007;178:90–97. doi: 10.1016/j.bbr.2006.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganel R, Ho T, Maragakis NJ, Jackson M, Steiner JP, Rothstein JD. Selective up-regulation of the glial Na+-dependent glutamate transporter GLT1 by a neuroimmunophilin ligand results in neuroprotection. Neurobiol Dis. 2006;21:556–567. doi: 10.1016/j.nbd.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Graybiel AM. The basal ganglia. Trends Neurosci. 1995;18:60–62. [PubMed] [Google Scholar]
- Gunawardena S, Goldstein LS. Polyglutamine diseases and transport problems: deadly traffic jams on neuronal highways. Arch Neurol. 2005;62:46–51. doi: 10.1001/archneur.62.1.46. [DOI] [PubMed] [Google Scholar]
- Harper PS. Huntington's Disease. London: W.B. Saunders; 1991. [Google Scholar]
- Hassel B, Tessler S, Faull RL, Emson PC. Glutamate Uptake is Reduced in Prefrontal Cortex in Huntington's Disease. Neurochem Res. 2007 doi: 10.1007/s11064-007-9463-1. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Klapstein GJ, Fisher RS, Zanjani H, Cepeda C, Jokel ES, Chesselet MF, Levine MS. Electrophysiological and morphological changes in striatal spiny neurons in R6/2 Huntington's disease transgenic mice. J Neurophysiol. 2001;86:2667–2677. doi: 10.1152/jn.2001.86.6.2667. [DOI] [PubMed] [Google Scholar]
- Lada MW, Vickroy TW, Kennedy RT. Evidence for neuronal origin and metabotropic receptor-mediated regulation of extracellular glutamate and aspartate in rat striatum in vivo following electrical stimulation of the prefrontal cortex. J Neurochem. 1998;70:617–625. doi: 10.1046/j.1471-4159.1998.70020617.x. [DOI] [PubMed] [Google Scholar]
- Laforet GA, Sapp E, Chase K, McIntyre C, Boyce FM, Campbell M, Cadigan BA, Warzecki L, Tagle DA, Reddy PH, Cepeda C, Calvert CR, Jokel ES, Klapstein GJ, Ariano MA, Levine MS, DiFiglia M, Aronin N. Changes in cortical and striatal neurons predict behavioral and electrophysiological abnormalities in a transgenic murine model of Huntington's disease. J Neurosci. 2001;21:9112–9123. doi: 10.1523/JNEUROSCI.21-23-09112.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb HM, Ormrod D, Scott LJ, Figgitt DP. Ceftriaxone: an update of its use in the management of community-acquired and nosocomial infections. Drugs. 2002;62:1041–1089. doi: 10.2165/00003495-200262070-00005. [DOI] [PubMed] [Google Scholar]
- Li L, Murphy TH, Hayden MR, Raymond LA. Enhanced striatal NR2B-containing N-methyl-D-aspartate receptor-mediated synaptic currents in a mouse model of Huntington disease. J Neurophysiol. 2004;92:2738–2746. doi: 10.1152/jn.00308.2004. [DOI] [PubMed] [Google Scholar]
- Lievens JC, Woodman B, Mahal A, Spasic-Boscovic O, Samuel D, Kerkerian-Le GL, Bates GP. Impaired glutamate uptake in the R6 Huntington's disease transgenic mice. Neurobiol Dis. 2001;8:807–821. doi: 10.1006/nbdi.2001.0430. [DOI] [PubMed] [Google Scholar]
- Lipski J, Wan CK, Bai JZ, Pi R, Li D, Donnelly D. Neuroprotective potential of ceftriaxone in in vitro models of stroke. Neuroscience. 2007;146:617–629. doi: 10.1016/j.neuroscience.2007.02.003. [DOI] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- Melendez RI, Hicks MP, Cagle SS, Kalivas PW. Ethanol exposure decreases glutamate uptake in the nucleus accumbens. Alcohol Clin Exp Res. 2005;29:326–333. doi: 10.1097/01.alc.0000156086.65665.4d. [DOI] [PubMed] [Google Scholar]
- Menalled LB, Sison JD, Dragatsis I, Zeitlin S, Chesselet MF. Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington's disease with 140 CAG repeats. J Comp Neurol. 2003;465:11–26. doi: 10.1002/cne.10776. [DOI] [PubMed] [Google Scholar]
- Miele M, Berners M, Boutelle MG, Kusakabe H, Fillenz M. The determination of the extracellular concentration of brain glutamate using quantitative microdialysis. Brain Res. 1996;707:131–133. doi: 10.1016/0006-8993(95)01371-7. [DOI] [PubMed] [Google Scholar]
- Mineur YS, Picciotto MR, Sanacora G. Antidepressant-like effects of ceftriaxone in male C57BL/6J mice. Biol Psychiatry. 2007;61:250–252. doi: 10.1016/j.biopsych.2006.04.037. [DOI] [PubMed] [Google Scholar]
- Nagatomo K, Ueda Y, Doi T, Nakajima A. An acute dysfunction of the glutamate transport activity has been shown to generate free radicals and suppress the anti-oxidant ability in the hippocampus of rats. Neurosci Res. 2007;57:477–480. doi: 10.1016/j.neures.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Nau R, Prange HW, Muth P, Mahr G, Menck S, Kolenda H, Sorgel F. Passage of cefotaxime and ceftriaxone into cerebrospinal fluid of patients with uninflamed meninges. Antimicrob Agents Chemother. 1993;37:1518–1524. doi: 10.1128/aac.37.7.1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang YB, Voloboueva LA, Xu LJ, Giffard RG. Selective dysfunction of hippocampal CA1 astrocytes contributes to delayed neuronal damage after transient forebrain ischemia. J Neurosci. 2007;27:4253–4260. doi: 10.1523/JNEUROSCI.0211-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons LH, Justice JB., Jr Extracellular concentration and in vivo recovery of dopamine in the nucleus accumbens using microdialysis. J Neurochem. 1992;58:212–218. doi: 10.1111/j.1471-4159.1992.tb09298.x. [DOI] [PubMed] [Google Scholar]
- Rawls SM, Tallarida R, Robinson W, Amin M. The beta-lactam antibiotic, ceftriaxone, attenuates morphine-evoked hyperthermia in rats. Br J Pharmacol. 2007;151:1095–1102. doi: 10.1038/sj.bjp.0707309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebec GV, Barton SJ, Ennis MD. Dysregulation of ascorbate release in the striatum of behaving mice expressing the Huntington's disease gene. J Neurosci. 2002;22(RC202) doi: 10.1523/JNEUROSCI.22-02-j0006.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebec GV, Barton SJ, Marseilles AM, Collins K. Ascorbate treatment attenuates the Huntington behavioral phenotype in mice. Neuroreport. 2003;14:1263–1265. doi: 10.1097/00001756-200307010-00015. [DOI] [PubMed] [Google Scholar]
- Rebec GV, Conroy SK, Barton SJ. Hyperactive striatal neurons in symptomatic Huntington R6/2 mice: variations with behavioral state and repeated ascorbate treatment. Neuroscience. 2006;137:327–336. doi: 10.1016/j.neuroscience.2005.08.062. [DOI] [PubMed] [Google Scholar]
- Rebec GV, Pierce RC. A vitamin as neuromodulator: ascorbate release into the extracellular fluid of the brain regulates dopaminergic and glutamatergic transmission. Prog Neurobiol. 1994;43:537–565. doi: 10.1016/0301-0082(94)90052-3. [DOI] [PubMed] [Google Scholar]
- Rebec GV, Witowski SR, Sandstrom MI, Rostand RD, Kennedy RT. Extracellular ascorbate modulates cortically evoked glutamate dynamics in rat striatum. Neurosci Lett. 2005;378:166–170. doi: 10.1016/j.neulet.2004.12.027. [DOI] [PubMed] [Google Scholar]
- Robinson MB. The family of sodium-dependent glutamate transporters: a focus on the GLT-1/EAAT2 subtype. Neurochem Int. 1998;33:479–491. doi: 10.1016/s0197-0186(98)00055-2. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, Dykes HM, Vidensky S, Chung DS, Toan SV, Bruijn LI, Su ZZ, Gupta P, Fisher PB. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–77. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- Sandstrom MI, Rebec GV. Extracellular ascorbate modulates glutamate dynamics: role of behavioral activation. BMC Neurosci. 2007;832 doi: 10.1186/1471-2202-8-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JY, Fang ZH, Yu ZX, Wang CE, Li SH, Li XJ. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J Cell Biol. 2005;171(6):1001–1012. doi: 10.1083/jcb.200508072. 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shou M, Ferrario CR, Schultz KN, Robinson TE, Kennedy RT. Monitoring dopamine in vivo by microdialysis sampling and on-line CE-laser-induced fluorescence. Anal Chem. 2006;78:6717–6725. doi: 10.1021/ac0608218. [DOI] [PubMed] [Google Scholar]
- Shou M, Smith AD, Shackman JG, Peris J, Kennedy RT. In vivo monitoring of amino acids by microdialysis sampling with on-line derivatization by naphthalene-2,3-dicarboxyaldehyde and rapid micellar electrokinetic capillary chromatography. J Neurosci Methods. 2004;138:189–197. doi: 10.1016/j.jneumeth.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Smith AD, Justice JB. The effect of inhibition of synthesis, release, metabolism and uptake on the microdialysis extraction fraction of dopamine. J Neurosci Methods. 1994;54:75–82. doi: 10.1016/0165-0270(94)90161-9. [DOI] [PubMed] [Google Scholar]
- Timmerman W, Westerink BH. Brain microdialysis of GABA and glutamate: what does it signify? Synapse. 1997;27:242–261. doi: 10.1002/(SICI)1098-2396(199711)27:3<242::AID-SYN9>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Trotti D, Danbolt NC, Volterra A. Glutamate transporters are oxidant-vulnerable: a molecular link between oxidative and excitotoxic neurodegeneration. Trends Pharmacol Sci. 1998;19:328–334. doi: 10.1016/s0165-6147(98)01230-9. [DOI] [PubMed] [Google Scholar]
- Tzingounis AV, Wadiche JI. Glutamate transporters: confining runaway excitation by shaping synaptic transmission. Nat Rev Neurosci. 2007;8:935–947. doi: 10.1038/nrn2274. [DOI] [PubMed] [Google Scholar]
- Wood DA, Buse JE, Wellman CL, Rebec GV. Differential environmental exposure alters NMDA but not AMPA receptor subunit expression in nucleus accumbens core and shell. Brain Res. 2005;1042:176–183. doi: 10.1016/j.brainres.2005.02.029. [DOI] [PubMed] [Google Scholar]
- Zeron MM, Hansson O, Chen N, Wellington CL, Leavitt BR, Brundin P, Hayden MR, Raymond LA. Increased sensitivity to N-methyl-D-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington's disease. Neuron. 2002;33:849–860. doi: 10.1016/s0896-6273(02)00615-3. [DOI] [PubMed] [Google Scholar]