

Summary
Synaptic activity induces changes in the number of dendritic spines. Here, we report a pathway of regulated endocytosis triggered by arcadlin, a protocadherin induced by electroconvulsive and other excitatory stimuli in hippocampal neurons. The homophilic binding of extracellular arcadlin domains activates TAO2β, a splice variant of the thousand and one amino acid protein kinase 2, cloned here by virtue of its binding to the arcadlin intracellular domain. TAO2β is a MAPKKK that activates the MEK3 MAPKK, which phosphorylates the p38 MAPK. Activation of p38 feeds-back on TAO2β, phosphorylating a key serine required for triggering endocytosis of N-cadherin at the synapse. Arcadlin knockout increases the number of dendritic spines, and the phenotype is rescued by siRNA knockdown of N-cadherin. This pathway of regulated endocytosis of N-cadherin via protocadherin/TAO2β/MEK3/p38 provides a molecular mechanism for transducing neuronal activity into changes in synaptic morphologies.
Introduction
Various cellular events have been correlated with synaptic plasticity, a mechanism that is believed to underlie learning and memory. Cell-adhesion molecules are among the proteins responsible for these cellular events (Bailey et al., 1992; Manabe et al., 2000; Sytnyk et al., 2006). Among them, N-cadherin, a classical cadherin cell-adhesion molecule abundantly expressed in hippocampal excitatory synaptic junctions, has been most intensely correlated with synaptic plasticity (Bozdagi et al., 2000; Murase et al., 2002; Okamura et al., 2004; Tanaka et al., 2000; Tang et al., 1998). In a previous study, we found that an activity-regulated synaptic cell-adhesion molecule, arcadlin, is also essential for synaptic plasticity (Yamagata et al., 1999). Arcadlin, the rat ortholog of human protocadherin-8 (pcdh8) and of mouse, Xenopus, and zebrafish paraxial protocadherin (PAPC), is a unique member of the protocadherin superfamily and has a distinct cytoplasmic region (Strehl et al., 1998; Yamagata et al., 1999; Yamamoto et al., 2000). In Xenopus and zebrafish, arcadlin/PAPC plays an important role in homophilic cell adhesion and gastrulation movements of the paraxial mesoderm (Kim et al., 1998; Yamamoto et al., 2000). Recently, Xenopus arcadlin/PAPC has been shown to downregulate the adhesion activity of C-cadherin, a classical cadherin of developing Xenopus (Chen and Gumbiner, 2006). The interacting partners of arcadlin/PAPC in the CNS have not been identified.
The activity of cell-adhesion molecules in synaptic membranes is regulated in part by endocytosis. ApCAM, an Aplysia neural cell-adhesion molecule, for example, becomes internalized in presynaptic membrane upon the acquisition of long-term potentiation (LTP) (Bailey et al., 1992). Clathrin-coated pits and vesicles have been found in postsynaptic dendritic spines (Cooney et al., 2002; Racz et al., 2004). One of the mechanisms that regulate the endocytosis involves p38 MAPK (Johnson and Lapadat, 2002; Zhu et al., 2002), a serine/threonine kinase that responds to various stress-related stimuli (Tibbles and Woodgett, 1999). TAO2, a MAP kinase kinase kinase (MAPKKK) initially isolated as a mammalian homolog of Ste20p in S. cerevisiae (Chen et al., 1999), serves as a regulator of the p38 MAPK. Extracellular stimuli, such as those of certain G protein-coupled receptors or insulin receptor, are transduced via TAO kinases to p38 MAPK in nonneural tissues (Chen et al., 2003). The p38 MAPK is highly expressed in the brain, where it is thought to be involved in synaptic plasticity (Thomas and Huganir, 2004). The functions of TAO kinases and their upstream receptors in the CNS are not known.
In this study, we have found that N-cadherin, the most abundant classical cadherin in hippocampal excitatory synapses (Benson and Tanaka, 1998), serves as a target of arcadlin/PAPC in the mammalian nervous system. Arcadlin is induced by neural stimulation and transported to the synaptic membrane, where it binds to and internalizes N-cadherin. During our investigations into the mechanism of this internalization, we cloned a spliced form of TAO2 kinase (TAO2β) that binds to the arcadlin intracellular domain. Homophilic interactions of arcadlin on the cell surface activate p38 MAPK through the activation of TAO2β. In turn, active p38 MAPK feeds-back on TAO2β, phosphorylating its carboxy-terminal domain at a specific serine. This triggers the coendocytosis of an N-cadherin-arcadlin complex. Furthermore, knockout of arcadlin in the mouse leads to an increase in dendritic spines in cultured neurons, and siRNA knockdown of N-cadherin recovered the spine density. We propose that endocytosis regulated by this signal transduction pathway involving a protocadherin, a MAPKKK that binds to its intracellular domain, a MAPK, and a classical cadherin regulates the adhesiveness of synaptic membranes and hence the number of spines in an activity-dependent way.
Results
Arcadlin Interacts with N-Cadherin
Arcadlin is an activity-regulated cell-adhesion molecule whose expression level is very low in the resting brain but is vigorously induced by neural stimulation and recruited to the dendritic spine (Yamagata et al., 1999). Upon neural stimulation, such as maximal electroconvulsive seizure (MECS), arcadlin immunoreactivity increased and displayed a punctate distribution in the stratum lucidum of the hippocampal CA3 region, in which N-cadherin is also found (Figures 1A and 1B; Fannon and Colman, 1996).
Figure 1. Arcadlin Is Associated with N-Cadherin at Hippocampal Synaptic Puncta.
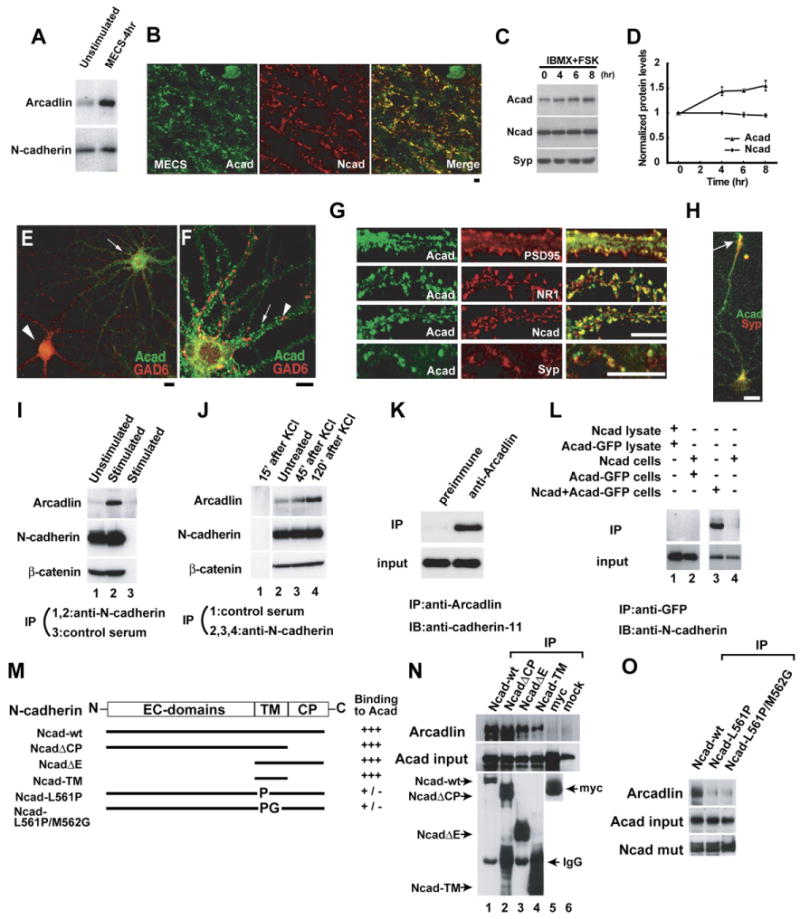
(A) Hippocampal extracts from unstimulated (control) and electroconvulsive shocked (MECS-4hr) rats were immunoblotted for arcadlin and N-cadherin.
(B) CA3 stratum lucidum doubly immunolabeled for arcadlin (Acad, green) and N-cadherin (Ncad, red) 4 hr after MECS. Significant colocalization confirmed (r = 0.51).
(C) Elevation of cAMP by IBMX and forskolin (FSK) induces the expression of arcadlin in cultured hippocampal neurons as examined by immunoblots. Syp, synaptophysin.
(D) The graph shows the relative band intensities of arcadlin and N-cadherin normalized with the bands for synaptophysin (mean ± SEM).
(E) Hippocampal neurons treated with IBMX and forskolin for 4 hr and immunostained for arcadlin (green) and GAD6 (red).
(F) An excitatory neuron double labeled for arcadlin (green) and GAD6 (red).
(G) Neurons double labeled for arcadlin (green) and PSD-95 (red; r = 0.43), NR1 (red), N-cadherin (red; r = 0.36), or synaptophysin (red).
(H) Hippocampal neuron (3 DIV) stained for arcadlin (green) and synaptophysin (red).
(I) Immunoprecipitation of N-cadherin from unstimulated (lane 1) and MECS-treated (lane 2) rat hippocampal extracts, immunoblotted for arcadlin (top), N-cadherin (middle), and β-catenin (bottom). Lane 3, control preimmune serum.
(J) Immunoprecipitation of N-cadherin from cultured neurons stimulated with 25 mM KCl for 1 min and further cultured for 45–120 min, immunoblotted for arcadlin (top), N-cadherin (middle), and β-catenin (bottom). Lane 1, control preimmune serum.
(K) Immunoprecipitation of arcadlin from MECS-treated rat brain extracts, immunoblotted for cadherin-11. Left, control preimmune serum.
(L) COS7 cells were transfected with arcadlin-EGFP and/or N-cadherin and subjected to immunoprecipitation with anti-GFP serum followed by immunoblot with anti-N-cadherin antibody (top). Bottom, input. Lane 1, the lysate of N-cadherin-transfected cells was mixed in vitro with the lysate of arcadlin-transfected cells. Lane 2, arcadlin-expressing cells and N-cadherin-expressing cells were cocultured. Lane 3, arcadlin and N-cadherin were cotransfected. Lane 4, a control without transfecting arcadlin-EGFP. The lack of coimmunoprecipitation in lanes 1 and 2 suggested that these molecules do not interact in vitro or in trans, but associate laterally in the same membrane.
(M) N-cadherin mutants including wild-type (Ncad-wt), cytoplasmic domain deleted (NcadΔCP), extracellular domain deleted (NcadΔE), transmembrane segment alone (Ncad-TM), and point mutated in the middle of transmembrane region (Ncad-L561P, Ncad-L561P/M562G) were examined for the binding to full-length arcadlin. N-cadherin mutants fused with myc-tag were cotransfected with arcadlin into COS7 cells and immunoprecipitated with anti-myc antibody.
(N) Top, blot of arcadlin coimmunoprecipitated with the indicated N-cadherin mutants (lanes 1–4) or controls (lanes 5 and 6). In lane 2, abundant NcadΔCP (invisible) pushes the arcadlin band down to the lower molecular size position. Middle, arcadlin input. Bottom, immunoprecipitated N-cadherin mutants (myc probed).
(O) Top, blot of arcadlin coimmunoprecipitated with the indicated N-cadherin point mutants. Middle, arcadlin input. Bottom, immunoprecipitated N-cadherin mutants (myc probed).
Scale bars, 10 μm.
In cultured hippocampal neurons, spontaneous synaptic activity causes detectable expression of arcadlin, which was increased by brief treatment with glutamate or elevation in cAMP with isobutyl methylxanthine (IBMX) and forskolin (Figures 1C and 1D and data not shown). Although N-cadherin is also known to be induced by elevating cAMP level in acute hippocampal slices (Bozdagi et al., 2000), there was no significant induction of N-cadherin in our culture (Figures 1C and 1D). This discrepancy is presumably due to the lack of glial support and the higher spontaneous activity of neurons in dispersed culture. The expression of arcadlin in cultures treated with IBMX and forskolin was confined to glutamate decarboxylase 65 (GAD6)-negative, non-GABAergic neurons (Figure 1E, arrow). In such neurons, arcadlin showed punctate distribution in dendrites (Figure 1F, arrow). The arcadlin puncta were not colocalized or apposed to the GAD6-puncta, which correspond to inhibitory axonal termini (Figure 1F, arrowhead). In contrast, the arcadlin-puncta colocalized with postsynaptic markers for excitatory synapses, such as PSD-95 and the NMDA receptor subunit NR1, as well as N-cadherin (Figure 1G). Arcadlin was expressed in developing axonal growth cones of young neurons, showing that arcadlin is also present in developing presynaptic membranes (Figure 1H, arrow).
We discovered the association of arcadlin with N-cadherin fortuitously during the course of immunoprecipitation studies of N-cadherin in hippocampal lysates. Arcadlin was coimmunoprecipitated with N-cadherin in dissected rat hippocampi 4 hr after electroconvulsions, but very little was found in unstimulated hippocampi (Figure 1I; note that the amount of β-catenin bound to N-cadherin is not affected). Coimmunoprecipitation was detectable in resting cultured neurons, which was increased by neural stimulation, such as depolarization by KCl (Figure 1J). Another synaptic classical cadherin, cadherin-11, was also found to be associated with the induced arcadlin in the brain (Figure 1K). The fact that arcadlin is targeted to multiple classical cadherins is reminiscent of the notion that Xenopus arcadlin/PAPC downregulates the adhesion activity of C-cadherin (Chen and Gumbiner, 2006). In this study, we focused on N-cadherin as the most abundant example of these cadherins. Coimmnoprecipitation experiments from cocultured cell lines transfected with N-cadherin and arcadlin independently (single transfections) or simultaneously (cotransfection) revealed that they associated laterally in the same membrane (Figure 1L). Consistently, we were able to localize their interaction domains to their transmembrane segments by deletion mutant analyses (Figures 1M and 1N). The affinity of N-cadherin to arcadlin was significantly reduced by a point mutation L561P or L561P/M562G in the middle of the transmembrane α-helix of N-cadherin (Figures 1M and 1O). The corresponding amino acid of E-cadherin plays a pivotal role in homophilic cis dimerization (Huber et al., 1999).
Arcadlin Induces the Internalization of N-Cadherin
To test whether arcadlin had any effect on the adhesive activity of N-cadherin, we performed cell-aggregation assays using L929 cells (Takeichi, 1977). Although arcadlin itself has a homophilic adhesive activity, it is too weak to be detected in the aggregation assay optimized for classical cadherins (Yamagata et al., 1999). We found that arcadlin downregulated the homophilic adhesiveness of N-cadherin (Figures 2A and 2B). The inhibitory effect was not attributable to either the expression level of N-cadherin (Figure 2C) or the intracellular molecules associated with N-cadherin, such as catenins (Figure 2D). We therefore hypothesized that the downregulation of N-cadherin activity could involve the internalization of N-cadherin by analogy to the case of apCAM (Bailey et al., 1992) and L1 (Kamiguchi and Lemmon, 2000). Recently, a key role for endocytosis in the disassembly of E-cadherin cell-cell adhesion has been reported (Troyanovsky et al., 2006).
Figure 2. Arcadlin Inhibits the Adhesive Activity of N-Cadherin.
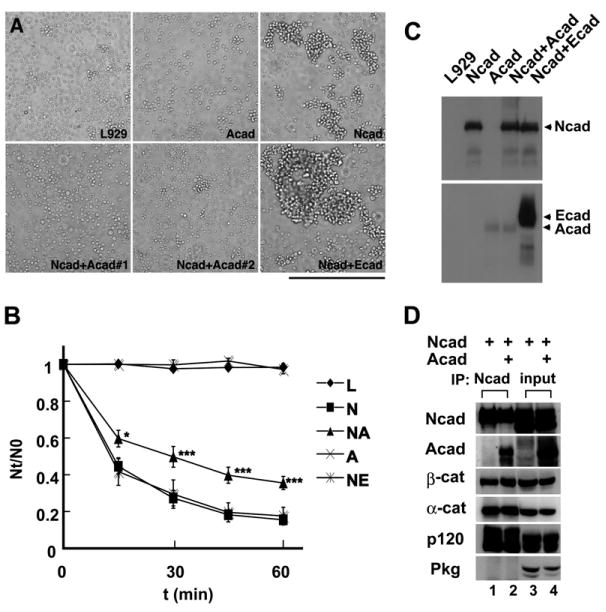
(A) Cell-aggregation assay was performed using L929 cells stably transfected with N-cadherin (Ncad), arcadlin (Acad), and both (Ncad + Acad #1 and Ncad + Acad #2; two independent transfected cell lines). Scale bar, 500 μm.
(B) Adhesive activity was quantified by counting the number of aggregates (mean ± SEM) at indicated times (Nt). *p < 0.05, ***p < 0.001, compared to Ncad.
(C) N-cadherin expression level of each cell type was examined by western blot.
(D) COS7 cells were doubly transfected with N-cadherin and arcadlin (lanes 2 and 4) or N-cadherin and mock (lanes 1 and 3), immunoprecipitated with the antibody against N-cadherin, and subjected to immunoblot (lanes 1–2). Lanes 3–4, inputs.
To examine whether arcadlin enhances the internalization of N-cadherin, we first quantified surface N-cadherin level by labeling neuronal proteins on the extracellular surface with biotin. The biotin-labeled surface proteins were isolated with avidin-sepharose beads and immunoblotted for N-cadherin (Figure 3A). Membrane depolarization with KCl and elevating cAMP level, both of which induced endogenous arcadlin (Figures 1C, 1D, and 1J), resulted in significant reduction in surface N-cadherin levels (Figures 3A and 3B), whereas surface levels of neuroligin, as a control, did not change (Figures 3C and 3D).
Figure 3. Depolarization or Elevated cAMP Level Increases N-Cadherin Internalization through an Arcadlin-Dependent Pathway.
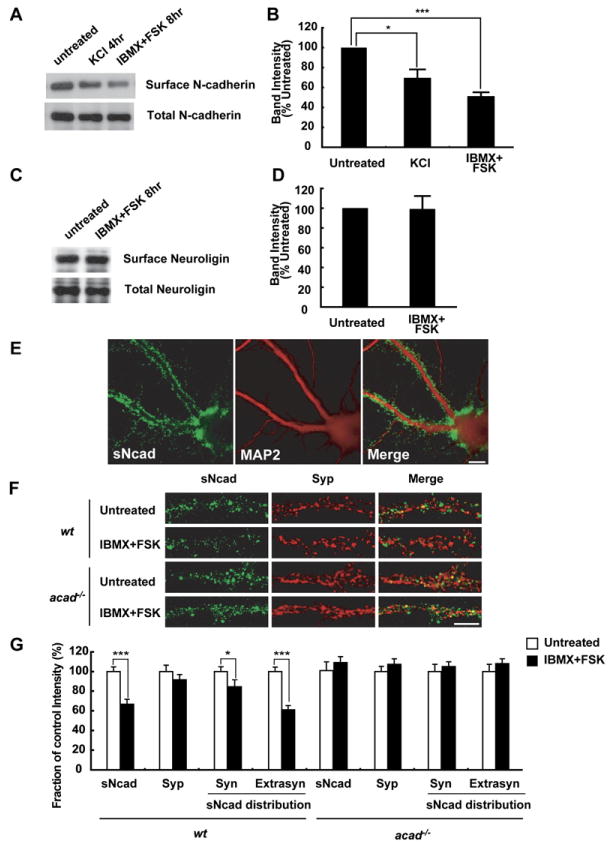
(A) Neuronal cell surface protein biotinylated and isolated with avidin-sepharose was immunoblotted for N-cadherin. Bottom, total protein.
(B) Densitometric quantification of surface N-cadherin levels. Surface protein levels were normalized with total protein levels and shown as proportion to the control experiment (%, mean ± SEM). n = 6.
(C) Neuronal cell surface protein was immunoblotted for neuroligin. Bottom, total protein.
(D) Densitometric quantification of surface neuroligin levels. Surface protein levels were normalized with total protein levels and shown as proportion to the control experiment (%, mean ± SEM). n = 5.
(E) Live neurons were incubated with anti-N-cadherin extracellular domain antibody (MT79) at 4°C for 30 min. Surface N-cadherin (sNcad) was labeled with Alexa-488-conjugated secondary antibody without membrane permeabilization (green). MAP2 was subsequently labeled after the permeabilization (red).
(F) Pretreatment of the neurons with IBMX and forskolin resulted in a decrease in surface N-cadherin intensity (green). Red, synaptophysin (Syp). Note that the change in surface N-cadherin intensity is not obvious in acad/papc−/− neurons.
(G) Quantification of surface N-cadherin intensity (mean ± SEM). The change in the surface N-cadherin mean intensity was observed in both the synaptophysin-overlapping fraction (Syn) and synaptophysin-nonoverlapping fraction (Extrasyn) populations. n = 40–50 dendrites from 4–5 independent experiments.
*p < 0.05, ***p < 0.001. Scale bars, 10 μm.
Next, we quantified the amount of surface-associated N-cadherin microscopically. We generated a new polyclonal antibody, MT79, which recognizes the extracellular domain of N-cadherin (see Figure S1 in the Supplemental Data available with this article online). Mouse hippocampal neurons at age 14–17 days were incubated in medium containing anti-N-cadherin MT79 antibody at 4°C for 30 min. Surface-associated N-cadherins were then labeled with the secondary antibody without permeabilization of plasma membrane (Figure 3E, green). To analyze the synaptic population, we examined the N-cadherin signal that overlapped with synaptophysin (Figure 3G; synaptic versus extrasynaptic). A 33.4% ± 5.1% decrease in the mean intensity of total surface N-cadherin in the IBMX + forskolin treatment group relative to control was observed (Figures 3F and 3G). (The mean intensity of synaptophysin did not differ between groups.) The decrease in the surface N-cadherin mean intensity was observed in both the synaptic (synaptophysin-overlapping fraction) and extrasynaptic (synaptophysin-nonoverlapping fraction) populations.
To show that the cAMP (IBMX + forskolin)-induced internalization of N-cadherin was indeed mediated by arcadlin, we utilized arcadlin (acad/papc)−/− mice (Yamamoto et al., 2000). The acad−/− mice were apparently normal, and the gross expression level of N-cadherin appeared the same as wild-type in brain sections and cultured neurons (data not shown). Neurons cultured from acad−/− mice extended dendrites and axons normally. The surface N-cadherin intensity of acad−/− neurons was slightly higher than that of acad+/+ neurons (9.8% ± 3.6% increase in biotinylation assay, n = 5) and of acad+/− neurons (37.9 ± 1.3 [n = 100] versus 33.1 ± 0.9 [n = 100], arbitrary fluorescence units in microscopic analysis, data collected from ten independent experiments). In these acad−/− neurons, there was no significant change in the mean intensity of total surface N-cadherin in the IBMX + forskolin treatment group relative to control (Figures 3F and 3G). The data indicate that the IBMX + forskolin treatment-induced N-cadherin internalization is mediated by arcadlin.
Identification of an Isoform of TAO2 Kinase as a Signal Transducer of Arcadlin
To dissect the molecular mechanism of endocytosis of N-cadherin by arcadlin, we searched for an intracellular binding partner of arcadlin. A splice form of TAO2 kinase was cloned in a yeast two-hybrid screen using a cDNA library prepared from electroconvulsed rat hippocampi and the cytoplasmic domain of arcadlin as bait (Figure 4A and Figure S2). This isoform of TAO2 kinase (named TAO2β) of 1056 amino acids shares the common serine/threonine protein kinase catalytic domain and the MEK binding domain with the original TAO2 kinase (renamed as TAO2α hereafter) but has a unique carboxy-terminal regulatory domain that shows no apparent homology to any known protein motif (Figure 4A and Figure S2B). The mRNA portion corresponding to the C-terminal domain of TAO2α is transcribed from only one exon, whereas that encoding the TAO2β C terminus is derived partly from the same exon, and mostly from three downstream exons, suggesting that tao2α and -β mRNAs are alternative splicing products from the same gene (Figure S2A).
Figure 4. TAO2β Binds to the Intracellular Domain of Arcadlin.
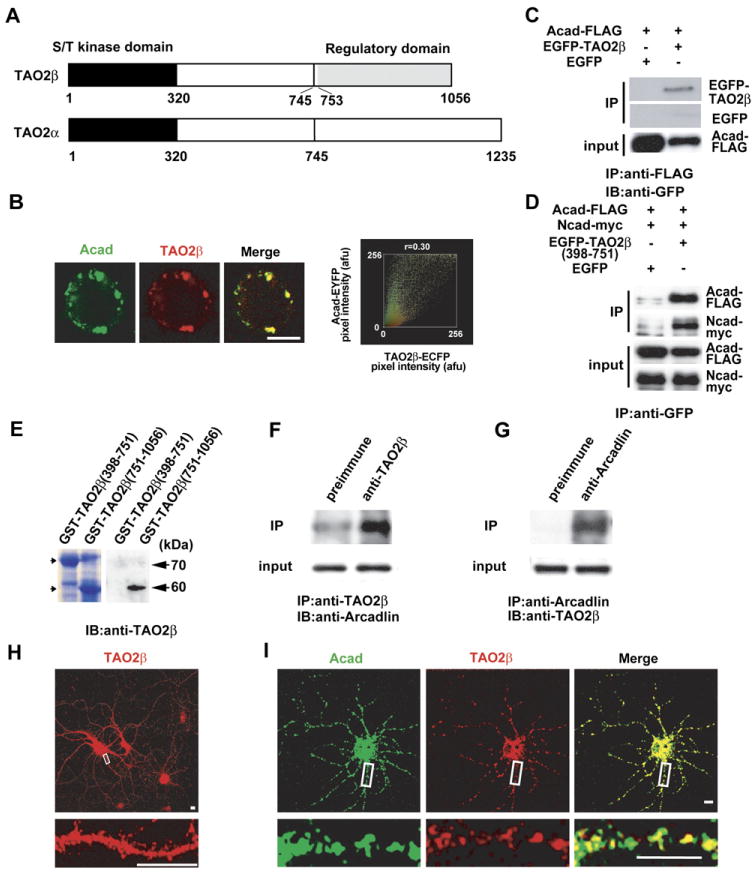
(A) Diagram of TAO2 kinases. An isoform of TAO2 (TAO2β) was identified as an intracellular binding partner of arcadlin. The original TAO2 was renamed as TAO2α. Most of the C-terminal regulatory domain of TAO2β (aa 753–1056) is distinct from that of TAO2α.
(B) Typical fluorescence image (left) and scatterplot (right) of HEK293T cells coexpressing TAO2β-ECFP and arcadlin-EYFP. Significant colocalization confirmed (r = 0.30).
(C) Arcadlin-FLAG was immunoprecipitated (IP) from HEK293T cells transfected with arcadlin-flag and EGFP-tao2β and immunoblotted (IB) with anti-GFP serum.
(D) EGFP-TAO2β(398-751) or control EGFP was immunoprecipitated with anti-GFP serum (IP) from HEK293T cells transfected with arcadlin-flag, N-cadherin-myc, and EGFP-tao2β(398–751) and immunoblotted for arcadlin (anti-FLAG) and N-cadherin (anti-myc). Left, transfected with EGFP instead of EGFP-tao2β(398–751).
(E) The affinity purified anti-TAO2β antibody recognized the unique regulatory domain specific for the β isoform. Left, CBB protein staining.
(F) Hippocampal extract immunoprecipitated with anti-TAO2β antibody and immunoblotted for arcadlin.
(G) Hippocampal extract immunoprecipitated with anti-arcadlin antibody and immunoblotted for TAO2β.
(H) Cultured neurons were immunostained for TAO2β.
(I) Confocal live imaging of arcadlin-EYFP (green) and TAO2β-ECFP (red) expressed in dendrites.
Scale bars, 10 μm.
A recombinant TAO2β tagged at its carboxyl terminus with enhanced cyan fluorescent protein (TAO2β-ECFP) colocalized with arcadlin-EYFP (enhanced yellow fluorescent protein) in HEK293T cells (Figure 4B). The molecular interaction between these proteins was confirmed by coimmunoprecipitation (Figure 4C). The formation of a trimeric complex of TAO2β, arcadlin, and N-cadherin was confirmed in triple-transfected HEK293T cells (Figure 4D). The association of EGFP-TAO2β (398–751), the central domain common to both α and β isoforms, suggested that TAO2α can also bind to arcadlin (Figure 4D). In order to examine in vivo interactions of TAO2β, we produced and purified a polyclonal antibody recognizing the carboxy-terminal domain specific for the β isoform of TAO2 kinase (Figure 4E). Coimmunoprecipitation of endogenous arcadlin protein with the anti-TAO2β antibody (Figure 4F) and, reciprocally, of TAO2β with anti-arcadlin antibody (Figure 4G) confirmed that these molecules associate in vivo in MECS-treated rat hippocampi. Immunolocalization of TAO2β showed puncta in dendrites (Figure 4H). Arcadlin-EYFP and TAO2β-ECFP transfected into cultured hippocampal neurons colocalized in dendrites (Figure 4I). We conclude that the arcadlin protocadherin and the TAO2β MAPKKK interact in hippocampal neurons.
Arcadlin Homophilic Interaction Triggers Activation of p38 MAPK and Internalization
The arcadlin/PAPC extracellular domain mediates homophilic binding (Chen and Gumbiner, 2006; Kim et al., 1998; Yamagata et al., 1999). In addition, arcadlin is a transiently expressed protein in hippocampal neurons, whose protein level peaks at 4 hr after the synaptic stimulation and largely disappears within 8 hr (Yamagata et al., 1999). During this period, the arcadlin protein is transported to pre- and postsynaptic membranes and rapidly turned over (Yamagata et al., 1999). In HEK293T cells cotransfected with arcadlin-EGFP and arcadlin-flag, arcadlin-arcadlin lateral interactions in the same membrane were readily detectable (Figure 5A, lane3). To analyze trans interactions specifically, we utilized arcadlin-L, a splice variant of arcadlin containing a 98 amino acid insertion in its cytoplasmic region. Arcadlin-L-EGFP-expressing cells and arcadlin-L-FLAG-expressing cells were cocultured so that these two types of cells attached to each other. Immunoprecipitation of arcadlin-L-EGFP with anti-flag antibody indicated that there is significant binding activity in trans (Figure 5A, lane 1). Application of a soluble extracellular fragment of recombinant arcadlin protein (Acad-EC, purified via a His-tag) into the culture medium competed trans-association, indicating that Acad-EC binds to the extracellular domain of arcadlin in trans (Figure 5A, lane 2). Although cis interaction may be also involved, Acad-EC at this concentration was not sufficient to replace the lateral oligomerization (data not shown).
Figure 5. Homophilic Interaction of Arcadlin Causes Internalization via the Activation of Arcadlin-TAO2β-p38 MAPK Signaling Pathway.
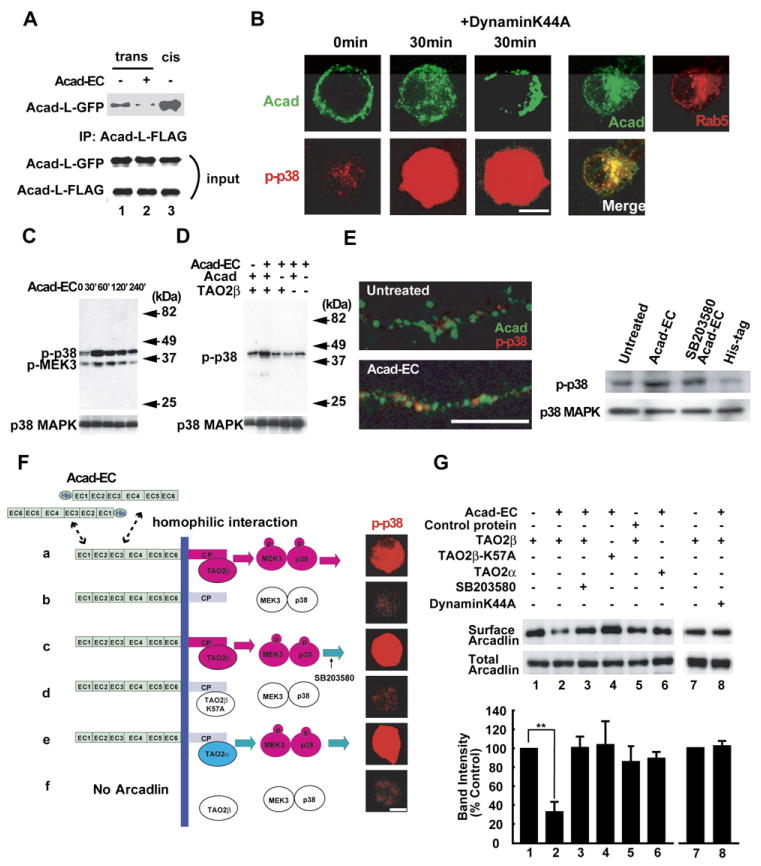
(A) Arcadlin-L-GFP or arcadlin-L-FLAG were expressed on different cell surfaces independently and allowed to interact across the cell-cell junction in mixed cell culture and were immunoprecipitated with anti-FLAG antibody followed by immunoblot for GFP (top). Bottom, input. Acad-EC protein added in the culture medium inhibited the trans-interaction (lane 2, compared with lane 1). Lane 3, cotransfection of arcadlin-l-EGFP and arcadlin-l-flag, which allows cis-interaction.
(B) HEK293T cells transfected with arcadlin, tao2β, MEK3, and p38 MAPK were treated with Acad-EC (10 μg/ml, 30 min) and immunostained. dynaminK44A (center) or EGFP-rab5 (right) was additionally cotransfected. Surface and cytoplasmic localizations were revealed by optical sectionings by a confocal microscope.
(C) Extracts of HEK293T cells transfected with arcadlin, tao2β, MEK3, and p38 MAPK and treated with Acad-EC were immunoblotted for phospho-p38 MAPK and phospho-MEK3.
(D) Extracts of HEK293T cells with or without transfection of arcadlin and tao2β were immunoblotted for p38 phosphorylation upon the addition of Acad-EC to culture medium.
(E) Cultured hippocampal neurons at 18 DIV pretreated with IBMX and forskolin were treated with Acad-EC (10 μg/ml) for 30 min followed by immunostaining (left) for arcadlin (green) and phospho-p38 MAPK (red); corresponding immunoblot shown to the right.
(F) Reconstitution of the arcadlin-TAO2β-p38 MAPK pathway in HEK293T cells transfected as described. Cells in (C) were pretreated with 10 μM SB203580. Cells were treated with Acad-EC for 30 min and immunostained for phospho-p38 MAPK.
(G) Quantification of surface arcadlin level by biotinylation method in HEK293T cells transfected and treated as indicated. Densitometric quantification from five separate experiments is summarized in histogram (mean ± SEM).
**p < 0.01. Scale bars, 10 μm.
Arcadlin molecules expressed in HEK293T cells abundantly localized to the cell surface. There was also detectable fraction of arcadlin in intracellular vesicles (Figure 5B, 0 min). Homophilic interaction of arcadlin on the cell surface with Acad-EC added to the culture medium triggered the rapid translocation of arcadlin from the periphery to the center of HEK293T cells cotransfected as described below (Figure 5B). This shift is mediated by endocytosis, because the moved arcadlin colocalized with EGFP-rab5 as a marker for endosomes and because the internalization of arcadlin was blocked by a coexpression of a dominant-negative form of dynamin, as shown in Figures 5B and 5G. A similar endocytic response was observed upon the application of the antibody against the extracellular region of arcadlin (data not shown). Therefore, a binding of the extracellular domain is sufficient to enhance the endocytosis of arcadlin. We next used this Acad-EC reagent to investigate the signal transduction mechanism that triggers endocytosis. It should be noted that a detectable level of background arcadlin endocytosis before the addition of Acad-EC may be triggered by the cis homophilic interaction of the transfected arcadlin (Figure 5B, 0 min).
Because TAO2β forms a molecular complex with the arcadlin intracellular domain, we asked whether p38 MAPK, a main target kinase of the TAO2α-MAPKKK pathway bridged by MEK3 (MAPKK-3) (Chen et al., 2003), was activated by the arcadlin signal. HEK293T cells cotransfected with arcadlin, tao2β, MEK3, and p38 MAPK were treated by addition of purified Acad-EC (10 μg/ml) to the culture medium. Immunostaining and immunoblot of the phosphorylated forms of p38 MAPK and MEK3 revealed that the phosphorylation levels of both kinases were enhanced within 30 min of the application of Acad-EC (Figures 5B and 5C). The p38 MAPK phosphorylation is mediated by arcadlin and TAO2β, because Acad-EC did not exert any response in HEK293T cells lacking either arcadlin or tao2β transfection (Figure 5D). Importantly, addition of Acad-EC protein triggered the activation of endogenous p38 MAPK in the dendritic shaft of primary cultures of rat hippocampal neurons (Figure 5E). Taken together, the results suggest that the arcadlin extracellular domain activates p38 MAPK via TAO2β. In cultured neurons, activation of p38 was detected specifically in the dendritic shaft after addition of protocadherin extracellular domain.
TAO2β Is Required for p38 Activation and Endocytosis of Arcadlin
We then addressed whether TAO2β is necessary for the phosphorylation of p38 MAPK and the endocytosis of arcadlin. We reconstituted the arcadlin-TAO2β-MEK3-p38 MAPK signaling pathway in HEK293T cells by cotransfecting arcadlin with tao2β, tao2βK57A (catalytically defective TAO2 kinase; Chen et al., 2003), or tao2α. After the treatment with Acad-EC for 30 min, cells were analyzed for the phosphorylation of p38 MAPK (Figure 5F) and endocytosis of arcadlin by the surface biotinylation assay (Figure 5G). The phosphorylation of p38 MAPK was significantly increased, and surface arcadlin levels were significantly reduced in cells expressing wild-type TAO2β (Figures 5F and 5G). In mock or tao2βK57A-transfected cells, neither the endocytosis of arcadlin nor the phosphorylation of p38 MAPK was observed (Figures 5F and 5G). Cells expressing TAO2α displayed full activation of p38 MAPK but were deficient in the internalization of arcadlin (Figures 5F and 5G). We conclude from these data that the kinase activity of TAO2α and -β that resides in their common catalytic domains is sufficient for the phosphorylation and activation of p38 MAPK. The endocytosis of arcadlin, however, depends exclusively on TAO2β, suggesting that the unique carboxy-terminal domain of TAO2β is required for the endocytosis of the arcadlin protocadherin.
A TAO2β-p38 MAPK Feed-Back Loop Mediates the Endocytosis of Arcadlin
We next investigated the requirement of p38 MAPK for the endocytosis of arcadlin. We found that SB203580, a p38 MAPK inhibitor, blocked the endocytosis of arcadlin (Figure 5G, compare lanes 2 and 3). This suggested that a feed-back loop, in which p38 MAPK regulates the endocytosis of arcadlin, might exist. We first postulated that arcadlin itself was a direct substrate of p38 MAPK. However, arcadlin was not phosphorylated by p38 MAPK (data not shown). We then tested whether p38 MAPK phosphorylates TAO2β. Because the experiments above indicated that the function of TAO2β responsible for the endocytosis of arcadlin resided in the carboxy-terminal domain, the unique carboxy-terminal region (751–1056) of TAO2β was fused to glutathione S-transferase (GST) and subjected to an in vitro kinase reaction with purified p38 MAPK. GST-TAO2β (751–1056) was indeed phosphorylated by activated p38 MAPK (Figure 6A, lane 2). Within this region, there were two sites encoding a serine preceded by a proline in positions 951 and 1010 as possible substrate sites for MAPK family members (Figure 6B; Kyriakis and Avruch, 2001), but their mutation into phosphorylation-resistant alanines did not affect the incorporation of 32P (Figure 6A, lane 3 and data not shown). A detailed deletion mutant analysis (data not shown) then revealed that the main target of p38 MAPK was localized within the region between positions 1036 and 1056 (Figure 6B). We generated Ser to Ala point mutations on positions 1038, 1040, 1042, and 1045 and found that incorporation of 32P diminished in TAO2βS1038A, indicating that Ser1038 is the main target of p38 MAPK (Figures 6A and 6B, lane 4).
Figure 6. Feed-Back Phosphorylation of TAO2β at Ser1038 by p38 MAPK Is Essential for the Endocytosis of Arcadlin.
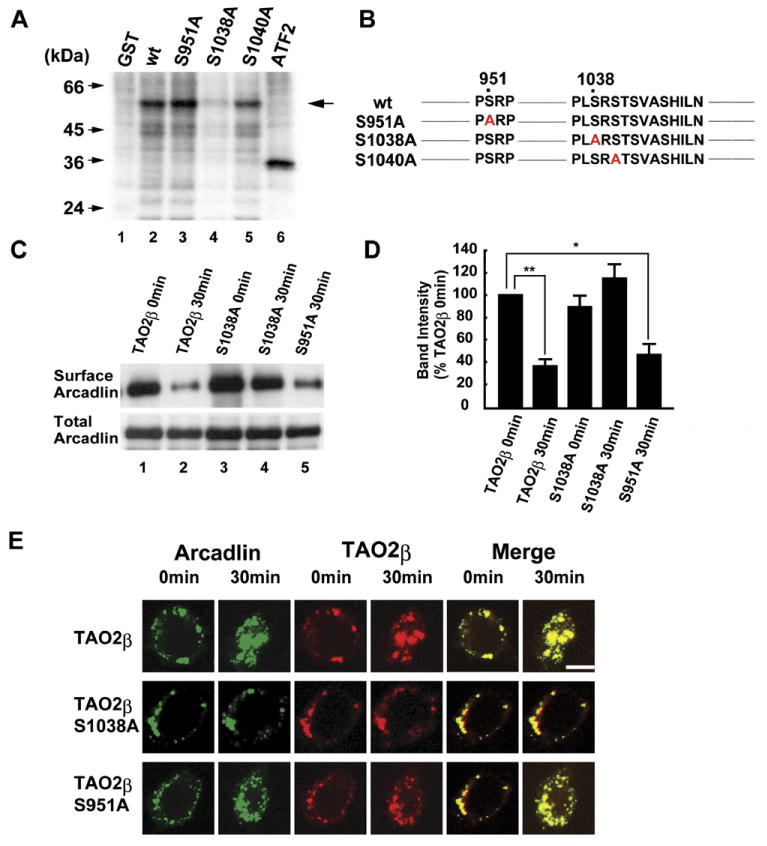
(A) GST-TAO2β, -TAO2βS951A, -TAO2βS1038A, or -TAO2βS1040A were incubated with activated p38 MAPK and [γ-32P]ATP and subjected to SDS-PAGE followed by autoradiography. The activated p38 MAPK was prepared by immunoprecipitation from Acad-EC-treated HEK293T cells transfected with arcadlin, tao2β, MEK3, and p38 MAPK. Arrow indicates 32P-incorporated GST-TAO2β mutant proteins. ATF2, a positive control.
(B) Mutations of TAO2β examined as substrates of p38 MAPK.
(C) Surface-biotinylation assay of arcadlin in HEK293T cells transfected with arcadlin, MEK3, p38 MAPK, and tao2β (or tao2βS1038A or tao2βS951A) before and 30 min after the addition of Acad-EC into culture medium.
(D) Quantification histogram of three independent results of (C) (mean ± SEM).
(E) Time-lapse confocal images of live cells expressing arcadlin-EYFP and TAO2β-ECFP (top), phosphorylation-resistant TAO2βS1038A-ECFP (middle), or control TAO2βS951A-ECFP (bottom) before and 30 min after Acad-EC treatment.
*p < 0.05, **p < 0.01. Scale bar, 10 μm.
We next investigated whether phosphorylation of Ser1038 was required for the endocytosis of arcadlin. Cells expressing TAO2βS951A, used here as a control, exhibited normal endocytosis of arcadlin, whereas the phosphorylation-resistant TAO2βS1038A remained in the surface after addition of Acad-EC protein (Figure 6C, lanes 1–4; see Figure 6D for quantification). Time-lapse images of HEK293T cells expressing arcadlin-EYFP and TAO2βS1038A-ECFP or control TAO2βS951A-ECFP confirmed that TAO2βS1038A fails to induce the endocytosis of arcadlin in living cells (Figure 6E).
These results indicate that the p38 MAPK that is activated by the arcadlin-TAO2β-MEK3 signaling pathway in turn phosphorylates TAO2β on Ser1038, resulting in the formation of a feed-back signaling loop. Phosphorylation of Ser1038 of TAO2β seems to be essential to activate the endocytic machinery following homophilic interaction of arcadlin. Thus, it appears we have identified a molecular pathway for protocadherin-mediated endocytosis.
The Arcadlin-TAO2β-p38 MAPK Pathway Regulates N-Cadherin Endocytosis
Finally, we tested whether the arcadlin-induced internalization of N-cadherin is mediated by the arcadlin-TAO2β-p38 MAPK molecular pathway defined above. The quantification of neuronal surface proteins by biotinylation showed that treatment with Acad-EC caused the reduction in surface arcadlin/N-cadherin levels in neurons (Figure 7A; see Figure 7B for quantification). This reduction was inhibited by SB203580, suggesting that the internalization was mediated by the p38 MAPK pathway (Figures 7A and 7B). Although arcadlin also binds to cadherin-11 (see Figure 1K), there was no significant endocytosis of cadherin-11 upon the treatment with Acad-EC (Figures 7C and 7D). It seems that arcadlin is targeted to multiple species of classical cadherins, but not all of them undergo the endocytosis through this pathway. Individual classical cadherins might code for distinct subsets of synapses.
Figure 7. The Arcadlin-TAO2β-p38 MAPK Pathway Mediates the Internalization of N-Cadherin.
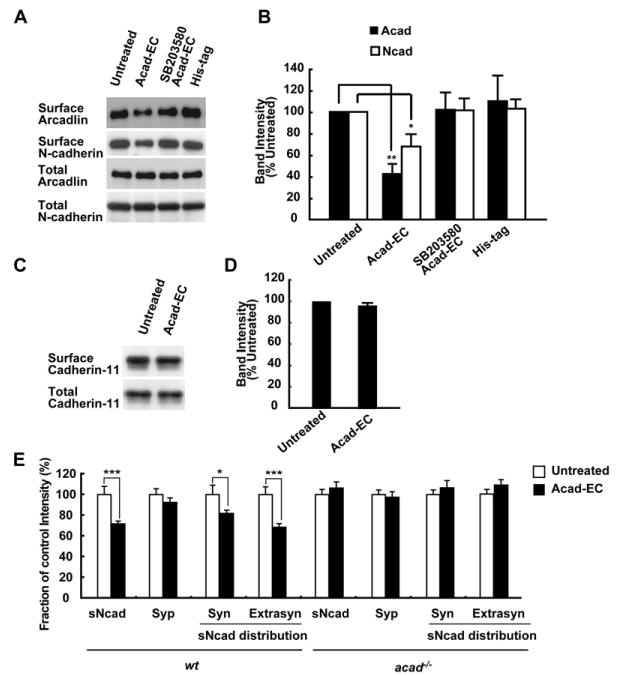
(A) Internalization of N-cadherin upon treatment of hippocampal neurons with Acad-EC analyzed by labeling surface proteins with biotin. His-tag peptide used as a control.
(B) Quantification of six independent experiments (A) (mean ± SEM).
(C) Surface (top) and total (bottom) cadherin-11 levels in hippocampal neurons with or without Acad-EC treatment were analyzed by immunoblot following the surface labeling with biotin.
(D) Quantification of five independent experiments (C) (mean ± SEM).
(E) Live neurons were incubated with or without Acad-EC prior to the surface N-cadherin labeling. Treatment of neurons with Acad-EC for 30 min resulted in a significant decrease in the surface N-cadherin mean intensity (mean ± SEM) in both the synaptophysin-overlapping fraction (Syn) and synaptophysin-nonoverlapping fraction (Extrasyn) populations. Note that the surface N-cadherin intensity of acad/papc−/− neurons did not change. n = 40–50 dendrites from 4–5 independent experiments.
*p < 0.05, **p < 0.01, ***p < 0.001.
Similar results were obtained in the microscopic quantification of N-cadherin internalization. A 28.3% ± 2.7% decrease in the mean intensity of total surface N-cadherin in the Acad-EC treatment group (30 min) relative to control was observed. The decrease in the surface N-cadherin mean intensity was observed in both the synaptic and extrasynaptic populations (Figure 7E). In acad−/− neurons, there was no significant change in the mean intensity of total surface N-cadherin in the Acad-EC treatment group relative to control (Figure 7E).
In HEK293T cells cotransfected with arcadlin, N-cadherin, and tao2β, addition of Acad-EC protein also triggered endocytosis (Figure S3). Arcadlin and N-cadherin were cointernalized in the presence of TAO2β but were retained on the plasma membrane in its absence (Figure S3). Taken together, these data indicate that the internalization of N-cadherin is mediated by the arcadlin-TAO2β-p38 MAPK molecular pathway and triggered by homophilic interactions between arcadlin protocadherin extracellular domains.
Arcadlin Mutation Increases Dendritic Spine Density
What is the consequence of the arcadlin-induced internalization of N-cadherin in the dendritic spine membrane? To address this question, we examined the morphology and the number of spines of hippocampal neurons. Cultured hippocampal neurons derived from acad−/− mice protruded a significantly larger number of spines than wild-type neurons (Figure 8B). This phenotype was rescued by the transfection of arcadlin cDNA (Figures 8A and 8B and Figure S4A). The rescuing effect was more prominent where the axons of transfected neurons were attached to the transfected dendrites (Figure 8A, square bracket). The other splice variant arcadlin-L did not recover the spine number (Figures 8A and 8B and Figure S4A), indicating that only arcadlin, not arcadlin-L, regulates spine density.
Figure 8. The Effects of Arcadlin on Dendritic Spine Density.
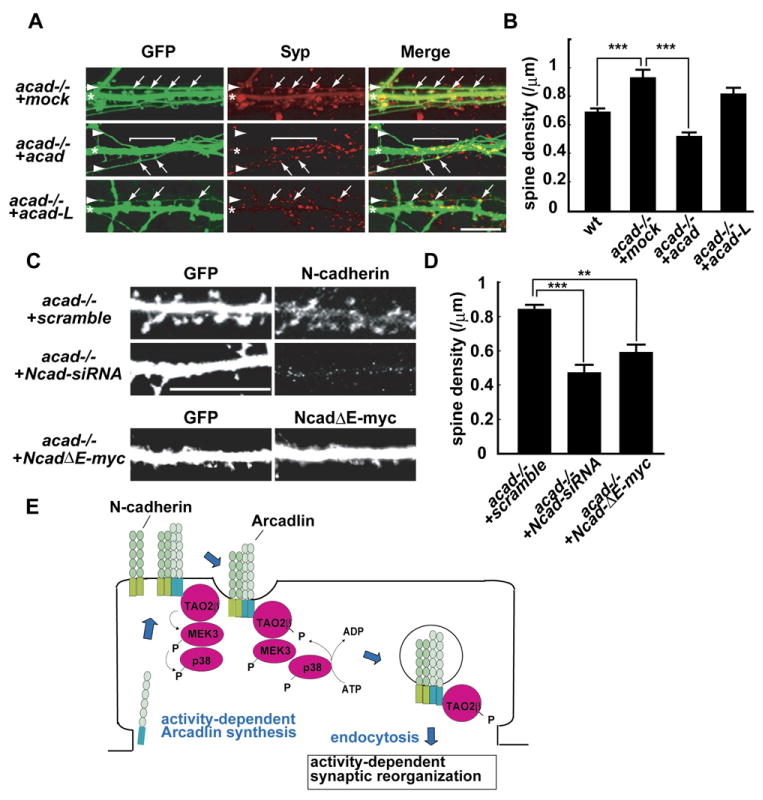
(A) Cultured acad−/− neurons were doubly transfected with EGFP and mock, arcadlin, or arcadlin-L at 6 DIV and visualized with EGFP at 15 DIV. The expressions of arcadlin and arcadlin-L were confirmed by immunocytochemistry (Figure S4A). Arrowheads, axons; asterisks, dendrites; arrows, spine heads attached to axons.
(B) Quantification of dendritic spine density (mean ± SEM) of WT and acad−/− neurons transfected with mock, arcadlin, or arcadlin-L. n = 30 independent neurons from 10 independent experiments.
(C) Cultured acad−/− neurons were doubly transfected with EGFP and N-cadherin-siRNA (Ncad-siRNA), scramble-siRNA, or NcadΔE-myc at 6 DIV and visualized with GFP at 15 DIV. Expression of N-cadherin was examined by immunocytochemistry. Coexpression of NcadΔE was confirmed by immunostaining for the myc-epitope. The suppression of N-cadherin protein level with the N-cadherin-siRNA was confirmed in mouse N-cadherin-GFP-transfected HEK293T cells (Figure S4B).
(D) Quantification of dendritic spine density (mean ± SEM) of neurons transfected with siRNAs or NcadΔE. n = 30 independent neurons from 10 independent experiments.
(E) A model for arcadlin-TAO2β-p38-mediated endocytosis of N-cadherin.
**p < 0.01, ***p < 0.001. Scale bars, 10 μm.
We then asked whether the N-cadherin endocytosis caused the arcadlin-induced change in spine density. There are several studies showing the relationship between N-cadherin activity and spine number. The spine number of N-cadherin KO neurons is maintained (Jungling et al., 2006; Kadowaki et al., 2007); a sustained period of N-cadherin loss in these neurons might allow other synaptic cell adhesion molecules to compensate for N-cadherin. In contrast, spine number is suppressed in neurons whose N-cadherin is knocked down by RNAi techniques (Saglietti et al., 2007). Consistently, an expression of a dominant-negative form of N-cadherin reduces the number of synaptic puncta (Togashi et al., 2002). In the present study, the spine density of acad−/− neurons was reduced by siRNA knockdown of N-cadherin (Figures 8C and 8D and Figure S4B). Moreover, a similar effect was observed by the expression of a dominant-negative form of N-cadherin (NcadΔE) (Figures 8C and 8D). The data suggest that the arcadlin-induced change in spine number is due at least in part to the N-cadherin endocytosis.
Discussion
Our results are consistent with a molecular pathway in which N-cadherin endocytosis is regulated in dendrites by the following sequence of events. (1) Neural activity increases expression of the protocadherin arcadlin in excitatory synapses. (2) Homophilic interactions between extracellular domains of arcadlin molecules activate the phosphorylation of the catalytic domain of the MAPKKK TAO2β, which is constitutively bound to the arcadlin intracellular domain. (3) TAO2β activates p38 MAPK via the MEK3 MAPKK. (4) Activated p38 MAPK feeds-back on TAO2β, phosphorylating it at Ser1038 of its specialized carboxy-terminal regulatory domain. (5) Phosphorylated TAO2β bound to arcadlin initiates the endocytosis of both arcadlin and N-cadherin. The striking effects of mutated or overexpressed arcadlin suggests an inhibitory role in regulating the activity of N-cadherin, one of the key molecules involved in spine remodeling (Okamura et al., 2004; Togashi et al., 2002). We propose that this pathway may provide a molecular explanation for how cell-adhesion molecules may transduce synaptic activity into changes in the morphology of dendritic spine membranes (Figure 8E).
Arcadlin Is an Upstream Receptor that Activates the p38 MAPK Pathway
Arcadlin is a member of protocadherin superfamily of cell-adhesion proteins characterized by ∼110 amino acid extracellular repeats that possess homophilic binding affinity. Indeed, homophilic cell-adhesion activity of arcadlin/PAPC has been demonstrated (Kim et al., 1998; Yamagata et al., 1999). Our present data also suggests that arcadlin is not a static adhesion protein, but rather plays a highly dynamic role during the remodeling of synaptic spine membrane. Homophilic interactions in trans between arcadlin extracellular domains trigger a newly discovered signal transduction cascade connected to p38 MAPK.
The p38 MAPK pathway has been studied extensively (Johnson and Lapadat, 2002). Only recently have neural roles for p38 MAPK in synaptic plasticity been demonstrated (Thomas and Huganir, 2004). To our knowledge, however, there is only one molecule identified as a p38 MAPK regulator in neurons: dual leucine zipper-bearing kinase (DLK), which is a serine/threonine kinase activated by calcium entry at synaptic terminals (Mata et al., 1996). We have now identified TAO2β as another regulator of p38 MAPK in neural tissue. TAO2β is a signal transducer poised close to the inner plasma membrane via association with the intracellular domain of the arcadlin protocadherin. In this sense, the arcadlin protocadherin serves as receptor, rather than an adhesion protein. The observation that the expression of arcadlin is induced after a few hours of synaptic stimulation (Yamagata et al., 1999) suggests that arcadlin serves as a receptor for a late-onset signal transduction pathway resulting in p38 MAPK activation.
The Cytoplasmic Region of Arcadlin Confers Specificity on Signal Transduction
A multitude of protocadherins are expressed in mammalian brain (Wu and Maniatis, 1999). Rat arcadlin is the ortholog of human and mouse pcdh8, as well as Xenopus and zebrafish papc. The arcadlin cDNA, however, lacks a 294 base pair exon encoding the cytoplasmic region of pcdh8/papc, resulting in its gene product having a uniquely short cytoplasmic region (Yamagata et al., 1999). The long and short forms of arcadlin/PAPC/pcdh8 are differentially expressed in early embryos, implying distinct functions of these isoforms (Makarenkova et al., 2005). The signal transduction pathway transduced by the short form of arcadlin was found here to involve TAO2β, p38 MAPK, and endocytosis. This pathway appears to depend on the shorter cytoplasmic tail of arcadlin, as our unpublished results indicate that the long form of rat arcadlin/PAPC/pcdh8 does not induce p38 MAPK activation or endocytosis.
Endocytosis of Synaptic Cadherin May Lead to the Disruption of Synaptic Adherens Junction
Studies during the past several years have revealed the importance of intracellular trafficking as a means of regulating the functions of classical cadherins (Bryant and Stow, 2004; Chen and Gumbiner, 2006). In epithelial tissues, E-cadherin is a key cell-cell adhesion molecule at adherens junctions and undergoes endocytosis when adherens junctions are disrupted by the action of extracellular signals (Troyanovsky et al., 2006). The dynamin-dependent endocytosis of synaptic N-cadherin triggered by arcadlin may also lead to the disruption of synaptic adherens junctions.
Puncta adherentia are synaptic adherens junctions that reside in the periphery of the synaptic active zone (Uchida et al., 1996). Because the expression of N-cadherin in mature excitatory synapses in hippocampal neurons is concentrated in puncta adherentia, the arcadlin-mediated endocytosis is likely to take place in their vicinity. In fact, the arcadlin immunoreactivity was often found in puncta adherentia in MECS-treated brains (unpublished data). The perisynaptic zone that includes puncta adherentia has recently been recognized as a hot spot of membrane remodeling during synaptic plasticity. AMPA glutamate receptors, for example, undergo activity-dependent delivery to (Shi et al., 1999) and removal from (Zhu et al., 2002) perisynaptic membranes. Synaptic activity-induced activation of the arcadlin-TAO2β-MEK3-p38 signaling pathway could regulate the adhesiveness of synaptic membrane at puncta adherentia and also the activity of neurotransmitter receptors.
Arcadlin-Triggered N-Cadherin Endocytosis Is a Late-Onset Event
Recently, Tai et al. (2007) reported that N-cadherin internalization is surprisingly high under the unstimulated conditions (about 50% of the surface N-cadherin is internalized after 100 min). They have shown that N-cadherin endocytosis is reduced by activating NMDA receptor, and the surface-localized N-cadherins in spines are stabilized with the recruitment of β-catenin into spines. This phenomenon is consistent with the stabilization of surface N-cadherin in response to NMDAR stimulation, as demonstrated by an enhanced resistance to trypsin (Tanaka et al., 2000). Such an enhanced adhesivity of the synaptic membranes, which occurs within a few minutes after the synaptic stimulation, does not require protein synthesis, providing a possible explanation for the short-term remodeling of spine structure (Okamura et al., 2004). On the other hand, the arcadlin-mediated N-cadherin internalization does not operate during these early stages after synaptic stimulation, because the arcadlin-induced internalization of N-cadherin begins at least 4 hr after the stimulation and requires protein synthesis (Figures 1 and 3). Whether or not these arcadlin-dependent and -independent mechanisms share a common endocytic pathway remains to be investigated. Thus, the arcadlin-mediated N-cadherin endocytosis is a late-onset event during the recovery phase following the synaptic stimulation and may provide a homeostatic mechanism balancing total tone and complexity of the neural network.
There is another processing system in which a physiological concentration of glutamate induces a rapid cleavage of N-cadherin (becomes obvious within 15 min) by presenilin-1/γ-secretase, resulting in the generation of cytoplasmic fragment (CTF2) (Marambaud et al., 2003; Uemura et al., 2006). We have confirmed the existence of a 35 kD CTF2 (4.3% ± 1.5% of full-length N-cadherin, n = 5), which increased to 15.6% ± 2.3% (n = 5) after treating with KCl (data not shown). Our experiments showed that the CTF2 amount was not significantly different between 2 and 4 hr after KCl treatment, suggesting that the CTF2 production was completed within 2 hr after neuronal stimulation. On the other hand, our present finding of full-length N-cadherin internalization became evident only after the arcadlin induction at least 4 hr after the stimulation, and occupied as much as 33.8% ± 1.7% of surface N-cadherin (Figure 3B). The internalization of N-cadherin, therefore, is distinct from the presenilin-1/γ-secretase pathway. These two different systems might operate sequentially to regulate surface N-cadherin amount during the activity-dependent processes.
Inhibitory Role of Arcadlin in Dendritic Spine Number
Activity-induced changes in synaptic morphology, including the enlargement of synaptic apposition zones and the emergence of new synapses, have been correlated with long-term potentiation (LTP) (Colicos et al., 2001; Muller et al., 2000; Nagerl et al., 2004). Conversely, long-term depression has been correlated with the pruning of synaptic spines (Nagerl et al., 2004; Zhou et al., 2004). We first thought that arcadlin would be a “positive” regulator of synaptic remodeling, because arcadlin accumulates at the synaptic junction where LTP takes place, and neutralizing antibodies indicated that it is required for the establishment of electrophysiological LTP (Yamagata et al., 1999). Unexpectedly, however, arcadlin loss of function caused an increase in the number of synaptic spines (Figure 8). We observed that the arcadlin neutralizing antibody triggered internalization of surface arcadlin, suggesting that the antibody treatment might block LTP by accelerating the cointernalization with N-cadherin. In addition, it has been shown that other neural activity-regulated molecules, such as MEF2 and SNK, suppress the number of spines (Flavell et al., 2006; Pak and Sheng, 2003). Our findings suggest that activity-inducible proteins may dampen synaptic function after elevated activity by negatively regulating the spine numbers. In the future, it will be interesting to investigate how arcadlin protocadherin mutations affect learning and memory in the mouse.
In conclusion, the signaling pathway presented here implicates the adhesive apparatus of synaptic spine membranes as a focal point of vigorous synaptic activity-dependent remodeling. The arcadlin-TAO2β-p38 pathway of activity-regulated endocytosis may provide a possible relationship between the modulation of the N-cadherin adhesive machinery and the insertion and removal of neurotransmitter receptors.
Experimental procedures
Molecular Cloning and Constructing Plasmids
Expression vectors and other plasmids were constructed as described in Supplemental Data. Yeast two-hybrid screening was performed as described previously (Irie et al., 2000). In summary, rat hippocampal cDNAs were subcloned in the SalI/NotI sites of the pPC86 vector, which contains the GAL4 activation domain, and the cytoplasmic region of arcadlin was subcloned in-frame in the SalI/NotI sites of the pPC97 vector, which contains the GAL4 DNA binding domain. The plasmids were used to transform PCY2 cells, and positive clones were selected on double-minus plates (Leu−, Trp−) and assayed for β-galactosidase activity.
Cell Culture and Transfection
Details of culture and transfection of COS7, HEK293T cells, and hippocampal neurons from rats (E18) and mice (P0) are provided as Supplemental Data. Inhibition of N-cadherin expression by siRNA was performed as described (Paik et al., 2004). The siRNAs were N-cadherin siRNA (UGUCAAUGGGGUUCUCCACdTdT and GUGGAGAACCCCAUUGACAdTdT) and scrambled siRNA (CAUGCGGAUUCGGAUUUUCdTdT and GAAAAUCCGAAUCCGCAUGdTdT). Oligonucleotides were synthesized by Dharmacon Research Inc., deprotected, and duplexed as described. The target sequence was common for mouse and rat N-cadherin, and their suppression was confirmed in mouse and rat neurons by immunostaining.
Cell-Aggregation Assay
Cell-aggregation assay was performed as described (Takeichi, 1977). Briefly, single cells were prepared by harvesting from monolayer cells with 0.01% crystalline trypsin (type I, Sigma) in HCMF (HEPES-buffered Ca2+- and Mg2+-free HBSS) containing 0.1 mM CaCl2 for 15 min at 37°C followed by supplementation with 0.01% soybean trypsin inhibitor (type I-S, Sigma). 1 × 106 cells suspended in 3 ml HCMF were put into each well (2.8 cm × 1.5 cm) and incubated at 37°C on a gyratory shaker at 80 rpm. The total particle number in cell suspension at each time point was counted with a Coulter counter with 100 μm aperture.
Immunostaining
Rat brain sections, cultured hippocampal neurons, and HEK293T cells were fixed with methanol or 2% paraformaldehyde, blocked, and permeabilized with BL solution (5% normal goat serum, 0.1% Triton X-100, 0.02% sodium azide in PBS), and incubated overnight at 4°C with primary antibodies: anti-arcadlin (rabbit, 1:200; Yamagata et al., 1999), anti-N-cadherin (rabbit, 1:200; Okamura et al., 2004; Transduction lab, mouse, 12.5 μg/ml), anti-synaptophysin (Zymed, rabbit, 1:40), anti-PSD-95 (Upstate biotech, mouse, 1:200), anti-GAD6 (Developmental Studies Hybridoma Bank, mouse, 1 μg/ml), anti-NMDA receptor (NR1) (PharMingen, mouse, 1:500), anti-phospho-p38 MAPK (Sigma, mouse, 1:200), anti-GFP (Molecular Probe, rabbit, 1:400), anti-myc (Calbiochem, mouse, 2 μg/ml; Upstate biotech, rabbit, 1:1000), and anti-FLAG (M2) (Sigma, mouse, 1:200). Immunoreactivity was visualized using species-specific fluorochrome-conjugated secondary antibodies.
Immunoprecipitation
Rat hippocampi, cultured neurons, COS7 cells, and HEK293T cells were homogenized with lysis buffers optimized for each tissue and centrifuged to obtain clear protein extracts. The samples were incubated overnight at 4°C with packed 10 μl of protein A Sepharose (GE Healthcare) and specific antibodies: anti-arcadlin (rabbit, 5 μl), anti-TAO2β (rabbit, 5 μl), anti-N-cadherin (Sigma, rabbit, 10 μl), anti-myc (Calbiochem, mouse, 2 μg), and anti-GFP (Molecular Probe, rabbit, 2 μl). The precipitated immune complex was eluted with SDS sample buffer and immunoblotted with specific antibodies: anti-arcadlin (rabbit, 1:2000), anti-TAO2β (rabbit, 1:2000), anti-N-cadherin (Transduction lab, mouse, 1:3000), anti-cadherin-11 (Zymed, mouse, 1:800), anti-β-catenin (Zymed, rabbit, 1:1000), anti-α-catenin (Transduction lab, mouse, 1:250), anti-p120 (Transduction lab, mouse, 1:1000), anti-plakoglobin (Transduction lab, mouse, 1:2000), and anti-myc (Calbiochem, mouse, 1:1000). Details of immunoprecipitation are provided as Supplemental Data.
Characterization of the Association Modes of Arcadlin and N-Cadherin
Association modes of arcadlin-arcadlin and N-cadherin-arcadlin interactions were determined by cell-mix coimmunoprecipitation experiments (Nuriya and Huganir, 2006). The mixture of cells was prepared by transfecting arcadlin-EGFP and arcadlin-flag (or N-cadherin) into two separate dishes of cells, which were once harvested by trypsinization after 24–30 hr and then mixed together in a new dish to allow these two discrete populations of cells to grow in contact with each other. Other groups of cells that were cotransfected with arcadlin-EGFP and arcadlin-flag (or N-cadherin) were treated in the same way. 48 to 50 hr after the transfection, Acad-EC was added to arcadlin-flag-expressing cells for 30 min to dissociate the arcadlin-arcadlin interaction. Then, the cells were harvested and subjected to coimmunoprecipitation analyses.
In Vitro Kinase Assay
Activated p38 MAPK was prepared in HEK293T cells transfected with arcadlin, tao2β, MEK3, and p38 MAPK. After 48 hr of transfection, cells were exposed to Acad-EC for 30 min and lysed in lysis buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM sodium orthovanadate, 1 mg/ml leupeptin, 1 mM PMSF). Cell lysates were incubated with immobilized phospho-p38 MAPK monoclonal antibody (Cell Signaling Technology). To examine which amino acid residue of TAO2β can be phosphorylated by p38 MAPK, substrate (GST-TAO2β751-1056, -TAO2βS951A, -TAO2βS1038A, -TAO2βS1040A, or ATF2) was incubated with immobilized phospho-p38 MAPK in kinase buffer (25 mM Tris-HCl [pH 7.5], 5 mM β-glycerophosphate, 2 mM dithiothreitol, 0.1 mM sodium orthovanadate, 10 mM magnesium chloride) containing 100 μM [γ-32P]ATP at 30°C for 30 min. The phosphorylated proteins were resolved by SDS-PAGE and analyzed by autoradiography.
Surface Biotinylation Assay
After various treatments and washing twice with ice-cold PBS, HEK293T cells and neurons were incubated with EZ-Link Sulfo-NHS-SS-Biotin for 30 min at 4°C. The biotinylation reaction was terminated with three washes with cold TBS. Cells were lysed with RIPA buffer (20 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.5% deoxycholate, 0.1% SDS, 10 μg/ml leupeptin, 10 μg/ml pepstatin A, 10 μg/ml antipain, 1 mM PMSF) and centrifuged at 10,000 × g at 4°C for 2 min. Immobilized NeutrAvidin Gel (Pierce) was added to the supernatants and incubated at 4°C for 3 hr. After being washed with RIPA buffer, the bound biotinylated proteins were subjected to SDS-PAGE followed by western blot with anti-N-cadherin (Transduction lab, mouse, 1:3000), anti-arcadlin (1:1000), or anti-neuroligin (Synaptic systems, mouse, 1:1000) antibodies. The densitometrically quantified data were statistically analyzed by unpaired two-tailed Student's t test.
Surface N-Cadherin Labeling
Surface N-cadherin labeling was performed as described (Tai et al., 2007) with modifications. Neurons (14–17 DIV) after various treatments were incubated for 30 min on ice in the presence of N-cadherin surface antibody (MT79, 42 μg/ml in ice-cold conditioned medium supplied with 10 mM HEPES-NaOH [pH7.4]). Neurons were rinsed three times with ice-cold HBSS, fixed with 4% paraformaldehyde for 30 min, rinsed three times with PBS, and then incubated with a secondary antibody for 1 hr at RT. After rinsing with PBS and permeabilization with BL, neurons were incubated with primary and secondary antibodies against the intracellular proteins sequentially for 1 hr each at RT. After wash with PBS, neurons were mounted for imaging. Surface N-cadherin staining was imaged with a Zeiss 510 Meta confocal microscope with a 40× objective (NA = 1.3). Each stack was maximal Z-projected into a single stack. Individual neuron within each projected image was registered by using the RegisterROI plug-in of ImageJ software (W. Rasband, National Institutes of Health, Bethesda, MD; http://rsb.info.nih.gov/ij/). Two dendrites were straightened per cell. Average fluorescence intensities were computed separately for spines and shafts in each dendrite. Total dendritic signals represent the sum of spine and shaft signals. Separation of synaptophysin-overlapping fraction from synaptophysin-nonoverlapping fraction populations was performed with the ColocalizeRGB plug-in of ImageJ. The intensity of the overlapping fraction was normalized with the relative intensity for synaptophysin.
Supplementary Material
The Supplemental Data for this article can be found online at http://www.neuron.org/cgi/content/full/56/3/456/DC1/.
Acknowledgments
We thank Naomasa Miki, Takahiro Fujimoto, and Kirsten Arndt for valuable discussions; Masumi Ichikawa and Junko Kuroda-Kimura for confocal microscopy; Hideru Togashi for advice with culturing mouse neurons; Machiko Kamimoto, Tomoyuki Sugimoto, and Takehisa Uchiyama for statistics; Greg Phillips, Weisong Shan, and David Colman for reagents; Kyoko Suzuki, Hong Shi, and Atsunori Ohnishi for technical assistance. This work was supported by KAKENHIs (12053278, 15300130, and 18209037 to K.Y.; 13210093, 14017063, and 17500253 to H.T.) Y.Y. is supported by National Institute of Health (NS20147). E.M.D.R. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Accession Number: We have deposited the sequence for the new isoform of TAO2 kinase (=TAO2β) into DDBJ (http://www.ddbj.nig.ac.jp/). The accession number is AB290408.
References
- Bailey CH, Chen M, Keller F, Kandel ER. Serotonin-mediated endocytosis of apCAM: an early step of learning-related synaptic growth in Aplysia. Science. 1992;256:645–649. doi: 10.1126/science.1585177. [DOI] [PubMed] [Google Scholar]
- Benson DL, Tanaka H. N-cadherin redistribution during synaptogenesis in hippocampal neurons. J Neurosci. 1998;18:6892–6904. doi: 10.1523/JNEUROSCI.18-17-06892.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozdagi O, Shan W, Tanaka H, Benson DL, Huntley GW. Increasing numbers of synaptic puncta during late-phase LTP: N-cadherin is synthesized, recruited to synaptic sites, and required for potentiation. Neuron. 2000;28:245–259. doi: 10.1016/s0896-6273(00)00100-8. [DOI] [PubMed] [Google Scholar]
- Bryant DM, Stow JL. The ins and outs of E-cadherin trafficking. Trends Cell Biol. 2004;14:427–434. doi: 10.1016/j.tcb.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Chen X, Gumbiner BM. Paraxial protocadherin mediates cell sorting and tissue morphogenesis by regulating C-cadherin adhesion activity. J Cell Biol. 2006;174:301–313. doi: 10.1083/jcb.200602062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Hutchison M, Cobb MH. Isolation of the protein kinase TAO2 and identification of its mitogen-activated protein kinase/extracellular signal-regulated kinase kinase binding domain. J Biol Chem. 1999;274:28803–28807. doi: 10.1074/jbc.274.40.28803. [DOI] [PubMed] [Google Scholar]
- Chen Z, Raman M, Chen L, Lee SF, Gilman AG, Cobb MH. TAO (thousand-and-one amino acid) protein kinases mediate signaling from carbachol to p38 mitogen-activated protein kinase and ternary complex factors. J Biol Chem. 2003;278:22278–22283. doi: 10.1074/jbc.M301173200. [DOI] [PubMed] [Google Scholar]
- Colicos MA, Collins BE, Sailor MJ, Goda Y. Remodeling of synaptic actin induced by photoconductive stimulation. Cell. 2001;107:605–616. doi: 10.1016/s0092-8674(01)00579-7. [DOI] [PubMed] [Google Scholar]
- Cooney JR, Hurlburt JL, Selig DK, Harris KM, Fiala JC. Endosomal compartments serve multiple hippocampal dendritic spines from a widespread rather than a local store of recycling membrane. J Neurosci. 2002;22:2215–2224. doi: 10.1523/JNEUROSCI.22-06-02215.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fannon AM, Colman DR. A model for central synaptic junctional complex formation based on the differential adhesive specificities of the cadherins. Neuron. 1996;17:423–434. doi: 10.1016/s0896-6273(00)80175-0. [DOI] [PubMed] [Google Scholar]
- Flavell SW, Cowan CW, Kim TK, Greer PL, Lin Y, Paradis S, Griffith EC, Hu LS, Chen C, Greenberg ME. Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science. 2006;311:1008–1012. doi: 10.1126/science.1122511. [DOI] [PubMed] [Google Scholar]
- Huber O, Kemler R, Langosch D. Mutations affecting transmembrane segment interactions impair adhesiveness of E-cadherin. J Cell Sci. 1999;112:4415–4423. doi: 10.1242/jcs.112.23.4415. [DOI] [PubMed] [Google Scholar]
- Irie Y, Yamagata K, Gan Y, Miyamoto K, Do E, Kuo CH, Taira E, Miki N. Molecular cloning and characterization of Amida, a novel protein which interacts with a neuron-specific immediate early gene product arc, contains novel nuclear localization signals, and causes cell death in cultured cells. J Biol Chem. 2000;275:2647–2653. doi: 10.1074/jbc.275.4.2647. [DOI] [PubMed] [Google Scholar]
- Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- Jungling K, Eulenburg V, Moore R, Kemler R, Lessmann V, Gottmann K. N-Cadherin transsynaptically regulates short-term plasticity at glutamatergic synapses in embryonic stem cell-derived neurons. J Neurosci. 2006;26:6968–6978. doi: 10.1523/JNEUROSCI.1013-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadowaki M, Nakamura S, Machon O, Krauss S, Radice GL, Takeichi M. N-cadherin mediates cortical organization in the mouse brain. Dev Biol. 2007;304:22–33. doi: 10.1016/j.ydbio.2006.12.014. [DOI] [PubMed] [Google Scholar]
- Kamiguchi H, Lemmon V. Recycling of the cell adhesion molecule L1 in axonal growth cones. J Neurosci. 2000;20:3676–3686. doi: 10.1523/JNEUROSCI.20-10-03676.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Yamamoto A, Bouwmeester T, Agius E, De Robertis EM. The role of paraxial protocadherin in selective adhesion and cell movements of the mesoderm during Xenopus gastrulation. Development. 1998;125:4681–4690. doi: 10.1242/dev.125.23.4681. [DOI] [PubMed] [Google Scholar]
- Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- Makarenkova H, Sugiura H, Yamagata K, Owens G. Alternatively spliced variants of protocadherin 8 exhibit distinct patterns of expression during mouse development. Biochim Biophys Acta. 2005;1681:150–156. doi: 10.1016/j.bbaexp.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Manabe T, Togashi H, Uchida N, Suzuki SC, Hayakawa Y, Yamamoto M, Yoda H, Miyakawa T, Takeichi M, Chisaka O. Loss of cadherin-11 adhesion receptor enhances plastic changes in hippocampal synapses and modifies behavioral responses. Mol Cell Neurosci. 2000;15:534–546. doi: 10.1006/mcne.2000.0849. [DOI] [PubMed] [Google Scholar]
- Marambaud P, Wen PH, Dutt A, Shioi J, Takashima A, Siman R, Robakis NK. A CBP binding transcriptional repressor produced by the PS1/[epsilon]-cleavage of N-cadherin is inhibited by PS1 FAD mutations. Cell. 2003;114:635–645. doi: 10.1016/j.cell.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Mata M, Merritt SE, Fan G, Yu GG, Holzman LB. Characterization of dual leucine zipper-bearing kinase, a mixed lineage kinase present in synaptic terminals whose phosphorylation state is regulated by membrane depolarization via calcineurin. J Biol Chem. 1996;271:16888–16896. doi: 10.1074/jbc.271.28.16888. [DOI] [PubMed] [Google Scholar]
- Muller D, Toni N, Buchs PA. Spine changes associated with long-term potentiation. Hippocampus. 2000;10:596–604. doi: 10.1002/1098-1063(2000)10:5<596::AID-HIPO10>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Murase S, Mosser E, Schuman EM. Depolarization drives beta-Catenin into neuronal spines promoting changes in synaptic structure and function. Neuron. 2002;35:91–105. doi: 10.1016/s0896-6273(02)00764-x. [DOI] [PubMed] [Google Scholar]
- Nagerl UV, Eberhorn N, Cambridge SB, Bonhoeffer T. Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron. 2004;44:759–767. doi: 10.1016/j.neuron.2004.11.016. [DOI] [PubMed] [Google Scholar]
- Nuriya M, Huganir RL. Regulation of AMPA receptor trafficking by N-cadherin. J Neurochem. 2006;97:652–661. doi: 10.1111/j.1471-4159.2006.03740.x. [DOI] [PubMed] [Google Scholar]
- Okamura K, Tanaka H, Yagita Y, Saeki Y, Taguchi A, Hiraoka Y, Zeng LH, Colman DR, Miki N. Cadherin activity is required for activity-induced spine remodeling. J Cell Biol. 2004;167:961–972. doi: 10.1083/jcb.200406030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik J-H, Skoura A, Chae S-S, Cowan AE, Han DK, Proia RL, Hla T. Sphingosine 1-phosphate receptor regulation of N-cadherin mediates vascular stabilization. Genes Dev. 2004;18:2392–2403. doi: 10.1101/gad.1227804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak DTS, Sheng M. Targeted protein degradation and synapse remodeling by an inducible protein kinase. Science. 2003;302:1368–1373. doi: 10.1126/science.1082475. [DOI] [PubMed] [Google Scholar]
- Racz B, Blanpied TA, Ehlers MD, Weinberg RJ. Lateral organization of endocytic machinery in dendritic spines. Nat Neurosci. 2004;7:917–918. doi: 10.1038/nn1303. [DOI] [PubMed] [Google Scholar]
- Saglietti L, Dequidt C, Kamieniarz K, Rousset M-C, Valnegri P, Thoumine O, Beretta F, Fagni L, Choquet D, Sala C, et al. Extracellular interactions between GluR2 and N-cadherin in spine regulation. Neuron. 2007;54:461–477. doi: 10.1016/j.neuron.2007.04.012. [DOI] [PubMed] [Google Scholar]
- Shi SH, Hayashi Y, Petralia RS, Zaman SH, Wenthold RJ, Svoboda K, Malinow R. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science. 1999;284:1811–1816. doi: 10.1126/science.284.5421.1811. [DOI] [PubMed] [Google Scholar]
- Strehl S, Glatt K, Liu QM, Glatt H, Lalande M. Characterization of two novel protocadherins (PCDH8 and PCDH9) localized on human chromosome 13 and mouse chromosome 14. Genomics. 1998;53:81–89. doi: 10.1006/geno.1998.5467. [DOI] [PubMed] [Google Scholar]
- Sytnyk V, Leshchyns'ka I, Nikonenko AG, Schachner M. NCAM promotes assembly and activity-dependent remodeling of the postsynaptic signaling complex. J Cell Biol. 2006;174:1071–1085. doi: 10.1083/jcb.200604145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai CY, Mysore SP, Chiu C, Schuman EM. Activity-regulated N-cadherin endocytosis. Neuron. 2007;54:771–785. doi: 10.1016/j.neuron.2007.05.013. [DOI] [PubMed] [Google Scholar]
- Takeichi M. Functional correlation between cell adhesive properties and some cell surface proteins. J Cell Biol. 1977;75:464–474. doi: 10.1083/jcb.75.2.464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H, Shan W, Phillips GR, Arndt K, Bozdagi O, Shapiro L, Huntley GW, Benson DL, Colman DR. Molecular modification of N-cadherin in response to synaptic activity. Neuron. 2000;25:93–107. doi: 10.1016/s0896-6273(00)80874-0. [DOI] [PubMed] [Google Scholar]
- Tang L, Hung CP, Schuman EM. A role for the cadherin family of cell adhesion molecules in hippocampal long-term potentiation. Neuron. 1998;20:1165–1175. doi: 10.1016/s0896-6273(00)80497-3. [DOI] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL. MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci. 2004;5:173–183. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- Tibbles LA, Woodgett JR. The stress-activated protein kinase pathways. Cell Mol Life Sci. 1999;55:1230–1254. doi: 10.1007/s000180050369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togashi H, Abe K, Mizoguchi A, Takaoka K, Chisaka O, Takeichi M. Cadherin regulates dendritic spine morphogenesis. Neuron. 2002;35:77–89. doi: 10.1016/s0896-6273(02)00748-1. [DOI] [PubMed] [Google Scholar]
- Troyanovsky RB, Sokolov EP, Troyanovsky SM. Endocytosis of cadherin from intracellular junctions is the driving force for cadherin adhesive dimer disassembly. Mol Biol Cell. 2006;17:3484–3493. doi: 10.1091/mbc.E06-03-0190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida N, Honjo Y, Johnson KR, Wheelock MJ, Takeichi M. The catenin/cadherin adhesion system is localized in synaptic junctions bordering transmitter release zones. J Cell Biol. 1996;135:767–779. doi: 10.1083/jcb.135.3.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura K, Kihara T, Kuzuya A, Okawa K, Nishimoto T, Bito H, Ninomiya H, Sugimoto H, Kinoshita A, Shimohama S. Activity-dependent regulation of β-catenin via ∈-cleavage of N-cadherin. Biochem Biophys Res Commun. 2006;345:951–958. doi: 10.1016/j.bbrc.2006.04.157. [DOI] [PubMed] [Google Scholar]
- Wu Q, Maniatis T. A striking organization of a large family of human neural cadherin-like cell adhesion genes. Cell. 1999;97:779–790. doi: 10.1016/s0092-8674(00)80789-8. [DOI] [PubMed] [Google Scholar]
- Yamagata K, Andreasson KI, Sugiura H, Maru E, Dominique M, Irie Y, Miki N, Hayashi Y, Yoshioka M, Kaneko K, et al. Arcadlin is a neural activity-regulated cadherin involved in long term potentiation. J Biol Chem. 1999;274:19473–19479. doi: 10.1074/jbc.274.27.19473. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Kemp C, Bachiller D, Geissert D, De Robertis EM. Mouse paraxial protocadherin is expressed in trunk mesoderm and is not essential for mouse development. Genesis. 2000;27:49–57. [PubMed] [Google Scholar]
- Zhou Q, Homma KJ, Poo MM. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron. 2004;44:749–757. doi: 10.1016/j.neuron.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Zhu JJ, Qin Y, Zhao M, Van Aelst L, Malinow R. Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell. 2002;110:443–455. doi: 10.1016/s0092-8674(02)00897-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The Supplemental Data for this article can be found online at http://www.neuron.org/cgi/content/full/56/3/456/DC1/.