

Abstract
The Escherichia coli clamp loader, γ complex (γ3δδ′λψ), catalyzes ATP-driven assembly of β clamps onto primer-template DNA (p/tDNA), enabling processive replication. The mechanism by which γ complex targets p/tDNA for clamp assembly is not resolved. According to previous studies, charged/polar amino acids inside the clamp loader chamber interact with the double-stranded (ds) portion of p/tDNA. We find that dsDNA, not ssDNA, can trigger a burst of ATP hydrolysis by γ complex and clamp assembly, but only at far higher concentrations than p/tDNA. Thus, contact between γ complex and dsDNA is necessary and sufficient, but not optimal, for the reaction, and additional contacts with p/tDNA likely facilitate its selection as the optimal substrate for clamp assembly. We investigated whether a conserved sequence—HRVW279QNRR—in δ subunit contributes to such interactions, since Tryptophan-279 specifically cross-links to the primer-template junction. Mutation of δ-W279 weakens γ complex binding to p/tDNA, hampering its ability to load clamps and promote proccessive DNA replication, and additional mutations in the sequence (δ-R277, δ-R283) worsen the interaction. These data reveal a novel location in the C-terminal domain of the E. coli clamp loader that contributes to DNA binding and helps define p/tDNA as the preferred substrate for the reaction.
INTRODUCTION
Clamps and clamp loaders enhance the ability of DNA polymerase to extend a primer from a few nucleotides to several thousand nucleotides per primer-template binding event, and therefore serve as essential accessory proteins in DNA replication. Clamps are ring-shaped, two or three subunit-containing proteins that encircle the double helix and can move along its length, forming mobile tethers for the polymerase and enabling processive DNA synthesis (1). Clamp loaders are claw-shaped, multi-subunit protein complexes that use ATP to catalyze assembly of clamps onto primed DNA templates for use by the polymerase (2–4). In addition to their role in DNA replication, clamps and clamp loaders play important roles in other processes of DNA metabolism, including DNA repair and recombination, as well as in mechanisms of cell cycle control (5,6). Not surprisingly, these proteins are found in all cells. The structures and mechanisms of action of these proteins from model organisms, such as Escherichia coli (γ complex and β clamp), Saccharomyces cerevisiae, Pyrococcus furiosus (RFC complex and PCNA clamp) and the T4 bacteriophage (gp44/62 complex and gp45 clamp), are under active investigation.
In E. coli, the β clamp comprises two identical subunits that bind head-to-tail to fashion a ring (7). The clamp loader comprises three γ/τ subunits and one each of the δ, δ′, χ and ψ subunits; τ is a longer version of γ, and its extra domain serves to hold core polymerases in a holoenzyme at the replication fork (8); however, a complex containing three γ subunits is a fully functional clamp loader as well (9–12). The γ/τ, δ, and δ′ proteins form a minimal, functional clamp loader (13); the χ subunit helps coordinate the actions of the clamp loader, primase and single-stranded DNA-binding protein at the replication fork (14,15); the ψ subunit, whose function has been rather inscrutable until lately, facilitates productive interactions of the clamp loader with both clamp and DNA during clamp assembly (16). γ/τ, δ and δ′ are arranged in a circle that is closed completely at the C-terminus and open at the N-terminus (17). These proteins are members of the large AAA+ family (ATPases Associated with diverse cellular Activities), which contain two signature N-terminal domains that catalyze ATP binding and hydrolysis (18,19), although only γ and τ are functional ATPases in the E. coli clamp loader (20–22). In addition to the Walker A and B motifs, each γ/τ and δ′ subunit also contains the conserved SRC motif, which has an ‘arginine finger’ that participates in the ATPase active site of the neighboring subunit. This arginine finger is necessary for catalysis, and also helps mediate ATP binding and hydrolysis-coupled conformational changes in the clamp loader that drive clamp assembly on primer-template DNA (p/tDNA) (23,24).
The clamp assembly reaction, as currently understood, involves an ordered series of events (Figure 1): (i) the clamp loader binds ATP; (ii) consequently, changes in the clamp loader conformation facilitate binding to the clamp and clamp opening, followed by binding to p/tDNA and positioning it within the open clamp; (iii) p/tDNA binding triggers rapid ATP hydrolysis by the γ/τ subunits; and (iv) the resultant change in clamp loader conformation facilitates release of the clamp and DNA, linked topologically to each other. While the clamp loader can interact with and act on each of its three substrates—ATP, β clamp, DNA—individually, one of the more intriguing features of the reaction mechanism is how each substrate modulates clamp loader interactions/actions with the other substrates in a coordinated fashion, resulting in catalysis of clamp assembly on DNA (25). Past studies of γ complex in the presence of ATP, β, and/or DNA have clarified both its intrinsic and substrate-mediated structure–function capabilities. For example, in the absence of ATP, γ complex can bind the β clamp, but with very low affinity, whereas in the presence of ATP, the interaction occurs with a tight KD ∼ 20 nM (16,26,27). This observation is consistent with ATP stabilizing a γ complex conformation that binds optimally to β. Also, γ complex has a basal ATPase rate that increases dramatically following DNA binding (26,28). This occurs both in the absence and presence of β. In the absence of β, however, the reaction is inefficient in that only a fraction of γ complex appears to achieve a conformation in which the γ subunits catalyze rapid ATP hydrolysis, whereas in the presence of β, all the γ subunits catalyze rapid ATP hydrolysis upon DNA binding (23,29,30). This observation is consistent with β stabilizing a γ complex conformation that binds optimally to DNA, which in turn leads to maximal stimulation of its ATPase activity, and release of the clamp assembled on DNA.
Figure 1.

γ complex clamp loader structure and function. (A) Minimal steps in the mechanism of γ complex-catalyzed β assembly on p/tDNA. (B) Crystal structure of γ complex with key residues of the conserved HRVWQNRR loop in δ subunit highlighted (R277, W279, R283).
All the evidence thus far indicates that p/tDNA with a 5′ single-stranded overhang—the appropriate target for clamp assembly in DNA replication—specifically stimulates the maximal ATPase activity of γ complex (30). When p/tDNA enters the central chamber of the clamp loader, γ and δ′ subunits present conserved lysine, arginine and polar residues for interaction with the double-stranded (ds) portion of the DNA (31,32). Mutation of these residues impacts both DNA binding and DNA-dependent ATPase activities of γ complex, affirming their important role in recognizing the dsDNA and coupling this step with the ATP hydrolysis and clamp/DNA release steps in the reaction (33). The single-stranded (ss) template is thought to exit the clamp loader via a gap between the δ and δ′ subunits (Figure 1) (31,33,34), but it is not clear whether there are sites on the clamp loader that specifically recognize this portion of p/tDNA. In an earlier study, we discovered a Tryptophan residue in the δ subunit (δ-W279) that cross-links specifically at the ss/ds junction of p/tDNA (35). Although the binding site for dsDNA on the clamp loader has been well defined, thus far δ-W279 is the only lead to a site that might interact with the ss/dsDNA junction or ssDNA template. If so, this residue should facilitate binding and selection of p/tDNA as the substrate for clamp assembly.
In order to investigate the hypothesis that δ-W279 is involved in γ complex binding to p/tDNA, and subsequent steps in the reaction, we examined the effects of mutating this residue on clamp loader function. Furthermore, δ-W279 is part of a highly conserved sequence in bacterial clamp loaders with several charged and polar residues that might interact with DNA (HRVW279QNRR; Figure 1). We therefore examined the effects of mutating additional residues in the vicinity of δ-W279, in order to determine whether this previously undefined motif plays a role in binding p/tDNA. The data provide useful insights into the clamp assembly mechanism of γ complex and related clamp loaders.
MATERIALS AND METHODS
Proteins, DNA and other reagents
Escherichia coli Pol III core proteins (α,ε,θ) (36) and the minimal (γδδ′) and complete (γ,δ,δ′, χ, ψ) γ complexes (10) were reconstituted from pure subunits and then purified from unassembled subunits as described. The β clamp was purified as described (7). Mutations in the holA gene encoding δ [W279A, W279Y and R277A, R282A, R283A (−/+W279A)], were made by QuikChange (Stratagene, La Jolla, CA) site-directed mutagenesis and confirmed by sequencing the entire holA gene. The mutant proteins were expressed, purified and reconstituted into γ complex in the same manner as wild-type δ. Protein concentrations were determined by Bradford assay or absorbance at 280 nm in 6 M guanidinium-HCl, 20 mM potassium phosphate, pH 6.5. Phosphate-binding protein (PBP) was purified and labeled with 7-diethylamino-3-((((2-maleimidyl)ethyl)amino)carbonyl) coumarin (MDCC) as described previously, except the final Mono-Q column chromatography step was omitted (37).
Short synthetic DNAs were purchased from Integrated DNA Technologies (Integrated DNA Technologies, Coralville, IA): 30-nt primer, 5′-CTG GTA ATA TCC AGA ACA ATA TTA CCG CCA-3′; 55-nt template, 5′-(NH2) TGA GCG TTT TTC CTG TTG CAA TGG CTG GCG GTA ATA TTG TTC TGG ATA TTA CCA G-3′; 105-nt template, 5′-GAG CGT CAA AAT GTA GGT ATT TCC ATG AGC GTT TTT CCT GTT GCA ATG GCT GGC GGT AAT ATT GTT CTG GAT ATT ACC AGC AAG GCC GAT AGT TTG AGT TCT TCT-3′; 37 nt, 5′– ATT TCC TTC AGC AGA TAG GAA CCA TAC TGA TTC ACA T-3′ (−/+NH2); 37-nt complement, 5′-ATG TGA ATC AGT ATG GTT CCT ATC TGC TGA AGG AAA T-3′; the 55- and 37-nt DNAs were modified with an amino linker for labeling with 5-(6)-carboxytetramethylrhodamine (TAMRA; Invitrogen, Carlsbad, CA). The DNAs were purified by denaturing PAGE on 20% acrylamide/6 M urea gels, followed by electro-elution and ethanol precipitation, and then labeled with TAMRA as described previously, with some modifications (38). Briefly, 800 μg of TAMRA was dissolved in dimethylsulfoxide and incubated with 400 μg DNA in 0.1 M sodium tetraborate, pH 8.5 (final volume 400 μl), for 16 h at 25°C, in the dark. Excess TAMRA in the reaction was extracted by several washes with water-saturated butanol, followed by gel-filtration on a 5-ml P6 resin column (Bio-Rad, Hercules, CA) in 10 mM Tris–HCl, pH 8.0, 0.1 mM EDTA. Fractions containing labeled DNA were identified by absorbance at 260 and 555 nm and pooled, and the DNA was purified further by denaturing PAGE, eluted by diffusion into 10 mM Tris–HCl, pH 8.0 and concentrated by ethanol precipitation. DNA concentrations were determined by absorbance at 260 nm (TAMRA contribution to DNA absorbance at 260 nm was <10%). Primer-template and duplex DNA stocks were prepared by annealing complementary single strands (1:1.15 ratio; labeled:unlabeled) with heating to 95°C for 2 min and slow cooling to 25°C in buffer (20 mM Tris–HCl, pH 7.5, 100 mM NaCl), and analyzed by nondenaturing PAGE to confirm >95% annealed product. α32P-ATP and γ32P-ATP were purchased from (Perkin Elmer Life Sciences, Waltham, M), and ATP and purine nucleoside phosphorylase (PNPase) were purchased from Sigma-Aldrich (Sigma-Aldrich, St. Louis, MO). dNTPs were purchased from GE Healthcare (GE Healthcare, Piscataway, NJ), and 7-methylguanosine (7-MEG) was purchased from RI Chemical, Inc. (RI Chemical, Inc., Orange, CA). PEI cellulose-F TLC plates were purchased from EM Science (EM Science, Gibbstown, NJ).
DNA replication assays
DNA replication assays were performed with 30 nt/105 nt primer/template DNA at 37°C, in a manner similar to that described previously (39); a template with at least 25 nt ssDNA overhangs on either side of the primer supports processive DNA replication following β assembly. Briefly, the primer was labeled with 32P using T4 polynucleotide kinase (New England Biolabs, Beverly, MA), purified from free γ32P-ATP and then annealed with the template strand. Next, a 20-μl reaction containing 2.6 nM p/tDNA, 0.7 mM ATP, 80 μM dNTPs in buffer (20 mM Tris–HCl, pH 7.5, 8 mM MgCl2, 5 mM DTT, 40 μg/ml BSA, 5% glycerol, varying NaCl) was prepared by addition of 40 nM SSB, followed by a mix of 20 nM β dimer and 18 nM core, the reaction initiated by addition of 6 nM wild-type or mutant γ complex (or γδδ′) and quenched after 30 s by addition of 30 μl of 20 mM EDTA, 95% formamide (control reactions were performed similarly except without the clamp loader). The products were analyzed by denaturing PAGE on 10% acrylamide/8 M Urea gels and visualized by a PhosphorImager (GE Healthcare).
DNA-binding assays
Anisotropy measurements of γ complex binding to TAMRA-labeled 30 nt/55 nt p/tDNA, 37 nt ssDNA and 37 nt dsDNAs were performed in a manner similar to that described previously (30,40); a 55-nt template was used instead of 105 nt to have lower anisotropy of free DNA. Briefly, 50 nM DNA was titrated with 0–1 μM wild-type or mutant γ complex (or γδδ′) in buffer (20 mM Tris–HCl, pH 7.5, 8 mM MgCl2, 5 mM DTT, 40 μg/ml BSA, varying NaCl) containing 200 μM ATPγS and 1.5 μM β dimer. Binding was initiated by mixing two stock solutions, one containing γ complex plus DNA and the other containing DNA, in different ratios to vary γ complex concentration (200 μl final volume). The mixtures were incubated for 5 min at 15°C, and assayed on a FluoroMax-3 fluorometer (Jobin-Yvon Horiba Group; Edison, NJ, USA) using vertically polarized light at λEX = 555 nm. The anisotropy was calculated from emitted vertical and horizontal polarized fluorescence intensities at λEM = 580 nm. The binding isotherms obtained from plots of observed anisotropy versus γ complex (or γδδ′) concentration were fit to a quadratic equation describing a 1:1 ligand:macromolecule interaction.
ATPase assays
Pre-steady-state assays using MDCC-labeled PBP to measure phosphate (Pi) release following ATP hydrolysis were performed on a KinTek SF-2001 stopped-flow instrument (KinTek Corp.; Austin, TX, USA) at 25°C in a manner similar to that described previously (29,41). Briefly, 0.75-μM wild type or mutant γ complex (or γδδ′), preincubated with 3 μM β dimer, was mixed in a 1:1 ratio with 1.5 mM ATP for 1 s, followed by further mixing with 1.2, 4.2 or 21 μM DNA (either 30 nt/105 nt p/tDNA, 37 nt/37 nt complement dsDNA or 37 nt ssDNA) and 18 μM MDCC-PBP in a 2:1 ratio, in buffer (20 mM Tris–HCl, pH 7.5, 8 mM MgCl2, 5 mM DTT, 40 μg/ml BSA, 5% glycerol, varying NaCl or NaOAc, 200 μM 7-MEG and 0.1 U/ml PNPase), and the change in fluorescence upon MDCC-PBP binding to Pi was measured over time by excitation at 425 nm and emission at >450 nm (final concentrations: 0.25 μM γ complex, 1 μM β, 0.5 mM ATP, 0.4, 1.4 or 7 μM DNA, and 6 μM MDCC-PBP). Along with each experiment, a Pi calibration curve relating MDCC-PBP fluorescence to Pi concentration was generated under the same conditions (using phosphate standard solution; Sigma–Aldrich). Raw data averaged from at least five traces were divided by the slope from the Pi calibration curve to determine the molar amount of Pi generated during the reaction. Assays of γ complex activity in the absence of β were performed by mixing 0.5 μM γ complex in a 1:1 ratio with 1 mM ATP, 0.8 μM p/tDNA and 12 μM MDCC-PBP followed by measurement of Pi release over time (final concentrations: 0.25 μM γ complex, 0.5 mM ATP, 0.4 μM DNA and 6 μM MDCC-PBP).
Steady-state assays of wild-type and mutant γ complex (or γδδ′) ATPase activity were performed at 24°C with 0.25 μM γ complex and 1 mM ATP, in the absence or presence of 0.5 μM β and/or 0.4 μM p/t DNA at different NaCl or NaOAc concentrations. Reactions were quenched at varying times with 0.5 M EDTA, pH 8.0 and formation of α32P-ADP from α32P-ATP was analyzed by thin layer chromatography on PEI-TLC plates with 0.6 M potassium phosphate buffer, pH 3.4, and quantified using a PhosphorImager.
RESULTS
dsDNA is necessary and sufficient to trigger ATP hydrolysis by γ complex, but p/tDNA is the substrate of choice
Studies in the past few years have demonstrated that DNA binding to γ complex stimulates its ATPase activity (in the absence or presence of β clamp) (16,29). The p/tDNA triggers rapid ATP hydrolysis by all three γ subunits in the complex, most efficiently in the presence of β (30), and this hydrolysis step is coupled to completion of the clamp assembly reaction with release of the clamp·p/tDNA complex (25). We are interested in the question of how clamp loaders target p/tDNA as the substrate for clamp assembly, and therefore initiated this study by surveying the interaction of γ complex with various DNAs. Figure 2A shows data for γ complex binding to p/tDNA, ssDNA, and dsDNA under conditions that favor the interaction; i.e. in the presence of ATPγS, β clamp and at low salt concentration (50–60 mM NaCl). The assay measures changes in anisotropy upon protein binding to 30 nt/55 nt p/tDNA (labeled with TAMRA on the 5′ end of the template strand), or 37 nt ssDNA and dsDNA (labeled with TAMRA on the 3′ end of one strand). As increasing concentrations of γ complex were added to a solution containing 50 nM of DNA (plus β and ATPγS), the anisotropy of p/tDNA rose to saturation and yielded an apparent KD of 60 nM for the interaction (Figure 2A). In contrast, barely any change in anisotropy was detected for both the ssDNA and dsDNA, indicating no interaction or an unfavorable equilibrium for interaction between these DNAs and γ complex. These data reveal that the γ complex has a strong preference for p/tDNA over other DNA structures—even the ss and ds components of p/tDNA.
Figure 2.

Requirements for DNA binding to γ complex and stimulation of its ATPase activity. (A) Fluorescence anisotropy of TAMRA-labeled 30 nt/55 nt p/tDNA, 37 nt dsDNA and 37 nt ssDNA (50 nM) was used to assay their interaction with γ complex in the presence of β and ATPγS, at 60 mM NaCl. The KD is 60 ± 10 nM for p/tDNA and no binding is detectable for dsDNA and ssDNA. In pre-steady-state assays, an ATPase reporter assay measuring Pi release shows that (B) γ complex incubated with β and ATP prior to mixing with 30 nt/105 nt p/tDNA catalyzes a rapid burst of ATP hydrolysis by all three γ subunits, followed by slower step(s) and the steady-state phase (the flat line is the basal ATPase activity of γ complex in the absence of DNA). (C) Duplex DNA (37 nt) also stimulates burst ATPase kinetics by γ complex, except only at very high DNA concentration, whereas (D) ssDNA (37 nt) is unable to do so even at high concentration (final concentrations: 0.25 μM γ complex, 1 μM β, 500 μM ATP, 1.4–7 μM DNA and 6 μM MDCC-PBP).
Next, in order to assess the influence of these DNAs on γ complex function, we measured their ability to stimulate its ATPase activity. We used a reporter assay for ATP hydrolysis that is based on the change in fluorescence of MDCC-labeled PBP when it binds Pi. This assay has been used effectively to measure the kinetics of Pi release following ATP hydrolysis in the millisecond time scale (37), and it reveals the rate of ATP hydrolysis when Pi release is a fast step in the reaction. As reported previously, when γ complex is preincubated with β and ATP for 1 s prior to addition of p/tDNA (30 nt/105 nt), DNA binding triggers a rapid burst of ATP hydrolysis and Pi release by the three γ subunits in the first catalytic turnover, which is followed by slower step(s) and steady state (Figure 2B; final concentrations: 0.25 μM γ complex and 1.4 μM p/tDNA) (29). The burst of ATP hydrolysis is associated with release of the clamp on p/tDNA (29) (in the absence of β and DNA, or in the presence of β only, γ complex has very low basal ATPase activity; Figure 2B). When p/tDNA was replaced by dsDNA (37/37 nt), we found that at 1.4-μM concentration it stimulates γ complex ATPase activity over the basal level, but a burst phase is barely detectable. Only at much higher dsDNA concentrations did we detect a significant burst of ATP hydrolysis, although even at 7-μM dsDNA the amplitude and the rate were still lower than with p/tDNA (Figure 2C). A 37-nt ssDNA, however, does not stimulate rapid ATP hydrolysis even at 7-μM concentration (Figure 2D). Together, the data suggest that the dsDNA portion of p/tDNA is an essential feature for its productive interaction with γ complex. And, while the ssDNA portion itself appears to be ineffective, the combination of dsDNA and ssDNA, and possibly the ss/dsDNA junction, favors selection of p/tDNA as the substrate for clamp assembly. In order to understand the basis for this selectivity, we decided to find out if there are sites on γ complex, other than the known dsDNA binding sites, which might contribute to its high-affinity interaction with p/tDNA.
δ-W279 contributes to the p/t DNA-binding activity of γ complex
In an earlier study, a conserved Tryptophan residue (W279) in the δ subunit of γ complex was found to cross-link p/tDNA, specifically to a 5-bromo-2-deoxyuridine base located on the template strand adjacent to the recessed 3′-OH primer-template junction (35). This finding presented δ-W279, possibly along with conserved neighboring cationic residues, as a promising p/tDNA-binding site in γ complex; therefore, we mutated it to alanine and tyrosine to assess its role in the clamp loader DNA binding and clamp assembly mechanism. The mutant proteins, δW279A and δW279Y, were overexpressed and purified in the same manner as wild-type δ, and reconstituted into full (δW279A-γ complex, δW279Y-γ complex) and minimal (δW279A-γδδ′, δW279Y-γδδ′) clamp loader complexes. During the reconstitution and purification process, the mutant complexes exhibited the same properties as wild-type complexes (Supplementary Figure S1A). Moreover, treatment of δW279A and δW279Y with Elastase enzyme yielded the same partial proteolysis pattern as wild-type δ (Supplementary Figure S1B), indicating that these mutations do not cause gross changes in protein structure.
We tested the hypothesis that δ-W279 is involved in clamp loader–DNA interaction by comparing binding of the wild-type and mutant γ complexes to TAMRA-labeled p/tDNA. As shown previously in Figure 2A, increasing concentrations of γ complex mixed with 50 nM of p/tDNA, plus β and ATPγS, increased the anisotropy of p/tDNA and yielded a KD of 60 nM, at 60 mM NaCl. In contrast an apparent KD of 600 nM was measured for δW279A-γ complex, revealing a 10-fold reduction in the affinity of the alanine mutant for p/tDNA (Figure 3A). A KD of 160 nM was measured for δW279Y-γ complex, indicating that substitution of Tryptophan with a ‘like’ residue, Tyrosine, does not impact the protein–DNA interaction as severely (Figure 3A); the reason for somewhat lower amplitude of DNA binding with δW279Y-γ complex is unclear, and may reflect a fraction of inactive complex in the reaction.
Figure 3.

The p/tDNA-binding affinity of wild-type and mutant γ complexes. (A) Fluorescence anisotropy of TAMRA-labeled 30 nt/55 nt p/t DNA (50 nM) was measured following titration with γ complex, in the presence of β and ATPγS, at 60 mM NaCl. Wild-type γ complex binds p/tDNA with high affinity (KD = 60 ± 10 nM), δW279Y-γ complex exhibits slightly lower binding and weaker affinity (KD = 159 ± 15 nM) and δW279A-γ complex has 10-fold lower affinity for p/t DNA (KD = 590 ± 80 nM). (B) At 100 mM NaCl, interaction between wild-type γ complex and p/t DNA is weaker (KD = 498 ± 12 nM) and barely detectable for δW279A-γ complex.
We also observed that at higher NaCl concentration (100 mM), wild-type γ complex binding to p/tDNA is weakened (KD ∼ 500 nM; Figure 3B). It is likely that the salt interferes with electrostatic contacts between charged/polar residues on the clamp loader and DNA, thereby weakening the interaction. This result is consistent with our hypothesis that the duplex portion is essential for high-affinity binding of p/tDNA to γ complex, which in turn leads to rapid ATP hydrolysis and clamp assembly. Under these conditions, δW279A-γ complex binding to p/tDNA is barely detectable (a KD value cannot be determined from the data; Figure 3B), while δW279Y-γ complex binding is weakened to a similar extent as wild-type γ complex (KD ∼ 800 nM; Figure 3B). Thus, in addition to contacts between the clamp loader and dsDNA, the presence of δ-W279 (or at least an aromatic residue) near the primer-template junction appears important for γ complex–p/tDNA interaction. A similar result is obtained with the minimal γδδ′ complex, except that even at 60 mM NaCl wild-type γδδ′ binds p/tDNA with a KD of 160 nM and δW279A-γδδ′ binding to p/tDNA is barely detectable (Supplementary Figure S2A).
δ-W279 facilitates p/tDNA-dependent stimulation of γ complex ATPase activity
The next set of experiments was performed to examine the effect of δ-W279 mutation on the p/tDNA-dependent ATPase activity of γ complex, which drives the clamp assembly reaction. Initially, steady-state ATPase rates of wild-type and mutant proteins were measured in the absence and presence of the 30 nt/105 nt p/tDNA, at varying NaCl concentrations. The ATPase rate of γ complex alone is the same for all the proteins, kcat = 0.1 s−1 [Vmax/(γ3δδ′χψ)], and does not vary with NaCl concentration up to at least 300 mM, which serves as a control indicating that the DNA-independent activity of the clamp loader is not altered by mutation of δ-W279 (Figure 4A, inset). The ATPase rate of all the γ complexes is substantially higher in the presence of β and p/tDNA, at low salt concentration (kcat = 1.8–2.1 s−1). The rates drop with increasing NaCl concentration, especially sharply for δW279A-γ complex, and half-maximal activity is measured at ∼230 mM, 190 mM and 130 mM NaCl for wild-type γ complex, δW279Y-γ complex and δW279A-γ complex, respectively (Figure 4A). Thus, consistent with the DNA-binding data, the DNA-dependent ATPase activity of the clamp loader is reduced significantly in a salt-dependent manner by mutation of Tryptophan to Alanine.
Figure 4.

ATPase activity of wild type and mutant γ complexes. (A) Steady state ATPase assays show that the β + p/tDNA-stimulated ATPase rate (kcat) of γ3δδ′χψ varies similarly with NaCl for complexes containing δ (closed circle) and δW279Y (open square), whereas the complex containing δW279A (open triangle) exhibits lower activity (the inset in A shows similar basal activity of all three complexes in the absence of DNA). (B) In pre-steady state assays at 100 mM NaCl, wild type γ complex pre-incubated with β and ATP catalyzes a rapid burst of ATP hydrolysis on mixing with 30 nt/105 nt p/tDNA. Mutant δW279Y-γ complex exhibits a decrease in the burst rate and amplitude and, notably, for δW279A-γ complex the burst phase is not detectable (final concentrations: 0.25 μM γ complex, 1 μM β, 500 μM ATP, 0.4 μM p/t DNA, and 6 μM MDCC-PBP). (C) δW279A-γ complex activity is similar to that of γ complex in the presence of duplex DNA and slightly lower in the presence of ssDNA (7 μM DNA final). (D) Reducing NaCl to 60 mM (0.4 μM p/tDNA) or raising p/tDNA concentration to 1.4 μM (100 mM NaCl) restores δW279A-γ complex burst ATPase kinetics to the same level as wild type γ complex. (E) In the absence of β, p/t DNA can stimulate a burst of ATP hydrolysis by both γ complex and δW279A-γ complex at 60 mM NaCl, but not by δW279A-γ complex at 100 mM NaCl.
Next, we examined whether δ-W279 influences the p/tDNA-stimulated rapid ATP hydrolysis activity of γ complex. Pre-steady-state ATPase experiments measuring Pi release were performed initially under stringent conditions, at 100 mM NaCl and low DNA concentration (0.4 μM p/tDNA). Wild-type γ complex catalyzes a rapid burst of ATP hydrolysis when mixed with p/tDNA, following preincubation with β and ATP for one second (Figure 4B), similar to that observed at 60 mM NaCl and with 1.4 μM DNA (Figure 2B). In case of δW279A-γ complex, however, burst activity is barely detectable (Figure 4B). The δW279Y-γ complex still catalyzes rapid ATP hydrolysis, although the burst rate and amplitude are lower compared with wild-type γ complex (Figure 4B), which may reflect the fact that this mutant has intermediate affinity for p/tDNA (Figure 3). Data from experiments performed with the minimal clamp loader are also consistent with the above findings. The DNA-dependent steady-state ATPase rate of δW279A-γδδ′ is lower and more sensitive to NaCl concentration than that of wild-type γδδ′ and δW279Y-γδδ′ (Supplementary Figure S2B). Also, while the latter complexes catalyze rapid ATP hydrolysis in the presence of p/tDNA and β, δW279A-γδδ′ does not do so under the same conditions (Supplementary Figure S2C). Note: the lower amplitude of the burst phase for wild-type γδδ′ relative to γ complex (<3 ATP hydrolyzed per clamp loader) is thought to reflect some fraction of ‘inactive’ γδδ′ in the reaction or perhaps, intriguingly, asymmetry among γ subunits in the complex such that all three are activated for ATP hydrolysis only when the complex achieves a conformation enabled by the ψ subunit (16).
These results confirm that δ-W279 has a significant role in the γ complex–DNA interaction. More specifically, our hypothesis is that δ-W279 is important for γ complex binding to the primer-template junction and/or template strand; therefore, it follows that the δ-W279A mutation should not affect the dsDNA-dependent ATPase activity of the clamp loader. Indeed, as shown in Figure 4C, in the presence of excess dsDNA (7 μM), δW279A-γ complex catalyzes a burst of ATP hydrolysis comparable to wild-type γ complex. Interestingly, the ssDNA-stimulated ATPase rate of the mutant is lower than that of wild-type complex (albeit only by 2-fold), which is also consistent with a role for δ-W279 in γ complex interactions with the ss/dsDNA junction and/or ssDNA. Additionally, it is possible to restore the p/tDNA-dependent ATPase activity of δW279A-γ complex to wild-type levels under conditions that favor clamp loader binding to the ds portion of p/tDNA. As shown in Figure 4D, lowering the NaCl concentration to 60 mM NaCl (0.4 μM p/tDNA) or increasing p/tDNA concentration to 1.4 μM in the reaction (100 mM NaCl) stimulates δW279A-γ complex activity. The burst ATPase kinetics of the mutant now mirror those of wild-type γ complex at 0.4 μM p/tDNA (100 mM NaCl); δW279Y-γ complex activity also recovers to wild-type levels at lower NaCl and/or higher DNA concentrations (data not shown). Together, these findings suggest that δ-W279 does not participate directly in γ complex binding to dsDNA, but rather complements this interaction to facilitate high-affinity binding to p/tDNA, which in turn triggers ATP hydrolysis and leads to clamp assembly.
Several of the experiments described above were performed also in the presence of sodium acetate instead of sodium chloride (Supplementary Figure S3). As is the case with NaCl, the p/tDNA-stimulated ATPase activity of δW279A-γ complex is more sensitive to increasing NaOAc concentration than wild-type γ complex (Figure S3A). The defect in mutant complex activity becomes apparent at a higher range of NaOAc concentrations compared with NaCl, consistent with previous reports that changing the anion in the reaction can shift the concentration range of salt effects on protein–nucleic acid interactions and corresponding activities (42). At 100 mM NaOAc, both wild-type γ complex and δW279A-γ complex catalyze rapid ATP hydrolysis (Figure S3B), whereas at 200 mM NaOAc, unlike wild-type γ complex, δW279A-γ complex has barely detectable burst ATPase activity (Figure S3C). These data indicate that the inhibitory effect of the alanine mutation at δ-W279 is independent of the type of anion present in the reaction.
We performed an additional set of experiments to confirm that the effects of δ-W279 mutation reflect specifically its role in the p/tDNA-binding activity of γ complex. Thus far, all experiments demonstrating defects in the mutant clamp loaders have been performed in the presence of β; i.e. DNA binding (Figure 3) and ATPase (Figure 4) assays. It is known that β influences both the DNA binding and ATPase activities of the clamp loader, thus the question remains whether substitution of δ-W279 with alanine adversely affects γ complex interaction with β, which in turn could impact its DNA binding, ATPase and, ultimately, its clamp loading activity (26,43,44). We consider this an unlikely possibility given that δ-W279 is located virtually at the other end of δ from the β binding site (43) (Figure 1); also, all the experiments have been performed with several-fold excess β over γ complex. We can, however, address the caveat directly because p/tDNA can trigger rapid ATP hydrolysis by γ complex even in the absence of β, although the burst rate and amplitude are not optimal (29). The data in Figure 4E show that when it is mixed with ATP and p/tDNA in the absence of β, γ complex hydrolyzes ATP with burst kinetics, at both 60 mM and 100 mM NaCl concentrations. δW279A-γ complex activity is similar to that of the wild-type complex at 60 mM NaCl, but no burst of ATP hydrolysis is detectable at the higher salt concentration (Figure 4E); the steady-state rate is slightly faster, which may reflect more rapid release of products. Thus, δ-W279 does directly affect γ complex binding to p/tDNA and, as a result, subsequent steps in the clamp assembly reaction.
Mutation of δ-W279 in γ complex impedes processive DNA replication
The significance of δ-W279 for clamp loader function was tested in DNA replication assays, by measuring extension of a 32P-labeled primer by E. coli Pol III core DNA polymerase. In the absence of clamp and clamp loader proteins, the core polymerase exhibits distributive activity on a 30-nt/105-nt p/tDNA substrate and is unable to extend the primer to the end of the template (80 nt product) (Figure 5, lane 1). The presence of wild type γ complex and β facilitates processive core polymerase activity, and now it can synthesize the full-length product (Figure 5, lane 2). At 50 mM NaCl concentration, both δW279A-γ complex and δW279Y-γ complex can support processive DNA synthesis (Figure 5, lanes 3 and 4). However, reactions containing δW279A-γ complex display salt sensitivity, as revealed by the decrease in full-length product formation at 100 mM NaCl (Figure 5, lanes 5–7); the activity of δW279Y-γ complex appears comparable to that of the wild-type clamp loader (Figure 5, lane 7). A similar result is obtained with γδδ′ complexes, except the absence of χ/ψ subunits makes the clamp loaders more sensitive to salt, and the difference between wild-type γδδ′ and δW279A-γδδ′ function is detectable even at 80 mM NaCl concentration (Supplementary Figure S2D). These data confirm the hypothesis that Tryptophan 279 of the δ subunit contributes toward optimal functioning of the clamp loader in DNA replication.
Figure 5.

A role for δ-W279 in clamp loader function during DNA replication. Extension of 5′ 32P-labeled 30-nt primer on a 105-nt template by E. coli Pol III core polymerase in the presence of wild-type and mutant clamp loaders, at different NaCl concentrations. Reactions containing p/tDNA, SSB, ATP, β and dNTPs were initiated with core polymerase in the absence of clamp loader (lane 1) or with the clamp loader, and DNA products analyzed on a denaturing gel. Reactions containing γ complex (lanes 2 and 5), δW279A-γ complex (lanes 3 and 6) and δW279Y-γ complex (lanes 4 and 7) reveal loss of processive DNA replication with the mutant δW279A-γ complex at 100 mM NaCl. The markers are 32P-labeled 30- and 81-nt DNAs.
Additional residues in the conserved motif neighboring δ-W279 are involved in γ complex–p/tDNA interaction
According to the crystal structure, the HRVW279QNRR sequence in δ forms a loop-like structure with charged and polar residues that appear to be in a favorable position for interaction with DNA as it exits the clamp loader (Figure 1). In order to determine whether this motif contributes to the clamp loader mechanism beyond the function(s) associated with δ-W279, we examined the effects of mutating the three positively charged residues (Arginine) in the motif to Alanine. R277 and R283 were chosen because their cationic side chains are exposed and accessible for interaction with DNA, and their interesting bookend positions in the motif, while R282 was chosen as a potential negative control, since according to the crystal structure its side chain is buried and should not be accessible for interaction with DNA (17). We made these changes individually and in conjunction with the already well-characterized δ-W279A mutation, as this allowed us to assess the role of the Arginine residues under two different conditions that are more (Figure 4B) or less (Figure 4D) stringent for clamp loader activity.
The δ mutants were purified and subjected first to Elastase treatment. The δ-R277A and δ-R283A single and double (with δ-W279A) mutants yielded the same pattern of proteolysis as wild-type and δW279A proteins, suggesting that these mutations do not effect gross changes in δ structure (Supplementary Figure S4A). In contrast, the δ-R282A mutation appears to alter the protein structure quite significantly, which is consistent with its buried position in the protein where it hydrogen bonds with other residues in the C-terminal domain. The mutant proteins were then incorporated into γ complexes and tested for DNA binding and ATPase activities; the δR282A mutant did not form a complex with the other clamp loader subunits (Supplementary Figure S4B). The steady-state ATPase rates of the mutant complexes were the same as that of wild-type and δW279A-γ complex in the absence of DNA, indicating that the mutations did not affect the DNA-independent activities of the clamp loader; kcat = 0.08–0.12 s−1 in the absence of β and kcat = 0.015–0.025 s−1 in the presence of β (data not shown).
Next, we performed pre-steady-state experiments to measure the p/tDNA-stimulated ATPase activities of the mutant proteins. Under stringent conditions, low DNA (0.4 μM) and high NaCl (100 mM) concentrations, δR277A-γ complex maybe exhibits a slight decrease in burst ATPase activity (Figure 6A) compared with wild-type γ complex, while δR283A-γ complex exhibits a more significant decrease in activity (Figure 6B); however, neither mutant is as defective as δW279A-γ complex. Under the same conditions, the double mutants, δW279A/R277A-γ complex and δW279A/R283A-γ complex, show no detectable burst ATPase activity and are clearly even more defective than δW279A-γ complex (Figure 6A and B). We also did not detect any interaction between these double mutants and p/tDNA in fluorescence anisotropy-based assays (data not shown), unlike the weak interaction detected with δW279A-γ complex (Figure 3). These data indicate that clamp loaders carrying δ double mutants, δW279A/R277A and δW279A/R283A, have greater DNA-binding defects than the single mutant δ-W279A.
Figure 6.

The effect of δ-R277A and δ-R283A mutations on γ complex function. (A, B) At 0.4 μM p/tDNA and 100 mM NaCl, the p/tDNA-stimulated ATPase activity of δR277A-γ complex is comparable to γ complex, and δR283A-γ complex activity is slightly lower, but in the case of both double mutants, δW279A/R277A-γ complex and δW279A/R283A-γ complex, the burst phase is lost. (C, D) Under less stringent conditions (1.4 μM p/tDNA and 60 mM NaCl), the ATPase activity of the double mutants recovers somewhat (δW279A/R277A-γ complex more so than δW279A/R283A-γ complex), while the single mutants are indistinguishable from wild-type γ complex.
Under less stringent conditions, higher DNA (1.4 μM) and lower NaCl (60 mM), the activities of the single mutants, δR277A-γ complex and δR283A-γ complex, are indistinguishable from wild-type γ complex (Figure 6C and D). We also detect recovery of δW279A/R277A-γ complex ATPase activity, although the burst rate and amplitude are still lower compared with the single mutants (Figure 6C). The activity of δW279A/R283A-γ complex also increases slightly under these conditions (Figure 6D). As is the case with wild-type γ complex and dsDNA (Figure 2C), the burst amplitude and rate of ATP hydrolysis catalyzed by δW279A/R283A-γ complex rise further with increasing DNA concentrations, but the full burst is not achieved even at 7-μM p/tDNA (data not shown), suggesting that this double mutant suffers a severe loss in p/tDNA-binding ability. Together, these data affirm that a conserved sequence in the C-terminal domain of δ contributes to the p/tDNA-binding activity of γ complex, and indicate that multiple residues in the motif participate in the interaction and are important for subsequent steps in the clamp loading reaction.
DISCUSSION
In the clamp·clamp loader complex, β is positioned underneath γ complex, docked on the N-terminal surfaces of the γ, δ and δ′ subunits. Of these proteins, δ is primarily responsible for binding and opening β clamp for assembly on primed DNA templates. The three γ subunits provide the ATPase activity, and along with δ′, also contribute to β binding and coordination of ATP binding and hydrolysis with β opening and closing around DNA (3). In addition, both γ and δ′ subunits are known to play a prominent role in binding DNA. Earlier structure–function analyses of both E. coli γ complex and S. cerevisiae RFC revealed conserved charged and polar amino acids on the inner surface of the clamp loader that interact with the minor groove of dsDNA (31–33). Mutations of these residues in the γ and δ′ subunits cause defects in the DNA binding, DNA-dependent ATPase, and clamp release activities of the clamp loader (33). Thus far, no specific site has been identified on clamp loaders that can interact with the single-stranded template or the primer-template junction. One possible location is a cleft in the C-terminal portion of the δ subunit that has a width of about 12 Å, enough to allow entry of ssDNA but not dsDNA (17). A loop-like structure extends out over the top of the cleft, and a Tryptophan residue (δ-W279) in this loop specifically forms a zero-length cross-link with the first or second template base from the 3′-OH primer-template junction (Figure 1) (35). This finding suggests that in addition to binding and opening the clamp, the δ subunit participates in binding to DNA—perhaps the ss/dsDNA junction and/or ssDNA template of p/tDNA.
In the clamp assembly reaction, γ complex binds ATP and β and then, upon interaction with DNA, catalyzes a burst of ATP hydrolysis and releases the β·DNA complex. Until now, only p/tDNA was known to elicit this rapid ATP hydrolysis response, and given our finding that δ-W279 lies close to the ss/dsDNA junction (35) and is a highly conserved residue in bacterial clamp loaders (∼95% W, ∼3% Y in aligned sequences), we decided to investigate its possible function in selective interaction of the clamp loader with p/tDNA. The results of our study reveal that while δ-W279 is not essential for γ complex·β binding to p/tDNA, it is necessary for high-affinity interaction between the two, at salt concentrations considered comparable to physiological conditions (45), which in turn is necessary for the clamp loader to achieve a conformation that is ‘activated’ for rapid ATP hydrolysis and clamp assembly (Figures 4 and 5). Consistent with the sequence conservation data, mutation of δ-W279 to Tyrosine has much lower impact on γ complex activity compared with the Alanine mutation (Figure 3), suggesting that an aromatic side chain is important at this location. The data reveal that δ-W279 does not participate directly in the interactions between γ complex and the duplex portion of p/tDNA, rather it complements these contacts by interacting with the ss/dsDNA junction and/or ssDNA template (possible stacking with bases), and thus facilitates selection of p/tDNA as the appropriate substrate for clamp assembly.
A function that can be considered analogous to that of δ-W279 has been discovered recently for ψ subunit of γ complex. The minimal complex of γδδ′ is deficient in p/tDNA binding, which is likely an underlying reason why it is not as active a clamp loader as γ complex (γδδ′χψ), especially under high NaCl concentrations. Addition of ψ to γδδ′ restores full DNA binding and corresponding burst ATPase activity (even though ψ is not known to bind DNA directly), suggesting that the presence of this subunit also favors the ‘active’ conformation of the clamp loader (16). Addition of the β clamp to γδδ′ also brings its activity closer to that of full γ complex (16). The subtle effects of so many factors on γ complex function are consistent with the idea that the clamp loader exists in different conformations in equilibrium, and that several events in the reaction, including appropriate interactions with ATP, β and DNA, shift the equilibrium toward a conformation that is optimal for catalysis of clamp assembly on DNA.
The C-terminal sequence of the δ subunit is not conserved among clamp loaders from diverse organisms (46). Among bacterial clamp loaders, however, the sequence neighboring δ-W279 is highly conserved, with cationic and polar residues projecting out from a loop-like structure that could participate in electrostatic and hydrogen bonding interactions between γ complex and p/tDNA (HRVW279QNRR; Figure 1). We tested this hypothesis by mutation of two exposed cationic residues in the loop, δ-R277 and δ-R283, to alanine, individually and in conjunction with δ-W279A mutation, followed by analysis of the corresponding γ complexes. We also mutated δ-R282 to alanine, hoping it could serve as a negative control as its side chain appears buried in the C-terminal domain unlike the other arginines. It turned out that the mutation alters the protein structure, suggesting that δ-R282 plays a role in maintaining the structural integrity of δ (perhaps R282 helps anchor the otherwise exposed loop in the δ C-terminal domain). The single mutants, δR277A-γ complex and δR283A-γ complex, have slightly lower p/tDNA-stimulated ATPase activity than γ complex, but their defects are mild compared with δW279A-γ complex. The double mutants, δW279A/R277A-γ complex and δW279A/R283A-γ complex, have lower p/tDNA-stimulated ATPase activity than δW279A-γ complex, indicating that the mutations have an additive effect, consistent with their contributing to the same aspect of γ complex structure–function. In the absence of any structural data on a clamp loader·DNA complex, it is not clear whether residues in the loop make direct contact with ssDNA or affect the interaction in an indirect manner, although the former is entirely possible given the positive electrostatic potential of this conserved motif in the δ subunit.
It is also noteworthy that while γ complex·β does not exhibit a burst of ATP hydrolysis in the presence of low concentrations of dsDNA or ssDNA, in both cases its ATPase activity is still substantially greater than in the absence of DNA. Also, we found that higher concentrations of dsDNA, but not ssDNA, restore burst ATPase kinetics (Figure 2). Thus, it appears that a duplex DNA structure is necessary and sufficient for DNA-mediated activation of γ complex·β for rapid ATP hydrolysis; our finding that blunt-ended dsDNA can trigger rapid ATP hydrolysis by γ complex is consistent with a previous report that blunt-ended dsDNA can stimulate release of the β clamp from γ complex (33). Together, these data support the existence of more than one γ complex·β conformation in the reaction, depicted below with γATP·β as the species whose ATPase activity is stimulated to some extent on binding DNA (N) and γ*ATP·β as the species that catalyzes a burst of ATP hydrolysis on binding DNA.
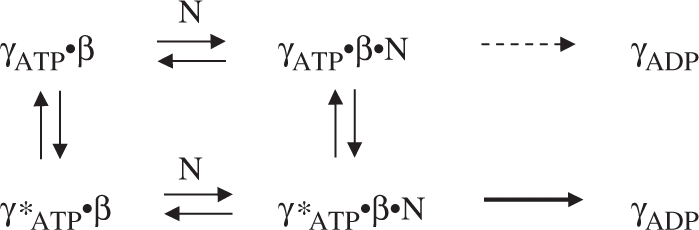
According to this scheme, if p/tDNA binds γ*ATP·β with high affinity, low concentrations of the DNA would stabilize this conformation and lead to rapid ATP hydrolysis, as seen in Figure 2B. Given mutations that lower p/tDNA-binding affinity, higher concentrations of the DNA would be required to shift the equilibrium from γATP·β to γ*ATP·β and produce the same effect, as seen in Figure 6. Similarly, if dsDNA binds γ*ATP·β with low affinity, high concentrations of this DNA would be necessary to produce the same effect, as seen in Figure 2C. In the case of ssDNA, we do not observe any burst of ATP hydrolysis at the highest concentration tested (7 μM; Figure 2D), which suggests that ssDNA is severely lacking in structural features necessary for high-affinity binding to the γ*ATP·β conformation.
In summary, we began this investigation in order to understand more clearly the basis of clamp loader selectivity for primed DNA templates as substrates for clamp assembly. We found that the dsDNA portion of p/tDNA appears to be essential for productive interaction between DNA and γ complex·β, which leads to ATP hydrolysis and β assembly on DNA. However, dsDNA alone is a poor substrate for clamp assembly, because of its low binding affinity for the ‘active’ conformation of γ complex·β (γ*ATP·β in the scheme above). Other features of p/tDNA, such as the 3′-OH primer/template junction and the ssDNA template enhance the interaction, likely via contacts with additional sites on γ complex beyond those that bind dsDNA. Results of our mutational analysis make a strong case that a conserved motif in the C-terminal domain of δ is one such site helping γ complex select p/tDNA as a bona fide substrate for clamp assembly.
Since the C-terminal region of δ is not conserved among analogous clamp loader subunits in other organisms, it is not clear whether aromatic and charged/polar residues help impose DNA substrate selectivity in these proteins as well. Thus far, it appears that the S. cerevisiae RFC clamp loader is more promiscuous than γ complex both in its ability to bind a variety of DNA structures (47) and the related rapid ATP hydrolysis activity (Chen,S., Hingorani,M.M., unpublished data). Such promiscuity may be necessary for RFC to function on DNA substrates in DNA replication as well as in other processes such as bidirectional DNA mismatch repair (48); therefore, it is possible that a γ complex-like mechanism for highly selective p/tDNA binding does not exist in RFC. Alternative eukaryotic clamp loaders, e.g. Rad17-RFC (Rad24-RFC in S. cerevisiae) (49,50), Ctf18-RFC (51,52) and Elg1-RFC (53), that function in DNA damage checkpoints and sister chromatid cohesion, among other processes, apparently bind yet other types of DNA structures (54). We expect that comparative structure–function analyses will likely reveal customized features in these clamp loaders that enable their function on distinct DNA substrates.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We are grateful for the experiments performed by Nina Yao (The Rockefeller University) that made important contributions to this study. This work was supported by National Institutes of Health (GM64514 to M.M.H. and GM38839 to M.O’D.). Funding to pay the Open Access publication charges for this article was provided by National Institutes of Health (GM64514).
Conflict of interest statement. None declared.
REFERENCES
- 1.Georgescu RE, Kim SS, Yurieva O, Kuriyan J, Kong XP, O’Donnell M. Structure of a sliding clamp on DNA. Cell. 2008;132:43–54. doi: 10.1016/j.cell.2007.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O’Donnell M, Kuriyan J. Clamp loaders and replication initiation. Curr. Opin. Struct. Biol. 2006;16:35–41. doi: 10.1016/j.sbi.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 3.Indiani C, O’Donnell M. The replication clamp-loading machine at work in the three domains of life. Nat. Rev. Mol. Cell Biol. 2006;7:751–761. doi: 10.1038/nrm2022. [DOI] [PubMed] [Google Scholar]
- 4.Johnson A, O’Donnell M. Cellular DNA replicases: components and dynamics at the replication fork. Annu. Rev. Biochem. 2005;74:283–315. doi: 10.1146/annurev.biochem.73.011303.073859. [DOI] [PubMed] [Google Scholar]
- 5.Majka J, Burgers PM. The PCNA-RFC families of DNA clamps and clamp loaders. Prog. Nucleic Acid Res. Mol. Biol. 2004;78:227–260. doi: 10.1016/S0079-6603(04)78006-X. [DOI] [PubMed] [Google Scholar]
- 6.Maga G, Hubscher U. Proliferating cell nuclear antigen (PCNA): a dancer with many partners. J. Cell Sci. 2003;116:3051–3060. doi: 10.1242/jcs.00653. [DOI] [PubMed] [Google Scholar]
- 7.Kong XP, Onrust R, O’Donnell M, Kuriyan J. Three-dimensional structure of the β subunit of E. coli DNA polymerase III holoenzyme: a sliding DNA clamp. Cell. 1992;69:425–437. doi: 10.1016/0092-8674(92)90445-i. [DOI] [PubMed] [Google Scholar]
- 8.Studwell-Vaughan PS, O’Donnell M. Constitution of the twin polymerase of DNA polymerase III holoenzyme. J. Biol. Chem. 1991;266:19833–19841. [PubMed] [Google Scholar]
- 9.Pritchard AE, Dallmann HG, Glover BP, McHenry CS. A novel assembly mechanism for the DNA polymerase III holoenzyme DnaX complex: association of δδ′ with DnaX(4) forms DnaX(3)δδ′. EMBO J. 2000;19:6536–6545. doi: 10.1093/emboj/19.23.6536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Onrust R, Finkelstein J, Naktinis V, Turner J, Fang L, O’Donnell M. Assembly of a chromosomal replication machine: two DNA polymerases, a clamp loader, and sliding clamps in one holoenzyme particle. I. Organization of the clamp loader. J. Biol. Chem. 1995;270:13348–13357. doi: 10.1074/jbc.270.22.13348. [DOI] [PubMed] [Google Scholar]
- 11.Onrust R, Finkelstein J, Turner J, Naktinis V, O’Donnell M. Assembly of a chromosomal replication machine: two DNA polymerases, a clamp loader, and sliding clamps in one holoenzyme particle. III. Interface between two polymerases and the clamp loader. J. Biol. Chem. 1995;270:13366–13377. doi: 10.1074/jbc.270.22.13366. [DOI] [PubMed] [Google Scholar]
- 12.McHenry CS. Chromosomal replicases as asymmetric dimers: studies of subunit arrangement and functional consequences. Mol. Microbiol. 2003;49:1157–1165. doi: 10.1046/j.1365-2958.2003.03645.x. [DOI] [PubMed] [Google Scholar]
- 13.Onrust R, O’Donnell M. DNA polymerase III accessory proteins. II. Characterization of δ and δ′. J. Biol. Chem. 1993;268:11766–11772. [PubMed] [Google Scholar]
- 14.Yuzhakov A, Kelman Z, O’Donnell M. Trading places on DNA–a three-point switch underlies primer handoff from primase to the replicative DNA polymerase. Cell. 1999;96:153–163. doi: 10.1016/s0092-8674(00)80968-x. [DOI] [PubMed] [Google Scholar]
- 15.Glover BP, McHenry CS. The χ ψ subunits of DNA polymerase III holoenzyme bind to single-stranded DNA-binding protein (SSB) and facilitate replication of an SSB-coated template. J. Biol. Chem. 1998;273:23476–23484. doi: 10.1074/jbc.273.36.23476. [DOI] [PubMed] [Google Scholar]
- 16.Anderson SG, Williams CR, O’Donnell M, Bloom LB. A function for the ψ subunit in loading the Escherichia coli DNA polymerase sliding clamp. J Biol Chem. 2007;282:7035–7045. doi: 10.1074/jbc.M610136200. [DOI] [PubMed] [Google Scholar]
- 17.Jeruzalmi D, O’Donnell M, Kuriyan J. Crystal structure of the processivity clamp loader γ complex of E. coli DNA polymerase III. Cell. 2001;106:429–441. doi: 10.1016/s0092-8674(01)00463-9. [DOI] [PubMed] [Google Scholar]
- 18.Erzberger JP, Berger JM. Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu. Rev. Biophys. Biomol. Struct. 2006;35:93–114. doi: 10.1146/annurev.biophys.35.040405.101933. [DOI] [PubMed] [Google Scholar]
- 19.Hanson PI, Whiteheart SW. AAA+ proteins: have engine, will work. Nat. Rev. Mol. Cell. Biol. 2005;6:519–529. doi: 10.1038/nrm1684. [DOI] [PubMed] [Google Scholar]
- 20.Tsuchihashi Z, Kornberg A. ATP interactions of the τ and γ subunits of DNA polymerase III holoenzyme of Escherichia coli. J. Biol. Chem. 1989;264:17790–17795. [PubMed] [Google Scholar]
- 21.Xiao H, Naktinis V, O’Donnell M. Assembly of a chromosomal replication machine: two DNA polymerases, a clamp loader, and sliding clamps in one holoenzyme particle. IV. ATP-binding site mutants identify the clamp loader. J. Biol. Chem. 1995;270:13378–13383. doi: 10.1074/jbc.270.22.13378. [DOI] [PubMed] [Google Scholar]
- 22.Podobnik M, Weitze TF, O’Donnell M, Kuriyan J. Nucleotide-induced conformational changes in an isolated Escherichia coli DNA polymerase III clamp loader subunit. Structure. 2003;11:253–263. doi: 10.1016/s0969-2126(03)00027-3. [DOI] [PubMed] [Google Scholar]
- 23.Snyder AK, Williams CR, Johnson A, O’Donnell M, Bloom LB. Mechanism of loading the Escherichia coli DNA polymerase III sliding clamp: II. Uncoupling the β and DNA binding activities of the γ complex. J. Biol. Chem. 2004;279:4386–4393. doi: 10.1074/jbc.M310430200. [DOI] [PubMed] [Google Scholar]
- 24.Johnson A, O’Donnell M. Ordered ATP hydrolysis in the γ complex clamp loader AAA+ machine. J. Biol. Chem. 2003;278:14406–14413. doi: 10.1074/jbc.M212708200. [DOI] [PubMed] [Google Scholar]
- 25.Bloom LB. Dynamics of loading the Escherichia coli DNA polymerase processivity clamp. Crit. Rev. Biochem. Mol. Biol. 2006;41:179–208. doi: 10.1080/10409230600648751. [DOI] [PubMed] [Google Scholar]
- 26.Turner J, Hingorani MM, Kelman Z, O’Donnell M. The internal workings of a DNA polymerase clamp-loading machine. EMBO J. 1999;18:771–783. doi: 10.1093/emboj/18.3.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Naktinis V, Onrust R, Fang L, O’Donnell M. Assembly of a chromosomal replication machine: two DNA polymerases, a clamp loader, and sliding clamps in one holoenzyme particle. II. Intermediate complex between the clamp loader and its clamp. J. Biol. Chem. 1995;270:13358–13365. [PubMed] [Google Scholar]
- 28.Hingorani MM, Bloom LB, Goodman MF, O’Donnell M. Division of labor–sequential ATP hydrolysis drives assembly of a DNA polymerase sliding clamp around DNA. EMBO J. 1999;18:5131–5144. doi: 10.1093/emboj/18.18.5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Williams CR, Snyder AK, Kuzmic P, O’Donnell M, Bloom LB. Mechanism of loading the Escherichia coli DNA polymerase III sliding clamp: I. Two distinct activities for individual ATP sites in the γ complex. J. Biol. Chem. 2004;279:4376–4385. doi: 10.1074/jbc.M310429200. [DOI] [PubMed] [Google Scholar]
- 30.Ason B, Handayani R, Williams CR, Bertram JG, Hingorani MM, O’Donnell M, Goodman MF, Bloom LB. Mechanism of loading the Escherichia coli DNA polymerase III β sliding clamp on DNA. Bona fide primer/templates preferentially trigger the γ complex to hydrolyze ATP and load the clamp. J. Biol. Chem. 2003;278:10033–10040. doi: 10.1074/jbc.M211741200. [DOI] [PubMed] [Google Scholar]
- 31.Bowman GD, Goedken ER, Kazmirski SL, O’Donnell M, Kuriyan J. DNA polymerase clamp loaders and DNA recognition. FEBS Lett. 2005;579:863–867. doi: 10.1016/j.febslet.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 32.Bowman GD, O’Donnell M, Kuriyan J. Structural analysis of a eukaryotic sliding DNA clamp-clamp loader complex. Nature. 2004;429:724–730. doi: 10.1038/nature02585. [DOI] [PubMed] [Google Scholar]
- 33.Goedken ER, Kazmirski SL, Bowman GD, O’Donnell M, Kuriyan J. Mapping the interaction of DNA with the Escherichia coli DNA polymerase clamp loader complex. Nat. Struct. Mol. Biol. 2005;12:183–190. doi: 10.1038/nsmb889. [DOI] [PubMed] [Google Scholar]
- 34.Miyata T, Suzuki H, Oyama T, Mayanagi K, Ishino Y, Morikawa K. Open clamp structure in the clamp-loading complex visualized by electron microscopic image analysis. Proc. Natl Acad. Sci. USA. 2005;102:13795–13800. doi: 10.1073/pnas.0506447102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Magdalena Coman M, Jin M, Ceapa R, Finkelstein J, O’Donnell M, Chait BT, Hingorani MM. Dual functions, clamp opening and primer-template recognition, define a key clamp loader subunit. J. Mol. Biol. 2004;342:1457–1469. doi: 10.1016/j.jmb.2004.07.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Studwell-Vaughan PS, O’Donnell M. DNA polymerase III accessory proteins. V. θ encoded by holE. J. Biol. Chem. 1993;268:11785–11791. [PubMed] [Google Scholar]
- 37.Brune M, Hunter JL, Howell SA, Martin SR, Hazlett TL, Corrie JE, Webb MR. Mechanism of inorganic phosphate interaction with phosphate binding protein from Escherichia coli. Biochemistry. 1998;37:10370–10380. doi: 10.1021/bi9804277. [DOI] [PubMed] [Google Scholar]
- 38.Perez-Howard GM, Weil PA, Beechem JM. Yeast TATA binding protein interaction with DNA: fluorescence determination of oligomeric state, equilibrium binding, on-rate, and dissociation kinetics. Biochemistry. 1995;34:8005–8017. doi: 10.1021/bi00025a006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bloom LB, Turner J, Kelman Z, Beechem JM, O’Donnell M, Goodman MF. Dynamics of loading the β sliding clamp of DNA polymerase III onto DNA. J. Biol. Chem. 1996;271:30699–30708. doi: 10.1074/jbc.271.48.30699. [DOI] [PubMed] [Google Scholar]
- 40.Ason B, Bertram JG, Hingorani MM, Beechem JM, O’Donnell M, Goodman MF, Bloom LB. A model for Escherichia coli DNA polymerase III holoenzyme assembly at primer/template ends. DNA triggers a change in binding specificity of the γ complex clamp loader. J. Biol. Chem. 2000;275:3006–3015. doi: 10.1074/jbc.275.4.3006. [DOI] [PubMed] [Google Scholar]
- 41.Bertram JG, Bloom LB, Hingorani MM, Beechem JM, O’Donnell M, Goodman MF. Molecular mechanism and energetics of clamp assembly in Escherichia coli. The role of ATP hydrolysis when γ complex loads β on DNA. J. Biol. Chem. 2000;275:28413–28420. doi: 10.1074/jbc.M910441199. [DOI] [PubMed] [Google Scholar]
- 42.Leirmo S, Harrison C, Cayley DS, Burgess RR, Record M.T., Jr. Replacement of potassium chloride by potassium glutamate dramatically enhances protein-DNA interactions in vitro. Biochemistry. 1987;26:2095–2101. doi: 10.1021/bi00382a006. [DOI] [PubMed] [Google Scholar]
- 43.Jeruzalmi D, Yurieva O, Zhao Y, Young M, Stewart J, Hingorani M, O’Donnell M, Kuriyan J. Mechanism of processivity clamp opening by the δ subunit wrench of the clamp loader complex of E. coli DNA polymerase III. Cell. 2001;106:417–428. [PubMed] [Google Scholar]
- 44.Leu FP, O’Donnell M. Interplay of clamp loader subunits in opening the β sliding clamp of Escherichia coli DNA polymerase III holoenzyme. J. Biol. Chem. 2001;276:47185–47194. doi: 10.1074/jbc.M106780200. [DOI] [PubMed] [Google Scholar]
- 45.Record MT, Jr, Courtenay ES, Cayley S, Guttman HJ. Biophysical compensation mechanisms buffering E. coli protein-nucleic acid interactions against changing environments. Trends Biochem. Sci. 1998;23:190–194. doi: 10.1016/s0968-0004(98)01207-9. [DOI] [PubMed] [Google Scholar]
- 46.Bullard JM, Pritchard AE, Song MS, Glover BP, Wieczorek A, Chen J, Janjic N, McHenry CS. A three-domain structure for the δ subunit of the DNA polymerase III holoenzyme δ domain III binds δ′ and assembles into the DnaX complex. J. Biol. Chem. 2002;277:13246–13256. doi: 10.1074/jbc.M108708200. [DOI] [PubMed] [Google Scholar]
- 47.Hingorani MM, Coman MM. On the specificity of interaction between the Saccharomyces cerevisiae clamp loader replication factor C and primed DNA templates during DNA replication. J. Biol. Chem. 2002;277:47213–47224. doi: 10.1074/jbc.M206764200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Iyer RR, Pluciennik A, Burdett V, Modrich PL. DNA mismatch repair: functions and mechanisms. Chem. Rev. 2006;106:302–323. doi: 10.1021/cr0404794. [DOI] [PubMed] [Google Scholar]
- 49.Lindsey-Boltz LA, Bermudez VP, Hurwitz J, Sancar A. Purification and characterization of human DNA damage checkpoint Rad complexes. Proc. Natl Acad. Sci. USA. 2001;98:11236–11241. doi: 10.1073/pnas.201373498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Green CM, Erdjument-Bromage H, Tempst P, Lowndes NF. A novel Rad24 checkpoint protein complex closely related to replication factor C. Curr. Biol. 2000;10:39–42. doi: 10.1016/s0960-9822(99)00263-8. [DOI] [PubMed] [Google Scholar]
- 51.Naiki T, Kondo T, Nakada D, Matsumoto K, Sugimoto K. Chl12 (Ctf18) forms a novel replication factor C-related complex and functions redundantly with Rad24 in the DNA replication checkpoint pathway. Mol. Cell. Biol. 2001;21:5838–5845. doi: 10.1128/MCB.21.17.5838-5845.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mayer ML, Gygi SP, Aebersold R, Hieter P. Identification of RFC(Ctf18p, Ctf8p, Dcc1p): an alternative RFC complex required for sister chromatid cohesion in S. cerevisiae. Mol. Cell. 2001;7:959–970. doi: 10.1016/s1097-2765(01)00254-4. [DOI] [PubMed] [Google Scholar]
- 53.Kanellis P, Agyei R, Durocher D. Elg1 forms an alternative PCNA-interacting RFC complex required to maintain genome stability. Curr. Biol. 2003;13:1583–1595. doi: 10.1016/s0960-9822(03)00578-5. [DOI] [PubMed] [Google Scholar]
- 54.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.