


Abstract
Systematic identification of binding partners for modular domains such as Src homology 2 (SH2) is important for understanding the biological function of the corresponding SH2 proteins. We have developed a worldwide web-accessible computer program dubbed SMALI for scoring matrix-assisted ligand identification for SH2 domains and other signaling modules. The current version of SMALI harbors 76 unique scoring matrices for SH2 domains derived from screening oriented peptide array libraries. These scoring matrices are used to search a protein database for short peptides preferred by an SH2 domain. An experimentally determined cut-off value is used to normalize an SMALI score, therefore allowing for direct comparison in peptide-binding potential for different SH2 domains. SMALI employs distinct scoring matrices from Scansite, a popular motif-scanning program. Moreover, SMALI contains built-in filters for phosphoproteins, Gene Ontology (GO) correlation and colocalization of subject and query proteins. Compared to Scansite, SMALI exhibited improved accuracy in identifying binding peptides for SH2 domains. Applying SMALI to a group of SH2 domains identified hundreds of interactions that overlap significantly with known networks mediated by the corresponding SH2 proteins, suggesting SMALI is a useful tool for facile identification of signaling networks mediated by modular domains that recognize short linear peptide motifs.
INTRODUCTION
Phosphorylation by protein kinases is a central paradigm in signal transduction and it regulates almost all essential cellular functions such as proliferation, differentiation, migration and survival (1). Deregulated phosphorylation of proteins is often associated with an abnormal state of a cell and can result in malignant transformation (2). The human genome encodes ∼518 protein kinases, of which 90 are tyrosine kinases and another 43 are tyrosine kinase-like (3). By adding a phosphate moiety to the hydroxyl group of a Tyr residue, protein-tyrosine kinases can directly modulate the activity of the target protein, alter its subcellular localization and/or promote the formation of specific signaling complexes. The latter function of tyrosine phosphorylation is mediated by protein modules, such as the Src homology 2 (SH2) and phosphotyrosine-binding (PTB) domains, which recognize pTyr-containing peptides (4,5). Binding of an SH2 or a PTB domain to a phosphotyrosyl sequence provides a general mechanism for the formation of specific protein complexes in intracellular signal transduction, which serves to propagate and regulate a signal emanated from a protein-tyrosine kinase.
The importance of tyrosine phosphorylation in normal cellular function is also highlighted by the great number of SH2 and PTB domains identified in metazoa (6,7). The human genome encodes 120 SH2 domains distributed in 110 distinct proteins, which constitutes the largest family of modular domains capable of recognizing a phosphotyrosine (7). Although the pTyr residue is indispensable for SH2-binding in the majority of cases (8), the specificity of a given SH2 domain is typically determined by a few residues C-terminal to the pTyr (5). Identifying the specific phosphotyrosyl peptide motif recognized by an SH2 domain is a key to understand the function of the corresponding SH2-containing protein. On a larger scale, comprehensive knowledge about the specificity of all mammalian SH2 and PTB domains would make it possible to gauge, in principle, the phosphotyrosine cellular signaling network mediated by these domains. As a first step towards this lofty goal, we recently determined the phosphotyrosyl motifs selected, respectively, by 76 human SH2 domains using an oriented peptide array library (OPAL) approach (13). The parent library consisted of the degenerated sequence XX-pY-XXXX, where X denotes a mixture of 19 naturally occurring amino acids except Cys, and screening of the OPAL yielded selectivity for positions −2 to +4 with respect to the pTyr. This specificity information is necessary for future exploration of SH2 domain function and for the identification of SH2-mediated protein–protein interactions. To take advantage of the OPAL screen data, we generated position-specific scoring matrices (PSSM) for 76 SH2 domains and developed a world-wide web-based (WWW) computer program called scoring matrix-assisted ligand identification (SMALI) for facile identification of linear peptides preferred by an SH2 domain from searching a protein database. Although SMALI is similar to the motif-scanning method, Scansite, developed by Yaffe, Cantley and colleagues (9), SMALI contains PSSMs for 76 SH2 domains in contrast to 14 employed by the latter. Moreover, an SMALI PSSM incorporates selectivity information for six positions (from −2 to +4 relative to the pTyr) of a peptide, whereas most of the PSSMs for SH2 domains used in Scansite were derived from earlier studies that addressed the selectivity from pTyr+1 through pTyr+3 (10,11). To restrict the return from a search to target proteins that have a high probability to be physiologically relevant, SMALI contains an optional filter for phosphorylated peptides. The physiological relevance of a predicted interaction may be further enhanced by applying two additional filters namely signal transduction and subcellular colocalizations (of the query and subject proteins). These novel features make SMALI a useful approach besides Scansite to identify phosphotyrosine-mediated binding events. Here, we describe the usage of the SMALI program and an experimental approach by which to determine the cut-off value for a prediction. We evaluated the performance of SMALI against Scansite for predicting binding peptides for the NCK, CRK and FGR SH2 domains, and applied SMALI to a representative group of 12 SH2 domains in order to identify the corresponding protein–protein interaction (PPI) network. The SMALI-derived PPI network overlaps significantly with known interactions for these SH2-containing proteins, suggesting that SMALI can recapitulate known interactions and identify novel PPIs. The SMALI program, accessible via http://lilab.uwo.ca/SMALI.htm, is frequently updated to include more modular interaction domains and the corresponding PSSMs. To maximize the usage of these matrices, we are also making them available to other bioinformatic programs such as the Scansite and NetPhorest (Linding et al. unpublished results) that aim at identifying protein-binding events and/or signaling pathways according to the principle of domain-short linear motif recognition.
MATERIALS AND METHODS
Derivation of position-specific scoring matrices
The OPAL membrane was scanned and quantified on a BioRad FluoroImager. A selectivity value Xi, p is assigned to each amino acid i at position p in the peptide based on an OPAL result, by subtracting the background signal of the membrane from each data spot. The Xi, p is used to calculate a score Si, p, defined as an element of the query SH2 domain scoring matrix, by the formula Si, p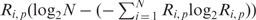 , where N is the number of residue types in the OPAL array (N = 19 except for Cys) and
, where N is the number of residue types in the OPAL array (N = 19 except for Cys) and 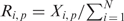 . In this formula, the term
. In this formula, the term 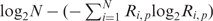 represents information content of all residues at position p and Si, p denotes information content of residue i at this position. Information content of Cys, which was not included in the OPAL, is set equal to the mean Si, p value at a given position. A peptide score Sm, or SMALI score, is calculated using the formula
represents information content of all residues at position p and Si, p denotes information content of residue i at this position. Information content of Cys, which was not included in the OPAL, is set equal to the mean Si, p value at a given position. A peptide score Sm, or SMALI score, is calculated using the formula 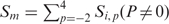 , assuming entropy independence between positions. A peptide with a larger SMALI score is considered to have a greater propensity for binding to the query SH2 domain. A relative score is defined as the ratio of SMALI score over a cut-off value, corresponding to the score at that separates the top 4.5% of peptides from the remaining Tyr-containing peptides taken from all human proteins in the Swiss-Prot database, with the exception of BRDG1 SH2 (3.5%) and GRB2 SH2 (5.5%).
, assuming entropy independence between positions. A peptide with a larger SMALI score is considered to have a greater propensity for binding to the query SH2 domain. A relative score is defined as the ratio of SMALI score over a cut-off value, corresponding to the score at that separates the top 4.5% of peptides from the remaining Tyr-containing peptides taken from all human proteins in the Swiss-Prot database, with the exception of BRDG1 SH2 (3.5%) and GRB2 SH2 (5.5%).
Peptide array synthesis and probing
Peptide arrays were synthesized following established protocols (12). To determine the ability of the peptides on the array to bind an SH2 domain, the SH2 domain was expressed as GST-fusion and purified to homogeneity on a glutathione affinity column and fast-performance liquid chromatography (FPLC) column. The same procedures used for OPAL screening (13) was used to probe the array for binding to the GST-SH2 protein (applied at 1 μM). Finally, the peptide array was scanned and quantified on a BioRad FluoroImager and the background signal was subtracted from each peptide spot.
Differentiation of binding and nonbinding peptides in an array
While in most cases the spot value will provide the quantitative information about the strength of binding for a peptide on the array, the line between binding and nonbinding peptides becomes blurred when the binding signal is weak. We used the distribution pattern on spot values on an array to determine a cut-off value by which to differentiate binding from nonbinding peptides. When the numbers of binding and nonbinding peptides are comparable, the distribution of spot values follows a bimodal pattern where the peak at a large spot value represents binding, while the peak at the small value represents nonbinding peptides. In this case, the transition point between the two peaks is selected as the cut-off. When the signals are extremely biased, the distribution of spot values can be unimodal, and therefore no apparent transition is detected. This is the case with the BRDG1 SH2 peptide array for which an overwhelming number of peptides showed binding. In this case, we define a nonbinding peptide as one with a spot value smaller than the average spot value across the entire array subtracted by 1.5× SD. Based on the earlier definition, peptides with spot values >1.3 are considered binding peptides for the BRDG1 SH2 domain (Table S1), >1.8 for the GRB2 SH2 (Table S2), >0.8 for NCK SH2 (Table S3), >0.7 for CRK SH2 (Table S4) and >0.4 for FGR SH2 (Table S5). The five peptide arrays together contained 16 known binding peptides for different SH2 domains. All are correctly classified, suggesting the classification scheme outline above is a reasonable representation of the true binding data.
RESULTS
Overview of the SMALI program
The derivation of PSSM based on the experimental data from OPAL screens was described elsewhere (13). Briefly, the OPAL-binding profile for an SH2 domain was obtained and quantified for signal strength at each peptide spot on the array (Figure 1A). The information-entropy algorithm was applied to the signals to generate the corresponding scoring matrix (Figure 1B). The current version of the SMALI program includes two modules, peptide scan and domain scan. The peptide scan module is used to identify short peptides that have a high propensity to bind a modular interaction domain such as SH2. In contrast, the domain scan module is used to identify domains that are preferred by a query protein. To predict peptide ligands for a query SH2 domain, all Tyr-containing peptides in the Swiss-Prot database (14) are retrieved and scored using PSSM for that SH2 domain. Peptides are ranked in a descending order based on the SMALI scores, and a peptide with a larger score is considered to have a greater tendency to bind the query SH2 domain. Inside the peptide scan module, a user could select one of the 76 SH2 domains currently covered by the SMALI site. After selecting a protein database (the Swiss-Prot database is used as a default in SMALI), one can choose to run the program without filters or with filters to restrict the proteins to be included in the output file (Figure 1D). Because the Swiss-Prot database contains over 200 000 tyrosines from human proteins, it is necessary to limit the output size of a SMALI prediction by parsing the output through a number of filters.
Figure 1.

Schematic representation of the SMALI program. (A) An OPAL-SH2 binding profile (shown here for the BRDG1 SH2 domain) was used to generate a position specific scoring matrix (PSSM) (B). (C) The PSSM was used to search a protein database for tyrosine-containing peptides that are preferred by a query SH2 domain. (D) Selected peptides are ranked according to their SMALI scores and put out either unfiltered or filtered through one or more filters as shown. (E) The output file size can be selected. A sample output file is shown (see text for detail).
Three filters were therefore implemented that may be used individually or in combination. The ‘phosphorylation potential’ filter selects only peptides whose phosphorylation has been experimentally verified. This information is taken directly from the databases PhosphoSite (15) and Phospho.ELM (16). Because SH2 domains bind specifically to pTyr-containing sequences, those that are not phosphorylated on Tyr are unlikely to be of physiological relevance even when they produce large SMALI scores. The application of phosphorylation filter reduces the candidate peptides from over 200 000 to ∼8000 (15). The second filter, signaling transduction, limits proteins returned from a search to those involved in signal transduction processes. Because most SH2 domains are involved in cellular signal transduction, the identification of signaling proteins that bind to SH2 domains may have a greater potential to be physiologically relevant. Signaling proteins are identified according to the PFAM domain database and Gene Ontology (GO) terms (17–19). Specifically, a subject is classified as a signaling protein if it contains one or more of the 116 signaling domains defined in the PFAM and/or SMART databases (20,21), or if it is annotated with one or more of the following GO terms or their child terms: signal transduction, signal transducer activity, protein kinase activity, phosphoprotein phosphatase activity and protein amino acid dephosphorylation. The third filter is created to keep in an output only those proteins that colocalize with the query SH2 protein in specific subcellular compartments as annotated in Swiss-Prot. The following compartments are used with this filter: (i) cytoplasm, (ii) nucleus, (iii) mitochondrion, (iv) golgi apparatus, (v) endoplasmic reticulum and (vi) endosome. Approximately 34% of human proteins in Swiss-Prot are annotated with a role in signal transduction, while 71% assigned to specific cellular compartments. To date, 63 SH2 domain-containing proteins have been annotated by subcellular localization, some of which are identified in more than one cellular compartment (e.g. ABL1 exists in either cytoplasm or nucleus). In cases where different regions of a protein are assigned to distinct subcellular locations (i.e. membrane proteins), the region containing the putative binding site(s) for the query SH2 is considered. For instance, the cytoplasmic region (residues 323–428) of the membrane protein NACHR alpha 10 (Swiss-Prot ID: Q9GZZ6) is scanned if a query SH2 is annotated with cytoplasmic localization.
Typical output format of the peptide scan module is shown in Figure 1E. The output size can be set by a user to 100, 250 or 500. The first two columns of the output file report a SMALI score of the peptide target and its relative score calculated by normalizing the raw SMALI score against a cut-off value (defined as the score corresponding to the top 4.5% of peptides ranked by SMALI, see subsequent sections for detail). A relative score of >1.0 suggests a strong potential for binding. The output file also includes information about the peptide sequence, the position of Tyr residue in the subject protein, gene name, protein name, GeneBank identification (ID), Swiss-Prot ID, molecular weight of the subject protein and localizations if available. To match the prediction with known interactions, the last two columns of the output list interactions between the query and subject proteins that have been curated in PPI databases or in domain-peptide interaction databases such as Phospho.Elm (16). Two PPI databases are currently linked to SMALI: the IntAct database where interactions are derived from experiments (22,23), and the I2D database that combines literature-derived human PPIs with those inferred from other species (24). IntAct contains over 400 interactions that may involve SH2 domains and the I2D collects ∼2000 potential SH2-mediated interactions. The confidence level of an SH2 domain-ligand interaction predicted by SMALI is greater if the corresponding PPI is also listed in a database.
In contrast to the peptide scan module that identify peptide targets for a query SH2 domain/protein, the domain scan module of SMALI is used to identify SH2 domains preferred by a query protein that harbors one or more Tyr phosphorylation sites. A query protein can be specified by its Swiss-Prot/TrEMBL ID or its complete or partial sequence entered in FASTA format in the space provided (Figure 2A). Prior to activate a search, the user has the option of selecting one, a subgroup or all SH2 domains (default). The output file of a domain scan lists the query protein sequence with all tyrosine residues highlighted. In a separate panel, the Tyr-containing peptides are listed along with a group of SH2 domains preferred by them (Figure 2B). The numbers in the parenthesis besides an SH2 domain denotes its relative SMALI score for a given Tyr site. An SH2 domain with a larger relative score has a greater tendency to bind to a Tyr site. The output file lists only those SH2 domains that have a relative SMALI score >1.0 (see next section for the derivation of relative SMALI score).
Figure 2.

Sample output of the domain-scan module in SMALI. (A) A query protein can be entered with an ID or by typing in the sequence in the space provided. Partial sequence is also acceptable. One or more SH2 domains in the pull-down menu may be selected for the prediction. (B) Tabulated results showing the query protein name, sequence, locations of Tyr residues and SH2 domains predicted to bind a particular Tyr site (assuming the site is phosphorylated). A relative SMALI score is given in parenthesis beside a selected SH2 domain. Only SH2 domains with a relative score of >1.0 are listed.
Experimental determination of SMALI cut-off values
While it is reasonable to assume that a peptide with a larger SMALI score has a greater tendency to bind a query SH2 domain, this assumption has to be verified experimentally. In addition, a cut-off value is needed to limit the size of the output file and to identify interactions that have a high probability to occur. Moreover, a given peptide may produce different SMALI scores for different SH2 domains, and it would be impossible to determine which SH2 domain is preferred by the peptide based on the raw SMALI scores. Therefore, it is necessary to derive a relative SMALI score that allows for direct comparison between SH2 domains. To this end, we applied SMALI to predict peptide ligands for the BRDG1 and GRB2 SH2 domains, respectively and synthesized these peptides in an array format to test their binding to the two SH2 domains. These two SH2 domains represent two extreme cases since few physiological targets have been identified for the BRDG1 SH2 domain (25), whereas a dozen or so have been characterized for the GRB2 SH2 domain. To gauge the repertoire of peptides that potentially bind the BRDG1 SH2 domain, we searched the Swiss-Prot human protein database and retrieved1488 peptides ranked in the top 5% by SMALI (Table S1). These peptides were then synthesized as an array and screened for binding to the purified BRDG1 SH2 domain following established procedures (12,13). As shown in Figure 3A, while the majority of peptides belonging to the top two-thirds of list displayed binding to the BRDG1 SH2 domain, only a small fraction of the bottom third exhibited binding, suggesting that the ability of a peptide to bind BRDG1 SH2 domain correlates grossly with the raw SMALI score. Because only a small fraction of all Tyr residues contained in the Swiss-Prot database is expected to be phosphorylated in vivo, we performed a more targeted binding assay for the GRB2 SH2 domain on a set of peptides selected from the Phosphosite database. We selected a total of 720 peptides of which 360 corresponded to the peptides with large SMALI scores (upper half in Figure 3B) and the remaining 360 were taken randomly from the Phosphosite database (Table S2). While most peptides predicted by SMALI indeed exhibited binding to the GRB2 SH2 domain, only a small fraction of the randomly chosen peptides (lower half in Figure 3B) showed detectable binding.
Figure 3.

Validation of SMALI predicted interactions by peptide array and derivation of cut-off SMALI values. (A) Binding profile of the BRDG1 SH2 domain to an array of 1488 top-ranked phosphotyrosine-containing peptides selected by SMALI from the Swiss-Prot human protein database. (B) Binding of the GRB2 SH2 domain to 720 phosphopeptides taken from the Phosphosite database (15). The first 360 peptides (upper portion) was based on SMALI prediction, whereas the second half (lower portion) was randomly chosen from the database. Dark spots indicate positive binding. (C and D) Distribution of binding peptides over SMALI scores for the BRDG1 (C) and GRB2 SH2 (D) domains. The histograms show ‘hit rate’, defined as the percentage of binding peptides, at a given SMALI score range (in increments of 0.1 and 0.2, respectively for C and D). (E and F) An optimal SMALI cut-off value is arbitrarily defined as the SMALI score that produces the greatest F-measure. F-measure = 2 × precision × recall/(precision + recall), where precision = binding peptides correctly predicted/binding peptides predicted and recall = binding peptides correctly predicted/real binding peptides. For the BRDG1 SH2 domain, the SMALI score 1.4 produced the largest F-measure 0.84 (E). Coincidently, this SMALI value corresponds to a hit-rate of ∼50%. For the GRB2 SH2 domain, the cut-off SMALI score is 1.6. (F and G) Distribution of all Tyr-containing peptides (total 203 494) in Swiss-Prot human database according to SMALI scores calculated using PSSM for BRDG1 (G) or the GRB2 SH2 (H) domain. The SMALI cut-off of 1.4 for the BRDG1 SH2 domain corresponds to the top 3.5% scoring peptides located to the right of the cut-off value (G). For GRB2 SH2, the cut-off corresponds to the top 5.5% peptides ranked according to SMALI.
To correlate the peptide array results with the SMALI score, we calculated the experimentally observed ‘hit-rates’ of peptide-domain interactions and graphed them against the corresponding SMALI scores (at 0.1 or 0.2 intervals). It is apparent from Figure 3C and D that a larger SMALI score generally corresponds to a greater hit rate for either the BRDG1 or the GRB2 SH2 domain. To generate a cut-off value for SMALI prediction, we next calculated the F-measure and plotted it against the SMALI score (Figure 3E and F). We arbitrarily defined a SMALI cut-off as the score corresponding to the greatest F-measure value, which represents the best compromise between precision of prediction and the rate of recall. For the BRDG1 SH2 domain, the cut-off of 1.4 corresponds to peptides ranked in the top 3.5% by SMALI (Figure 3G). In the peptide screening, 82% of the peptides with the score >1.4 are true binders. In a previous study, we synthesized 22 peptides and measured their respective dissociation constants (Kd) for the BRDG1 SH2 domain in solution (13). Half of these peptides have a SMALI score >1.4, whereas the remaining half has scores below the cut-off. For the first half, 10 (or 91%) displayed strong binding in solution. In contrast, 9 (or 82%) of the second group of peptides exhibited weak or no binding to the BRDG1 SH2 domain. These results suggest that the cut-off is suitable for identifying authentic binding partners for BRDG1.
Analysis of the F-measure led to a SMALI cut-off value of 1.65 for the GRB2 SH2 domain, which corresponds to the top 5.5% of all Tyr-containing peptides collected in the Swiss-Prot human protein database (Figure 3H). Interestingly, all 13 known ligands of the GRB2 SH2 domain have scores greater than the cut-off, were correctly identified by SMALI, and showed strong binding in the peptide array screen (Table 1). Therefore, the experimentally determined cut-off value is suitable for identifying physiological binding partners for GRB2.
Table 1.
Known GRB2 SH2-peptide interactions re-examined in the peptide array experiment
| SH2 Protein (Alias)a | Description | pY site | pY-peptide | SMALI score | Peptide arrayb | References |
|---|---|---|---|---|---|---|
| BCR_HUMAN (Bcr) | Breakpoint cluster region protein | 177 | KPFpYVNVEF | 2.67 | + | (34) |
| IRS1_RAT (Irs1) | Insulin receptor substrate 1 | 895 | PGEpYVNIEF | 2.61 | + | (35,36) |
| FAK2_HUMAN (PYK2) | Focal adhesion kinase 2 | 881 | DLVpYLNVME | 2.53 | + | (37) |
| ERBB2_HUMAN (ErbB2) | Receptor tyrosine-protein kinase erbB-2 | 1139 | QPEpYVNQPD | 2.51 | + | (38) |
| FAK1_HUMAN (FAK) | Focal adhesion kinase | 925 | DKVpYENVTG | 2.43 | + | (39) |
| SHC1_HUMAN (Shc) | SHC-transforming protein 1 | 427 | DPSpYVNVQN | 2.42 | + | (40) |
| VGFR1_HUMAN (VEGFR-1) | Vascular endothelial growth factor receptor 1 | 1213 | DVRpYVNAFK | 2.41 | + | (41) |
| PGFRB_HUMAN (PDGFR-β) | Beta-type platelet-derived growth factor receptor | 716 | AELpYSNALP | 2.40 | + | (42) |
| LAT_MOUSE (LAT) | Linker for activation of T-cells family member 1 | 175 | IDDpYVNVPE | 2.38 | + | (43) |
| TIE2_HUMAN (TIE2) | Angiopoietin-1 receptor | 1102 | RKTpYVNTTL | 2.35 | + | (44) |
| LAT_MOUSE (LAT) | Linker for activation of T-cells family member 1 | 235 | APDpYENLQE | 2.24 | + | (43) |
| PTN11_HUMAN (Ptpn11) | Tyrosine-protein phosphatase non-receptor type 11 | 546 | GHEpYTNIKY | 1.94 | + | (45) |
| SHC1_HUMAN (Shc) | SHC-transforming protein 1 | 349 | DHQpYYNDFP | 1.86 | + | (46) |
aProtein names are according to Swiss-Prot convention with the commonly used alias given in parenthesis.
bPeptides showing positive binding in the array (Figure 3B) are identified with ‘+’. See Methods section for details of experimentation.
While in principle one could carry out similar experiments for the remaining SH2 domains in order to determine the corresponding cut-off values, the amount of work involved would be enormous. Nevertheless, from the binding data obtained for the BRDG1 and GRB2 SH2 domain, it is reasonable to assume that the top 4.5% (average cut-off value for the BRDG1 and GRB2 SH2 domains) of peptides ranked by SMALI have a high probability to bind a query SH2 domain. We have therefore set the SMALI score that separate the top 4.5% of peptides from the remainder (except for the BRDG1 and GRB2 SH2s) as the reference point for an SMALI prediction. The cut-off value was used as a common denominator to normalize the raw SMALI score. This produces the relative SMALI score listed in Figure 1E, which serves as a measure of propensity for a peptide to bind a query SH2 domain. A relative score of >1.0 indicates high potential, whereas a score smaller than 1.0 indicates a low potential for binding. The assignment of a relative SMALI score also makes it possible to compare and rank different SH2 domains for their propensity to bind a given peptide ligand in the ‘domain scan’ module of the SMALI program.
Comparison between SMALI and Scansite
Scansite is a web-based program capable of identifying domain-binding peptides or kinase substrates using PSSMs derived from screening peptide libraries synthesized chemically or displayed on bacteriophages (26,27). Scansite incorporates three threshold values—‘high’, ‘medium’ or ‘low’ stringency—to determine the accuracy of prediction. For instance, a peptide is reported as a ‘high stringency’ hit if its Sf score falls within the top 0.2% of all peptides in the same group (i.e. Tyr-containing). Scansite currently incorporates PSSMs for 14 SH2 domains from ABL1, CRK, FGR, FYN, GRB2, ITK, LCK, NCK, SRC, SHIP, SHIP, PIK3R1, PLCG1_N and PLCG1_C, respectively. All matrices have counterparts in SMALI except for the PLCG1_N SH2 domain.
Since both SMALI and Scansite can be used to predict SH2–ligand interactions, we next compared their performance in predicting targets for SH2 domains from NCK, CRK and FGR. For each SH2 domain, the top 336 candidate peptides selected by either Scansite or SMALI were synthesized on a membrane and tested for binding to the SH2 domain. The sequences and ranking orders of the peptides by either SMALI or Scansite are listed in Tables S3–S5. Results of screening the peptide arrays with the corresponding SH2 domain are shown in Figure 4. For peptides predicted by SMALI to bind an SH2 domain, 40% are found real for NCK, 90% for CRK and 98% for FGR. In contrast, 15% of peptides identified by Scansite as binders for NCK were real, while 32 and 87% were real for CRK and FGR SH2-binding, respectively (Table 2). Interestingly, neither program predicted NCK SH2 ligands with a >50% accuracy. We speculate that other factors, such as negative selection and position-dependence, which are not accounted for in a PSSM, may play a ‘dominant negative’ role in some SH2–ligand interactions. We calculated the average SMALI score of the Scansite-predicted peptides and found it to be smaller than the average score for the SMALI-predicted peptides. This agrees with our observation that peptides with larger SMALI scores have greater propensities to bind a query SH2 domain (Table 2). Taken together, SMALI exhibited improved accuracy than Scansite in identifying peptide ligands for the three SH2 domains examined herein. Nevertheless, we also observed that the combination of the two programs identified more binding peptides for an SH2 domain than did either alone. Therefore, the integration of SMALI and Scansite should facilitate the identification of SH2 domain–ligand interactions.
Figure 4.

Validation of peptide ligands for the SH2 domains of CRK (A), NCK (B) and FGR (C), respectively as identified by SMALI (upper half of each peptide array) or Scansite (bottom half). For each SH2 domain, a total of 336 peptides were examined, of which the first 168 was identified as top binders by SMALI and the last 168 by the Scansite. The sequences of the peptides and their respective ranking orders on SMALI or Scansite are provided in Tables S3–S5. See also Table 2 for a summary of the result.
Table 2.
Accuracy of prediction for SH2-binding peptides by SMALI or Scansitea
| SH2 domain | SMALI score cut-off | SMALI | Scansite | ||
|---|---|---|---|---|---|
| SMALI score (average, SD) | Hit rate (%) | SMALI score (average, SD) | Hit rate (%) | ||
| NCK1 | 1.40 | 2.02, 0.11 | 40 | 1.73, 0.29 | 15 |
| CRK | 1.65 | 2.19, 0.08 | 90 | 1.64, 0.28 | 32 |
| FGR | 1.35 | 1.84, 0.10 | 98 | 1.49, 0.30 | 87 |
aPeptides with spot values >0.8 are defined as binding peptides for the NCK1 SH2 domain, >0.7 for the CRK SH2 domain and >0.4 for FGR SH2 domain, based on the distribution of spot values in a peptide array experiment (see Materials and Methods section for details; see also Figure 4 and Tables S3–S5 for experimental data).
Predicting SH2 signaling network by SMALI
The determination of specificity of two-thirds of human SH2 domains makes it possible to gauge the signaling space involving the SH2 domain. To interrogate whether SMALI can aid in the identification of authentic SH2–ligand interactions in a larger scale than described earlier, we applied it to a group of 12 SH2 domains with the phosphorylation filter and identified all peptides with a relative SMALI score >1.0. These SH2 domains were selected to represent the major specificity groups I (motif poYξξφ, where ξ denotes a hydrophilic residue and φ is a hydrophobic residue) and II (motif poYxxφ, where x denotes any residue) (13). The corresponding SH2-containing proteins have also been studied extensively by either conventional or proteomic approaches such that a number of interactions involving them have been reported in the literature. As seen in Table 3, each SH2 domain could potentially interact with hundreds of target proteins, suggesting that other factors such as protein expression and localization must play a role in dictating which interactions occur in vivo. To assess the accuracy of the prediction, we examined the overlap between the predicted interactions and those curated in comprehensive PPI databases such as I2D (24) and IntAct (22). We found that the overlap between the predicted and known interactions ranging from 20.3% for Fyn to 49.3% for PIK3. This overlap is significantly greater than expected by chance (P < 0.006), confirming that SMALI is an efficient method to recapitulate authentic SH2–target interactions. The overlap would have been more extensive if we had knowledge on which interactions listed in a PPI database indeed involve an SH2 domain and discounted those that are not directly mediated by the query SH2 domain. It should also be noted that the intersection between the PPI space and corresponding SMALI space for a given SH2 protein is rather small (with the exception of Grb2, Table 3), suggesting that many authentic SH2–target interactions awaits identification or experimental validation.
Table 3.
Overlap between SMALI-predicted SH2-ligand interactions and those listed in PPI databases
| SH2 domain classificationa | SH2-containing proteins | SH2-interacting proteins predicted by SMALIb | SH2-interacting proteins included in PPI databasesc | Intersection between SMALI and PPI spaced | Overlap of SMALI network with PPI databases (%)e | Statistical significance of overlapf (P-value) |
|---|---|---|---|---|---|---|
| IA | SRC | 298 | 104 | 63 | 15 (23.8) | <0.0004 |
| IA | LYN | 204 | 69 | 50 | 13 (26.0) | <0.00001 |
| IA | ABL1 | 253 | 63 | 46 | 11 (23.9) | <0.0006 |
| IA | FYN | 313 | 99 | 69 | 14 (20.3) | <0.006 |
| IB | CRK | 395 | 44 | 35 | 14 (40.0) | <0.00005 |
| IB | CRKL | 274 | 40 | 30 | 9 (30) | <0.0006 |
| IC | GRB2 | 420 | 383 | 250 | 68 (27.2) | <0.00001 |
| IC | GRAP2 | 308 | 27 | 18 | 7 (38.9) | <0.0009 |
| IIA | PIK3R1 | 317 | 98 | 73 | 35 (49.3) | <0.00001 |
| IIA | PTPN11 | 288 | 67 | 59 | 20 (33.9) | <0.00001 |
| IIA | VAV1 | 170 | 54 | 45 | 13 (28.9) | <0.00001 |
| IIB | SHC1 | 275 | 98 | 77 | 22 (28.6) | <0.00001 |
aThe SH2 domain classification is based on (13). Group IA has a common motif poY−−φ, IB has poYxxφ, IC has poYxNx, IIA has poYφxφ and IIB has the motif poY[E/D/x]xφ, where ‘−’ denotes a negatively charged residue, φ denotes a hydrophobic residue and x is any type of residues.
bNumber of proteins predicted to bind to a specific SH2 domain-containing protein in the table by SMALI with a relative score >1.0. The Phosphorylation filter is applied with the prediction.
cNumber of binding proteins for a specific SH2-containing protein according to PPI databases I2D (24) and IntAct (22).
dSMALI space is the number of proteins (3253) used in the prediction. These include all proteins listed in the PhosphoSite and Phospho.ELM databases that contain a pTyr. Intersection is defined as the protein space covered by both SMALI and the PPI databases.
eNumber of common interactions shared between the PPI databases and SMALI prediction for a given SH2-containing protein. The percentage of overlap (in parenthesis) is calculated by dividing this number by the intersected space between PPI and SMALI.
fObserved overlap over that expected by chance.
DISCUSSION
It is clear from comparative genomic analyses that signaling domains have undergone a drastic expansion in multicellular organisms (28,29). Taking the SH2 domain for example, in contrast to yeast that contains no functional SH2 domain, a human cell harbors over a hundred such domains. The same pattern of domain expansion is also observed for other signaling modules such as SH3, PTB and PDZ, to name just a few. The abundance of these interaction modules in the human genome suggests that they play important roles in regulating normal cellular function (30). Because a number of prevalent signaling domains promote PPIs by binding to short linear motifs present in other proteins, delineating the specificity of these domains provides an effective means to decipher the multitude of protein interactions mediated by them. Additionally, the specificity information allows for ready identification of potential binding partners for an interaction domain. In this regard, Scansite was developed to capitalize on the knowledge of domain and kinase specificity, and has become an essential tool in the toolbox of signal transduction (26,27). The SMALI method described herein utilizes the same principles as those guided the Scansite, but is distinguishable from the latter in the following. First, the current version of SMALI contains specificity information and the corresponding scoring matrices for 76 human SH2 domains, making it possible now to predict phosphotyrosine peptide–SH2 domain interactions at or near proteome scale. The origin of the PSSMs (13) dictates that SMALI is dedicated to the prediction of human or other PPIs. Second, the scoring matrices employed by SMALI contain experimentally defined selectivity information for positions −2 through +4 with respect to the invariant pTyr. In comparison, most SH2 matrices employed by Scansite contain experimentally derived selectivity information on the C-terminal residues only (10,11). Although we have not subjected it to rigorous tests yet, the inclusion of N-terminal selectivity may enhance the accuracy of prediction since it allows distinctions to be made between two peptides that may contain an identical C-terminal sequence. Moreover, some SH2 domains, including those from BRDG1, PLCγ1 and SHP2, have shown selectivity beyond P+3. Third, SMALI is imbedded with several filters to limit the return from a search to proteins that are most likely to be of physiologically relevance. Of particular usage is the phosphorylation filter, since it limits the output to proteins whose phosphorylation has been experimentally verified. Fourth, the threshold value for a SMALI prediction is inferred from experiments and the resulting normalized SMALI score can be used as a direct measure of binding propensity of a peptide to a query SH2 domain. The normalized propensity score also eliminates the difference in the range of SMALI scores for different SH2 domains and allow for direct comparison of two SH2 domains for propensity to bind a given peptide.
A useful bioinformatic program should not only be capable of recapitulating known knowledge but also predict novel biology. We have put SMALI to rigorous tests on both functions. SMALI faithfully recapitulated all known interactions mediated by the GRB2 SH2 domain and predicted novel interactions involving the BRDG1 SH2 domain (12). Our network analysis on a set of 12 SH2 domains also revealed a significant overlap between SMALI predicted SH2–ligand interactions and known interactions that involve the corresponding SH2 proteins. Since the specificity of the SH2 domain is tightly coupled to the specificity of tyrosine kinases (31), SMALI may play a role in identifying signaling networks initiated by protein-tyrosine kinases. In this regard, we attempted to identify a kinase-SH2 signaling network involving a group of SH2 domains by combining SMALI with NetworKIN (32), a web-based program that was developed recently to identify phosphorylation sites and the corresponding kinases based on linear motif-recognition and network context (32,33). The predicted PTK–substrate–SH2 network not only recapitulates many known interactions, but reveals a number of novel signaling pathways (Li and Li, unpublished data). This exercise suggests that by combining SMALI with existing programs on kinase specificity and/or network analysis, novel signaling pathways can be uncovered.
To make full use of the OPAL-derived scoring matrices, we have made them available to other bioinformatic programs such as NetPhorest (Linding et al. unpublished data). We will also make our matrices available to Scansite and related programs that predict PPIs based on linear motifs. Moreover, the SMALI site will be updated regularly to include more scoring matrices derived from OPAL or other experiments. Because the OPAL approach can be applied in principle to any modular domains, including kinases and phosphatases that recognize short linear peptide motifs, we anticipate SMALI will be expanded to the prediction of interactions mediated by a variety of interaction domains and for the identification of kinase substrates in a similar manner as described here. Despite the usefulness of SMALI or Scansite in identifying peptide ligands for an SH2 domain, it should be realized that the physiological relevance of a prediction remains to be established by experiments. An in vitro binding event does not always correspond to an in vivo interaction because other factors such as protein expression, phosphorylation, localization and/or scaffolding may dictate whether a given interaction will indeed occur in a cell.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
This work was supported by grants from Genome Canada (to S.S.-C.L.) through the Ontario Genome Institute, the Canadian Institute of Health Research (to S.S.-C.L.) and the Canadian Cancer Society (to S.S.-C.L.). S.S.-C.L. holds a Canada Research Chair in Functional Genomics and Cellular Proteomics. Funding to pay the Open Access publication charges for this article was provided by Genome Canada.
Conflict of interest statement. None declared.
REFERENCES
- 1.Johnson SA, Hunter T. Kinomics: methods for deciphering the kinome. Nat. Methods. 2005;2:17–25. doi: 10.1038/nmeth731. [DOI] [PubMed] [Google Scholar]
- 2.Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355–365. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- 3.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 4.Pawson T, Scott JD. Protein phosphorylation in signaling - 50 years and counting. Trends Biochem. Sci. 2005;30:286–290. doi: 10.1016/j.tibs.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 5.Pawson T. Specificity in signal transduction: from phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell. 2004;116:191–203. doi: 10.1016/s0092-8674(03)01077-8. [DOI] [PubMed] [Google Scholar]
- 6.Smith MJ, Hardy WR, Murphy JM, Jones N, Pawson T. Screening for PTB domain binding partners and ligand specificity using proteome-derived NPXY peptide arrays. Mol. Cell. Biol. 2006;26:8461–8474. doi: 10.1128/MCB.01491-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu BA, Jablonowski K, Raina M, Arce M, Pawson T, Nash PD. The human and mouse complement of SH2 domain proteins—establishing the boundaries of phosphotyrosine signaling. Mol. Cell. 2006;22:851–868. doi: 10.1016/j.molcel.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 8.Hwang PM, Li C, Morra M, Lillywhite J, Muhandiram DR, Gertler F, Terhorst C, Kay LE, Pawson T, Forman-Kay JD, et al. A “three-pronged” binding mechanism for the SAP/SH2D1A SH2 domain: structural basis and relevance to the XLP syndrome. EMBO J. 2002;21:314–323. doi: 10.1093/emboj/21.3.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Obenauer JC, Cantley LC, Yaffe MB. Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003;31:3635–3641. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Songyang Z, Shoelson SE, Chaudhuri M, Gish G, Pawson T, Haser WG, King F, Roberts T, Ratnofsky S, Lechleider RJ, et al. SH2 domains recognize specific phosphopeptide sequences. Cell. 1993;72:767–778. doi: 10.1016/0092-8674(93)90404-e. [DOI] [PubMed] [Google Scholar]
- 11.Songyang Z, Shoelson SE, McGlade J, Olivier P, Pawson T, Bustelo XR, Barbacid M, Sabe H, Hanafusa H, Yi T, et al. Specific motifs recognized by the SH2 domains of Csk, 3BP2, fps/fes, GRB-2, HCP, SHC, Syk, and Vav. Mol. Cell. Biol. 1994;14:2777–2785. doi: 10.1128/mcb.14.4.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu C, Ma MH, Brown KR, Geisler M, Li L, Tzeng E, Jia CY, Jurisica I, Li SS. Systematic identification of SH3 domain-mediated human protein-protein interactions by peptide array target screening. Proteomics. 2007;7:1775–1785. doi: 10.1002/pmic.200601006. [DOI] [PubMed] [Google Scholar]
- 13.Huang H, Li L, Wu C, Schibli D, Colwill K, Ma S, Li C, Roy P, Ho K, Songyang Z, et al. Defining the specificity space of the human src-homology 2 domain. Mol. Cell. Proteomics. 2007;7:768–784. doi: 10.1074/mcp.M700312-MCP200. [DOI] [PubMed] [Google Scholar]
- 14.Bairoch A, Apweiler R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000;28:45–48. doi: 10.1093/nar/28.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hornbeck PV, Chabra I, Kornhauser JM, Skrzypek E, Zhang B. PhosphoSite: a bioinformatics resource dedicated to physiological protein phosphorylation. Proteomics. 2004;4:1551–1561. doi: 10.1002/pmic.200300772. [DOI] [PubMed] [Google Scholar]
- 16.Diella F, Cameron S, Gemund C, Linding R, Via A, Kuster B, Sicheritz-Ponten T, Blom N, Gibson TJ. Phospho.ELM: a database of experimentally verified phosphorylation sites in eukaryotic proteins. BMC Bioinform. 2004;5:79. doi: 10.1186/1471-2105-5-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Camon E, Magrane M, Barrell D, Lee V, Dimmer E, Maslen J, Binns D, Harte N, Lopez R, Apweiler R. The gene ontology annotation (GOA) database: sharing knowledge in uniprot with gene Ontology. Nucleic Acids Res. 2004;32:D262–D266. doi: 10.1093/nar/gkh021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Finn RD, Mistry J, Schuster-Bockler B, Griffiths-Jones S, Hollich V, Lassmann T, Moxon S, Marshall M, Khanna A, Durbin R, et al. Pfam: clans, web tools and services. Nucleic Acids Res. 2006;34:D247–D251. doi: 10.1093/nar/gkj149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Letunic I, Copley RR, Pils B, Pinkert S, Schultz J, Bork P. SMART 5: domains in the context of genomes and networks. Nucleic Acids Res. 2006;34:D257–D260. doi: 10.1093/nar/gkj079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schultz J, Milpetz F, Bork P, Ponting CP. SMART, a simple modular architecture research tool: identification of signaling domains. Proc. Natl Acad. Sci. USA. 1998;95:5857–5864. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hermjakob H, Montecchi-Palazzi L, Lewington C, Mudali S, Kerrien S, Orchard S, Vingron M, Roechert B, Roepstorff P, Valencia A, et al. IntAct: an open source molecular interaction database. Nucleic Acids Res. 2004;32:D452–D455. doi: 10.1093/nar/gkh052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kerrien S, Alam-Faruque Y, Aranda B, Bancarz I, Bridge A, Derow C, Dimmer E, Feuermann M, Friedrichsen A, Huntley R, et al. IntAct—open source resource for molecular interaction data. Nucleic Acids Res. 2007;35:D561–D565. doi: 10.1093/nar/gkl958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown KR, Jurisica I. Online predicted human interaction database. Bioinformatics. 2005;21:2076–2082. doi: 10.1093/bioinformatics/bti273. [DOI] [PubMed] [Google Scholar]
- 25.Ohya K-i, Kajigaya S, Kitanaka A, Yoshida K, Miyazato A, Yamashita Y, Yamanaka T, Ikeda U, Shimada K, Ozawa K, et al. Molecular cloning of a docking protein, BRDG1, that acts downstream of the Tec tyrosine kinase. PNAS. 1999;96:11976–11981. doi: 10.1073/pnas.96.21.11976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Obenauer JC, Cantley LC, Yaffe MB. Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003;31:3635–3641. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yaffe MB, Leparc GG, Lai J, Obata T, Volinia S, Cantley LC. A motif-based profile scanning approach for genome-wide prediction of signaling pathways. Nat. Biotechnol. 2001;19:348–353. doi: 10.1038/86737. [DOI] [PubMed] [Google Scholar]
- 28.Rubin GM, Yandell MD, Wortman JR, Gabor Miklos GL, Nelson CR, Hariharan IK, Fortini ME, Li PW, Apweiler R, Fleischmann W, et al. Comparative genomics of the eukaryotes. Science. 2000;287:2204–2215. doi: 10.1126/science.287.5461.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anantharaman V, Iyer LM, Aravind L. Comparative genomics of protists: new insights into the evolution of eukaryotic signal transduction and gene regulation. Annu. Rev. Microbiol. 2007;61:453–475. doi: 10.1146/annurev.micro.61.080706.093309. [DOI] [PubMed] [Google Scholar]
- 30.Pawson T, Nash P. Assembly of cell regulatory systems through protein interaction domains. Science. 2003;300:445–452. doi: 10.1126/science.1083653. [DOI] [PubMed] [Google Scholar]
- 31.Songyang Z, Cantley LC. Recognition and specificity in protein tyrosine kinase-mediated signalling. Trends Biochem. Sci. 1995;20:470–475. doi: 10.1016/s0968-0004(00)89103-3. [DOI] [PubMed] [Google Scholar]
- 32.Linding R, Jensen LJ, Ostheimer GJ, van Vugt MA, Jorgensen C, Miron IM, Diella F, Colwill K, Taylor L, Elder K, et al. Systematic discovery of in vivo phosphorylation networks. Cell. 2007;129:1415–1426. doi: 10.1016/j.cell.2007.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Linding R, Jensen LJ, Pasculescu A, Olhovsky M, Colwill K, Bork P, Yaffe MB, Pawson T. NetworKIN: a resource for exploring cellular phosphorylation networks. Nucleic Acids Res. 2008;36:D695–D699. doi: 10.1093/nar/gkm902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma G, Lu D, Wu Y, Liu J, Arlinghaus RB. Bcr phosphorylated on tyrosine 177 binds Grb2. Oncogene. 1997;14:2367–2372. doi: 10.1038/sj.onc.1201053. [DOI] [PubMed] [Google Scholar]
- 35.Sun XJ, Crimmins DL, Myers M.G., Jr., Miralpeix M, White MF. Pleiotropic insulin signals are engaged by multisite phosphorylation of IRS-1. Mol. Cell. Biol. 1993;13:7418–7428. doi: 10.1128/mcb.13.12.7418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu B, Bird VG, Miller WT. Substrate specificities of the insulin and insulin-like growth factor 1 receptor tyrosine kinase catalytic domains. J. Biol. Chem. 1995;270:29825–29830. doi: 10.1074/jbc.270.50.29825. [DOI] [PubMed] [Google Scholar]
- 37.Chauhan D, Pandey P, Hideshima T, Treon S, Raje N, Davies FE, Shima Y, Tai YT, Rosen S, Avraham S, et al. SHP2 mediates the protective effect of interleukin-6 against dexamethasone-induced apoptosis in multiple myeloma cells. J. Biol. Chem. 2000;275:27845–27850. doi: 10.1074/jbc.M003428200. [DOI] [PubMed] [Google Scholar]
- 38.Dankort D, Jeyabalan N, Jones N, Dumont DJ, Muller WJ. Multiple ErbB-2/Neu phosphorylation sites mediate transformation through distinct effector proteins. J. Biol. Chem. 2001;276:38921–38928. doi: 10.1074/jbc.M106239200. [DOI] [PubMed] [Google Scholar]
- 39.Schlaepfer DD, Hanks SK, Hunter T, van der Geer P. Integrin-mediated signal transduction linked to Ras pathway by GRB2 binding to focal adhesion kinase. Nature. 1994;372:786–791. doi: 10.1038/372786a0. [DOI] [PubMed] [Google Scholar]
- 40.Ogura K, Tsuchiya S, Terasawa H, Yuzawa S, Hatanaka H, Mandiyan V, Schlessinger J, Inagaki F. Solution structure of the SH2 domain of Grb2 complexed with the Shc-derived phosphotyrosine-containing peptide. J. Mol. Biol. 1999;289:439–445. doi: 10.1006/jmbi.1999.2792. [DOI] [PubMed] [Google Scholar]
- 41.Ito N, Wernstedt C, Engstrom U, Claesson-Welsh L. Identification of vascular endothelial growth factor receptor-1 tyrosine phosphorylation sites and binding of SH2 domain-containing molecules. J. Biol. Chem. 1998;273:23410–23418. doi: 10.1074/jbc.273.36.23410. [DOI] [PubMed] [Google Scholar]
- 42.Arvidsson AK, Rupp E, Nanberg E, Downward J, Ronnstrand L, Wennstrom S, Schlessinger J, Heldin CH, Claesson-Welsh L. Tyr-716 in the platelet-derived growth factor beta-receptor kinase insert is involved in GRB2 binding and Ras activation. Mol. Cell. Biol. 1994;14:6715–6726. doi: 10.1128/mcb.14.10.6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang W, Trible RP, Zhu M, Liu SK, McGlade CJ, Samelson LE. Association of Grb2, Gads, and phospholipase C-gamma 1 with phosphorylated LAT tyrosine residues. Effect of LAT tyrosine mutations on T cell angigen receptor-mediated signaling. J. Biol. Chem. 2000;275:23355–23361. doi: 10.1074/jbc.M000404200. [DOI] [PubMed] [Google Scholar]
- 44.Jones N, Master Z, Jones J, Bouchard D, Gunji Y, Sasaki H, Daly R, Alitalo K, Dumont DJ. Identification of Tek/Tie2 binding partners. Binding to a multifunctional docking site mediates cell survival and migration. J. Biol. Chem. 1999;274:30896–30905. doi: 10.1074/jbc.274.43.30896. [DOI] [PubMed] [Google Scholar]
- 45.Bennett AM, Tang TL, Sugimoto S, Walsh CT, Neel BG. Protein-tyrosine-phosphatase SHPTP2 couples platelet-derived growth factor receptor beta to Ras. Proc. Natl Acad. Sci. USA. 1994;91:7335–7339. doi: 10.1073/pnas.91.15.7335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Velazquez L, Gish GD, van Der Geer P, Taylor L, Shulman J, Pawson T. The shc adaptor protein forms interdependent phosphotyrosine-mediated protein complexes in mast cells stimulated with interleukin 3. Blood. 2000;96:132–138. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.