

Abstract
A number of proteinases are expressed on the surface of leukocytes including members of the serine, metallo-, and cysteine proteinase superfamilies. Some proteinases are anchored to the plasma membrane of leukocytes by a transmembrane domain or a glycosyl phosphatidyl inositol (GPI) anchor. Other proteinases bind with high affinity to classical receptors, or with lower affinity to integrins, proteoglycans, or other leukocyte surface molecules. Leukocyte surface levels of proteinases are regulated by: 1) cytokines, chemokines, bacterial products, and growth factors which stimulate synthesis and/or release of proteinase by cells; 2) the availability of surface binding sites for proteinases; and/or 3) internalization or shedding of surface-bound proteinases. The binding of proteinases to leukocyte surfaces serves many functions including: 1) concentrating the activity of proteinases to the immediate pericellular environment; 2) facilitating pro-enzyme activation; 3) increasing proteinase stability and retention in the extracellular space; 4) regulating leukocyte function by proteinases signaling through cell surface binding sites or other surface proteins; and 5) protecting proteinases from inhibition by extracellular proteinase inhibitors. There is strong evidence that membrane-associated proteinases on leukocytes play critical roles in wound healing, inflammation, extracellular matrix remodeling, fibrinolysis, and coagulation. This review will outline the biology of membrane-associated proteinases expressed by leukocytes and their roles in physiologic and pathologic processes.
Keywords: proteinase, cell surface binding, pericellular proteolysis, metalloproteinase, serine proteinase
Introduction
Leukocytes and resident tissue cells produce a diverse array of proteinases that contribute to physiologic processes such as extracellular matrix (ECM) remodeling, wound healing, inflammation, coagulation, fibrinolysis, host defense against infection, and various pathologic processes. Until recently, there has been little information available about the mechanisms by which cells use and control their proteinases to degrade extracellular proteins in vivo. Proteinases must circumvent the effects of high-affinity, extracellular proteinase inhibitors in order to cleave extracellular proteins. Inflammatory cells bathed in fluids containing physiologic proteinase inhibitors are associated with pericellular proteolysis (Figure 1). One mechanism that enables cells to cleave or degrade proteins in their immediate environment is the localization of proteinases on cell surfaces. In vertebrates, serine, metallo-, and cysteine proteinases are expressed on the surfaces of various cell types including leukocytes, fibroblasts, epithelial cells, endothelial cells, and tumor cells (Table 1). These enzymes include members of the serine proteinase superfamily such as the leukocyte serine proteinases, proteinases involved in fibrinolysis, the type II transmembrane serine-protease (TTSP) family (Szabo et al., 2003; Qiu, Owen, Gray, Bass, & Ellis, 2007a), and the kallikrein/kinin system [reviewed in (Schmaier & McCrae, 2007)]. Serine proteases involved in the coagulation cascades can also bind to endothelial, platelet, and leukocyte cell surfaces [reviewed in (Doshi & Marmur, 2002; Bouchard & Tracy, 2001) and Table 1]. Metalloproteinases and lysosomal cysteine proteinase also function as cell membrane-associated enzymes (Cavallo-Medved & Sloane, 2003). Space limitations of the journal preclude comprehensive coverage of all proteinases known to be expressed on the surface of all types of cells. Because of my interest in leukocyte-proteinase mediated tissue injury, I will focus this review on proteinases expressed on the surface of leukocytes that play important roles in leukocyte biology. I will review the biology of leukocyte surface-bound proteinases, the advantages that cell surface binding confers upon individual proteinases, and the roles of leukocyte membrane-associated proteinases in physiologic and pathologic processes.
Figure 1. PMN pericellular proteolysis.
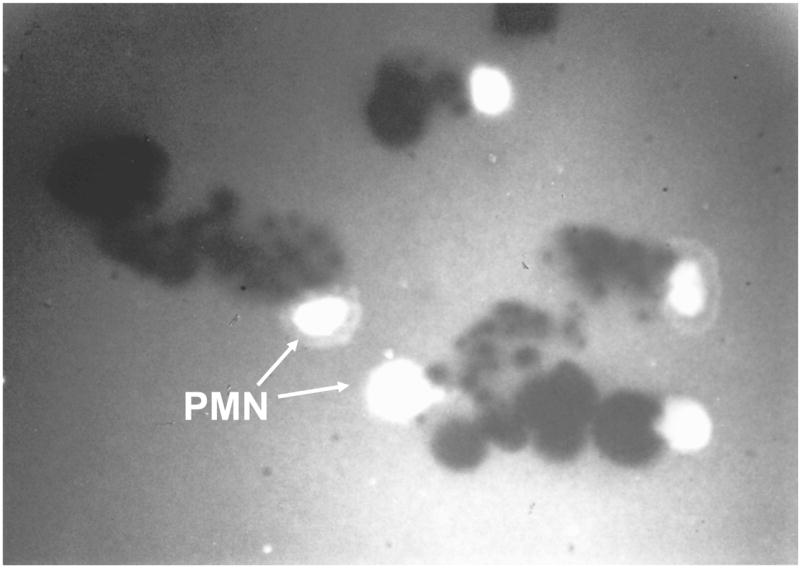
PMNs were incubated for 45 min. at 37°C on FITC-conjugated fibronectin which had been coated on tissue culture plates, and then opsonized. PMNs were bathed in 100% autologous serum which contains micromolar concentrations of TIMPs and serine proteinase inhibitors. Note that PMNs degrade fibronectin substrate as they migrate over it. However, fibronectin degradation is localized to the pericellular environment of the migrating PMNs by the inhibitors present in the bathing medium (arrows). Thus, physiologic proteinase inhibitors present in serum cannot block PMN pericellular proteolytic activity. When cells are bathed in inhibitor free buffers, the FITC-conjugated FN is completely degraded (not shown). One mechanism leading to this inhibitor-resistant pericellular proteolysis is expression of proteinases on the PMN surface in inhibitor-resistant forms which has been demonstrated for several serine and metallo-proteinases families.
Table 1.
Mechanisms of cell surface binding of proteinases
| Binding mechanism | Proteinases bound | Cell types | Proteolytic Activities |
|---|---|---|---|
| Transmembrane Domain | MT1-MMP
MT2-MMP MT3-MMP MT5-MMP |
Epithelial cells
Tumor cells Fibroblasts Macrophages |
ECM degradation, tumor invasiveness
Pro-MMP-2 &-13 binding and activation, TNF-α activation, proteinase inhibitor degradation |
| ADAMs | Epithelial cells
Endothelial cells Fibroblasts Smooth muscle cells Inflammatory cells Tumor cells |
Shedding of membrane- associated cytokines, apoptosis ligands, growth factors and receptors for these molecules
Pro-urokinase activation |
|
| Type II serine proteases(matriptase) | Monocytes, B lymphocytes | ||
| GPI anchor | uPAR and urokinase | PMN mononuclear phagocytes
Fibroblasts Epithelial cells Smooth muscle cells |
Fibrinolysis Activation of latent growth factors & proMMPs |
| MT4-MMP | CNS eosinophils | ECM degradation proMMP-2 activation | |
| MT6-MMP | PMN | ECM degradation | |
| Proteoglycans | Pro- and active MMP-2, -7, -9, and -13 | Uterine epithelial cells | ECM degradation Host defense |
| NE, CG, PR3 | PMN, macrophages | Wound healing Inflammation | |
| Integrins | β2: Pro-MMP-9 & -8, NE, CG | PMN | Unknown |
| α2β1: MMP-1 | Keratinocytes | Cell migration | |
| αvβ3: MMP-2 | Endothelial cells & melanoma cells | Tumor invasiveness and neovascularization | |
| TIMP-2 | Pro-MMP-2 and proMMP-13 | Fibroblasts Endothelium
Tumor cells |
ECM degradation
Tumor invasiveness |
| CD44 | MMP -2 & MMP-9 | Tumor cells & keratinocytes | ECM degradation, activation of latent TGF-β |
| MMP-7 | Tumor cells & epithelial cells | Post partum uterine involution, and lactation through HB-EGF shedding | |
| Tissue factor | Factor VII | Monocytes/macrophages | Activation of actor VII and other serine proteases involved in thrombosis |
| High-molecular- weight Kininogen | Prekallirein | Endothelial cells | Factor XI activation leading to thrombosis
Bradykinin generation Pro-urokinase activation |
| P11 | Cathepsin B | Tumor cells | Tumor growth and metastasis |
| Unknown | Cathepsin B | Macrophages, CTLs | Elastin degradation, degradation of perforins |
Legend for Table 1. ECM, extracellular matrix; PMN, polymorphonuclear neutrophils; TNF-α, tumor necrosis factor- α; GPI, glycosyl phosphatidyl inositol; CNS, central nervous system; NE, neutrophil elastase; CG, cathepsin G; PR3, proteinase 3; TGF-β, transforming growth factor- β, HB-EGF, heparin binding epidermal growth factor; CTLs, cytotoxic T lymphocytes
Serine Proteinases
Serine proteinases were among the earliest proteinases shown to be expressed on the surface of leukocytes. In 1985, urokinase type plasminogen activator (uPA or urokinase) was shown to bind to a surface receptor on monocytes and U937 cells (Vassalli, Baccino, & Belin, 1985). In 1990, proteinase 3 was identified on the surface of polymorphonuclear neutrophils [PMNs (Csernok, Ludemann, Gross, & Bainton, 1990)], and in the mid 1990s, neutrophil elastase (NE) and cathepsin G (CG) were also localized on the surface of activated PMNs (Owen, Campbell, Sannes, Boukedes, & Campbell, 1995b; Bangalore & Travis, 1994b). More recently, a new family of membrane-associated serine proteinases was identified: type II transmembrane serine proteases (TTSP). In 2006, a member of the TTSP family, matriptase, was identified on the monocyte surface (Kilpatrick et al., 2006).
Plasminogen Activators
Plasminogen activators are serine proteinases which convert plasminogen (an abundant extracellular protein) to plasmin, another serine proteinase which plays critical roles in degrading fibrin in blood clots and the provisional matrix deposited at sites of tissue injury. There are two plasminogen activators which differ in the structure of their non-catalytic domains (Blasi & Carmeliet, 2002; Medcalf, 2007). Tissue-type plasminogen activator (tPA), and urokinase-type plasminogen activator (uPA or urokinase). Both enzymes function as both soluble and cell-associated proteinases, but active tPA is primarily a clot-associated protease. Urokinase is expressed by leukocytes. Tissue-type plasminogen activator is not expressed by leukocytes, but binds to CD11b/CD18 integrin on macrophages to promote macrophage adhesion to fibrin and macrophage migration in vitro (Cao et al., 2006).
Structure, expression, and regulation of urokinase
Urokinase is produced and released by cells as an inactive, single chain pro-enzyme (pro-urokinase). This inactive pro-enzyme is cleaved at a single locus by a wide range of serine proteinases including plasmin, kallikrein, factor XIIa, matriptase, tryptase epsilon, human T cell-associated proteinase-1, and hepsin which is a TTSP (Blasi et al., 2002; Kilpatrick et al., 2006; Yasuda et al., 2005; Brunner, Simon, & Kramer, 1990; Moran et al., 2006). Several non-serine proteinases such as cathepsin B, cathepsin L, matrix metalloproteinase-3 (MMP-3) and the bacterial metalloproteinases, thermolysin also activate pro-urokinase (Kobayashi et al., 1993a; Goretzki et al., 1992; Orgel et al., 1998; Marcotte & Henkin, 1993). Cleavage of pro-urokinase by these enzymes generates a disulfilde-linked, two-chain, active enzyme (Blasi et al., 2002; Kilpatrick et al., 2006). Urokinase has three functional domains: 1) an NH2-terminal epidermal growth factor (EGF)-like domain which binds the enzyme to its cell surface receptor; 2) a kringle domain; and 3) a COOH-terminal catalytic domain with the His-Ser-Asp catalytic triad typical of the serine proteinase family. Pro-urokinase binds with high affinity (KD ~1 nM) to a specific receptor (uPA receptor or uPAR) which is a GPI-anchored protein on leukocytes and many other cells (Blasi et al., 2002; Yasuda et al., 2005; Ragno, 2006). However, pro-urokinase also binds to CD11b/CD18 integrin on PMNs which accelerates plasminogen activation and fibrinolysis on the PMN surface (Pluskota, Soloviev, Bdeir, Cines, & Plow, 2004).
Urokinase is expressed by PMNs, monocytes, macrophages, and many other types of cells (Table 1). However, the biology of urokinase differs in PMNs compared to other urokinase-expressing cell types. PMNs do not synthesize urokinase de novo. Preformed urokinase is stored in the specific granules of PMNs along with uPAR. Unstimulated PMNs express minimal cell surface uPAR or urokinase, but both proteins translocate rapidly from the specific granules to the PMN surface when PMNs are activated to degranulate by phorbol esters, chemoattractants, and cytokines (Plesner et al., 1994; Heiple & Ossowski, 1986). In monocytes, macrophages, and other cells, urokinase expression is regulated at the transcriptional level by pro-inflammatory mediators and growth factors (Medcalf, 2007). During the chemotactic response, receptor-bound uPA becomes rapidly polarized to the leading edge of these phagocytes where it regulates cell adherence and migration (Blasi et al., 2002; Mondino & Blasi, 2004). The main inhibitor of urokinase in plasma is plasminogen activator inhibitor-1 (PAI-1), a member of the serine proteinase inhibitor (serpin) family which is secreted by many cell types including endothelial cells. Other less efficient inhibitors of urokinase include PAI-2, and protease nexin I (Blasi et al., 2002; Mondino et al., 2004). These inhibitors inhibit urokinase by forming irreversible covalent complexes with the enzyme.
The binding of pro-urokinase to its surface receptor not only promotes urokinase and plasmin activation on the cell surface (Blasi et al., 2002), but also protects both enzymes from inhibition, and regulates urokinase surface levels by inducing urokinase-uPAR endocytosis. Urokinase bound to uPAR has a 40% reduction in its association rate constant for PAI-1 and PAI-2 when compared to soluble urokinase (Ellis, Wun, Behrendt, Ronne, & Dano, 1990). Urokinase-mediated cleavage of cell membrane-bound plasminogen generates membrane-bound plasmin which is also resistant to inhibition by α2-plasmin inhibitor which is an effective inhibitor of soluble plasmin (Ellis et al., 1990). Although binding of PAI-1 to surface-bound urokinase is reduced, when this happens, it is followed by endocytosis of the proteinase-inhibitor complex by clathrin-coated pits and members of the low density lipoprotein receptor family. The enzyme-inhibitor complex dissociates from uPAR, and the inhibitor-enzyme complex is degraded in the lysosomes, but uPAR is recycled back to the cell surface to bind additional urokinase (Blasi et al., 2002; Mondino et al., 2004). However, cleavage and shedding of uPAR is currently regarded as a major regulatory process for this receptor (Blasi et al., 2002).
In vitro activities of urokinase and plasmin
Urokinase and tPA both have critical roles in fibrinolysis by generating active plasmin (Carmeliet et al., 1994; Blasi et al., 2002). The urokinase-uPAR system was initially thought to simply concentrate plasmin-mediated fibrinolysis at cell surfaces during tissue injury. However, plasmin has numerous other functions including cleaving proteins other than fibrin and activating cells. For example, plasmin cleaves and activates latent growth factors (Taipale, Koli, & Keski-Oja, 1992) and latent pro-MMPs (Parks, Wilson, & Lopez-Boado, 2004). Thus, plasmin may inhibit fibrotic responses to injury by clearing the provisional fibrin matrix at sites of injury and by activating pro-MMPs, or promote tissue fibrosis by activating latent transforming growth factor-β (TGF-β) which stimulates (myo)fibroblasts to deposit collagen in tissues. Plasmin also binds with low affinity (likely via its lysine binding sites) to plasma membrane sites of leukocytes, platelets, and endothelial cells and activates these cells by an active site-dependent manner (Syrovets & Simmet, 2004). This leads to homotypic aggregation of PMNs, platelet degranulation, release of arachidonic acid from endothelial cells, release of pro-inflammatory mediators from monocytes, and induction of monocyte chemotaxis (Syrovets et al., 2004). Plasmin activates platelets by cleaving and activating platelet protease activated receptor-4 [PAR-4 (Quinton, Kim, Derian, Jin, & Kunapuli, 2004)], but has no effect on platelet PAR-1. However, plasmin activates PAR-1 on the surface of macrophages, which increases macrophage MMP-12 production (Churg et al., 2007; Raza, Nehring, Shapiro, & Cornelius, 2000). Thus by generating plasmin, urokinase stimulates not only fibrinolysis, but also regulates ECM turnover and inflammation in tissues.
Urokinase also stimulates intracellular signaling to promote adhesion and migration of leukocytes and other cells by binding to uPAR even though this receptor lacks a cytoplasmic domain. After binding urokinase, uPAR signals by binding to other transmembrane proteins (including integrins and G protein coupled receptors) or to ECM proteins to promote cell adhesion, migration, apoptosis, and cell proliferation [reviewed in (Blasi et al., 2002; Mondino et al., 2004; Crippa, 2007; Ragno, 2006; Blasi, 2001). For example, uPAR stimulates chemotaxis of leukocytes after urokinase binds to uPAR which unmasks a chemotactic epitope in uPAR. The uncovering of this normally cryptic epitope in intact uPAR is caused by endoproteolytic removal of the amino-terminal domain of uPAR by active proteinases including those resulting from the biologic activity of uPAR [such as uPA, plasmin, and several active MMPs (Blasi et al., 2002)]. This signal is transduced by uPAR binding to a G protein-coupled fMLP receptor (Resnati et al., 2002; Blasi, 2001). The urokinase receptor also binds with high affinity to α3β1 and α5β1 integrins, and with lower affinity to β2 and β3 integrins, to promote adhesion and spreading of leukocytes and tumor cells on ECM proteins (Blasi et al., 2002; Sidenius & Blasi, 2003; Wei et al., 1996). Urokinase-uPAR binding also regulates cell survival and apoptosis. For example, urokinase activates and releases various growth factors which promote cell survival and proliferation (Hildenbrand et al., 2008). Urokinase-uPAR intracellular signaling also modulates the cell proliferation/apoptosis ratio by regulating cell-matrix interactions and the expression of anti-apoptotic proteins of the Bcl family which protect cells from apoptosis (Alfano, Laccarino, & Stoppelli, 2006; Hildenbrand et al., 2008).
In vivo studies of urokinase and plasmin
Studies of humans and mice with altered expression of components of the plasminogen system confirm critical roles for this system not only in fibrinolysis, but also in wound healing, inflammation, angiogenesis, and fibrotic responses to tissue injury. This system also plays critical roles in promoting tumor cell growth and metastasis [reviewed in (Blasi et al., 2002; Laufs, Schumacher, & Allgayer, 2006; Pillay, Dass, & Choong, 2007; Aguirre-Ghiso, 2007; Montuori, Visconte, Rossi, & Ragno, 2005)].
Fibrinolysis
Human subjects with severe homozygous type I plasminogen deficiency develop ligneous conjunctivitis, a rare and unusual form of chronic pseudomembranous conjunctivitis, and also develop additional pseudo-membranous lesions of other mucous membranes (Mingers, Heimburger, Zeitler, Kreth, & Schuster, 1997). These lesions are caused by massive fibrin depositions within the extravascular space of mucous membranes due to the lack of clearance of these depositions by plasmin. Mice genetically deficient in plasminogen (plasminogen−/− mice) also develop ligneous conjunctivitis (Drew et al., 1998). Plasminogen−/− mice are also predisposed to developing spontaneous severe thrombosis in multiple organ systems due to impaired fibrinolysis, and also suffer retarded growth, reduced fertility, and decreased survival (Ploplis et al., 1995; Bugge, Flick, Daugherty, & Degen, 1995). Mice singly deficient in tPA, urokinase, or uPAR are healthy and have normal life span in the unchallenged state, but tPA−/− mice have impaired clot lysis, and urokinase−/− mice have occasional hepatic fibrin deposits (Carmeliet et al., 1994; Bugge et al., 1996a). However, mice with combined deficiency of tPA and uPA suffer extensive spontaneous fibrin deposition and have impaired growth, reduced fertility, and decreased survival similar to that occurring in plasminogen−/− mice (Carmeliet et al., 1994). Mice deficient in urokinase, tPA and plasminogen die from generalized thrombosis and inflammation (Carmeliet et al., 1994). The inflammation in these mice is secondary to thrombosis and extravascular fibrin deposition (Bugge et al., 1996b; Drew et al., 1998). These data indicate that in mice there is no significant alternative fibrinolytic pathway to tPA and urokinase.
Wound healing
Studies of urokinase−/− mice also demonstrate that urokinase-mediated activation of plasmin plays an important role in healing skin wounds in mice by clearing fibrin (Bugge et al., 1996a; Romer et al., 1996). Urokinase also plays a critical role in the repair of the ischemic myocardium in mice by promoting the migration of fibroblasts into the injured myocardium, activating TGF-β, and promoting angiogenesis by inducing migration of vascular smooth muscle cells and endothelial cells into the injured myocardium (Heymans et al., 1999). In these wound healing models, urokinase does not signal through uPAR since uPAR−/− mice have minimal abnormalities in these model systems (Carmeliet et al., 1997; Levi et al., 2001). Likely, urokinase binds to ECM proteins in the pericellular environment in lieu of uPAR during wound healing.
Leukocyte migration
The role of uPAR in regulating leukocyte migration in vivo is controversial. Some studies of uPAR−/− mice have found no role for uPAR in regulating leukocyte recruitment into various organs (Dewerchin et al., 1996; Cao et al., 2006), whereas other studies report that uPAR promotes PMN and lymphocyte recruitment into inflamed peritoneum and into the lung in murine models of lung inflammation and infection, possibly by uPAR signaling through β2 integrins, leading to decreased clearance of pathogens and impaired host survival (May et al., 1998; Gyetko et al., 2000; Gyetko et al., 2001; Rijneveld et al., 2002).
Fibrosis
Plasminogen activators and their inhibitors regulate lung fibrotic responses to injury. Transgenic mice over-expressing PAI-1 have increased lung fibrosis compared to control mice in the bleomycin-mediated lung fibrosis (Eitzman et al., 1996), whereas PAI−1−/− mice and transgenic mice over-expressing urokinase in the lung in an inducible manner are protected in this model (Eitzman et al., 1996; Sisson et al., 2002). Likely, urokinase protects against lung fibrotic responses to injury by generating plasmin which digests fibrin in the provisional matrix generated during bleomycin-mediated lung injury, and activates pro-MMPs leading to increased removal of collagen and other ECM proteins deposited in the lung.
Neutrophil elastase (NE), Cathepsin G (CG) and Proteinase 3 (PR3)
Structure, expression, and regulation
NE, CG and PR3 are serine proteinases comprised of a single chain glycoprotein with ~200 amino acid residues (Pham, 2006). All three enzymes are all highly cationic with CG being the most and PR3 the least cationic. These proteinases are predominantly expressed by PMNs, but are not synthesized de novo by mature blood PMNs. Instead, they are synthesized at the promyelocyte stage of PMN development in the bone marrow and stored at millimolar concentrations as active enzymes in PMN azurophil granules (Borregaard & Cowland, 1997). A subpopulation of monocytes with a PMN-like pro-inflammatory phenotype (P monocytes) also store preformed NE, CG and PR3 in their primary granules (Kargi, Campbell, & Kuhn, III, 1990; Owen, Campbell, Boukedes, Stockley, & Campbell, 1994). Macrophages do express serine proteinases under physiologic conditions but can be induced to synthesize NE under some pathologic conditions (Dollery et al., 2003).
The major inhibitors of NE, CG, and PR3 are serpins which comprise about 10% of all plasma proteins, and include α1-proteinase inhibitor (α1-PI) and α1-antichymotrypsin (α1-Ach), which are synthesized and secreted by hepatocytes. The universal inhibitor, α2-macroglobulin (α2-M), also inhibits these enzymes (Carrell, 1986). Other inhibitors of these enzymes are secretory leukocyte proteinase inhibitor (SLPI) and elafin which are produced by epithelial cells and found in a variety of glandular secretions such as the upper and lower respiratory tract and synovial fluid (Kramps, Rudolphus, Stolk, Willems, & Dijkman, 1991; Sallenave, Silva, Marsden, & Ryle, 1993).
Membrane binding of serine proteinases
Under some circumstances, NE, CG, and PR3 are freely released from the azurophilic granules of PMNs and the primary granules of P monocytes when cells are activated to degranulate. In contrast to other proteinase-containing granules and vesicles in PMNs which translocate to and fuse with the plasma membrane when PMNs are activated, the azurophilic granules translocate to the PMN plasma membrane but do not fuse with it (Borregaard et al., 1997). Serine proteinases are released from PMNs into the extracellular space in large amounts in vitro only when the cells are exposed to pharmacologic agonists that potently induce degranulation such as cytochalasin B, phorbol esters, and calcium ionophores. Activation of PMNs with physiologically relevant stimuli such as cytokines, chemokines, and bacterial products induces more modest free release of these enzymes [less than 2% of the cellular content in vitro (Owen, Campbell, Boukedes, & Campbell, 1995a; Owen, Campbell, Boukedes, & Campbell, 1997; Campbell, Campbell, & Owen, 2000)]. However, significant free release of serine proteinases from PMNs may occur in vivo during extensive or frustrated phagocytosis (Liszt, Schnittker-Schulze, Stuhlsatz, & Greiling, 1991) or in diseases in which macrophage clearance of apoptotic PMNs is either impaired or inadequate due to excessive influx of PMNs into tissues. This occurs in the airways of COPD patients during acute infective disease exacerbations or in the airways and lungs of patients with cystic fibrosis (Naylor et al., 2007; Vandivier et al., 2002; Matthay & Zimmerman, 2005). In these diseases, PMNs undergo necrosis and PMN proteinases are discharged into the extracellular space where they contribute to tissue destruction.
All three serine proteinases are also expressed on the surface of PMNs and membrane-bound NE, CG, and PR3 are likely to be the most important forms of the enzymes during physiologic and pathologic processes. Unstimulated PMNs express minimal amounts of membrane-bound NE and CG. Activation of PMNs with cytokines, bacterial products, chemoattractants, and fMLP, a synthetic bacterial-like peptide induces up to 20-fold increases in PMN surface expression of NE, CG, and PR3 [(Owen et al., 1995b; Owen et al., 1997; Owen et al., 1995a; Campbell et al., 2000) and (Figure 2)]. Under these conditions, 6-fold more NE and CG binds to the PMN plasma membrane than is freely released by cells.
Figure 2. Neutrophil elastase is expressed on the surface of activated PMNs.

Human PMNs were activated for 30 min. at 37°C with 10−7M fMLP and then fixed and immunostained with rabbit anti-NE (top panel) or non-immune rabbit IgG (bottom panel) followed by a secondary antibody conjugated to a red fluorophore. Cells were examined using Normaski objective (left panel) and confocal miscroscopy (right panel). Note the intense staining for NE on the surface of activated PMNs. Most NE (and CG) bind to HSPG and CSPG on the PMN surface by an active site-independent manner (Campbell et al., 2007). However, a small proportion of NE and CG bind via their active sites to CD11b/CD18 integrins on PMNs to regulate PMN adhesion to extracellular matrix proteins (Cai et al., 1996).
There are several notable differences between PR3 versus NE and CG which may explain, in part, why among these enzymes only PR3 has been implicated in the pathogenesis of Wegener’s granulomatosis (vide infra). First, the subcellular localization of PR3 differs from that of NE and CG. NE and CG are only stored in the azurophil granules of PMNs, but PR3 is also present in the membrane of secretory vesicles which translocate to the plasma membrane much more readily than the azurophil granules (Witko-Sarsat et al., 1999a). Second, PR3 moves also to the plasma membrane during apoptosis in the absence of degranulation (Kantari et al., 2007). Third, in contrast to NE and CG significant PR3 is expressed on the surface of unstimulated PMNs (Witko-Sarsat et al., 1999b).
Biologic roles of membrane-bound serine proteinases
Soluble NE, CG, and PR3 have diverse activities including bacterial killing, and cleaving diverse proteins including ECM proteins, inflammatory mediators, and cell surface receptors, and activating various types of cells (Pham, 2006; Owen & Campbell, 1999). Membrane-bound NE, CG and PR3 on PMNs have similar catalytic activity and efficiency as the soluble forms of the proteinases since membrane-bound NE and PR3 degrade ECM components (Owen et al., 1995b; Campbell et al., 2000), membrane-bound NE and CG activate coagulation proteins on monocyte surfaces (Allen & Tracy, 1995) and membrane-bound CG potently converts the biologically inactive peptide angiotensin I to angiotensin II (Owen & Campbell, 1998) which increases vascular smooth muscle tone and permeability, and stimulates mononuclear cell chemotaxis. Membrane-bound NE and CG are also potent inducers of goblet cell degranulation (Takeyama et al., 1998). However, membrane-bound NE, CG and PR3 have been shown to be resistant to inhibition by physiologic inhibitors including α1-PI and α1-Ach, and membrane-bound CG activates angiotensin I even when cells are bathed in undiluted serum which contains high concentrations of physiologic inhibitors of CG (Owen et al., 1995b; Owen et al., 1998; Campbell et al., 2000; Bangalore & Travis, 1994a). Recently, surface-bound NE on PMNs was shown to be inhibited by α1-PI in bronchoalveolar lavage (BAL) samples from patients with pneumonia and acute lung injury, but PMN surface-bound CG and PR3 were substantially resistant to inhibition by α1-PI (Korkmaz, Attucci, Jourdan, Juliano, & Gauthier, 2005). It is not clear why differences in susceptibility of membrane-bound NE to inhibition by α1-PI have been reported. One possibility is that in the study of Korkmaz et al, much lower concentrations of membrane-bound NE were assayed for their susceptibility to inhibition than in the earlier published studies. It is possible that when low concentrations of membrane-bound NE are tested in vitro, α1-PI binds with much higher affinity to membrane-bound NE than to membrane-bound CG or PR3. Under these conditions, α1-PI –membrane-bound NE interactions are sufficient to produce substantial inhibition of membrane-bound NE. However, during inflammatory reactions in vivo, high concentrations of membrane-bound NE on activated PMNs are likely to be present in tissues which may not be efficiently inhibited by α1-PI present in extracellular fluids leading to NE-mediated tissue injury.
Surface receptors for serine proteinases on PMNs
The first receptor reported for serine proteinases on PMNs was CD11b/CD18 integrin. NE binds via its active site to this integrin which leads to detachment of PMNs from fibrinogen-coated surfaces (Cai & Wright, 1996). However, CD11b/CD18 integrins bind only a small fraction of the total number of NE molecules expressed on the surface of activated PMNs. More recently, we showed that NE and CG bind by an active site-independent but charge-dependent manner to high-capacity, low-affinity binding sites (~107 sites per cell and KD ~ 10−7M) which are the negatively charged sulfate groups in heparan sulfate- and chondroitin sulfate-containing proteoglycans (HSPG and CSPG) in PMN plasma membranes (Campbell & Owen, 2007). Likely, most of the NE and CG molecules expressed on the PMN surface bind by their positively charged external residues to HSPG and CSPG which form a reservoir which sequesters the large amounts of these enzymes released during PMN degranulation. A small proportion of serine proteinases bound to HSPG and CSPG may bind subsequently to CD11b/CD18 integrins to regulate PMN adhesion. Macrophages also express high-capacity, low-affinity binding sites for NE and CG, which are also likely to be HSPG and CSPG (Campbell, 1982). NE and CG released by PMNs bind to these macrophage binding sites and are subsequently internalized by macrophages (Campbell, White, Senior, Rodriguez, & Kuhn, III, 1979). CG also binds to and activates a high affinity fMLP receptor on PMNs and monocytes which stimulates migration of these cells (Sun et al., 2004; Chertov et al., 1997).
The binding sites for PR3 on PMNs are less clear, but PR3 competes with NE and CG for HSPG and CSPG bindings sites on PMNs (Campbell et al., 2007). PR3 also binds to lipid components in PMN membranes (Goldmann, Niles, & Arnaout, 1999) including the phospholipid, scramblase-1 (Kantari et al., 2007). CD177 (also called NB1), a GPI-anchored glycoprotein on PMNs may also serve as a receptor for PR3 (Bauer et al., 2007; von Vietinghoff et al., 2007). Thus, although NE and PR3 are homologs, there are differences in the mechanisms by which they bind to the surface of PMNs which suggests that they may play different roles in regulating acute inflammatory processes.
Biologic roles of membrane-bound NE, CG, and PR3
Based upon their catalytic activities membrane-bound NE, CG, and PR3 are well equipped to contribute to PMN pericellular proteolysis. While there is little evidence to support a role for PMN proteinases in degrading ECM proteins during PMN migration through tissues (Hirche, Atkinson, Bahr, & Belaaouaj, 2004; MacIvor et al., 1999), membrane-bound serine proteinases on PMNs may clear tissue debris and regulate the biologic activities of inflammatory mediators during wound healing and inflammatory responses to injury (Abbott et al., 1998; Pham, 2006). Membrane-bound serine proteinases on PMNs may also contribute directly to lung ECM destruction in COPD and inflammatory arthritis (Shapiro et al., 2003; Adkison, Raptis, Kelley, & Pham, 2002) and mucus hyper-secretion in COPD (Takeyama et al., 1998).
Wegener’s granulomatosis
Membrane-bound PR3 plays a critical role in the pathogenesis of Wegener’s granulomatosis which is a systemic, autoimmune vasculitis affecting the kidneys and the upper respiratory tract. Circulating anti-neutrophil cytoplasmic autoantibodies (cANCA) are detected in ~95% of patients with active Wegener’s granulomatosis (Gross, Csernok, & Flesch, 1993). These autoantibodies are directed against PR3 and play a direct role in vascular injury in this disease (Bosch et al., 1993; Gross, Trabandt, & Csernok, 1998). Circulating PMNs and monocytes from patients with Wegener’s granulomatosis express PR3 on their surface and cANCA bind via their F(ab)2 component to membrane-bound PR3 (Csernok, Schmitt, Ernst, Bainton, & Gross, 1993; Csernok et al., 1990). Ligation of Fcγ receptors on leukocytes by the Fc component of cANCA is a potent stimulus for leukocyte degranulation and activation of the respiratory burst (Kettritz, Jennette, & Falk, 1997; Ralston, Marsh, Lowe, & Wewers, 1997). Proteinases and oxidants released by PMNs activated in this way cause the vascular inflammation and injury characteristic of this syndrome.
Individuals differ in percentage of circulating PMNs that constitutively express membrane-bound PR3. This percentage is genetically determined and is now considered as a risk factor for Wegener’s granulomastosis and other inflammatory diseases. For example, individuals that have a large subset of neutrophils expressing membrane PR3 at baseline are at increased risk for developing vasculitis and rheumatoid arthritis (Witko-Sarsat et al., 1999b; Schreiber et al., 2005; Schreiber, Busjahn, Luft, & Kettritz, 2003; van Rossum, Limburg, & Kallenberg, 2003). The differences in subcellular localization and mobilization of PR3 to the PMN surface compared to that of NE and CG (vide supra) may explain, in part, why PR3 rather than NE or CG plays a critical role in the pathogenesis of Wegener’s granulomastosis.
Type II Transmembrane Serine Proteinases (TTSPs)
TTSPs are a large family of serine proteinases anchored to the surface of diverse cell types by a transmembrane domain [reviewed in (Szabo et al., 2003)]. All members have a short hydrophobic signal anchor located at their amino terminus and an extracellular serine proteinase domain at their carboxy terminus. These domains are separated by a stem region that may contain different additional protein domains. Hespin was the first identified TTSP in 1998, and at least 17 mammalian TTSP have been identified in humans. Evidence is emerging that these enzymes play important roles in embryonic development and in tumor growth and metastasis (Szabo et al., 2003; List, Bugge, & Szabo, 2006). One member of this family, matriptase is expressed by leukocytes.
Membrane-type serine protease-1 (MT1-SP or matriptase)
Expression and localization
This enzyme was first described in 1993 and was found to be expressed predominantly by normal and malignant epithelial cells (Oberst et al., 2001). More recently, matriptase has been shown to be expressed by monocytes, B lymphocytes, and endothelial cells (Kilpatrick et al., 2006; Qiu, Owen, Gray, Bass, & Ellis, 2007b; Seitz et al., 2007). Matriptase is a 80–90 kDa protein which is synthesized as an inactive single chain zymogen. Matriptase activation is a complex process involving its non catalytic domains and its main inhibitor, Kunitz type inhibitor hepatocytes growth factor activator inhibitor (List et al., 2005). The zymogen is cleaved within its activation motif in the serine proteinase domain generating a two chain enzyme from the single chain zymogen.
Biologic functions
In epithelial cells, matriptase plays a critical role in epithelial differentiation, in part, by processing profilaggrin (List et al., 2006). Other substrates reported for matriptase are pro-urokinase, hepatocytes growth factor, and PAR-2 (Takeuchi et al., 2000; Lee, Dickson, & Lin, 2000). Matriptase is expressed at high levels on the surface of freshly isolated monocytes and B lymphocytes (Kilpatrick et al., 2006). Activation of these cells in vitro leads rapidly to loss of expression of matriptase from their surfaces. Surface-bound matriptase is co-localized with uPAR on the surface of monocytic cell lines where it potently activates pro-urokinase bound to uPAR, albeit 100-fold less efficiently than plasmin. Thus, monocyte-derived matriptase may contribute to the generation of plasmin on the surface of monocytes. Matriptase is also upregulated on the surface of monocytes adherent to endothelial cells in atherosclerotic lesions, and monocyte-derived matriptase may stimulate inflammation in these lesions by activating endothelial PAR-2 and increasing endothelial production of pro-inflammatory mediators (Seitz et al., 2007). However, it is not clear whether matriptase expressed by B lymphocytes function plays any role in physiologic or pathologic processes.
Metalloproteinases
Members of the matrix metalloproteinase and ADAM subfamilies of metalloproteinases are expressed on the surface of leukocytes.
Matrix metalloproteinases (MMPs)
Structure, expression, and activation of MMPs
This class of more than 20 proteinases in humans depend on Zn2+ ions for their activity (Puente, Sanchez, Overall, & Lopez-Otin, 2003; Parks et al., 2004). MMPs are 40–50% identical at the amino acid level and have common structural domains (Fig. 3). The domains include: 1) a pro-enzyme domain that maintains the enzyme in its latent form; 2) the catalytic domain containing the zinc binding consensus sequence HEXXHXXGXXH which binds the zinc molecule essential for catalysis; and 3) a carboxy-terminal hemopexin domain which binds MMPs to their substrates, cell membranes, and tissue inhibitors of MMPs (TIMPs). The gelatinases (MMP-2 and MMP-9) have additional domains (Fig. 3). The six membrane-type MMPs (MT-MMP) also have either a membrane-spanning domain or a GPI membrane anchor.
Figure 3. Domain structure of MMPs.
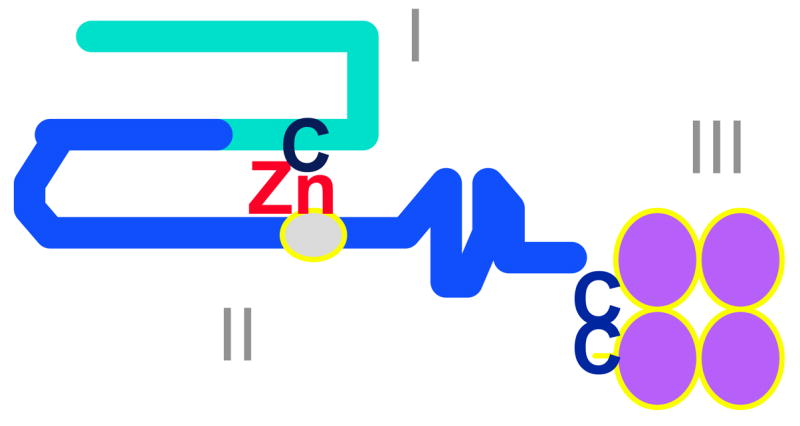
MMPs share common features including a pro-enzyme domain (I), a catalytic domain with the active site zinc bound to the HEXXHXXGXXH consensus sequence (II), and a C-terminal domain (III) which may regulate MMPs binding to their substrates and to tissue inhibitors of metalloproteinases. The catalytic zinc (Zn) atom interacts with a conserved cysteine (C) in domain I to maintain the pro-enzyme in an inactive conformation. The gelatinases have an additional domain similar to the fibronectin type II domain, which interrupts the catalytic domain. MMP-9 also has a region with homology to type V collagen (not shown).
MMPs are synthesized and secreted from cells as latent pro-enzymes (pro-MMPs). Activation of pro-MMPs can be achieved in vitro by various proteinases and reactive oxygen species which disrupt the interaction between the active site zinc atom in the catalytic domain and a conserved cysteine within the pro-enzyme domain (Murphy et al., 1999). Exposure of the zinc results in auto-cleavage of the pro-domain yielding the mature active enzyme. The mechanism of extracellular activation of pro-MMPs in vivo has not been determined, but reactive oxygen species inactivate rather than activate MMPs in the lung in vivo (Fu, Kassim, Parks, & Heinecke, 2003).
Regulation of MMPs
In most cells including leukocytes, the MMP genes are tightly regulated at the transcriptional level and their expression is induced during tissue remodeling, wound healing, inflammation and other processes by a variety of growth factors, cytokines, chemokines, bacterial products, and surfactant proteins (Campbell, Cury, Lazarus, & Welgus, 1987; Busiek, Baraji, Nehring, Parks, & Welgus, 1995; Cury, Campbell, Lazarus, Albin, & Welgus, 1988; Wahl & Corcoran, 1993; Trask et al., 2001). In mononuclear phagocytes, MMPs are synthesized and then rapidly secreted by cells rather than stored. PMN-derived MMP-8 and MMP-9 are notable exceptions to this rule, since they are stored in PMN secondary and tertiary granules, respectively, and rapidly released from these granules when PMNs are activated with degranulating agonists (Dewald, Bretz, & Baggiolini, 1982; Chatham, Heck, & Blackburn, 1992).
Inhibitors of MMPs
The activity of MMPs is controlled by the four members of the tissue inhibitors of metalloproteinase (TIMP) family and by α2-macroglobulin (Murphy et al., 2003). In addition, a transmembrane inhibitor, RECK, which inhibits MMPs-2 and -9 and MT1-MMP, has recently been shown to be essential for restricting MMP activity during embryonic development (Oh et al., 2001).
Activities of MMPs
MMPs can degrade all of the components of the ECM in vitro. Based on this, MMPs can be divided into groups (Shapiro, 1998) including: 1) the interstitial collagenases (MMPs-1 [human only], -8, -13) which cleave native triple helical interstitial collagens; 2) the gelatinases (MMP-2 and MMP-9) which degrade gelatins (denatured collagens), elastin and basement membrane proteins; 3) the stromelysins (MMP-3, MMP-10, and MMP-11) which have a broad spectrum of activity against ECM proteins; 4) the elastolytic MMP-7 and MMP-12 which also have a broad spectrum of susceptible ECM substrates including basement membrane components; and 5) the MT-MMP (see below).
The main function of MMPs in vivo was initially thought to be in ECM remodeling. Some studies support a role for MMPs in ECM degradation in vivo. For example, MMP-12 degrades lung elastin in cigarette smoke-exposed mice leading to pulmonary emphysema (Hautamaki, Kobayashi, Senior, & Shapiro, 1997). The lung elastin fragments generated by MMP-12 are chemotactic for blood monocytes and thereby amplify chronic lung inflammation and ECM destruction in the lungs of smoke-exposed mice (Houghton et al., 2006). Many MMPs are expressed by tumor cells, and tumor cell MMP-mediated ECM degradation plays a critical role in promoting tumor growth and metastasis in vivo [reviewed in (Stamenkovic, 2000)]. However, there is increasing evidence that MMPs play important roles in regulating inflammation, host defense, and angiogenesis by cleaving diverse molecules such as cytokines, chemokines, clotting factors, regulators of angiogenesis, defensins, to either increase or decrease their biologic activities. For example, MMP-7 sheds KC from syndecans on the surface of lung epithelial cells thereby promoting PMN influx into the lung during bleomycin-mediated ALI in mice (Li, Park, Wilson, & Parks, 2002), MMP-8 activates a PMN chemokine in murine skin inflammatory reactions (Balbin et al., 2003), and MMP-12 sheds and activates pro-TNF-α from macrophage surfaces in murine lung acutely exposed to cigarette smoke thereby increasing neutrophilic lung inflammation (Churg et al., 2003). Several MMPs inactivate pro-inflammatory mediators to dampen inflammation in tissues (McQuibban et al., 2002). MMP-7 cleaves and activates defensins to promote host defense in the murine gut (Wilson et al., 1999). MMP-12 cleaves plasminogen to generate angiostatin which inhibits angiogenesis and tumor growth (Cornelius et al., 1998; Balbin et al., 2003). Some MMPs activate PARs leading to activation of leukocytes, platelets, and tumor cells (Goerge et al., 2006; Pei, 2005; Chung et al., 2004).
Membrane binding of MMPs lacking transmembrane domains or GPI anchors
MMPs-1, 2, -7, -8, -9, and -13 lack a transmembrane domain and have been thought to function exclusively as soluble proteinases after their secretion from cells. However, these MMPs have recently been shown to bind to various molecules on the surfaces of leukocytes (vide infra) and other cells types (summarized in Table 1). Examples of MMPs that bind to cells other than leukocytes are active MMP-1 which binds to α2β1 integrin on keratinocytes during wound healing (Dumin et al., 2001), and active MMP-2 which binds to αvβ3 integrin on the surface of melanoma cells and angiogenic blood vessels and participates in pericellular proteolysis (Brooks et al., 1996). Pro- and active forms of MMP-2, -7, -9 and -13 are bound to HSPGs in the plasma membrane of rat uterine epithelial cells [Table 1, (Yu & Woessner, Jr., 2000)] where they may maintain the patency of the glandular lumen, promote host defense against bacteria by activating defensins, and activate or degrade cell surface proteins that regulate epithelial cell function. MMP-7 binds to CD44 on the surface of tumor cell lines, postpartum uterine and lactating mammary gland epithelia, and uterine smooth muscle cells where it forms a complex with and activates the HB-EGF precursor. The active HB-EGF thus generated, engages and activates its receptor, ErbB4, promoting epithelial cell survival (Yu, Woessner, Jr., McNeish, & Stamenkovic, 2002). CD44 is a docking molecule for active MMP-2 and -9 and on the surface of breast tumor cells, melanoma cells, and normal keratinocytes (Yu & Stamenkovic, 1999; Yu & Stamenkovic, 2000). MMP-9 bound to CD44 on tumor surfaces degrades collagen IV in the pericellular environment and promotes tumor cell invasion (Yu et al., 1999). MMP-2 and -9 bound to CD44 may also promote tissue remodeling by tumor cells by cleaving and activating latent TGF-β (Yu et al., 2000).
Binding of MMPs to leukocyte integrins
PMN-derived MMP-8 and MMP-9 are rapidly secreted by degranulating PMNs. Until recently, they have been thought to function exclusively as soluble enzymes. However, PMNs also express MMP-8 and -9 on their surface. Unstimulated cells have minimal cell-associated MMP-8 or -9, but activation of PMNs with cytokines, chemokines, and bacterial products leads rapidly to 10-fold increases in surface expression of these MMP due to translocation of the MMP-containing granules the PMN surface (Owen, Hu, Lopez-Otin, & Shapiro, 2004; Owen, Hu, Barrick, & Shapiro, 2003). Latent and active forms of these MMPs have been detected on the PMN surface, and active forms of membrane-bound MMP-8 and -9 on PMNs degrade ECM proteins as efficiently as the soluble forms of the proteinases (Owen et al., 2004; Owen et al., 2003). However, membrane-bound MMPs are resistant to inhibition by TIMPs. The pro-forms of these MMPs bind, at least in part, to a β2 integrin, CD11b/CD18 (Mac-1) on the PMN surface (Stefanidakis, Ruohtula, Borregaard, Gahmberg, & Koivunen, 2004). The pro-MMPs bind via negatively charged residues in their catalytic domain to the I domain of the CD11b chain on PMNs (Stefanidakis, Bjorklund, Ihanus, Gahmberg, & Koivunen, 2003). However, the mechanism by which MMP-8 and -9 are activated and subsequently retained on the PMN surface is not clear.
Biologic roles of membrane-bound MMP-8 and -9 on PMNs
Membrane-bound MMP-8 and -9 play no direct role in PMN migration through tissues since PMNs from MMP-8−/− and MMP-9−/− mice are able to migrate in vitro and into inflamed tissues in vivo (Betsuyaku, Shipley, Liu, & Senior, 1999; Owen et al., 2004). Surface-bound MMP-8 and -9 on activated PMNs may locally degrade the ECM, and remove tissue debris during wound healing or contribute to resolution of inflammatory responses since they cleave pro-inflammatory mediators in vitro, and MMP-8−/− mice have increased neutrophilic pulmonary inflammation in murine models of asthma and acute lung injury (Gueders et al., 2005; Owen et al., 2004).
Integral membrane MMPs (MT-MMPs)
Structure, activation, expression, and regulation
There are six members of this subfamily of metalloproteinases (Table 1). They share 30–50% sequence homology, the multi-domain structure typical of MMPs, and are expressed on cell surfaces by either a transmembrane domain spanning hydrophobic region or a GPI anchor (Itoh et al., 1999; Kang et al., 2001). Like MMPs, MT-MMPs are synthesized as inactive pro-enzymes, but unlike most MMPs, MT-MMPs are activated by furin-mediated cleavage on the pro-domain within the trans-Golgi network before they are transported to cell surfaces (Sato, Kinoshita, Takino, Nakayama, & Seiki, 1996; Pei, 1999; Imai et al., 1996).
The MT-MMP family is expressed by different cell types (Lehti, Lohi, Valtanen, & Keski-Oja, 1998) including leukocytes (Table 1). MT1-MMP is produced by macrophages, monocytes, and dendritic cells (Stawowy et al., 2005; Yang et al., 2006), MT4-MMP is expressed by eosinophils (Gauthier et al., 2003) and MT6-MMP is expressed only by PMNs (Kang et al., 2001). In most cells including mononuclear phagocytes, MT-MMP expression is generally upregulated at the steady state mRNA level by various cytokines and growth factors (Lohi, Lehti, Westermarck, Kahari, & Keski-Oja, 1996; Origuchi et al., 2000; Migita et al., 1996), or when cell adhere to ECM proteins (Ailenberg & Silverman, 1996). However, PMNs store MT6-MMP as a preformed proteinase within their gelatinase and secretory vesicles from where the enzyme translocates to the cell surface when PMNs are activated with degranulating agonists (Kang et al., 2001).
Inhibition
MT-MMPs vary in their susceptibility to inhibition by TIMPs. For example, TIMP-2, -3, and -4 inhibit MT1-MMP and MT2-MMP but TIMP-1 does not (D’Ortho et al., 1998; Hernandez-Barrantes, Shimura, Soloway, Sang, & Fridman, 2001; Butler, Will, Atkinson, & Murphy, 1997a). MT3-MMP is inhibited by TIMP-2 and -3 (Zhao et al., 2004). MT4-MMP is inhibited efficiently by TIMP-1 (Kolkenbrock, Essers, Ulbrich, & Will, 1999) and MT6-MMP is inhibited by TIMP-1 and -2 (Alon et al., 1994). TIMP-3 is sequestered at cell surfaces by binding to proteoglycans, and this may explain, in part, its greater activity against MT-MMP compared to other TIMPs which function as soluble inhibitors (Murphy et al., 2003). MT-MMP expression can also be controlled by ectodomain shedding MT3-MMP (Zhao et al., 2004).
Biologic roles
MT-MMPs contribute to ECM remodeling and regulate inflammation, angiogenesis, cell migration, and tumor invasiveness and metastasis. MT1-MMP, MT2-MMP, and MT3-MMP play critical roles in degrading interstitial collagens and denatured collagens. MT1-MMP deficiency in mice causes craniofacial dysmorphism, arthritis, osteopenia, dwarfism, and fibrosis of soft tissues due to loss of MT1-MMP-mediated collagenase activity which is essential for modeling of skeletal and extra-skeletal connective tissues (Holmbeck et al., 1999). By degrading interstitial collagen and other ECM proteins, MT1-MMP, MT2-MMP, and MT3-MMP independently confer tumor cells with the ability to degrade the basement membrane scaffolding which promotes transmigration of tumor cells during metastasis (Hotary, Allen, Punturieri, Yana, & Weiss, 2000). MT-MMPs also degrade basement membrane components and cartilage proteoglycans (D’Ortho et al., 1998; Ohuchi et al., 1997; D’Ortho et al., 1997; Kang et al., 2001) and degrade ECM proteins indirectly by activating pro-MMPs on cell surfaces (vide infra). MT-MMP may also regulate inflammation by cleaving and activating pro-tumor necrosis factor-α (TNF-α), and by degrading proteinase inhibitors (D’Ortho et al., 1997; English et al., 2000; Kang et al., 2001; Maquoi et al., 2000).
Activation of pro-MMPs on cell surfaces
MT-MMPs play critical roles in activating MMPs on cell surfaces to amplify pericellular proteolytic events. This was first reported for MT1-MMP activating pro-MMP-2 on tumor cells, fibroblasts, and endothelial cells (Strongin et al., 1995; Butler, Will, Atkinson, & Murphy, 1997b; Sato et al., 1996; Cao, Rehemtulla, Bahou, & Zucker, 1996; Cao, Sato, Takino, & Seiki, 1995). However, all members of the MT-MMP subfamily have now been shown to activate pro-MMP-2 on cell surfaces (Zhao et al., 2004; Wang, Johnson, Ye, & Dyer, 1999; Wang, Yi, Lei, & Pei, 1999; Butler et al., 1997a; Nie & Pei, 2003), including MT1-MMP on the surface of macrophages (Stawowy et al., 2005). Activation of pro-MMP2 involves the formation of a ternary complex of pro-MMP-2, TIMP-2 and MT1-MMP on cell surfaces (Fig. 4). The activation of pro-MMP-2 is a two step procedure in which pro-MMP-2 binds via its COOH terminal domain to the COOH terminal domain of TIMP-2 which itself binds via its NH2 terminal inhibitory domain to the active site of MT1-MMP. Adjacent TIMP-free MT1-MMP then cleaves the cell-associated pro-MMP-2 generating an intermediate form. This initial cleavage destabilizes the MMP-2 pro-domain leading to auto-proteolytic cleavage in an MMP-2-dependent-manner. It is also noteworthy that active MMP-2 also activates pro-MMP-13, thus MT-MMP activation of pro-MMP-2 can also lead to MMP-13 activation on cell surfaces. Many tumors express high levels of pro-MMP-2 and MT-MMP, and activation of pro-MMP-2 on the tumor surface permits invadopodia to locally degrade the ECM permitting tumor cell invasion and metastasis (Chen, 1996).
Figure 4. MT1-MMP activates pro-MMP-2 on cell surfaces.

Pro-MMP-2 and TIMP-2 form a ternary complex with MT1-MMP (or other MT-MMP family members) on the surface of fibroblasts, tumor cells, macrophages, and other cell types. TIMP-2 binds via its NH2 terminal inhibitory domain to MT-1-MMP. The COOH terminal domain of pro-MMP-2 binds to the COOH terminal domain of TIMP-2. Pro-MMP-2 is activated by adjacent free MT1-MMP which cleaves the NH2-terminal pro-domain of MMP-2, generating active MMP-2 anchored to the cell surface. Homotypic interactions between two adjacent MT1-MMP molecules facilitate pro-MMP-2 activation.
ADAMs
ADAMs are a family of at least 35 members of type I transmembrane proteins so called because they have a disintegrin and a metalloproteinase domain (Seals & Courtneidge, 2003). They belong to the adamalysin subfamily of metalloproteinases along with ADAM-TS enzymes. ADAM-TS proteinases are structurally similar to ADAMs, but differ from ADAMs in that they function as soluble proteinases and have one or more thrombospondin domains (Stone, Kroeger, & Sang, 1999; Primakoff & Myles, 2000). The first ADAMs identified in the early 1990s were the two subunits of the heterodimeric sperm protein, fertilin (ADAM-1 and -2), which induce sperm-oocyte fusion by binding to α6β1 integrin on oocytes (Wolfsberg et al., 1993; Myles, Kimmel, Blobel, White, & Primakoff, 1994). Interest in the ADAM family in the biomedical community soared in 1997 when metalloproteinase inhibitors were shown to prevent LPS-induced death by blocking TNF-α release from the surface of macrophages in experimental animals (Black et al., 1997). Characterization and cloning of the enzyme responsible for this activity led to the discovery of ADAM-17. Since then, other ADAMs have been shown to participate in diverse physiologic and pathologic processes.
Structure, expression, and regulation
Like the MMPs, ADAMs have a multi-domain structure (Fig. 5) including: 1) a pro-domain which maintains the metalloproteinase domain in a latent form; 2) a metalloproteinase catalytic domain; 3) a disintegrin domain which binds integrins; 4) a cysteine-rich region which may contain an epithelial growth factor (EGF)-like domain; 5) a transmembrane domain which anchors ADAMs to cell membranes; and 6) a carboxy-terminal cytoplasmic tail ( Seals & Courtneidge, 2003; Primakoff et al., 2000; Yamamoto et al., 1999). Although all ADAMs have a metalloproteinase domain, only ~50% carry the active sites Zn2+ atom and are predicted to be catalytically active. ADAMs are synthesized as latent enzymes and latency is maintained by an interaction between a conserved cysteine residue in the pro-domain and the active site zinc. Many of the ADAMs contain consensus sequences (RXXR) for furin and other pro-protein convertases and are activated by furin-mediated cleavage in the trans-Golgi before ADAMs are transported to cell surfaces (Yamamoto et al., 1999; Loechel, Overgaard, Oxvig, Albrechtsen, & Wewer, 1999; Roghani et al., 1999; Lum, Reid, & Blobel, 1998).
Figure 5. Structure and function of the ADAMs family members.

ADAMs have a multi domain structure including a pro-domain which maintains the metalloproteinase domain in a latent form by a cysteine residue in the pro-domain coordinating with the active site zinc atom in the metalloproteinase domain. Many ADAMs are activated by furin-mediated cleavage of the pro-domain in the trans-Golgi network. All ADAMs have a metalloproteinase (MP) domain, but only 50% have the active site zinc atom and are predicted to be catalytically active. ADAMs with an active MP domain can shed cytokines, growth factors, apoptosis ligands and receptors for these molecules to regulate many cellular processes. The disintegrin domain binds to integrins to increase or decrease cell adhesion and migration. The cysteine-rich region may contain an epithelial growth factor (EGF)-like domain which plays roles in cell adhesion and cell-cell fusion. The transmembrane domain anchors ADAMs to cell membranes. The COOH terminal cytoplasmic tail can regulate the sheddase activities of ADAMs and bind intracellular proteins and may play roles in intracellular signaling.
The expression of ADAMs varies widely in mammalian tissues. ADAMs known to be expressed by leukocytes include ADAM-8, -10, -15, -17, and -28 (Gomez-Gaviro et al., 2007; Armstrong, Godinho, Uppington, Whittington, & Millar, 2006; Li, Brazzell, Herrera, & Walcheck, 2006; Lum et al., 1998; Bridges et al., 2002). ADAM expression is generally regulated at the transcriptional level by various mediators. For example, ADAM-10 and -17, are up-regulated in many cell types in vitro by various agonists including phorbol esters, cytokines, chemokines, and growth factors (Walcheck et al., 2006; Bzowska, Jura, Lassak, Black, & Bereta, 2004; Bandyopadhyay et al., 2006) which generally regulate ADAM expression at the steady state mRNA level (Bzowska et al., 2004; Fujita et al., 2006). However, ADAM-8 is stored as a preformed proteinase in PMN specific and gelatinase granules and translocates rapidly to the surface when PMNs are activated with phorbol esters (Gomez-Gaviro et al., 2007).
Inhibitors
The activity of the metalloproteinase domain can be regulated by TIMPs, but ADAMs vary widely in their susceptibility to inhibition by TIMPs. For example, ADAM-17 is inhibited by TIMP-3, but not by TIMP-1 or -2 (Amour et al., 1998; Amour et al., 1999). ADAM-10 is inhibited by TIMP-1 and -3, but not by TIMP-2 and -4 (Amour et al., 2000). ADAM -8 and -9 are not inhibited by TIMP-1, -2 or -3 (Amour et al., 2002).
Biologic activities
The major functions of ADAMs are linked to their domain structure and include role for: 1) the metalloproteinase domain in shedding cell surface proteins; 2) the disintegrin domain in regulating cell adhesion and migration; 3) the cysteine-rich and disintegrin domains in promoting cell fusion; and 4) the cytoplasmic tail in intracellular signaling events.
Sheddase activities
ADAMs have been reported to cleave only a limited number of ECM proteins (Millichip, Dallas, Wu, Dale, & McKie, 1998) and likely play little role in ECM turnover. The critical function of the metalloproteinase domain is the shedding of a wide variety of transmembrane proteins from cell surfaces by juxtamembrane cleavage. The best known example is that of ADAM-17 (TNF-α convertase or TACE). TNF-α is expressed as an inactive 26 kDa protein on the surface of macrophages, PMNs, and other cells. Pro-TNF-α is cleaved by juxtacrine ADAM-17 on the cell surface releasing soluble, active 17 kDa TNF-α (Black et al., 1997). Together, ADAMs cleave a wide variety of membrane-associated proteins, but they lack unique consensus sequences and there is considerable overlap in substrates. Membrane-bound proteins cleaved by ADAMs include: 1) other cytokines and their receptors such as TRANCE, fractalkine, CXCL-16, CD40 ligand, TNF receptors, and IL-6 receptor (Lum et al., 1999; Garton et al., 2001; Gough et al., 2004; Amour et al., 2000; Althoff, Reddy, Voltz, Rose-John, & Mullberg, 2000); 2) epidermal growth factor (EGR) ligands and receptors such as TGF-α, EGF, HB-EGF, amphiregulin, betacellulin, amphicellulin, and Erb4/HER (Sahin et al., 2004; Rio, Buxbaum, Peschon, & Corfas, 2000); 3) adhesion molecules such as VCAM-1, L-selectin, N-cadherin, and CD44 (Garton et al., 2003; Walcheck, Alexander, St Hill, & Matala, 2003; Reiss et al., 2005; Vachon et al., 2002); 4) other receptors such as CD23, Notch, and ErbB4/HER (Fourie, Coles, Moreno, & Karlsson, 2003; Brou et al., 2000; Hartmann et al., 2002). As a result of these sheddase activities, ADAMs play roles in development and in regulating inflammation, cell adhesion, cell growth, proliferation, and survival.
The activity of ADAM sheddases can be regulated by non-transcriptional mechanisms. For example, G-protein coupled receptor agonists increase ADAM-17-mediated shedding of EGFR ligands by activating EGFR/MAP/ERK pathway (Schafer, Gschwind, & Ullrich, 2004). The cytoplasmic tail of ADAM-17 is phosphorylated by various intracellular kinases including Ras/Raf/MEK kinase leading to activation of ADAM-17 sheddase activities (Fan, Turck, & Derynck, 2003). Phorbol esters also regulate the activity of ADAM-17, but this does not involve the cytoplasmic tail of ADAM-17 since phorbol ester also activates an ADAM-17 mutant lacking the cytoplasmic tail (Doedens, Mahimkar, & Black, 2003). Cigarette smoke increases ADAM-17 shedding of EGFR ligands to regulate airway mucin production but the mechanism involved is unknown (Basbaum, Li, Gensch, Gallup, & Lemjabbar, 2002; Lemjabbar et al., 2003).
Disintegrin domain in regulating cell adhesion and migration
This domain enables ADAMs to regulate cell adhesion and migration. This domain is structurally similar to the snake venom disintegrins which have an RGD-binding motif which binds integrin αIIbβ3 to impair platelet aggregation leading to severe hemorrhage. However, mammalian ADAMs lack RGD sequences with the notable exception of human ADAM-15. ADAM-15 has an RGD-binding site through which it binds to αvβ3 and α5β1 integrins (Nath et al., 1999). However, ADAM-15 also contains Rx6DEVF sequences which mediate RGD-independent binding of many ADAMs to various integrins. By binding and regulating integrin activity ADAMs can either promote or inhibit cell adhesion, migration, and cell fusion. In addition to ADAM-1 and -2 binding to various integrins to promote sperm-oocyte fusion (Evans, 2001), ADAM-15 promotes cell-cell adhesion of fibroblasts to inhibit wound healing (Herren et al., 2001) and inhibits RGD-dependent αvβ3 adhesion and migration of ovarian tumor cells (Beck et al., 2005). Endothelial ADAM-15 binds to activated αIIbβ3 integrin on platelets to promote platelet adhesion and aggregation and thrombus formation (Langer, May, Bultmann, & Gawaz, 2005). ADAM-9 promotes fibroblast adhesion and motility by its disintegrin domain binding to α6β1 integrin (Nath et al., 2000). However, the roles of ADAMs in regulating leukocyte adhesion and migration have not been studied.
Cysteine-rich domain in regulating cell fusion and cell adhesion
This domain has been less well studied than the other ADAM domains, but has been implicated in cell fusion and adhesion. The cysteine rich domain of some ADAMs contains a sequence of amino acids resembling viral fusion peptides, and this domain plays roles in ADAM-12-mediated myoblast fusion and in ADAM-1- and ADAM-2-mediated sperm-oocyte fusion (Brzoska, Bello, Darribere, & Moraczewski, 2006; Blobel et al., 1992; Yagami-Hiromasa et al., 1995). The cysteine-rich domain of ADAM-2 also regulates cell adhesion by binding to the sulfate groups in the HSPG, syndecan-4, to promote cell adhesion which requires interactions between the cysteine-rich domain and activated β1 integrins (Iba et al., 2000). The disintegrin-cysteine rich domain of ADAM-9 binds to β1 integrins to promote keratinocyte adhesion and migration (Zigrino et al., 2007). However, the roles of this domain in leukocyte function have not been elucidated.
The cytoplasmic domain
This domain can regulate the sheddase activities of ADAMs or regulate intracellular signaling by binding to intracellular proteins or by being phosphorylated by intracellular kinases. For example, phosphorylation of the cytoplasmic tail of ADAM-17 by Ras/Raf/MEK kinase increases ADAM-17- mediated sheddase activities (Fan et al., 2003). The cytoplasmic domains of several ADAMs including ADAM-9, -12, and -15 have Src homology 3 (SH3)-binding motifs which binds Src kinases and other intracellular proteins (Nelson, Schlondorff, & Blobel, 1999; Howard, Nelson, Maciewicz, & Blobel, 1999). The cytoplasmic tails of ADAM-17 and -9 bind mitotic arrest deficient 2 (MAD2) and MAD2-β, respectively (Nelson et al., 1999). However, it is not clear whether the cytoplasmic tails of ADAM15 tranduce intracellular signaling events by binding intracellular proteins in leukocytes or other cells.
Biologic activities of ADAMs
Studies of mice or murine cells genetically deficient in ADAMs have confirmed roles for ADAMs in diverse physiologic processes (Table 2) including critical roles in morphogenesis for ADAM-10, -19 and -17. ADAM-10 is critical in early neural development by proteolytically activating Notch, which by binding to its receptors, promotes formation of different neural cell types in a spatially and temporally regulated manner (Hartmann et al., 2002). ADAM-17 deficient embryos have abnormal epithelial differentiation and growth in developing heart and lung which is likely due to defective EFGR ligand shedding (Peschon et al., 1998).
Table 2.
Phenotypes of mice genetically deficient in ADAMs
| Genotype | Phenotype |
|---|---|
| ADAM8−/− | Viable and fertile with no phenotype in the unchallenged state(Kelly et al., 2005). |
| ADAM9−/− | Viable and fertile with no phenotype in the unchallenged state(Weskamp et al., 2002). |
| ADAM10−/− | Embryonic lethal (E9.5). Defects in the heart and central nervous system development and vasculogenesis (Hartmann et Al, 2002). |
| ADAM12−/− | 30% embryonic lethal. Surviving mice have normal fertility. Minor brown fat and neck muscle hypertrophy (Kurisaki et al., 2003). |
| ADAM15−/− | Viable, fertile with and no phenotype in the unchallenged state. Reduced neovascularization in a murine model of retinopathy of prematurity (Horiuchi et al., 2003) |
| ADAM17−/− | Perinatal lethal. Epithelial dysplasia similar to that in TGF-α deficient mice with defective heart and lung development, and defective EGFR ligand shedding (Peschon et al., 1998; Zhao et Al., 2001; Shi et al., 2003). |
| ADAM19−/− | 80% post-natal lethality 1–3 days after birth with defective heart development (Zhou et al., 2004). |
Legands for table 2. TGF-α, transforming growth factor-α; EGFR, epidermal growth factor receptor.
ADAM expression is also dysregulated in various organ systems during pathologic processes. Ischemia up-regulates ADAM-17 expression in rat forebrain (Hurtado et al., 2001). ADAM-15 and -17 expression are enhanced on macrophages and fibroblasts in rheumatoid synovium (Bohm, Aigner, Blobel, Kalden, & Burkhardt, 2001; Ohta et al., 2001), and ADAM-8 is upregulated in neurons, astrocytes, and microglia in neurodegenerative processes in the murine brain (Schlomann, Rathke-Hartlieb, Yamamoto, Jockusch, & Bartsch, 2000). ADAM-33 has been identified as the first asthma gene in Caucasians (Van Eerdewegh et al., 2002). ADAM-8 expression is upregulated in the airway structural cells and inflammatory infiltrates in human asthma subjects (Foley et al., 2007). However, the mechanisms by which ADAM-33 and -8 contribute to asthma pathogenesis are not clear (Shapiro & Owen, 2002). Several ADAMs are upregulated in tumors and hematologic malignancies including ADAM-10, -12, and -15 (Kveiborg et al., 2005; Liu et al., 2006; Ortiz, Karkkainen, & Huovila, 2004; Kenny & Bissell, 2007; Wu, Croucher, & McKie, 1997). Several ADAM deficient mice have no abnormalities in the unchallenged state (Table 2), but it is likely that future studies of these mice in murine models of disease will uncover critical roles for these in pathologic processes in various organs.
Cysteine proteinases
Cysteine proteinases have a two-domain globular structure, a similar size (about 23–27 kDa), and an active-site cysteine which is critical for catalytic activity (Turk, Turk, & Turk, 1997). There are four cysteine proteinases present in human lysosomal granules: cathepsins B, H, L, and S which are members of the papain superfamily of cysteine proteinases. They are synthesized as pro-enzymes which are processed by limited proteolysis to the active forms in lysosomes. The main naturally-occurring inhibitors of cysteine proteinases in tissues are the cystatin superfamily. The kininogens and α2-macroglobulin are the major inhibitors of cysteine proteinases in plasma (Henskens et al., 1996).
The main role of lysosomal cysteine proteinases in to degrade intracellular proteins under the acidic conditions (pH 5–6.5) of the lysosomes (Henskens, Veerman, & Nieuw Amerongen, 1996). However, these enzymes have also been implicated in extracellular proteolytic events at or near the surfaces of leukocytes, leukocyte-derived cells, and tumor cells. For example, osteoclasts do not store cysteine proteinases in large quantities within their lysosome. Rather, the enzymes are targeted to the osteoclast surface/bone interface where strong adhesive attachments and proton secretion by osteoclasts creates an environment that favors the resorption of bone matrix proteins in the pericellular environment (Baron, Neff, Louvard, & Courtoy, 1985; Blair, Teitelbaum, Ghiselli, & Gluck, 1989).
Cysteine proteinases including cathepsins S, L, and B are also involved in the degradation of insoluble extracellular elastin by lung macrophages at neutral pH in vitro (Chapman, Jr., Munger, & Shi, 1994; Mason, Johnson, Barrett, & Chapman, Jr., 1986; Shi, Munger, Meara, Rich, & Chapman, 1992). Among these lysosomal cysteine proteinases, only cathepsin B has been confirmed to be expressed on cell surfaces.
Cathepsin B
This lysosomal cysteine proteinase was first localized on the surface of alveolar macrophages in 1984 (Chapman, Jr. & Stone, 1984). About 50% of the alveolar macrophage’s content of cathepsin B was reported to be expressed on the cell surface where it contributes to extracellular elastin degradation. However, the binding site(s) for cathepsin B on macrophages have not yet been identified.
More recently, cathepsin B has been shown to be expressed on the surface of various tumor cells (Cavallo-Medved, Mai, Dosescu, Sameni, & Sloane, 2005; Cavallo-Medved et al., 2003; Calkins, Sameni, Koblinski, Sloane, & Moin, 1998). The light chain of annexin II tetramer, p11, has been reported to be the receptor for this enzyme on some tumor cells (Mai, Waisman, & Sloane, 2000). Surface-bound cathepsin B on tumor cells is active and can degrade extracellular proteins including type IV collagen and activates pro-urokinase, pro-MMPs, and activate growth factors (Sameni, Moin, & Sloane, 2000; Kobayashi et al., 1993b; Guo, Mathieu, Linebaugh, Sloane, & Reiners, Jr., 2002). Thus, surface cathepsin B on tumor cells may play roles in tumor growth and invasiveness (Podgorski & Sloane, 2003).
Catalytically active cathepsin B has also been reported on the surface of cytotoxic T cells (CTLs). Unstimulated CTLs express very low levels of surface cathepsin B but when they are activated to degranulate, surface cathepsin B levels rapidly increase (Balaji, Schaschke, Machleidt, Catalfamo, & Henkart, 2002). The binding sites for cathepsin B on CTLs have not been identified. Surface bound cathepsin B on CTLs prevents lymphocyte cell death by cleaving and inactivating perforin molecules released along with cathepsin B during CTL degranulation, thereby preventing perforin molecules which diffuse back to the CTL surface from injuring CTLs (Balaji et al., 2002).
Conclusions
An increasing number of biologically important neutral and acidic proteinases have been shown to be expressed on the surface of leukocytes, and become associated with leukocyte plasma membranes by diverse mechanisms. Surface localization of proteinases likely focuses and restricts proteolysis to the leukocyte pericellular environment thereby keeping proteinase activity under close regulatory control by regulating cell surface levels of proteinases by the availability of binding sites and by internalization or shedding of proteinases from the cell surface. Localization of proteinases on leukocyte surfaces confers many advantages to proteinases including rendering them resistant to inhibition by physiologic processes, promoting and amplifying activation of proenzymes, and increasing the stability and half life of proteinases in the extracellular space. Cell surface binding of proteinases also enables them to participate in intracellular signaling in leukocytes via the cytoplasmic tails or transmembrane proteinases, or by binding of proteinases to their receptors or adjacent cell surface proteins such as integrins. Leukocyte cell-surface proteinases make critical contributions to extracellular proteolysis, and cell adhesion and migration during physiologic and pathologic processes. Future studies likely will uncover additional roles for leukocyte cell-associated proteinases in disease processes.
The binding of proteinases to leukocytes has important therapeutic implications. Surface-bound serine and metallo-proteinases on PMNs and other cell types are likely to be the major bioactive forms of the proteinases in vivo. In addition, many membrane-associated proteinases are resistant to inhibition by physiologic inhibitors indicating that augmentation of physiologic inhibitors may not have therapeutic efficacy in diseases characterized by excessive proteolysis. However, low-molecular-weight synthetic inhibitors are very effective against membrane-bound forms of serine proteinases and MMPs on PMNs (Owen et al., 1995b; Owen et al., 1998; Owen et al., 2003; Owen et al., 2004), and against members of the ADAMs family (Martin, Eynstone, Davies, Williams, & Steadman, 2002; Amour et al., 2002) suggesting that they may have therapeutic potential in diseases characterized by excessive activity of membrane-bound proteinases. The binding sites for proteinases on leukocytes also represent novel therapeutic targets. In this respect it is noteworthy that delivering heparin or other sulfated compounds to animals dislodges MMPs from HSPG on cell surfaces, and attenuates excessive tissue destruction associated with inflammation and reduces tumor metastasis, and angiogenesis (Rogachefsky, Dean, Howell, & Altman, 1993; Anees, 1996). More knowledge about the mechanisms by which cell surface proteinases bind to and are regulated on leukocyte surfaces may facilitate the development of new treatment strategies to control the deleterious activities of these enzymes in inflammatory diseases.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbott RE, Corral CJ, MacIvor DM, Lin X, Ley TJ, Mustoe TA. Augmented inflammatory responses and altered wound healing in cathepsin G-deficient mice. Arch Surg. 1998;133:1002–1006. doi: 10.1001/archsurg.133.9.1002. [DOI] [PubMed] [Google Scholar]
- Adkison AM, Raptis SZ, Kelley DG, Pham CT. Dipeptidyl peptidase I activates neutrophil-derived serine proteases and regulates the development of acute experimental arthritis. J Clin Invest. 2002;109:363–371. doi: 10.1172/JCI13462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer. 2007;7:834–846. doi: 10.1038/nrc2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ailenberg M, Silverman M. Cellular activation of mesangial gelatinase A by cytochalasin D is accompanied by enhanced mRNA expression of both gelatinase A and its membrane-associated gelatinase A activator (MT-MMP) Biochem J. 1996;313(Pt 3):879–884. doi: 10.1042/bj3130879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfano D, Iaccarino I, Stoppelli MP. Urokinase signaling through its receptor protects against anoikis by increasing BCL-xL expression levels. J Biol Chem. 2006;281:17758–17767. doi: 10.1074/jbc.M601812200. [DOI] [PubMed] [Google Scholar]
- Allen DH, Tracy PB. Human coagulation factor V is activated to the functional cofactor by elastase and cathepsin G expressed at the monocyte surface. J Biol Chem. 1995;270:1408–1415. doi: 10.1074/jbc.270.3.1408. [DOI] [PubMed] [Google Scholar]
- Alon R, Cahalon L, Hershkoviz R, Elbaz D, Reizis B, Wallach D, et al. TNF-α binds to the N-terminal domain of fibronectin and augments the β1-integrin-mediated adhesion of CD4+ T lymphocytes to the glycoprotein. J Immunol. 1994;152:1304–1313. [PubMed] [Google Scholar]
- Althoff K, Reddy P, Voltz N, Rose-John S, Mullberg J. Shedding of interleukin-6 receptor and tumor necrosis factor alpha. Contribution of the stalk sequence to the cleavage pattern of transmembrane proteins. Eur J Biochem. 2000;267:2624–2631. doi: 10.1046/j.1432-1327.2000.01278.x. [DOI] [PubMed] [Google Scholar]
- Amour A, Hutton M, Knauper V, Slocombe PM, Webster A, Butler M, et al. Inhibition of the metalloproteinase domain of mouse TACE. Ann NY Acad Sci. 1999;878:728–731. doi: 10.1111/j.1749-6632.1999.tb07774.x. [DOI] [PubMed] [Google Scholar]
- Amour A, Knight CG, English WR, Webster A, Slocombe PM, Knauper V, et al. The enzymatic activity of ADAM8 and ADAM9 is not regulated by TIMPs. FEBS Lett. 2002;524:154–158. doi: 10.1016/s0014-5793(02)03047-8. [DOI] [PubMed] [Google Scholar]
- Amour A, Knight CG, Webster A, Slocombe PM, Stephens PE, Knauper V, et al. The in vitro activity of ADAM-10 is inhibited by TIMP-1 and TIMP-3. FEBS Lett. 2000;473:275–279. doi: 10.1016/s0014-5793(00)01528-3. [DOI] [PubMed] [Google Scholar]
- Amour A, Slocombe PM, Webster A, Butler M, Knight CG, Smith BJ, et al. TNF-α converting enzyme (TACE) is inhibited by TIMP-3. FEBS Lett. 1998;435:39–44. doi: 10.1016/s0014-5793(98)01031-x. [DOI] [PubMed] [Google Scholar]
- Anees M. Location of tumour cells in colon tissue by Texas red labelled pentosan polysulphate, an inhibitor of a cell surface protease. J Enzyme Inhib. 1996;10:203–214. doi: 10.3109/14756369609030314. [DOI] [PubMed] [Google Scholar]
- Armstrong L, Godinho SI, Uppington KM, Whittington HA, Millar AB. Contribution of TNF-alpha converting enzyme and proteinase-3 to TNF-alpha processing in human alveolar macrophages. Am J Respir Cell Mol Biol. 2006;34:219–225. doi: 10.1165/rcmb.2005-0087OC. [DOI] [PubMed] [Google Scholar]
- Balaji KN, Schaschke N, Machleidt W, Catalfamo M, Henkart PA. Surface cathepsin B protects cytotoxic lymphocytes from self-destruction after degranulation. J Exp Med. 2002;196:493–503. doi: 10.1084/jem.20011836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balbin M, Fueyo A, Tester AM, Pendas AM, Pitiot AS, Astudillo A, et al. Loss of collagenase-2 confers increased skin tumor susceptibility to male mice. Nat Genet. 2003;35:252–257. doi: 10.1038/ng1249. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay S, Hartley DM, Cahill CM, Lahiri DK, Chattopadhyay N, Rogers JT. Interleukin-1alpha stimulates non-amyloidogenic pathway by alpha-secretase (ADAM-10 and ADAM-17) cleavage of APP in human astrocytic cells involving p38 MAP kinase. J Neurosci Res. 2006;84:106–118. doi: 10.1002/jnr.20864. [DOI] [PubMed] [Google Scholar]
- Bangalore N, Travis J. Comparison of properties of membrane bound versus soluble forms of human leukocyte elastase and cathepsin G. Biol Chem Hoppe-Seyler. 1994a;375:659–666. doi: 10.1515/bchm3.1994.375.10.659. [DOI] [PubMed] [Google Scholar]
- Bangalore N, Travis J. Comparison of properties of membrane bound versus soluble forms of human leukocytic elastase and cathepsin G. Biol Chem Hoppe Seyler. 1994b;375:659–666. doi: 10.1515/bchm3.1994.375.10.659. [DOI] [PubMed] [Google Scholar]
- Baron R, Neff L, Louvard D, Courtoy PJ. Cell-mediated extracellular acidification and bone resorption: Evidence for a low pH in resorbing lacunae and localization of a 100 kD lysosomal membrane protein at the osteoclast ruffled border. J Cell Biol. 1985;101:2210–2222. doi: 10.1083/jcb.101.6.2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basbaum C, Li D, Gensch E, Gallup M, Lemjabbar H. Mechanisms by which gram-positive bacteria and tobacco smoke stimulate mucin induction through the epidermal growth factor receptor (EGFR) Novartis Found Symp. 2002;248:171–176. [PubMed] [Google Scholar]
- Bauer S, Abdgawad M, Gunnarsson L, Segelmark M, Tapper H, Hellmark T. Proteinase 3 and CD177 are expressed on the plasma membrane of the same subset of neutrophils. J Leukoc Biol. 2007;81:458–464. doi: 10.1189/jlb.0806514. [DOI] [PubMed] [Google Scholar]
- Beck V, Herold H, Benge A, Luber B, Hutzler P, Tschesche H, et al. ADAM15 decreases integrin alphavbeta3/vitronectin-mediated ovarian cancer cell adhesion and motility in an RGD-dependent fashion. Int J Biochem Cell Biol. 2005;37:590–603. doi: 10.1016/j.biocel.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Betsuyaku T, Shipley JM, Liu Z, Senior RM. Gelatinase B deficiency does not protect against lipopolysaccharide-induced acute lung injury. Chest. 1999;116(1 Suppl):17S–18S. [PubMed] [Google Scholar]
- Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-α from cells. Nature. 1997;385:729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- Blair HC, Teitelbaum SL, Ghiselli R, Gluck S. Osteoclastic bone resorption by a polarized vacuolar proton pump. Science. 1989;245:855–857. doi: 10.1126/science.2528207. [DOI] [PubMed] [Google Scholar]
- Blasi F. u-PA and cell migration: urokinase receptor as a ligand for a chemotactic G-protein-coupled receptor. Haemostasis. 2001;31(Suppl 1):59. [PubMed] [Google Scholar]
- Blasi F, Carmeliet P. uPAR: a versatile signalling orchestrator. Nat Rev Mol Cell Biol. 2002;3:932–943. doi: 10.1038/nrm977. [DOI] [PubMed] [Google Scholar]
- Blobel CP, Wolfsberg TG, Turck CW, Myles DG, Primakoff P, White JM. A potential fusion peptide and an integrin ligand domain in a protein active in sperm-egg fusion. Nature. 1992;356:248–252. doi: 10.1038/356248a0. [DOI] [PubMed] [Google Scholar]
- Bohm BB, Aigner T, Blobel CP, Kalden JR, Burkhardt H. Highly enhanced expression of the disintegrin metalloproteinase MDC15 (metargidin) in rheumatoid synovial tissue. Arthritis Rheum. 2001;44:2046–2054. doi: 10.1002/1529-0131(200109)44:9<2046::AID-ART354>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Borregaard N, Cowland JB. Granules of the human neutrophilic polymorphonuclear leukocyte. Blood. 1997;89:3503–3521. [PubMed] [Google Scholar]
- Bosch X, Font J, Mirapeix E, Revert L, Urbano-Marquez A, Ingelmo M. Anti-neutrophil cytoplasmic autoantibodies (ANCA): antigenic specificities and clinical associations. Adv Exp Med Biol. 1993;336:281–286. doi: 10.1007/978-1-4757-9182-2_42. [DOI] [PubMed] [Google Scholar]
- Bouchard BA, Tracy PB. Platelets, leukocytes, and coagulation. Curr Opin Hematol. 2001;8:263–269. doi: 10.1097/00062752-200109000-00001. [DOI] [PubMed] [Google Scholar]
- Bridges LC, Tani PH, Hanson KR, Roberts CM, Judkins MB, Bowditch RD. The lymphocyte metalloprotease MDC-L (ADAM 28) is a ligand for the integrin alpha4beta1. J Biol Chem. 2002;277:3784–3792. doi: 10.1074/jbc.M109538200. [DOI] [PubMed] [Google Scholar]
- Brooks PC, Stromblad S, Sanders LC, von Schalscha TL, Aimes RT, Stetler-Stevenson WG, et al. Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin αvβ3. Cell. 1996;85:683–693. doi: 10.1016/s0092-8674(00)81235-0. [DOI] [PubMed] [Google Scholar]
- Brou C, Logeat F, Gupta N, Bessia C, LeBail O, Doedens JR, et al. A novel proteolytic cleavage involved in Notch signaling: the role of the disintegrin-metalloprotease TACE. Mol Cell. 2000;5:207–216. doi: 10.1016/s1097-2765(00)80417-7. [DOI] [PubMed] [Google Scholar]
- Brunner G, Simon MM, Kramer MD. Activation of pro-urokinase by the human T cell-associated serine proteinase HuTSP-1. FEBS Lett. 1990;260:141–144. doi: 10.1016/0014-5793(90)80087-y. [DOI] [PubMed] [Google Scholar]
- Brzoska E, Bello V, Darribere T, Moraczewski J. Integrin alpha3 subunit participates in myoblast adhesion and fusion in vitro. Differentiation. 2006;74:105–118. doi: 10.1111/j.1432-0436.2005.00059.x. [DOI] [PubMed] [Google Scholar]
- Bugge TH, Flick MJ, Danton MJ, Daugherty CC, Romer J, Dano K, et al. Urokinase-type plasminogen activator is effective in fibrin clearance in the absence of its receptor or tissue-type plasminogen activator. Proc Natl Acad Sci USA. 1996a;93:5899–5904. doi: 10.1073/pnas.93.12.5899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugge TH, Flick MJ, Daugherty CC, Degen JL. Plasminogen deficiency causes severe thrombosis but is compatible with development and reproduction. Genes Dev. 1995;9:794–807. doi: 10.1101/gad.9.7.794. [DOI] [PubMed] [Google Scholar]
- Bugge TH, Kombrinck KW, Flick MJ, Daugherty CC, Danton MJ, Degen JL. Loss of fibrinogen rescues mice from the pleiotropic effects of plasminogen deficiency. Cell. 1996b;87:709–719. doi: 10.1016/s0092-8674(00)81390-2. [DOI] [PubMed] [Google Scholar]
- Busiek DF, Baraji V, Nehring LC, Parks WC, Welgus HG. Matrilysin expression by human mononuclear phagocytes and its regulation by cytokines and hormones. J Immunol. 1995;154:6484–6491. [PubMed] [Google Scholar]
- Butler GS, Will H, Atkinson SJ, Murphy G. Membrane-type-2 matrix metalloproteinase can initiate the processing of progelatinase A and is regulated by the tissue inhibitors of metalloproteinases. Eur J Biochem. 1997a;244:653–657. doi: 10.1111/j.1432-1033.1997.t01-1-00653.x. [DOI] [PubMed] [Google Scholar]
- Butler GS, Will H, Atkinson SJ, Murphy G. Membrane-type-2 matrix metalloproteinase can initiate the processing of progelatinase A and is regulated by the tissue inhibitors of metalloproteinases. Eur J Biochem. 1997b;244:653–657. doi: 10.1111/j.1432-1033.1997.t01-1-00653.x. [DOI] [PubMed] [Google Scholar]
- Bzowska M, Jura N, Lassak A, Black RA, Bereta J. Tumour necrosis factor-alpha stimulates expression of TNF-alpha converting enzyme in endothelial cells. Eur J Biochem. 2004;271:2808–2820. doi: 10.1111/j.1432-1033.2004.04215.x. [DOI] [PubMed] [Google Scholar]
- Cai TO, Wright SD. Human leukocyte elastase is an endogenous ligand for the integrin CR3 (CD11b/CD18, Mac-1, αMβ2) and modulates polymorphonuclear leukocyte adhesion. J Exp Med. 1996;184:1213–1223. doi: 10.1084/jem.184.4.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calkins CC, Sameni M, Koblinski J, Sloane BF, Moin K. Differential localization of cysteine protease inhibitors and a target cysteine protease, cathepsin B, by immuno-confocal microscopy. J Histochem Cytochem. 1998;46:745–751. doi: 10.1177/002215549804600607. [DOI] [PubMed] [Google Scholar]
- Campbell EJ. Human leukocyte elastase, cathepsin G, and lactoferrin: A family of neutrophil granule glycoproteins which bind to an alveolar macrophage receptor. Proc Natl Acad Sci USA. 1982;79:6941–6945. doi: 10.1073/pnas.79.22.6941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell EJ, Campbell MA, Owen CA. Bioactive proteinase 3 on the cell surface of human neutrophils: quantification, catalytic activity, and susceptibility to inhibition. J Immunol. 2000;165:3366–3374. doi: 10.4049/jimmunol.165.6.3366. [DOI] [PubMed] [Google Scholar]
- Campbell EJ, Cury JD, Lazarus CJ, Welgus HG. Monocyte procollagenase and tissue inhibitor of metalloproteinases: Identification, characterization, and regulation of secretion. J Biol Chem. 1987;262:15862–15868. [PubMed] [Google Scholar]
- Campbell EJ, Owen CA. The sulfate groups of chondroitin sulfate- and heparan sulfate-containing proteoglycans in neutrophil plasma membranes are novel binding sites for human leukocyte elastase and cathepsin G. J Biol Chem. 2007;282:14645–14654. doi: 10.1074/jbc.M608346200. [DOI] [PubMed] [Google Scholar]
- Campbell EJ, White RR, Senior RM, Rodriguez RJ, Kuhn C., III Receptor-mediated binding and internalization of leukocyte elastase by alveolar macrophages in vitro. JCI. 1979;64:824–833. doi: 10.1172/JCI109530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao C, Lawrence DA, Li Y, Von Arnim CA, Herz J, Su EJ, et al. Endocytic receptor LRP together with tPA and PAI-1 coordinates Mac-1-dependent macrophage migration. EMBO J. 2006;25:1860–1870. doi: 10.1038/sj.emboj.7601082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Rehemtulla A, Bahou W, Zucker S. Membrane type matrix metalloproteinase 1 activates pro-gelatinase A without furin cleavage of the N-terminal domain. J Biol Chem. 1996;271:30174–30180. doi: 10.1074/jbc.271.47.30174. [DOI] [PubMed] [Google Scholar]
- Cao J, Sato H, Takino T, Seiki M. The C-terminal region of membrane type matrix metalloproteinase is a functional transmembrane domain required for progelatinase A activation. J Biol Chem. 1995;270:801–805. doi: 10.1074/jbc.270.2.801. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Moons L, Lijnen R, Baes M, Lemaitre V, Tipping P, et al. Urokinase-generated plasmin activates matrix metalloproteinases during aneurysm formation. Nat Genet. 1997;17:439–444. doi: 10.1038/ng1297-439. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Schoonjans L, Kieckens L, Ream B, Degen J, Bronson R, et al. Physiological consequences of loss of plasminogen activator gene function in mice. Nature. 1994;368:419–424. doi: 10.1038/368419a0. [DOI] [PubMed] [Google Scholar]
- Carrell RW. Alpha1-antitrypsin: Molecular pathology, leukocytes, and tissue damage. JCI. 1986;78:1427–1431. doi: 10.1172/JCI112731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallo-Medved D, Dosescu J, Linebaugh BE, Sameni M, Rudy D, Sloane BF. Mutant K-ras regulates cathepsin B localization on the surface of human colorectal carcinoma cells. Neoplasia. 2003;5:507–519. doi: 10.1016/s1476-5586(03)80035-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallo-Medved D, Mai J, Dosescu J, Sameni M, Sloane BF. Caveolin-1 mediates the expression and localization of cathepsin B, pro-urokinase plasminogen activator and their cell-surface receptors in human colorectal carcinoma cells. J Cell Sci. 2005;118:1493–1503. doi: 10.1242/jcs.02278. [DOI] [PubMed] [Google Scholar]
- Cavallo-Medved D, Sloane BF. Cell-surface cathepsin B: understanding its functional significance. Curr Top Dev Biol. 2003;54:313–341. doi: 10.1016/s0070-2153(03)54013-3. [DOI] [PubMed] [Google Scholar]
- Chapman HA, Jr, Munger JS, Shi GP. The role of thiol proteases in tissue injury and remodeling. Am J Respir Crit Care Med. 1994;150:S155–S159. doi: 10.1164/ajrccm/150.6_Pt_2.S155. [DOI] [PubMed] [Google Scholar]
- Chapman HA, Jr, Stone OL. Comparison of live human neutrophil and alveolar macrophage elastolytic activity in vitro. Relative resistance of macrophage elastolytic activity to serum and alveolar proteinase inhibitors. JCI. 1984;74:1693–1700. doi: 10.1172/JCI111586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatham WW, Heck LW, Blackburn WD. Ligand dependent release of activated neutrophil collagenase - a role for surface bound IgG in the degradation of articular collagens. Matrix Suppl. 1992;1:207–208. [PubMed] [Google Scholar]
- Chen WT. Proteases associated with invadopodia, and their role in degradation of extracellular matrix. Enzyme Protein. 1996;49:59–71. doi: 10.1159/000468616. [DOI] [PubMed] [Google Scholar]
- Chertov O, Ueda H, Xu LL, Tani K, Murphy WJ, Wang JM, et al. Identification of human neutrophil-derived cathepsin G and azurocidin/CAP37 as chemoattractants for mononuclear cells and neutrophils. J Exp Med. 1997;186:739–747. doi: 10.1084/jem.186.5.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung AW, Radomski A, Alonso-Escolano D, Jurasz P, Stewart MW, Malinski T, et al. Platelet-leukocyte aggregation induced by PAR agonists: regulation by nitric oxide and matrix metalloproteinases. Br J Pharmacol. 2004;143:845–855. doi: 10.1038/sj.bjp.0705997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churg A, Wang RD, Tai H, Wang X, Xie C, Dai J, et al. Macrophage metalloelastase mediates acute cigarette smoke-induced inflammation via tumor necrosis factor-alpha release. Am J Respir Crit Care Med. 2003;167:1083–1089. doi: 10.1164/rccm.200212-1396OC. [DOI] [PubMed] [Google Scholar]
- Churg A, Wang X, Wang RD, Meixner SC, Pryzdial EL, Wright JL. Alpha1-antitrypsin suppresses TNF-alpha and MMP-12 production by cigarette smoke-stimulated macrophages. Am J Respir Cell Mol Biol. 2007;37:144–151. doi: 10.1165/rcmb.2006-0345OC. [DOI] [PubMed] [Google Scholar]
- Cornelius LA, Nehring LC, Harding E, Bolanowski M, Welgus HG, Kobayashi DK, et al. Matrix metalloproteinases generate angiostatin: Effects on neovascularization. J Immunol. 1998;161:6845–6852. [PubMed] [Google Scholar]
- Crippa MP. Urokinase-type plasminogen activator. Int J Biochem Cell Biol. 2007;39:690–694. doi: 10.1016/j.biocel.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Csernok E, Ludemann J, Gross WL, Bainton DF. Ultrastructural localization of proteinase 3, the target antigen of anti-cytoplasmic antibodies circulating in Wegener’s granulomatosis. Am J Pathol. 1990;137:1113–1120. [PMC free article] [PubMed] [Google Scholar]
- Csernok E, Schmitt WH, Ernst M, Bainton DF, Gross WL. Membrane surface proteinase 3 expression and intracytoplasmic immunoglobulin on neutrophils from patients with ANCA-associated vasculitides. Adv Exp Med Biol. 1993;336:45–50. doi: 10.1007/978-1-4757-9182-2_5. [DOI] [PubMed] [Google Scholar]
- Cury JD, Campbell EJ, Lazarus CJ, Albin RJ, Welgus HG. Selective upregulation of human alveolar macrophage collagenase production by lipopolysaccharide and comparison to collagenase production by fibroblasts. J Immunol. 1988;141:4306–4312. [PubMed] [Google Scholar]
- D’Ortho MP, Stanton H, Butler M, Atkinson SJ, Murphy G, Hembry RM. MT1-MMP on the cell surface causes focal degradation of gelatin films. FEBS Lett. 1998;421:159–164. doi: 10.1016/s0014-5793(97)01555-x. [DOI] [PubMed] [Google Scholar]
- D’Ortho MP, Will H, Atkinson S, Butler G, Messent A, Gavrilovic J, et al. Membrane-type matrix metalloproteinases 1 and 2 exhibit broad-spectrum proteolytic capacities comparable to many matrix metalloproteinases. Eur J Biochem. 1997;250:751–757. doi: 10.1111/j.1432-1033.1997.00751.x. [DOI] [PubMed] [Google Scholar]
- Dewald B, Bretz U, Baggiolini M. Release of gelatinase from a novel secretory compartment of human neutrophils. JCI. 1982;70:518–525. doi: 10.1172/JCI110643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewerchin M, Nuffelen AV, Wallays G, Bouche A, Moons L, Carmeliet P, et al. Generation and characterization of urokinase receptor-deficient mice. J Clin Invest. 1996;97:870–878. doi: 10.1172/JCI118489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doedens JR, Mahimkar RM, Black RA. TACE/ADAM-17 enzymatic activity is increased in response to cellular stimulation. Biochem Biophys Res Commun. 2003;308:331–338. doi: 10.1016/s0006-291x(03)01381-0. [DOI] [PubMed] [Google Scholar]
- Dollery CM, Owen CA, Sukhova GK, Krettek A, Shapiro SD, Libby P. Neutrophil elastase in human atherosclerotic plaques: production by macrophages. Circulation. 2003;107:2829–2836. doi: 10.1161/01.CIR.0000072792.65250.4A. [DOI] [PubMed] [Google Scholar]
- Doshi SN, Marmur JD. Evolving role of tissue factor and its pathway inhibitor. Crit Care Med. 2002;30:S241–S250. doi: 10.1097/00003246-200205001-00012. [DOI] [PubMed] [Google Scholar]
- Drew AF, Kaufman AH, Kombrinck KW, Danton MJ, Daugherty CC, Degen JL, et al. Ligneous conjunctivitis in plasminogen-deficient mice. Blood. 1998;91:1616–1624. [PubMed] [Google Scholar]
- Dumin JA, Dickeson SK, Stricker TP, Bhattacharyya-Pakrasi M, Roby JD, Santoro SA, et al. Pro-collagenase-1 (matrix metalloproteinase-1) binds the alpha(2)beta(1) integrin upon release from keratinocytes migrating on type I collagen. J Biol Chem. 2001;276:29368–29374. doi: 10.1074/jbc.M104179200. [DOI] [PubMed] [Google Scholar]
- Eitzman DT, McCoy RD, Zheng X, Fay WP, Shen T, Ginsburg D, et al. Bleomycin-induced pulmonary fibrosis in transgenic mice that either lack or overexpress the murine plasminogen activator inhibitor-1 gene. J Clin Invest. 1996;97:232–237. doi: 10.1172/JCI118396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis V, Wun TC, Behrendt N, Ronne E, Dano K. Inhibition of receptor-bound urokinase by plasminogen-activator inhibitors. J Biol Chem. 1990;265:9904–9908. [PubMed] [Google Scholar]
- English WR, Puente XS, Freije JMP, Knauper V, Amour A, Merryweather A, et al. Membrane type 4 matrix metalloproteinase (MMP17) has tumor necrosis factor-α convertase activity but does not activate pro-MMP2. J Biol Chem. 2000;275:14046–14055. doi: 10.1074/jbc.275.19.14046. [DOI] [PubMed] [Google Scholar]
- Evans JP. Fertilin beta and other ADAMs as integrin ligands: insights into cell adhesion and fertilization. Bioessays. 2001;23:628–639. doi: 10.1002/bies.1088. [DOI] [PubMed] [Google Scholar]
- Fan H, Turck CW, Derynck R. Characterization of growth factor-induced serine phosphorylation of tumor necrosis factor-alpha converting enzyme and of an alternatively translated polypeptide. J Biol Chem. 2003;278:18617–18627. doi: 10.1074/jbc.M300331200. [DOI] [PubMed] [Google Scholar]
- Foley SC, Mogas AK, Olivenstein R, Fiset PO, Chakir J, Bourbeau J, et al. Increased expression of ADAM33 and ADAM8 with disease progression in asthma. J Allergy Clin Immunol. 2007;119:863–871. doi: 10.1016/j.jaci.2006.12.665. [DOI] [PubMed] [Google Scholar]
- Fourie AM, Coles F, Moreno V, Karlsson L. Catalytic activity of ADAM8, ADAM15, and MDC-L (ADAM28) on synthetic peptide substrates and in ectodomain cleavage of CD23. J Biol Chem. 2003;278:30469–30477. doi: 10.1074/jbc.M213157200. [DOI] [PubMed] [Google Scholar]
- Fu X, Kassim SY, Parks WC, Heinecke JW. Hypochlorous acid generated by myeloperoxidase modifies adjacent tryptophan and glycine residues in the catalytic domain of matrix metalloproteinase-7 (matrilysin): an oxidative mechanism for restraining proteolytic activity during inflammation. J Biol Chem. 2003;278:28403–28409. doi: 10.1074/jbc.M304739200. [DOI] [PubMed] [Google Scholar]
- Fujita T, Maesawa C, Oikawa K, Nitta H, Wakabayashi G, Masuda T. Interferon-gamma down-regulates expression of tumor necrosis factor-alpha converting enzyme/a disintegrin and metalloproteinase 17 in activated hepatic stellate cells of rats. Int J Mol Med. 2006;17:605–616. [PubMed] [Google Scholar]
- Garton KJ, Gough PJ, Blobel CP, Murphy G, Greaves DR, Dempsey PJ, et al. Tumor necrosis factor-alpha-converting enzyme (ADAM17) mediates the cleavage and shedding of fractalkine (CX3CL1) J Biol Chem. 2001;276:37993–38001. doi: 10.1074/jbc.M106434200. [DOI] [PubMed] [Google Scholar]
- Garton KJ, Gough PJ, Philalay J, Wille PT, Blobel CP, Whitehead RH, et al. Stimulated shedding of vascular cell adhesion molecule 1 (VCAM-1) is mediated by tumor necrosis factor-alpha-converting enzyme (ADAM 17) J Biol Chem. 2003;278:37459–37464. doi: 10.1074/jbc.M305877200. [DOI] [PubMed] [Google Scholar]
- Gauthier MC, Racine C, Ferland C, Flamand N, Chakir J, Tremblay GM, et al. Expression of membrane type-4 matrix metalloproteinase (metalloproteinase-17) by human eosinophils. Int J Biochem Cell Biol. 2003;35:1667–1673. doi: 10.1016/s1357-2725(03)00136-5. [DOI] [PubMed] [Google Scholar]
- Goerge T, Barg A, Schnaeker EM, Poppelmann B, Shpacovitch V, Rattenholl A, et al. Tumor-derived matrix metalloproteinase-1 targets endothelial proteinase-activated receptor 1 promoting endothelial cell activation. Cancer Res. 2006;66:7766–7774. doi: 10.1158/0008-5472.CAN-05-3897. [DOI] [PubMed] [Google Scholar]
- Goldmann WH, Niles JL, Arnaout MA. Interaction of purified human proteinase 3 (PR3) with reconstituted lipid bilayers. Eur J Biochem. 1999;261:155–162. doi: 10.1046/j.1432-1327.1999.00259.x. [DOI] [PubMed] [Google Scholar]
- Gomez-Gaviro M, Dominguez-Luis M, Canchado J, Calafat J, Janssen H, Lara-Pezzi E, et al. Expression and regulation of the metalloproteinase ADAM-8 during human neutrophil pathophysiological activation and its catalytic activity on L-selectin shedding. J Immunol. 2007;178:8053–8063. doi: 10.4049/jimmunol.178.12.8053. [DOI] [PubMed] [Google Scholar]
- Goretzki L, Schmitt M, Mann K, Calvete J, Chucholowski N, Kramer M, et al. Effective activation of the proenzyme form of the urokinase-type plasminogen activator (pro-uPA) by the cysteine protease cathepsin L. FEBS Lett. 1992;297:112–118. doi: 10.1016/0014-5793(92)80339-i. [DOI] [PubMed] [Google Scholar]
- Gough PJ, Garton KJ, Wille PT, Rychlewski M, Dempsey PJ, Raines EW. A disintegrin and metalloproteinase 10-mediated cleavage and shedding regulates the cell surface expression of CXC chemokine ligand 16. J Immunol. 2004;172:3678–3685. doi: 10.4049/jimmunol.172.6.3678. [DOI] [PubMed] [Google Scholar]
- Gross WL, Csernok E, Flesch BK. ‘Classic’ anti-neutrophil cytoplasmic autoantibodies (cANCA), ‘Wegener’s autoantigen’ and their immunopathogenic role in Wegener’s granulomatosis. J Autoimmun. 1993;6:171–184. doi: 10.1006/jaut.1993.1015. [DOI] [PubMed] [Google Scholar]
- Gross WL, Trabandt A, Csernok E. Pathogenesis of Wegener’s granulomatosis. Ann Med Interne. 1998;149:280–286. [PubMed] [Google Scholar]
- Gueders MM, Balbin M, Rocks N, Foidart JM, Gosset P, Louis R, et al. Matrix metalloproteinase-8 deficiency promotes granulocytic allergen-induced airway inflammation. The Journal of Immunology. 2005;175:2589–2597. doi: 10.4049/jimmunol.175.4.2589. [DOI] [PubMed] [Google Scholar]
- Guo M, Mathieu PA, Linebaugh B, Sloane BF, Reiners JJ., Jr Phorbol ester activation of a proteolytic cascade capable of activating latent transforming growth factor-betaL a process initiated by the exocytosis of cathepsin B. J Biol Chem. 2002;277:14829–14837. doi: 10.1074/jbc.M108180200. [DOI] [PubMed] [Google Scholar]
- Gyetko MR, Sud S, Kendall T, Fuller JA, Newstead MW, Standiford TJ. Urokinase receptor-deficient mice have impaired neutrophil recruitment in response to pulmonary Pseudomonas aeruginosa infection. The Journal of Immunology. 2000;165:1513–1519. doi: 10.4049/jimmunol.165.3.1513. [DOI] [PubMed] [Google Scholar]
- Gyetko MR, Sud S, Sonstein J, Polak T, Sud A, Curtis JL. Antigen-driven lymphocyte recruitment to the lung is diminished in the absence of urokinase-type plasminogen activator (uPA) receptor, but is independent of uPA. The Journal of Immunology. 2001;167:5539–5542. doi: 10.4049/jimmunol.167.10.5539. [DOI] [PubMed] [Google Scholar]
- Hartmann D, De Strooper B, Serneels L, Craessaerts K, Herreman A, Annaert W, et al. The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for alpha-secretase activity in fibroblasts. Hum Mol Genet. 2002;11:2615–2624. doi: 10.1093/hmg/11.21.2615. [DOI] [PubMed] [Google Scholar]
- Hautamaki RD, Kobayashi DK, Senior RM, Shapiro SD. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science. 1997;277:2002–2004. doi: 10.1126/science.277.5334.2002. [DOI] [PubMed] [Google Scholar]
- Heiple JM, Ossowski L. Human neutrophil plasminogen activator is localized in specific granules and is translocated to the cell surface by exocytosis. J Exp Med. 1986;164:826–840. doi: 10.1084/jem.164.3.826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henskens YMC, Veerman ECI, Nieuw Amerongen AVN. Cystatins in health and disease. Biol Chem Hoppe-Seyler. 1996;377:71–86. doi: 10.1515/bchm3.1996.377.2.71. [DOI] [PubMed] [Google Scholar]
- Hernandez-Barrantes S, Shimura Y, Soloway PD, Sang QA, Fridman R. Differential roles of TIMP-4 and TIMP-2 in pro-MMP-2 activation by MT1-MMP. Biochem Biophys Res Commun. 2001;281:126–130. doi: 10.1006/bbrc.2001.4323. [DOI] [PubMed] [Google Scholar]
- Herren B, Garton KJ, Coats S, Bowen-Pope DF, Ross R, Raines EW. ADAM15 overexpression in NIH3T3 cells enhances cell-cell interactions. Exp Cell Res. 2001;271:152–160. doi: 10.1006/excr.2001.5353. [DOI] [PubMed] [Google Scholar]
- Heymans S, Luttun A, Nuyens D, Theilmeier G, Creemers E, Moons L, et al. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat Med. 1999;5:1135–1142. doi: 10.1038/13459. [DOI] [PubMed] [Google Scholar]
- Hildenbrand R, Gandhari M, Stroebel P, Marx A, Allgayer H, Arens N. The urokinase-system--role of cell proliferation and apoptosis. Histol Histopathol. 2008;23:227–236. doi: 10.14670/HH-23.227. [DOI] [PubMed] [Google Scholar]
- Hirche TO, Atkinson JJ, Bahr S, Belaaouaj A. Deficiency in neutrophil elastase does not impair neutrophil recruitment to inflamed sites. Am J Respir Cell Mol Biol. 2004;30:576–584. doi: 10.1165/rcmb.2003-0253OC. [DOI] [PubMed] [Google Scholar]
- Holmbeck K, Bianco P, Caterina J, Yamada S, Kromer M, Kuznetsov SA, et al. MT1-MMP-deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell. 1999;99:81–92. doi: 10.1016/s0092-8674(00)80064-1. [DOI] [PubMed] [Google Scholar]
- Hotary K, Allen E, Punturieri A, Yana I, Weiss SJ. Regulation of cell invasion and morphogenesis in a three-dimensional type I collagen matrix by membrane-type matrix metalloproteinases 1, 2, and 3. J Cell Biol. 2000;149:1309–1323. doi: 10.1083/jcb.149.6.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houghton AM, Quintero PA, Perkins DL, Kobayashi DK, Kelley DG, Marconcini LA, et al. Elastin fragments drive disease progression in a murine model of emphysema. J Clin Invest. 2006;116:753–759. doi: 10.1172/JCI25617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard L, Nelson KK, Maciewicz RA, Blobel CP. Interaction of the metalloprotease disintegrins MDC9 and MDC15 with two SH3 domain-containing proteins, endophilin I and SH3PX1. J Biol Chem. 1999;274:31693–31699. doi: 10.1074/jbc.274.44.31693. [DOI] [PubMed] [Google Scholar]
- Hurtado O, Cardenas A, Lizasoain I, Bosca L, Leza JC, Lorenzo P, et al. Up-regulation of TNF-alpha convertase (TACE/ADAM17) after oxygen-glucose deprivation in rat forebrain slices. Neuropharmacology. 2001;40:1094–1102. doi: 10.1016/s0028-3908(01)00035-1. [DOI] [PubMed] [Google Scholar]
- Iba K, Albrechtsen R, Gilpin B, Frohlich C, Loechel F, Zolkiewska A, et al. The cysteine-rich domain of human ADAM 12 supports cell adhesion through syndecans and triggers signaling events that lead to beta1 integrin-dependent cell spreading. J Cell Biol. 2000;149:1143–1156. doi: 10.1083/jcb.149.5.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai K, Ohuchi E, Aoki T, Nomura H, Fujii Y, Sato H, et al. Membrane-type matrix metalloproteinase 1 is a gelatinolytic enzyme and is secreted in a complex with tissue inhibitor of metalloproteinases 2. Cancer Res. 1996;56:2707–2710. [PubMed] [Google Scholar]
- Itoh Y, Kajita M, Kinoh H, Mori H, Okada A, Seiki M. Membrane type 4 matrix metalloproteinase (MT4-MMP, MMP-17) is a glycosylphosphatidylinositol-anchored proteinase. J Biol Chem. 1999;274:34260–34266. doi: 10.1074/jbc.274.48.34260. [DOI] [PubMed] [Google Scholar]
- Kang T, Yi J, Guo A, Wang X, Overall C, Jiang W, et al. Subcellular distribution and cytokine-/chemokine-regulated secretion of leukolysin/MT6-MMP/MMP-25 in neutrophils. J Biol Chem. 2001;276:21960–21968. doi: 10.1074/jbc.M007997200. [DOI] [PubMed] [Google Scholar]
- Kantari C, Pederzoli-Ribeil M, Amir-Moazami O, Gausson-Dorey V, Moura IC, Lecomte MC, et al. Proteinase 3, the Wegener autoantigen, is externalized during neutrophil apoptosis: evidence for a functional association with phospholipid scramblase 1 and interference with macrophage phagocytosis. Blood. 2007;110:4086–4095. doi: 10.1182/blood-2007-03-080457. [DOI] [PubMed] [Google Scholar]
- Kargi HA, Campbell EJ, Kuhn C., III Elastase and cathepsin G of human monocytes. Heterogeneity and subcellular localization to peroxidase-positive granules. J Histochem Cytochem. 1990;38:1179–1186. doi: 10.1177/38.8.2164060. [DOI] [PubMed] [Google Scholar]
- Kenny PA, Bissell MJ. Targeting TACE-dependent EGFR ligand shedding in breast cancer. J Clin Invest. 2007;117:337–345. doi: 10.1172/JCI29518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettritz R, Jennette JC, Falk RJ. Crosslinking of ANCA-antigens stimulates superoxide release by human neutrophils. J Am Soc Nephrol. 1997;8:386–394. doi: 10.1681/ASN.V83386. [DOI] [PubMed] [Google Scholar]
- Kilpatrick LM, Harris RL, Owen KA, Bass R, Ghorayeb C, Bar-Or A, et al. Initiation of plasminogen activation on the surface of monocytes expressing the type II transmembrane serine protease matriptase. Blood. 2006;108:2616–2623. doi: 10.1182/blood-2006-02-001073. [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Moniwa N, Sugimura M, Shinohara H, Ohi H, Terao T. Effects of membrane-associated cathepsin B on the activation of receptor-bound prourokinase and subsequent invasion of reconstituted basement membranes. Biochim Biophys Acta. 1993a;1178:55–62. doi: 10.1016/0167-4889(93)90109-3. [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Moniwa N, Sugimura M, Shinohara H, Ohi H, Terao T. Increased cell-surface urokinase in advanced ovarian cancer. Jpn J Cancer Res. 1993b;84:633–640. doi: 10.1111/j.1349-7006.1993.tb02023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolkenbrock H, Essers L, Ulbrich N, Will H. Biochemical characterization of the catalytic domain of membrane-type 4 matrix metalloproteinase. Biol Chem. 1999;380:1103–1108. doi: 10.1515/BC.1999.137. [DOI] [PubMed] [Google Scholar]
- Korkmaz B, Attucci S, Jourdan ML, Juliano L, Gauthier F. Inhibition of neutrophil elastase by alpha1-protease inhibitor at the surface of human polymorphonuclear neutrophils. The Journal of Immunology. 2005;175:3329–3338. doi: 10.4049/jimmunol.175.5.3329. [DOI] [PubMed] [Google Scholar]
- Kramps JA, Rudolphus A, Stolk J, Willems LNA, Dijkman JH. Role of antileukoprotease in the lung. Ann N Y Acad Sci. 1991;624:97–108. doi: 10.1111/j.1749-6632.1991.tb17010.x. [DOI] [PubMed] [Google Scholar]
- Kveiborg M, Frohlich C, Albrechtsen R, Tischler V, Dietrich N, Holck P, et al. A role for ADAM12 in breast tumor progression and stromal cell apoptosis. Cancer Res. 2005;65:4754–4761. doi: 10.1158/0008-5472.CAN-05-0262. [DOI] [PubMed] [Google Scholar]
- Langer H, May AE, Bultmann A, Gawaz M. ADAM 15 is an adhesion receptor for platelet GPIIb-IIIa and induces platelet activation. Thromb Haemost. 2005;94:555–561. doi: 10.1160/TH04-12-0784. [DOI] [PubMed] [Google Scholar]
- Laufs S, Schumacher J, Allgayer H. Urokinase-receptor (u-PAR): an essential player in multiple games of cancer: a review on its role in tumor progression, invasion, metastasis, proliferation/dormancy, clinical outcome and minimal residual disease. Cell Cycle. 2006;5:1760–1771. doi: 10.4161/cc.5.16.2994. [DOI] [PubMed] [Google Scholar]
- Lee SL, Dickson RB, Lin CY. Activation of hepatocyte growth factor and urokinase/plasminogen activator by matriptase, an epithelial membrane serine protease. J Biol Chem. 2000;275:36720–36725. doi: 10.1074/jbc.M007802200. [DOI] [PubMed] [Google Scholar]
- Lehti K, Lohi J, Valtanen H, Keski-Oja J. Proteolytic processing of membrane-type-1 matrix metalloproteinase is associated with gelatinase A activation at the cell surface. Biochem J. 1998;334(Pt 2):345–353. doi: 10.1042/bj3340345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemjabbar H, Li D, Gallup M, Sidhu S, Drori E, Basbaum C. Tobacco smoke-induced lung cell proliferation mediated by tumor necrosis factor alpha-converting enzyme and amphiregulin. J Biol Chem. 2003;278:26202–26207. doi: 10.1074/jbc.M207018200. [DOI] [PubMed] [Google Scholar]
- Levi M, Moons L, Bouche A, Shapiro SD, Collen D, Carmeliet P. Deficiency of urokinase-type plasminogen activator-mediated plasmin generation impairs vascular remodeling during hypoxia-induced pulmonary hypertension in mice. Circulation. 2001;103:2014–2020. doi: 10.1161/01.cir.103.15.2014. [DOI] [PubMed] [Google Scholar]
- Li Q, Park PW, Wilson CL, Parks WC. Matrilysin shedding of syndecan-1 regulates chemokine mobilization and transepithelial efflux of neutrophils in acute lung injury. Cell. 2002;111:635–646. doi: 10.1016/s0092-8674(02)01079-6. [DOI] [PubMed] [Google Scholar]
- Li Y, Brazzell J, Herrera A, Walcheck B. ADAM17 deficiency by mature neutrophils has differential effects on L-selectin shedding. Blood. 2006;108:2275–2279. doi: 10.1182/blood-2006-02-005827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- List K, Bugge TH, Szabo R. Matriptase: potent proteolysis on the cell surface. Mol Med. 2006;12:1–7. doi: 10.2119/2006-00022.List. [DOI] [PMC free article] [PubMed] [Google Scholar]
- List K, Szabo R, Molinolo A, Sriuranpong V, Redeye V, Murdock T, et al. Deregulated matriptase causes ras-independent multistage carcinogenesis and promotes ras-mediated malignant transformation. Genes Dev. 2005;19:1934–1950. doi: 10.1101/gad.1300705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liszt F, Schnittker-Schulze K, Stuhlsatz HW, Greiling H. Composition of proteoglycan fragments from hyaline cartilage produced by granulocytes in a model of frustrated phagocytosis. Eur J Clin Chem Clin Biochem. 1991;29:123–130. [PubMed] [Google Scholar]
- Liu PC, Liu X, Li Y, Covington M, Wynn R, Huber R, et al. Identification of ADAM10 as a major source of HER2 ectodomain sheddase activity in HER2 overexpressing breast cancer cells. Cancer Biol Ther. 2006;5:657–664. doi: 10.4161/cbt.5.6.2708. [DOI] [PubMed] [Google Scholar]
- Loechel F, Overgaard MT, Oxvig C, Albrechtsen R, Wewer UM. Regulation of human ADAM 12 protease by the prodomain. Evidence for a functional cysteine switch. J Biol Chem. 1999;274:13427–13433. doi: 10.1074/jbc.274.19.13427. [DOI] [PubMed] [Google Scholar]
- Lohi J, Lehti K, Westermarck J, Kahari VM, Keski-Oja J. Regulation of membrane-type matrix metalloproteinase-1 expression by growth factors and phorbol 12-myristate 13-acetate. Eur J Biochem. 1996;239:239–247. doi: 10.1111/j.1432-1033.1996.0239u.x. [DOI] [PubMed] [Google Scholar]
- Lum L, Reid MS, Blobel CP. Intracellular maturation of the mouse metalloprotease disintegrin MDC15. J Biol Chem. 1998;273:26236–26247. doi: 10.1074/jbc.273.40.26236. [DOI] [PubMed] [Google Scholar]
- Lum L, Wong BR, Josien R, Becherer JD, Erdjument-Bromage H, Schlondorff J, et al. Evidence for a role of a tumor necrosis factor-alpha (TNF-alpha)-converting enzyme-like protease in shedding of TRANCE, a TNF family member involved in osteoclastogenesis and dendritic cell survival. J Biol Chem. 1999;274:13613–13618. doi: 10.1074/jbc.274.19.13613. [DOI] [PubMed] [Google Scholar]
- MacIvor DM, Shapiro SD, Pham CT, Belaaouaj A, Abraham SN, Ley TJ. Normal neutrophil function in cathepsin G-deficient mice. Blood. 1999;94:4282–4293. [PubMed] [Google Scholar]
- Mai J, Waisman DM, Sloane BF. Cell surface complex of cathepsin B/annexin II tetramer in malignant progression. Biochim Biophys Acta. 2000;1477:215–230. doi: 10.1016/s0167-4838(99)00274-5. [DOI] [PubMed] [Google Scholar]
- Maquoi E, Frankenne F, Baramova E, Munaut C, Sounni NE, Remacle A, et al. Membrane type 1 matrix metalloproteinase-associated degradation of tissue inhibitor of metalloproteinase 2 in human tumor cell lines. J Biol Chem. 2000;275:11368–11378. doi: 10.1074/jbc.275.15.11368. [DOI] [PubMed] [Google Scholar]
- Marcotte PA, Henkin J. Characterization of the activation of pro-urokinase by thermolysin. Biochim Biophys Acta. 1993;1161:105–112. doi: 10.1016/0167-4838(93)90203-4. [DOI] [PubMed] [Google Scholar]
- Martin J, Eynstone LV, Davies M, Williams JD, Steadman R. The role of ADAM 15 in glomerular mesangial cell migration. J Biol Chem. 2002;277:33683–33689. doi: 10.1074/jbc.M200988200. [DOI] [PubMed] [Google Scholar]
- Mason RW, Johnson DA, Barrett AJ, Chapman HA., Jr Elastinolytic activity of human cathepsin L. Biochem J. 1986;233:925–927. doi: 10.1042/bj2330925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthay MA, Zimmerman GA. Acute lung injury and the acute respiratory distress syndrome: four decades of inquiry into pathogenesis and rational management. Am J Respir Cell Mol Biol. 2005;33:319–327. doi: 10.1165/rcmb.F305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May AE, Kanse SM, Lund LR, Gisler RH, Imhof BA, Preissner KT. Urokinase receptor (CD87) regulates leukocyte recruitment via beta 2 integrins in vivo. J Exp Med. 1998;188:1029–1037. doi: 10.1084/jem.188.6.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuibban GA, Gong JH, Wong JP, Wallace JL, Clark-Lewis I, Overall CM. Matrix metalloproteinase processing of monocyte chemoattractant proteins generates CC chemokine receptor antagonists with anti-inflammatory properties in vivo. Blood. 2002;100:1160–1167. [PubMed] [Google Scholar]
- Medcalf RL. Fibrinolysis, inflammation, and regulation of the plasminogen activating system. J Thromb Haemost 5 Suppl. 2007;1:132–142. doi: 10.1111/j.1538-7836.2007.02464.x. [DOI] [PubMed] [Google Scholar]
- Migita K, Eguchi K, Kawabe Y, Ichinose Y, Tsukada T, Aoyagi T, et al. TNF-α-mediated expression of membrane-type matrix metalloproteinase in rheumatoid synovial fibroblasts. Immunology. 1996;89:553–557. doi: 10.1046/j.1365-2567.1996.d01-789.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millichip MI, Dallas DJ, Wu E, Dale S, McKie N. The metallo-disintegrin ADAM10 (MADM) from bovine kidney has type IV collagenase activity in vitro. Biochem Biophys Res Commun. 1998;245:594–598. doi: 10.1006/bbrc.1998.8485. [DOI] [PubMed] [Google Scholar]
- Mingers AM, Heimburger N, Zeitler P, Kreth HW, Schuster V. Homozygous type I plasminogen deficiency. Semin Thromb Hemost. 1997;23:259–269. doi: 10.1055/s-2007-996099. [DOI] [PubMed] [Google Scholar]
- Mondino A, Blasi F. uPA and uPAR in fibrinolysis, immunity and pathology. Trends Immunol. 2004;25:450–455. doi: 10.1016/j.it.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Montuori N, Visconte V, Rossi G, Ragno P. Soluble and cleaved forms of the urokinase-receptor: degradation products or active molecules? Thromb Haemost. 2005;93:192–198. doi: 10.1160/TH04-09-0580. [DOI] [PubMed] [Google Scholar]
- Moran P, Li W, Fan B, Vij R, Eigenbrot C, Kirchhofer D. Pro-urokinase-type plasminogen activator is a substrate for hepsin. J Biol Chem. 2006;281:30439–30446. doi: 10.1074/jbc.M605440200. [DOI] [PubMed] [Google Scholar]
- Murphy G, Knauper V, Lee MH, Amour A, Worley JR, Hutton M, et al. Role of TIMPs (tissue inhibitors of metalloproteinases) in pericellular proteolysis: the specificity is in the detail. Biochem Soc Symp. 2003:65–80. doi: 10.1042/bss0700065. [DOI] [PubMed] [Google Scholar]
- Murphy G, Stanton H, Cowell S, Butler G, Knauper V, Atkinson S, et al. Mechanisms for pro matrix metalloproteinase activation. APMIS. 1999;107:38–44. doi: 10.1111/j.1699-0463.1999.tb01524.x. [DOI] [PubMed] [Google Scholar]
- Myles DG, Kimmel LH, Blobel CP, White JM, Primakoff P. Identification of a binding site in the disintegrin domain of fertilin required for sperm-egg fusion. Proc Natl Acad Sci USA. 1994;91:4195–4198. doi: 10.1073/pnas.91.10.4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath D, Slocombe PM, Stephens PE, Warn A, Hutchinson GR, Yamada KM, et al. Interaction of metargidin (ADAM-15) with alphavbeta3 and alpha5beta1 integrins on different haemopoietic cells. J Cell Sci. 1999;112(Pt 4):579–587. doi: 10.1242/jcs.112.4.579. [DOI] [PubMed] [Google Scholar]
- Nath D, Slocombe PM, Webster A, Stephens PE, Docherty AJ, Murphy G. Meltrin gamma(ADAM-9) mediates cellular adhesion through alpha(6)beta(1 )integrin, leading to a marked induction of fibroblast cell motility. J Cell Sci. 2000;113(Pt 12):2319–2328. doi: 10.1242/jcs.113.12.2319. [DOI] [PubMed] [Google Scholar]
- Naylor EJ, Bakstad D, Biffen M, Thong B, Calverley P, Scott S, et al. Haemophilus influenzae induces neutrophil necrosis: a role in chronic obstructive pulmonary disease? Am J Respir Cell Mol Biol. 2007;37:135–143. doi: 10.1165/rcmb.2006-0375OC. [DOI] [PubMed] [Google Scholar]
- Nelson KK, Schlondorff J, Blobel CP. Evidence for an interaction of the metalloprotease-disintegrin tumour necrosis factor alpha convertase (TACE) with mitotic arrest deficient 2 (MAD2), and of the metalloprotease-disintegrin MDC9 with a novel MAD2-related protein, MAD2beta. Biochem J. 1999;343(Pt 3):673–680. [PMC free article] [PubMed] [Google Scholar]
- Nie J, Pei D. Direct activation of pro-matrix metalloproteinase-2 by leukolysin/membrane-type 6 matrix metalloproteinase/matrix metalloproteinase 25 at the asn(109)-Tyr bond. Cancer Res. 2003;63:6758–6762. [PubMed] [Google Scholar]
- Oberst M, Anders J, Xie B, Singh B, Ossandon M, Johnson M, et al. Matriptase and HAI-1 are expressed by normal and malignant epithelial cells in vitro and in vivo. Am J Pathol. 2001;158:1301–1311. doi: 10.1016/S0002-9440(10)64081-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, Takahashi R, Kondo S, Mizoguchi A, Adachi E, Sasahara RM, et al. The membrane-anchored MMP inhibitor RECK is a key regulator of extracellular matrix integrity and angiogenesis. Cell. 2001;107:789–800. doi: 10.1016/s0092-8674(01)00597-9. [DOI] [PubMed] [Google Scholar]
- Ohta S, Harigai M, Tanaka M, Kawaguchi Y, Sugiura T, Takagi K, et al. Tumor necrosis factor-alpha (TNF-alpha) converting enzyme contributes to production of TNF-alpha in synovial tissues from patients with rheumatoid arthritis. J Rheumatol. 2001;28:1756–1763. [PubMed] [Google Scholar]
- Ohuchi E, Imai K, Fujii Y, Sato H, Seiki M, Okada Y. Membrane type 1 matrix metalloproteinase digests interstitial collagens and other extracellular matrix macromolecules. J Biol Chem. 1997;272:2446–2451. doi: 10.1074/jbc.272.4.2446. [DOI] [PubMed] [Google Scholar]
- Orgel D, Schroder W, Hecker-Kia A, Weithmann KU, Kolkenbrock H, Ulbrich N. The cleavage of pro-urokinase type plasminogen activator by stromelysin-1. Clin Chem Lab Med. 1998;36:697–702. doi: 10.1515/CCLM.1998.123. [DOI] [PubMed] [Google Scholar]
- Origuchi T, Migita K, Nakashima T, Tominaga M, Nakamura H, Nakashima M, et al. IL-1-mediated expression of membrane type matrix-metalloproteinase in rheumatoid osteoblasts. Clin Exp Rheumatol. 2000;18:333–339. [PubMed] [Google Scholar]
- Ortiz RM, Karkkainen I, Huovila AP. Aberrant alternative exon use and increased copy number of human metalloprotease-disintegrin ADAM15 gene in breast cancer cells. Genes Chromosomes Cancer. 2004;41:366–378. doi: 10.1002/gcc.20102. [DOI] [PubMed] [Google Scholar]
- Owen CA, Campbell EJ. Angiotensin II generation at the cell surface of activated neutrophils: Novel cathepsin G-mediated catalytic activity that is resistant to inhibition. J Immunol. 1998;160:1436–1443. [PubMed] [Google Scholar]
- Owen CA, Campbell EJ. The cell biology of leukocyte-mediated proteolysis. J Leukoc Biol. 1999;65:137–150. doi: 10.1002/jlb.65.2.137. [DOI] [PubMed] [Google Scholar]
- Owen CA, Campbell MA, Boukedes SS, Campbell EJ. Inducible binding of cathepsin G to the cell surface of neutrophils: A mechanism for mediating extracellular proteolytic activity of cathepsin G. J Immunol. 1995a;155:5803–5810. [PubMed] [Google Scholar]
- Owen CA, Campbell MA, Boukedes SS, Campbell EJ. Cytokines regulate membrane-bound leukocyte elastase on neutrophils: A novel mechanism for effector activity. Am J Physiol (Lung Cell Mol Physiol ) 1997;272:L385–L393. doi: 10.1152/ajplung.1997.272.3.L385. [DOI] [PubMed] [Google Scholar]
- Owen CA, Campbell MA, Boukedes SS, Stockley RA, Campbell EJ. A discrete subpopulation of human monocytes expresses a neutrophil-like pro-inflammatory (P) phenotype. Am J Physiol (Lung Cell Mol Physiol 11) 1994;267:L775–L785. doi: 10.1152/ajplung.1994.267.6.L775. [DOI] [PubMed] [Google Scholar]
- Owen CA, Campbell MA, Sannes PL, Boukedes SS, Campbell EJ. Cell-surface-bound elastase and cathepsin G on human neutrophils. A novel, non-oxidative mechanism by which neutrophils focus and preserve catalytic activity of serine proteinases. J Cell Biol. 1995b;131:775–789. doi: 10.1083/jcb.131.3.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen CA, Hu Z, Barrick B, Shapiro SD. Inducible expression of tissue inhibitor of metalloproteinases-resistant matrix metalloproteinase-9 on the cell surface of neutrophils. Am J Resp Cell Mol Biol. 2003;29:283–294. doi: 10.1165/rcmb.2003-0034OC. [DOI] [PubMed] [Google Scholar]
- Owen CA, Hu Z, Lopez-Otin C, Shapiro SD. Membrane-bound matrix metalloproteinase-8 on activated polymorphonuclear cells is a potent, tissue inhibitor of metalloproteinase-resistant collagenase and serpinase. J Immunol. 2004;172:7791–7803. doi: 10.4049/jimmunol.172.12.7791. [DOI] [PubMed] [Google Scholar]
- Parks WC, Wilson CL, Lopez-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol. 2004;4:617–629. doi: 10.1038/nri1418. [DOI] [PubMed] [Google Scholar]
- Pei D. Identification and characterization of the fifth membrane-type matrix metalloproteinase MT5-MMP. J Biol Chem. 1999;274:8925–8932. doi: 10.1074/jbc.274.13.8925. [DOI] [PubMed] [Google Scholar]
- Pei D. Matrix metalloproteinases target protease-activated receptors on the tumor cell surface. Cancer Cell. 2005;7:207–208. doi: 10.1016/j.ccr.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC, et al. An essential role for ectodomain shedding in mammalian development. Science. 1998;282:1281–1284. doi: 10.1126/science.282.5392.1281. [DOI] [PubMed] [Google Scholar]
- Pham CT. Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol. 2006;6:541–550. doi: 10.1038/nri1841. [DOI] [PubMed] [Google Scholar]
- Pillay V, Dass CR, Choong PF. The urokinase plasminogen activator receptor as a gene therapy target for cancer. Trends Biotechnol. 2007;25:33–39. doi: 10.1016/j.tibtech.2006.10.011. [DOI] [PubMed] [Google Scholar]
- Plesner T, Ploug M, Ellis V, Ronne E, Hoyer-Hansen G, Wittrup M, et al. The receptor for urokinase-type plasminogen activator and urokinase is translocated from two distinct intracellular compartments to the plasma membrane on stimulation of human neutrophils. Blood. 1994;83:808–815. [PubMed] [Google Scholar]
- Ploplis VA, Carmeliet P, Vazirzadeh S, Van VI, Moons L, Plow EF, et al. Effects of disruption of the plasminogen gene on thrombosis, growth, and health in mice. Circulation. 1995;92:2585–2593. doi: 10.1161/01.cir.92.9.2585. [DOI] [PubMed] [Google Scholar]
- Pluskota E, Soloviev DA, Bdeir K, Cines DB, Plow EF. Integrin alphaMbeta2 orchestrates and accelerates plasminogen activation and fibrinolysis by neutrophils. J Biol Chem. 2004;279:18063–18072. doi: 10.1074/jbc.M310462200. [DOI] [PubMed] [Google Scholar]
- Podgorski I, Sloane BF. Cathepsin B and its role(s) in cancer progression. Biochem Soc Symp. 2003:263–276. doi: 10.1042/bss0700263. [DOI] [PubMed] [Google Scholar]
- Primakoff P, Myles DG. The ADAM gene family: surface proteins with adhesion and protease activity. Trends Genet. 2000;16:83–87. doi: 10.1016/s0168-9525(99)01926-5. [DOI] [PubMed] [Google Scholar]
- Puente XS, Sanchez LM, Overall CM, Lopez-Otin C. Human and mouse proteases: a comparative genomic approach. Nat Rev Genet. 2003;4:544–558. doi: 10.1038/nrg1111. [DOI] [PubMed] [Google Scholar]
- Qiu D, Owen K, Gray K, Bass R, Ellis V. Roles and regulation of membrane-associated serine proteases. Biochem Soc Trans. 2007a;35:583–587. doi: 10.1042/BST0350583. [DOI] [PubMed] [Google Scholar]
- Qiu D, Owen K, Gray K, Bass R, Ellis V. Roles and regulation of membrane-associated serine proteases. Biochem Soc Trans. 2007b;35:583–587. doi: 10.1042/BST0350583. [DOI] [PubMed] [Google Scholar]
- Quinton TM, Kim S, Derian CK, Jin J, Kunapuli SP. Plasmin-mediated activation of platelets occurs by cleavage of protease-activated receptor 4. J Biol Chem. 2004;279:18434–18439. doi: 10.1074/jbc.M401431200. [DOI] [PubMed] [Google Scholar]
- Ragno P. The urokinase receptor: a ligand or a receptor? Story of a sociable molecule. Cell Mol Life Sci. 2006;63:1028–1037. doi: 10.1007/s00018-005-5428-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralston DR, Marsh CB, Lowe MP, Wewers MD. Antineutrophil cytoplasmic antibodies induce monocyte IL-8 release. Role of surface proteinase-3, alpha1-antitrypsin, and Fcgamma receptors. J Clin Invest. 1997;100:1416–1424. doi: 10.1172/JCI119662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raza SL, Nehring LC, Shapiro SD, Cornelius LA. Proteinase activated receptor-1 regulation of macrophage elastase secretion by serine proteinases. J Biol Chem. 2000;52:41243–41250. doi: 10.1074/jbc.M005788200. [DOI] [PubMed] [Google Scholar]
- Reiss K, Maretzky T, Ludwig A, Tousseyn T, De Strooper B, Hartmann D, et al. ADAM10 cleavage of N-cadherin and regulation of cell-cell adhesion and beta-catenin nuclear signalling. EMBO J. 2005;24:742–752. doi: 10.1038/sj.emboj.7600548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnati M, Pallavicini I, Wang JM, Oppenheim J, Serhan CN, Romano M, et al. The fibrinolytic receptor for urokinase activates the G protein-coupled chemotactic receptor FPRL1/LXA4R. Proc Natl Acad Sci USA. 2002;99:1359–1364. doi: 10.1073/pnas.022652999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rijneveld AW, Levi M, Florquin S, Speelman P, Carmeliet P, van Der PT. Urokinase receptor is necessary for adequate host defense against pneumococcal pneumonia. The Journal of Immunology. 2002;168:3507–3511. doi: 10.4049/jimmunol.168.7.3507. [DOI] [PubMed] [Google Scholar]
- Rio C, Buxbaum JD, Peschon JJ, Corfas G. Tumor necrosis factor-alpha-converting enzyme is required for cleavage of erbB4/HER4. J Biol Chem. 2000;275:10379–10387. doi: 10.1074/jbc.275.14.10379. [DOI] [PubMed] [Google Scholar]
- Rogachefsky RA, Dean DD, Howell DS, Altman RD. Treatment of canine osteoarthritis with insulin-like growth factor-1 (IGF-1) and sodium pentosan polysulfate. Osteoarthritis Cartilage. 1993;1:105–114. doi: 10.1016/s1063-4584(05)80025-1. [DOI] [PubMed] [Google Scholar]
- Roghani M, Becherer JD, Moss ML, Atherton RE, Erdjument-Bromage H, Arribas J, et al. Metalloprotease-disintegrin MDC9: Intracellular maturation and catalytic activity. J Biol Chem. 1999;274:3531–3540. doi: 10.1074/jbc.274.6.3531. [DOI] [PubMed] [Google Scholar]
- Romer J, Bugge TH, Pyke C, Lund LR, Flick MJ, Degen JL, et al. Impaired wound healing in mice with a disrupted plasminogen gene. Nat Med. 1996;2:287–292. doi: 10.1038/nm0396-287. [DOI] [PubMed] [Google Scholar]
- Sahin U, Weskamp G, Kelly K, Zhou HM, Higashiyama S, Peschon J, et al. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J Cell Biol. 2004;164:769–779. doi: 10.1083/jcb.200307137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallenave JM, Silva A, Marsden ME, Ryle AP. Secretion of mucus proteinase inhibitor and elafin by Clara cell and type II pneumocyte cell lines. Am J Respir Cell Mol Biol. 1993;8:126–133. doi: 10.1165/ajrcmb/8.2.126. [DOI] [PubMed] [Google Scholar]
- Sameni M, Moin K, Sloane BF. Imaging proteolysis by living human breast cancer cells. Neoplasia. 2000;2:496–504. doi: 10.1038/sj.neo.7900116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato H, Kinoshita T, Takino T, Nakayama K, Seiki M. Activation of a recombinant membrane type 1-matrix metalloproteinase (MT1-MMP) by furin and its interaction with tissue inhibitor of metalloproteinases (TIMP)-2. FEBS Lett. 1996;393:101–104. doi: 10.1016/0014-5793(96)00861-7. [DOI] [PubMed] [Google Scholar]
- Schafer B, Gschwind A, Ullrich A. Multiple G-protein-coupled receptor signals converge on the epidermal growth factor receptor to promote migration and invasion. Oncogene. 2004;23:991–999. doi: 10.1038/sj.onc.1207278. [DOI] [PubMed] [Google Scholar]
- Schlomann U, Rathke-Hartlieb S, Yamamoto S, Jockusch H, Bartsch JW. Tumor necrosis factor alpha induces a metalloprotease-disintegrin, ADAM8 (CD 156): implications for neuron-glia interactions during neurodegeneration. J Neurosci. 2000;20:7964–7971. doi: 10.1523/JNEUROSCI.20-21-07964.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmaier AH, McCrae KR. The plasma kallikrein-kinin system: its evolution from contact activation. J Thromb Haemost. 2007;5:2323–2329. doi: 10.1111/j.1538-7836.2007.02770.x. [DOI] [PubMed] [Google Scholar]
- Schreiber A, Busjahn A, Luft FC, Kettritz R. Membrane expression of proteinase 3 is genetically determined. J Am Soc Nephrol. 2003;14:68–75. doi: 10.1097/01.asn.0000040751.83734.d1. [DOI] [PubMed] [Google Scholar]
- Schreiber A, Otto B, Ju X, Zenke M, Goebel U, Luft FC, et al. Membrane proteinase 3 expression in patients with Wegener’s granulomatosis and in human hematopoietic stem cell-derived neutrophils. J Am Soc Nephrol. 2005;16:2216–2224. doi: 10.1681/ASN.2004070609. [DOI] [PubMed] [Google Scholar]
- Seals DF, Courtneidge SA. The ADAMs family of metalloproteases: multidomain proteins with multiple functions. Genes Dev. 2003;17:7–30. doi: 10.1101/gad.1039703. [DOI] [PubMed] [Google Scholar]
- Seitz I, Hess S, Schulz H, Eckl R, Busch G, Montens HP, et al. Membrane-type serine protease-1/matriptase induces interleukin-6 and -8 in endothelial cells by activation of protease-activated receptor-2: potential implications in atherosclerosis. Arterioscler Thromb Vasc Biol. 2007;27:769–775. doi: 10.1161/01.ATV.0000258862.61067.14. [DOI] [PubMed] [Google Scholar]
- Shapiro SD. Matrix metalloproteinase degradation of extracellular matrix: Biological consequences. Curr Opin Cell Biol. 1998;10:602–608. doi: 10.1016/s0955-0674(98)80035-5. [DOI] [PubMed] [Google Scholar]
- Shapiro SD, Goldstein NM, Houghton AM, Kobayashi DK, Kelley D, Belaaouaj A. Neutrophil elastase contributes to cigarette smoke-induced emphysema in mice. Am J Pathol. 2003;163:2329–2335. doi: 10.1016/S0002-9440(10)63589-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro SD, Owen CA. ADAM-33 surfaces as an asthma gene. N Engl J Med. 2002;347:936–938. doi: 10.1056/NEJMcibr022144. [DOI] [PubMed] [Google Scholar]
- Shi GP, Munger JS, Meara JP, Rich DH, Chapman HA. Molecular cloning and expression of human alveolar macrophage cathepsin S, an elastinolytic cysteine protease. J Biol Chem. 1992;267:7258–7262. [PubMed] [Google Scholar]
- Sidenius N, Blasi F. The urokinase plasminogen activator system in cancer: recent advances and implication for prognosis and therapy. Cancer Metastasis Rev. 2003;22:205–222. doi: 10.1023/a:1023099415940. [DOI] [PubMed] [Google Scholar]
- Sisson TH, Hanson KE, Subbotina N, Patwardhan A, Hattori N, Simon RH. Inducible lung-specific urokinase expression reduces fibrosis and mortality after lung injury in mice. Am J Physiol Lung Cell Mol Physiol. 2002;283:L1023–L1032. doi: 10.1152/ajplung.00049.2002. [DOI] [PubMed] [Google Scholar]
- Stamenkovic I. Matrix metalloproteinases in tumor invasion and metastasis. Semin Cancer Biol. 2000;10:415–433. doi: 10.1006/scbi.2000.0379. [DOI] [PubMed] [Google Scholar]
- Stawowy P, Meyborg H, Stibenz D, Borges Pereira SN, Roser M, Thanabalasingam U, et al. Furin-like proprotein convertases are central regulators of the membrane type matrix metalloproteinase-pro-matrix metalloproteinase-2 proteolytic cascade in atherosclerosis. Circulation. 2005;111:2820–2827. doi: 10.1161/CIRCULATIONAHA.104.502617. [DOI] [PubMed] [Google Scholar]
- Stefanidakis M, Bjorklund M, Ihanus E, Gahmberg CG, Koivunen E. Identification of a negatively charged peptide motif within the catalytic domain of progelatinases that mediates binding to leukocyte beta 2 integrins. J Biol Chem. 2003;278:34674–34684. doi: 10.1074/jbc.M302288200. [DOI] [PubMed] [Google Scholar]
- Stefanidakis M, Ruohtula T, Borregaard N, Gahmberg CG, Koivunen E. Intracellular and cell surface localization of a complex between alphaMbeta2 integrin and promatrix metalloproteinase-9 progelatinase in neutrophils. The Journal of Immunology. 2004;172:7060–7068. doi: 10.4049/jimmunol.172.11.7060. [DOI] [PubMed] [Google Scholar]
- Stone AL, Kroeger M, Sang QX. Structure-function analysis of the ADAM family of disintegrin-like and metalloproteinase-containing proteins (review) J Protein Chem. 1999;18:447–465. doi: 10.1023/a:1020692710029. [DOI] [PubMed] [Google Scholar]
- Strongin AY, Collier I, Bannikov G, Marmer BL, Grant GA, Goldberg GI. Mechanism of cell surface activation of 72-kDa type IV collagenase: Isolation of the activated form of the membrane metalloprotease. J Biol Chem. 1995;270:5331–5338. doi: 10.1074/jbc.270.10.5331. [DOI] [PubMed] [Google Scholar]
- Sun R, Iribarren P, Zhang N, Zhou Y, Gong W, Cho EH, et al. Identification of neutrophil granule protein cathepsin G as a novel chemotactic agonist for the G protein-coupled formyl peptide receptor. The Journal of Immunology. 2004;173:428–436. doi: 10.4049/jimmunol.173.1.428. [DOI] [PubMed] [Google Scholar]
- Syrovets T, Simmet T. Novel aspects and new roles for the serine protease plasmin. Cell Mol Life Sci. 2004;61:873–885. doi: 10.1007/s00018-003-3348-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo R, Wu Q, Dickson RB, Netzel-Arnett S, Antalis TM, Bugge TH. Type II transmembrane serine proteases. Thromb Haemost. 2003;90:185–193. doi: 10.1160/TH03-02-0071. [DOI] [PubMed] [Google Scholar]
- Taipale J, Koli K, Keski-Oja J. Release of transforming growth factor-beta 1 from the pericellular matrix of cultured fibroblasts and fibrosarcoma cells by plasmin and thrombin. J Biol Chem. 1992;267:25378–25384. [PubMed] [Google Scholar]
- Takeuchi T, harris JL, huang W, Yan KW, Coughlin SR, Craik CS. Cellular localization of membrane-type serine protease 1 and identification of protease-activated receptor-2 and single-chain urokinase-type plasminogen activator as substrates. J Biol Chem. 2000;275:26333–26342. doi: 10.1074/jbc.M002941200. [DOI] [PubMed] [Google Scholar]
- Takeyama K, Agusti C, Ueki I, Lausier J, Cardell LO, Nadel JA. Neutrophil-dependent goblet cell degranulation: role of membrane-bound elastase and adhesion molecules. Am J Physiol. 1998;275:L294–L302. doi: 10.1152/ajplung.1998.275.2.L294. [DOI] [PubMed] [Google Scholar]
- Trask BC, Malone MJ, Lum EH, Welgus HG, Crouch EC, Shapiro SD. Induction of macrophage matrix metalloproteinase biosynthesis by surfactant protein D. J Biol Chem. 2001;276:37846–37852. doi: 10.1074/jbc.M102524200. [DOI] [PubMed] [Google Scholar]
- Turk B, Turk V, Turk D. Structural and functional aspects of papain-like cysteine proteinases and their protein inhibitors. Biol Chem. 1997;378:141–150. [PubMed] [Google Scholar]
- Vachon E, Bourbonnais Y, Bingle CD, Rowe SJ, Janelle MF, Tremblay GM. Anti-inflammatory effect of pre-elafin in lipopolysaccharide-induced acute lung inflammation. Biol Chem. 2002;383:1249–1256. doi: 10.1515/BC.2002.138. [DOI] [PubMed] [Google Scholar]
- Van Eerdewegh P, Little RD, Dupuis J, Del Mastro RG, Falls K, Simon J, et al. Association of the ADAM33 gene with asthma and bronchial hyperresponsiveness. Nature. 2002;418:426–430. doi: 10.1038/nature00878. [DOI] [PubMed] [Google Scholar]
- van Rossum AP, Limburg PC, Kallenberg CG. Membrane proteinase 3 expression on resting neutrophils as a pathogenic factor in PR3-ANCA-associated vasculitis. Clin Exp Rheumatol. 2003;21:S64–S68. [PubMed] [Google Scholar]
- Vandivier RW, Fadok VA, Hoffmann PR, Bratton DL, Penvari C, Brown KK, et al. Elastase-mediated phosphatidylserine receptor cleavage impairs apoptotic cell clearance in cystic fibrosis and bronchiectasis. J Clin Invest. 2002;109:661–670. doi: 10.1172/JCI13572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassalli JD, Baccino D, Belin D. A cellular binding site for the Mr 55,000 form of the human plasminogen activator, urokinase. J Cell Biol. 1985;100:86–92. doi: 10.1083/jcb.100.1.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Vietinghoff S, Tunnemann G, Eulenberg C, Wellner M, Cristina CM, Luft FC, et al. NB1 mediates surface expression of the ANCA antigen proteinase 3 on human neutrophils. Blood. 2007;109:4487–4493. doi: 10.1182/blood-2006-10-055327. [DOI] [PubMed] [Google Scholar]
- Wahl LM, Corcoran ML. Regulation of monocyte/macrophage metalloproteinase production by cytokines. J Periodontol. 1993;64:467–473. [PubMed] [Google Scholar]
- Walcheck B, Alexander SR, St Hill CA, Matala E. ADAM-17-independent shedding of L-selectin. J Leukoc Biol. 2003;74:389–394. doi: 10.1189/jlb.0403141. [DOI] [PubMed] [Google Scholar]
- Walcheck B, Herrera AH, St Hill C, Mattila PE, Whitney AR, Deleo FR. ADAM17 activity during human neutrophil activation and apoptosis. Eur J Immunol. 2006;36:968–976. doi: 10.1002/eji.200535257. [DOI] [PubMed] [Google Scholar]
- Wang X, Yi J, Lei J, Pei D. Expression, purification and characterization of recombinant mouse MT5-MMP protein products. FEBS Lett. 1999;462:261–266. doi: 10.1016/s0014-5793(99)01534-3. [DOI] [PubMed] [Google Scholar]
- Wang Y, Johnson AR, Ye QZ, Dyer RD. Catalytic activities and substrate specificity of the human membrane type 4 matrix metalloproteinase catalytic domain. J Biol Chem. 1999;274:33043–33049. doi: 10.1074/jbc.274.46.33043. [DOI] [PubMed] [Google Scholar]
- Wei Y, Lukashev M, Simon DI, Bodary SC, Rosenberg S, Doyle MV, et al. Regulation of integrin function by the urokinase receptor. Science. 1996;273:1551–1555. doi: 10.1126/science.273.5281.1551. [DOI] [PubMed] [Google Scholar]
- Wilson CL, Ouellette AJ, Satchell DP, Ayabe T, Lopez-Boado YS, Stratman JL, et al. Regulation of intestinal α-defensin activation by the metalloproteinase matrilysin in innate host defense. Science. 1999;286:113–117. doi: 10.1126/science.286.5437.113. [DOI] [PubMed] [Google Scholar]
- Witko-Sarsat V, Cramer EM, Hieblot C, Guichard J, Nusbaum P, Lopez S, et al. Presence of proteinase 3 in secretory vesicles: evidence of a novel, highly mobilizable intracellular pool distinct from azurophil granules. Blood. 1999a;94:2487–2496. [PubMed] [Google Scholar]
- Witko-Sarsat V, Lesavre P, Lopez S, Bessou G, Hieblot C, Prum B, et al. A large subset of neutrophils expressing membrane proteinase 3 is a risk factor for vasculitis and rheumatoid arthritis. J Am Soc Nephrol. 1999b;10:1224–1233. doi: 10.1681/ASN.V1061224. [DOI] [PubMed] [Google Scholar]
- Wolfsberg TG, Bazan JF, Blobel CP, Myles DG, Primakoff P, White JM. The precursor region of a protein active in sperm-egg fusion contains a metalloprotease and a disintegrin domain: structural, functional, and evolutionary implications. ProcNatlAcadSciUSA. 1993;90:10783–10787. doi: 10.1073/pnas.90.22.10783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu E, Croucher PI, McKie N. Expression of members of the novel membrane linked metalloproteinase family ADAM in cells derived from a range of haematological malignancies. Biochem Biophys Res Commun. 1997;235:437–442. doi: 10.1006/bbrc.1997.6714. [DOI] [PubMed] [Google Scholar]
- Yagami-Hiromasa T, Sato T, Kurisaki T, Kamijo K, Nabeshima Y, Fujisawa-Sehara A. A metalloprotease-disintegrin participating in myoblast fusion. Nature. 1995;377:652–656. doi: 10.1038/377652a0. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Higuchi Y, Yoshiyama K, Shimizu E, Kataoka M, Hijiya N, et al. ADAM family proteins in the immune system. Immunol Today. 1999;20:278–284. doi: 10.1016/s0167-5699(99)01464-4. [DOI] [PubMed] [Google Scholar]
- Yang MX, Qu X, Kong BH, Lam QL, Shao QQ, Deng BP, et al. Membrane type 1-matrix metalloproteinase is involved in the migration of human monocyte-derived dendritic cells. Immunol Cell Biol. 2006;84:557–562. doi: 10.1111/j.1440-1711.2006.01465.x. [DOI] [PubMed] [Google Scholar]
- Yasuda S, Morokawa N, Wong GW, Rossi A, Madhusudhan MS, Sali A, et al. Urokinase-type plasminogen activator is a preferred substrate of the human epithelium serine protease tryptase epsilon/PRSS22. Blood. 2005;105:3893–3901. doi: 10.1182/blood-2003-10-3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q, Stamenkovic I. Localization of matrix metalloproteinase 9 to the cell surface provides a mechanism for CD44-mediated tumor invasion. Genes Dev. 1999;13:35–48. doi: 10.1101/gad.13.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14:163–176. [PMC free article] [PubMed] [Google Scholar]
- Yu WH, Woessner JF., Jr Heparan sulfate proteoglycans as extracellular docking molecules for matrilysin (matrix metalloproteinase 7) J Biol Chem. 2000;275:4183–4191. doi: 10.1074/jbc.275.6.4183. [DOI] [PubMed] [Google Scholar]
- Yu WH, Woessner JF, Jr, McNeish JD, Stamenkovic I. CD44 anchors the assembly of matrilysin/MMP-7 with heparin-binding epidermal growth factor precursor and ErbB4 and regulates female reproductive organ remodeling. Genes Dev. 2002;16:307–323. doi: 10.1101/gad.925702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Bernardo MM, Osenkowski P, Sohail A, Pei D, Nagase H, et al. Differential inhibition of membrane type 3 (MT3)-matrix metalloproteinase (MMP) and MT1-MMP by tissue inhibitor of metalloproteinase (TIMP)-2 and TIMP-3 rgulates pro-MMP-2 activation. J Biol Chem. 2004;279:8592–8601. doi: 10.1074/jbc.M308708200. [DOI] [PubMed] [Google Scholar]
- Zigrino P, Steiger J, Fox JW, Loffek S, Schild A, Nischt R, et al. Role of ADAM-9 disintegrin-cysteine-rich domains in human keratinocyte migration. J Biol Chem. 2007;282:30785–30793. doi: 10.1074/jbc.M701658200. [DOI] [PubMed] [Google Scholar]