

Abstract
Accurate force fields are essential for the success of molecular dynamics simulations. In apparent contrast to the conformational preferences of most force fields, recent NMR experiments suggest that short polyalanine peptides in water populate the polyproline II structure almost exclusively. To investigate this apparent contradiction, with its ramifications for the assessment of molecular force fields and the structure of unfolded proteins, we performed extensive simulations of Ala5 in water (∼5 μs total time), using twelve different force fields and three different peptide terminal groups. Using either empirical or density-functional-based Karplus relations for the J-couplings, we find that most current force fields do overpopulate the α-region, with quantitative results depending on the choice of Karplus relation and on the peptide termini. Even after reweighting to match experiment, we find that Ala5 retains significant α- and β-populations. In fact, several force fields match the experimental data well before reweighting and have a significant helical population. We conclude that radical changes to the best current force fields are not necessary, based on the NMR data. Nevertheless, experiments on short peptides open the way toward the systematic improvement of current simulation models.
All-atom molecular dynamics (MD) simulations are a powerful tool for the exploration and mechanistic interpretation of biological phenomena at a molecular scale (1). Nonetheless, longer simulation times and interest in the unfolded states of proteins (2–4) and in natively unstructured peptides (5,6) are uncovering deficiencies in the commonly used energy functions, or “force fields” (7), stimulating recent refinements of peptide backbone potentials (8–11). Conventional force field development relies heavily on gas-phase quantum chemical calculations and spectroscopic and thermodynamic data for small molecules (7). Experimental data for short peptides in solution would be a valuable addition (12), even if those data are usually averaged over heterogeneous equilibrium ensembles of structures (13,14).
Polyalanine is a well-established model for studies of peptide conformation and helix formation (2,4,11,15–18). An elegant recent study by Graf et al. (19) used scalar couplings measured by NMR in a series of polyalanine peptides (Ala3 to Ala7) to probe the distributions of backbone (φ,ψ) angles. Scalar couplings calculated from MD simulations were in only modest agreement with experiment. However, the simulation data could be reweighted by adjusting the relative populations of the α-, β-, and polyproline-II (ppII) regions of the Ramachandran map, with the implicit assumption that the simulations give a reasonably accurate sampling within each state. Remarkably, the reweighted populations indicated that Ala3 to Ala7 almost exclusively populate the ppII region of Ramachandran space with a small population of the β-region and negligible α-population.
In view of the importance of this conclusion for future force field development and structure in unfolded proteins (2,4), we have revisited it, employing a large set of MD simulation trajectories of polyalanine in explicit solvent at 300 K (4.9 μs total) (20,21). Details of the simulations are available in Supplementary Material, Data S1.
Remarkably, the 12 force field variants employed differ widely in their sampling of the Ramachandran space. Based on the scalar coupling data, most force fields indeed have too large a helical propensity. However, the quantitative results are sensitive to i), the choice of Karplus relation; ii), the protonation state; and iii), terminal blocking. Overall, we conclude that the NMR data are consistent with force fields that give a small helical population for Ala5, and do not require exclusive formation of ppII structure.
We classified (φ,ψ) space into “α” (–160 < φ < −20 and −120 < ψ < 50), “β” (–180° < φ < −90° and 50° < ψ < 240°; or 160° < φ < 180° and 110° < ψ < 180°), “ppII” (–90° < φ < −20° and 50° < ψ < 240°), and “other” regions, based on a set of φ,ψ frequencies recently derived from the Protein Data Bank (see Fig. S1 in Data S1). The more restrictive definition of Graf et al. (19) truncates the α-region in particular (see Fig. S1 in Data S1). The fraction of residues in the “other” category was always <0.5% except for the Gromos force fields (∼3%) and was combined with ppII in the subsequent analysis (note that omitting it altogether has a negligible effect on the results).
We quantified the agreement between experimental J-couplings and those calculated from unweighted simulation trajectories via Karplus relations (22) using
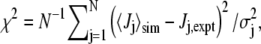 |
where  is the average of coupling j from the simulation. The set of N experimental couplings (19) Jj,expt includes 3JHNHα, 3JHNC', 3JHαC', 3JCC', 3JHNCβ (probing φ), 1JNCα, 2JNCα (probing ψ), and 3JHNCα (probing both φ and ψ) for each of the five residues. σj is the estimated systematic error in the couplings determined from the Karplus equation, arising mainly from the neglect of substituent effects (23) in the Karplus equation parameters (values and sources for σj given in Table S4 in Data S1). Sampling errors in
is the average of coupling j from the simulation. The set of N experimental couplings (19) Jj,expt includes 3JHNHα, 3JHNC', 3JHαC', 3JCC', 3JHNCβ (probing φ), 1JNCα, 2JNCα (probing ψ), and 3JHNCα (probing both φ and ψ) for each of the five residues. σj is the estimated systematic error in the couplings determined from the Karplus equation, arising mainly from the neglect of substituent effects (23) in the Karplus equation parameters (values and sources for σj given in Table S4 in Data S1). Sampling errors in 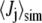 and experimental errors in Ji,expt are relatively small and so were not added to σj. We used the Karplus parameters of the original article (19), and two additional sets, with parameters determined from DFT calculations (24) on Ace-Ala-NMe (DFT1) and Ala-Ala-NH2 (DFT2). These may be more relevant to alanine, with its small side chain, than parameters fitted after pooling data from different residue types.
and experimental errors in Ji,expt are relatively small and so were not added to σj. We used the Karplus parameters of the original article (19), and two additional sets, with parameters determined from DFT calculations (24) on Ace-Ala-NMe (DFT1) and Ala-Ala-NH2 (DFT2). These may be more relevant to alanine, with its small side chain, than parameters fitted after pooling data from different residue types.
The α-helical content for different force fields varies between 13 and 98%, with high α-content resulting in poor agreement with experiment (Fig. 1, Table 1, and Table S5 in Data S1). However, a range of force fields with 10–30% α-content give a low χ2 ≤ 2; with the χ2 definition used here, this means the deviation from experiment is comparable to the error σj. In general, simulations of zwitterionic peptides or those with protonated C-termini resulted in lower χ2 than those for blocked peptides (see below).
FIGURE 1.

Secondary structure populations. Ternary diagrams on the left (A, C, and E) and right (B, D, and F) show the relative populations before and after reweighting for (A and B) the original Karplus parameters (19), (C and D) DFT1, and (E and F) DFT2 (24), respectively. Symbol areas are proportional to 1/χ2: (blue) blocked termini, (red) zwitterionic, (yellow) protonated C-terminus without ions, (green) protonated C-terminus with ions. Arrows indicate the direction in which each axis should be read, and colors indicate the corresponding scale and isolines.
TABLE 1.
Ramachandran populations for Ala5, before and after reweighting, for some representative force-fields (a full list is available in Data S1)
| Unweighted
|
Reweighted
|
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Time (ns) |
χ2
|
Populations
|
DFT1
|
DFT2
|
Orig
|
||||||||||
| FORCE-FIELD | DFT1 | DFT2 | ORIG | % α | % β | χ2 | % α | % β | χ2 | % α | % β | χ2 | % α | % β | |
| Amber03* | 120 | 1.8 | 1.5 | 1.6 | 33.0 | 30.7 | 1.4 | 11.0 | 39.9 | 0.8 | 8.0 | 24.5 | 0.3 | 6.6 | 5.3 |
| Amber99SB | 120 | 4.2 | 4.9 | 4.2 | 19.7 | 55.5 | 1.7 | 14.4 | 31.0 | 1.0 | 11.7 | 18.3 | 0.6 | 7.3 | 6.9 |
| AmberGS | 80 | 5.2 | 2.8 | 1.9 | 45.8 | 9.8 | 1.5 | 3.2 | 47.5 | 1.0 | 4.2 | 32.6 | 0.7 | 3.7 | 14.8 |
| CHARMM27/cmap* | 80 | 2.0 | 2.0 | 2.2 | 41.5 | 25.3 | 1.3 | 14.0 | 32.1 | 1.0 | 14.1 | 18.7 | 0.9 | 11.4 | 7.1 |
| OPLS-aa/L* | 80 | 1.8 | 2.0 | 2.0 | 30.9 | 33.5 | 1.5 | 10.0 | 42.0 | 1.1 | 7.7 | 22.3 | 0.4 | 5.2 | 3.9 |
| Gromos53a6 | 80 | 1.8 | 2.3 | 2.3 | 13.5 | 50.2 | 1.4 | 3.4 | 41.6 | 1.0 | 3.1 | 21.3 | 0.6 | 2.5 | 5.3 |
| Gromos43a1* | 80 | 1.4 | 1.4 | 1.6 | 14.0 | 41.2 | 1.2 | 2.4 | 40.8 | 0.7 | 2.3 | 23.7 | 0.5 | 2.6 | 8.6 |
In these simulations, the peptides were protonated at both termini (net charge +1).
Force fields with χ2 ≤ 2.25 before reweighting, corresponding to deviations from experiment up to 1.5 σj on average.
To determine the secondary structure populations that give the closest match with experiment, we adopt the procedure of Graf et al. (19). Each trajectory frame of a simulation was reweighted by a factor proportional to exp[−(nαɛα+nβɛβ)], where nα and nβ are the number of residues in the α-and β-regions in that frame, and ɛα and ɛβ are corresponding energy corrections (in kBT units relative to ppII), which are chosen to minimize χ2. The secondary structure content after reweighting is shown in Fig. 1 B, D, and F (optimal ɛα and ɛβ are given in Table S6 in Data S1). Using the Karplus parameters of Graf et al. (19), we also find almost exclusively ppII structure (Fig. 1 B). However, with the DFT1 and DFT2 parameter sets (24), there is a balance between β and ppII structure, with a small α-population (Fig. 1 D and F). Table 1 summarizes the Ramachandran populations before and after reweighting for a representative subset of the force fields.
Terminal effects on helical propensity are significant for short peptides (12), one reason being that the terminal charges at neutral pH oppose the helix dipole. We have quantified this effect by using pairs of simulations: one of a blocked peptide and the other of a C-terminally protonated peptide, both using the same force field. We optimize ɛα and ɛβ for the protonated molecule, which corresponds to experiment, and reweight the blocked molecule with the same values. The results (Fig. 2) show that blocking tends to favor α-structure over ppII; part of the reason for the low helicity reported by the NMR data is likely the use of uncapped peptides with protonated C-termini.
FIGURE 2.

Secondary structure populations for C-terminally protonated (blue) and blocked (red) peptides after re-weighting using (A) the original Karplus equation (19), (B) DFT1, and (C) DFT2 (24). Axes are as in Fig. 1.
The widely varying secondary structure propensities of polyalanine in different force fields (17) (Fig. 1) demonstrate the need for ongoing force field development. Although we find that, in general, current force fields overestimate α-structure, those that best match the experimental J-couplings have a significant α-helical fraction. For force fields for which χ2 ≤ 2 before reweighting (Table 1), the helical fraction varies between 14% and 33% (unweighted) and 2% and 11% (reweighted). We also find that terminal blocking significantly increases the α-content of Ala5. With the same weight factors as for the unblocked peptide, the helical population of blocked peptides is between 12% and 23%. Lastly, the choice of Karplus equation parameters affects the results. Residue-specific parameters for Karplus equations should improve the accuracy of structural information obtained from scalar couplings (24), which would make them useful in force field refinement.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Acknowledgments
This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health (NIH) and used the NIH Biowulf Linux cluster.
Robert B. Best's present address is Dept. of Chemistry, Lensfield Road, Cambridge CB2 1EW, UK.
Nicolae-Viorel Buchete's present address is School of Physics, University College Dublin, Belfield, Dublin 4, Ireland.
Editor: Kathleen B. Hall.
References
- 1.Karplus, M., and J. A. McCammon. 2002. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 9:646–652. [DOI] [PubMed] [Google Scholar]
- 2.Shi, Z., K. Chen, Z. Liu, and N. R. Kallenbach. 2006. Conformation of the backbone in unfolded proteins. Chem. Rev. 106:1877–1897. [DOI] [PubMed] [Google Scholar]
- 3.Merchant, K. A., R. B. Best, J. M. Louis, I. V. Gopich, and W. A. Eaton. 2007. Characterizing the unfolded states of proteins using single molecule FRET spectroscopy and molecular simulations. Proc. Natl. Acad. Sci. USA. 104:1528–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Makowska, J., S. Rodciewitz-Motowidlo, K. Baginska, J. A. Vila, A. Liwo, L. Chmurzynski, and H. A. Scheraga. 2006. Polyproline II conformation is one of many local conformational states and is not an overall conformation of unfolded peptides and proteins. Proc. Natl. Acad. Sci. USA. 103:1744–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bracken, C., L. M. Iakoucheva, P. R. Rorner, and A. K. Dunker. 2004. Combining prediction, computation and experiment for the characterization of protein disorder. Curr. Opin. Struct. Biol. 14:570–576. [DOI] [PubMed] [Google Scholar]
- 6.Yeh, I.-C., and G. Hummer. 2002. Peptide loop-closure kinetics from microsecond molecular dynamics simulations in explicit solvent. J. Am. Chem. Soc. 124:6563–6568. [DOI] [PubMed] [Google Scholar]
- 7.MacKerell, A. D. 2004. Empirical force fields for biological macromolecules: overview and issues. J. Comput. Chem. 25:1584–1604. [DOI] [PubMed] [Google Scholar]
- 8.Duan, Y., C. Wu, S. Chowdhury, M. C. Lee, G. Xiong, W. Zhang, R. Yang, P. Cieplak, R. Luo, T. Lee, J. Caldwell, J. Wang, and P. A. Kollman. 2003. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum chemical calculations. J. Comput. Chem. 24:1999–2012. [DOI] [PubMed] [Google Scholar]
- 9.MacKerell, A. D., M. Feig, and C. L. Brooks. 2004. Improved treatment of the protein backbone in empirical force fields. J. Am. Chem. Soc. 126:698–699. [DOI] [PubMed] [Google Scholar]
- 10.Hornak, V., R. Abel, A. Okur, B. Strockbine, A. Roitberg, and C. Simmerling. 2006. Comparison of multiple AMBER force-fields and development of improved protein backbone parameters. Proteins. 65:712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.García, A. E., and K. Y. Sanbonmatsu. 2002. α-Helical stabilization by side chain shielding of backbone hydrogen bonds. Proc. Natl. Acad. Sci. USA. 99:2782–2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muñoz, V., and L. Serrano. 1994. Elucidating the folding problem of helical peptides using empirical paramters. Nat. Struct. Biol. 1:399–409. [DOI] [PubMed] [Google Scholar]
- 13.Lindorff-Larsen, K., R. B. Best, M. A. Depristo, C. M. Dobson, and M. Vendruscolo. 2005. Simultaneous determination of protein structure and dynamics. Nature. 433:128–132. [DOI] [PubMed] [Google Scholar]
- 14.Peter, C., X. Daura, and W. F. van Gunsteren. 2001. Calculation of NMR-relaxation parameters for flexible molecules from molecular dynamics simulations. J. Biomol. NMR. 20:297–310. [DOI] [PubMed] [Google Scholar]
- 15.Young, W. S., and C. L. Brooks. 1996. A microscopic view of helix propagation: N and C-terminal helix growth in alanine helices. J. Mol. Biol. 259:560–572. [DOI] [PubMed] [Google Scholar]
- 16.Margulis, C. J., H. A. Stern, and B. J. Berne. 2002. Helix unfolding and intramolecular hydrogen bond dynamics in small α-helices in explicit solvent. J. Phys. Chem. B. 106:10748–10752. [Google Scholar]
- 17.Gnanakaran, S., and A. E. García. 2005. Helix-coil transition of alanine peptides in water: force-field dependence on the folded and unfolded structures. Proteins. 59:773–782. [DOI] [PubMed] [Google Scholar]
- 18.Zagrovic, B., G. Jayachandran, I. S. Millet, S. Doniach, and V. S. Pande. 2005. How large is an α-helix? Studies of the radii of gyration of helical peptides by small-angle x-ray scattering and molecular dynamics. J. Mol. Biol. 353:232–241. [DOI] [PubMed] [Google Scholar]
- 19.Graf, P., H. Nguyen, G. Stock, and H. Schwalbe. 2007. Structure and dynamics of the homologous series of alanine peptides: a joint molecular dynamics/NMR study. J. Am. Chem. Soc. 129:1179–1189. [DOI] [PubMed] [Google Scholar]
- 20.Buchete, N.-V., and G. Hummer. 2008. Coarse master equations for peptide folding dynamics. J. Phys. Chem. B. 112:6057–6069. [DOI] [PubMed] [Google Scholar]
- 21.Buchete, N.-V., and G. Hummer. 2008. Peptide folding kinetics from replica exchange molecular dynamics. Phys. Rev. E. 77:030902. [DOI] [PubMed] [Google Scholar]
- 22.Karplus, M. 1959. Contact electron-spin coupling of nuclear magnetic moments. J. Chem. Phys. 30:11–15. [Google Scholar]
- 23.Karplus, M. 1963. Vicinal proton coupling in nuclear magnetic resonance. J. Am. Chem. Soc. 85:2870–2871. [Google Scholar]
- 24.Case, D. A., C. Scheurer, and R. Brüschweiler. 2000. Static and dynamic effects on vicinal scalar J couplings in proteins and peptides: a MD/DFT analysis. J. Am. Chem. Soc. 122:10390–10397. [Google Scholar]