

Abstract
We perform atomistic simulations on a single collagen molecule to determine its intrinsic molecular strength. A tensile pull simulation to determine the tensile strength and Young's modulus is performed, and a simulation that separates two of the three helices of collagen examines the internal strength of the molecule. The magnitude of the calculated tensile forces is consistent with the strong forces of bond stretching and angle bending that are involved in the tensile deformation. The triple helix unwinds with increasing tensile force. Pulling apart the triple helix has a smaller, oscillatory force. The oscillations are due to the sequential separation of the hydrogen-bonded helices. The force rises due to reorienting the residues in the direction of the separation force. The force drop occurs once the hydrogen bond between residues on different helices break and the residues separate.
INTRODUCTION
Collagen is a major part of the extracellular matrix and connective tissue, including bone and cartilage (1). Because collagen has such a ubiquitous structural function, understanding the mechanical properties of collagen is important. Collagen structure is complex and diverse. Collagen fibers consists of a complex, hierarchical assembly, which determines its composite mechanical properties (2,3). A variety of different collagen molecules exist at both the molecular level due to amino acid sequence variety and the supramolecular level due to the formation of fibrils, networks, etc. In this article, we focus on examining the strength of the basic unit, an individual collagen molecule. The advent of single molecule experiments is enabling the investigation of the mechanical properties of individual molecules (4,5) We present results of molecular dynamics simulations of a collagen protein under two forms of strain. We calculate the forces as a function of strain and examine both the molecular rearrangement and the nature of the atomic interactions to gain insight on how the molecular structure yields the properties of this key structural protein.
The molecular collagen or tropocollagen is a triple helix made up of three polypeptide strands, each of which is a left-handed helix. The amino acid sequence typically has Gly repeated every third residue (-X-Y-Gly-). Between the helices there are some hydrogen bonds that promote stability. The helix diameter is 1.5 nm and in cells tropocollagen is typically ∼300 nm long.
Using modern experimental methods, the mechanical properties of collagen at the molecular level are now being probed (6-10). The cantilever in the atomic force microscope (AFM) has been used to measure the strength of collagen fibrils consisting of multiple collagen molecules (6). Interpretation of these measurements has questioned the packing structure of fibrils (8). The trimeric type I tropocollagen molecules was studied by atomic force microscopy, both topologically and by force spectroscopy (9,10) The use of optical tweezers to measure single molecule properties provides direct insight into molecular mechanics (11,12). Measurements on collagen I proteins obtained a persistence length of 14 ± 8 nm (6).
Some simulations of collagen have been performed. The role of hydroxylated prolines has been examined in molecular dynamics simulations of the collagen triple helix (13). The Young's modulus has been calculated (14). A bead-spring model has been developed to treat the mechanical properties of long tropocollagen molecules (15). For small deformation, they find a dominance of entropic elasticity. At larger deformation, they find a transition to elasticity characterized by first stretching and breaking of hydrogen bonds and then followed by deformation of covalent bonds in the protein backbone.
We have performed atomistic molecular dynamics simulations on a single collagen molecule to characterize the molecular strength. The tensile strength of the molecule is examined by applying tensile stress to stretch the molecule. The intrinsic cohesion of the triple helix is examined by separating a pair of strands of the triple helix. There are similarities to the single-molecule experiments separating the helices of DNA or RNA (4,5,11,12). Measurements on dsDNA have been able to examine the forces required to separate the two strands and measure sequence dependent force. Atomistic simulations of DNA stretching have also been performed(16-20). Although there are similarities, the molecular details between DNA and collagen are different. Collagen has less hydrogen bonds between the individual helices than DNA and does not have the base stacking interaction that stabilized DNA. One focus of this work is to understand the molecular strength of collagen and it relation with the molecular structure, e.g., hydrogen bonding.
In understanding the results of these simulations especially with respect to experimental results, it is necessary to know the connections between the deformation rate, the molecular timescales and the applied force magnitudes. We discuss these quantities for both simulations and experiments. A common feature of any atomistic simulation is that the pulling speeds are of the magnitude 1 m/s. This is simply a consequence of the time step (∼1 fs) and the total number of time steps or the total integrated time (1–10 ns). Within these parameters a total strain on the order of a nm is obtained. Especially given that collagen is a key component of structural tissue and that speeds of 1 m/s are common for the human body (e.g., running, throwing), this pulling speed is physically relevant, particularly with respect to the mechanical strength especially fracture. On the other hand, most experimental measurements performed by AFM or single molecule techniques have pulling speeds at orders of magnitude lower values. Simulations at pulling speeds much smaller than 1 m/s have much smaller strains that are negligible. For this reason, small pulling speeds are not viable and reaching 1 μ/s is out of the question. Direct comparison between simulations and such experiments is thus not possible. We view the measurements and calculations as probes at different velocities that provide complementary information about the molecule. This is especially relevant to collagen, which as a key structural component in biological systems, will undergo a wide range of deformation rates. More generally, the same basic physical properties will be determined in some circumstances allowing indirect comparison between experiment and simulation. Although in other cases, different physical properties will be shown. The fundamental factors that determine the different regimes are the molecular relaxation rates. For deformation rates slower than a dominant molecular relaxation, the dynamics of this molecular motion is not strongly perturbed. The applied forces will have a logarithmic dependence with respect to the deformation rate, but the qualitative nature of the dynamics at different rates will be similar. On the other hand, if the deformation rate is faster than the relaxation rate, then the relaxation dynamics is strongly perturbed (i.e., physically different), and the functional dependence of the applied force is typically different, (e.g., different scaling). With respect to the pulling simulations in this work, we are pulling on a short, rather stiff collagen molecule at a sufficiently slow speed that the dynamics of dihedrals, bonds, and similar few atom constructs can relax. The advantage of simulation is that both the structure and the forces can be determined.
The example of pulling on a simple hydrocarbon illustrates the ranges of physical parameters. The timescale of relaxation of the random walk configurations of a single long polyethylene polymer in solution is long in comparison to present simulation run times. A simulation that stretches the polymer by pulling on the ends at 1 m/s will distort the polymer faster than it can relax and will probe a different dynamics than a single molecule stretching experiment that pulls at 1 μm/s. If instead, a short alkane chain is considered, which has a conformational relaxation time less than a nanosecond due to dihedral dynamics, then the structural dynamics (e.g., dihedral motion) in a simulation will not differ fundamentally from a measurement at much lower speeds. This is not to say that the calculated forces will be the same as the measured values. The measured forces will be smaller, but the structural dynamics found in the simulations will match. Finally, if the long polyethylene polymer is pulled taut, then the dynamics once the polymer is sufficiently straight will be similar to a short alkane and the force-extension curves will have a correspondence with simulations.
The different intramolecular interactions that yield the polymer conformations not only yield the range of molecular relaxation times, but also the range of forces needed to strain molecular motion (21). In general, for single polymers in random walk conformations, entropic interactions are dominant. In particular, whenever the polymer contour length is much greater than the persistence length Lp, entropy determines the polymer conformation on length scales greater than Lp. In this case the magnitude of the forces is of order kBT/nm = 4 pN. If the chain is shorter than Lp, then larger forces are required to alter the polymer conformation. When separating molecular parts that interact through multiple van der Waals interactions, dihedral rotations or hydrogen bonds, the energy scale is ∼10 kBT and the work is done over a length of ∼0.4 nm. Thus, the forces are of magnitude 100 kBT. The carbon-carbon bond strength is ∼140 kBT. Bond breaking occurs over a smaller length scale (Å) so that the corresponding force is of order 5 nN. These regimes have been observed in measurements on collagen. The force measurements on collagen using optical tweezers are in the low force or entropic regime (6). AFM measurements have obtained forces from the entropic pN scale to the nN range (8,9).
Much of the experimental data has probed the first two of the regimes described in the previous paragraph. Recently, the high force or energetic regime was studied for a set of polymers using AFM measurements and for the corresponding monomers using ab initio quantum calculations (22). The force extension curves were calculated and measured for ssDNA, polyvinylamine, and various polypeptides for extensions just beyond the equilibrium contour length. The extension force ranges from 0.1 to 2.0 nN. The force extension curves were in agreement between the calculation and the experiments. This data is especially relevant for this simulation study on collagen.
METHODS
The protein databank entry 1BKV, a synthetic peptide containing a region from human type III collagen, serves as a starting configuration for the simulations of a single collagen molecule in solution (23). This particular collagen molecule consists of three identical helices containing 30 residues each. Acetic acid molecules, used for crystallization, are removed from the structure and missing hydrogen atoms are added to complete the peptide. This process results in a molecule consisting of 1116 atoms in total. To complete the system, the protein is solvated in an aqueous saline solution of 0.1 M sodium chloride and neutralized by addition of three extra chloride ions.
In the molecular dynamics simulations we use the CHARMM force field (24), with the addition of entries specific for hydroxy-proline, reflected by Table 1 and Table 2 corresponding to bonded force field representations. Simulations are carried out with the molecular dynamics code LAMMPS (25) after transforming a CHARMM input to a LAMMPS input (26). The resulting initial structure is relaxed for 2.0 ps using a time step of 1 fs.
TABLE 1.
Additional bond length and angle force field parameters for hydroxy-proline
| Type | id1 | id2 | id3 | kbond (kcal/mol/Å2), kangle (kcal/mol) | l (Å), θ (°) |
|---|---|---|---|---|---|
| Bond | OH1 | CP2 | 428.000 | 1.4300 | |
| Angle | H | OH1 | CP2 | 65.000 | 109.5000 |
| OH1 | CP2 | CP2 | 50.000 | 112.5000 | |
| OH1 | CP2 | CP3 | 50.000 | 112.2000 | |
| OH1 | CP2 | HA | 45.900 | 111.0000 |
TABLE 2.
Additional dihedral force field parameters for hydroxy-proline
| id1 | id2 | id3 | id4 | k1 (kcal/mol) | γ1 | k2 | γ2 | k3 | γ3 |
|---|---|---|---|---|---|---|---|---|---|
| H | OH1 | CP2 | CP2 | 1.30 | 0.00 | 0.30 | 0.00 | 0.42 | 0.00 |
| H | OH1 | CP2 | CP3 | 1.30 | 0.00 | 0.30 | 0.00 | 0.42 | 0.00 |
| X* | CP2 | OH1 | X* | 0.00 | 0.00 | 0.00 | 0.00 | 0.14 | 0.00 |
Acts as a wildcard and can be replaced by any possible id.
To determine the mechanical strength of a single collagen molecule, we stretch the molecule along its molecular axis. The collagen helix was aligned along the x-direction by applying a rotation to the PDB coordinates of just the protein. The water and ions were introduced subsequently into the simulation cell. The tensile stretch is performed by moving the terminal residues of each helix at constant velocity as depicted schematically in Fig. 1 with the two ends moving in opposite directions along the x axis. We moved whole terminal residues instead of a single atom within the terminal residue to reduce the effects of stronger fluctuations in the force on a single atom. We calculate the sum of forces f+ on all the atoms in the constrained residues moving in the +x-direction, and similarly calculate f− for the −x side. The tensile force Δf is defined as (f+ + f−)/2. To remove the noise of the fluctuations, we time averaged the force over a 100 ps window. The uncertainty is ∼100 pN. An initial configuration with simulation cell dimensions L = (15.27, 4.926, 4.926) nm is created according to the method described above and equilibrated for 500 ps. To accommodate the molecule's length, the simulation cell dimension is longer in this direction. Tensile pull is applied at a constant rate for a total of 2 ns, during which the molecule stretches an additional 2 nm. We define d as the distance the constrained, terminal residues are moved. The simulation cell's geometry remains unaltered during this molecular deformation.
FIGURE 1.

Schematic representation of the single-molecule simulations showing the single letter amino acid sequence. The letter X represents the hydroxyproline residue. The numbers give the indexing of the sequence. The residues in bold are pulled in the tensile or the separation simulations. In the tensile simulations, the terminal residue for each helix is pulled at constant velocity in opposite directions. For simulations separating the helices, the force is applied to two of the terminal Gly residues and perpendicular to the helical axis. In either case, the forces are summed over all the atoms in the residues being pulled at each end, and Δf = (f1 + f2)/2.
To separate two helices of the collagen protein, the N-terminal residues of two helices are pulled in opposite directions perpendicular to the primary molecular axis (Fig. 1). Because the helices have the same amino acid sequence, it does not matter which two helices were chosen. Separation is forced between the first and the second helix with residues numbered from 1–30 and 31–60 respectively. The third helix remains untouched. The force on the residues is calculated during the deformation in the same manner as the tensile simulations. An initial configuration with cell dimensions L = (14.29, 14.29, 3.94) nm is created as described earlier. An increase of the cell's length in the y-direction allows for pulling two helices in opposite y-directions. The pulling is performed at constant rate. In the 4 ns simulation, the total displacement is 8 nm. About 50% of the two helices are separated. If the separation would approach full separation, then end effects would appear. We only went to the halfway point to not encounter the end effects.
RESULTS AND DISCUSSION
Molecular tensile pull
The tensile force is plotted as a function of extension d in Fig. 2. The total extension is ∼2 nm beyond the initial 8 nm length of the collagen molecule. The total strain is large enough to stretch bonds. This collagen molecule is shorter than collagen's persistence length and is in a rod-like conformation. However, rod-like does not mean a rigid rod conformation; some flexibility exists and especially the ends of a molecule are in a more flexible state. The terminal residues are loosely bound to the triple helix. The initial part of the tensile strain removes this slack. Once this slack is removed the bonds are stretched and three-body angle bends occur. This results in the magnitude of the tensile forces being in the nN regime, as discussed in the Introduction. Because collagen is a triple helix, the tensile force is being applied to three molecules at once and the applied force increases by a factor of three. The maximum applied force per strand is then ∼1.7 nN, which is in the range for bond and angle stretching. The force curve rises linearly in the first half of the deformation. At d = 1 nm, the slope increases and the force rises nonlinearly. The slope of the force curve is related to the Young's modulus E, which the ratio of the stress to the strain. We calculated E using the first 1 nm of extension. To obtain the stress we divide the force by the cross-sectional area of collagen, using 1.5 nm as the collagen diameter. We obtain E = 6.1 GPa. This value is within the measured range for collagen fibrils (10) using nano indentation methods. In addition, the value is in agreement with previous tensile simulations (14).
FIGURE 2.

Force in tensile pull simulations as a function of distance over which pulled residues are translated. Dashed line is linear fit to first 1 nm.
There is a correlation between the change in linearity of the force and the structure of the deformed molecule, which we now address. Examinations near d = 1 nm of the triple helix conformations show that the helices are unwinding due to the applied force. At the end of the simulations the three strands are no longer wound around each other, but are parallel, straight strands. Visualization shows that at about d = 1 nm, the strands have unwound from each other. Larger extensions straighten the individual strands, which when separated still possessed some curvature from their helical conformation. Thus, the two regimes in the force-extension curve are due to unwinding of the triple helix and to straightening the unwound strands. To quantify the unwinding, we calculate an order parameter 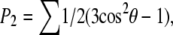 where the sum is over all angles θ between three adjacent alpha carbons. P2 is 1 for a straight, linear molecule. For a helix, the sum averages to zero. Fig. 3 shows that P2 increases from zero as the extension increases. The maximum value of P2 = 0.5 in the plot corresponds to an average θ = 145°, which is quite close to straight given the underlying zig-zag conformation that the angle terms demand.
where the sum is over all angles θ between three adjacent alpha carbons. P2 is 1 for a straight, linear molecule. For a helix, the sum averages to zero. Fig. 3 shows that P2 increases from zero as the extension increases. The maximum value of P2 = 0.5 in the plot corresponds to an average θ = 145°, which is quite close to straight given the underlying zig-zag conformation that the angle terms demand.
FIGURE 3.

Average order parameter P2 as a function of the tensile pull distance. Measurements, represented by points, straddle the moving average, represented by the solid line.
We can further examine the local structure to confirm the nature of the force magnitudes and its connection to structure. The unwinding of the triple helix involves altering the dihedral angles in the backbone. This will give forces of order 300 pN for the triple helix. For the linear force regime, this is a major contributor. As the strands become straight and more taut, stretching of bonds and angles in the backbone occurs. Over the full extension, the change in energy per bond in the backbone is 0.56 kcal/mol. Although this is not a large energy change, it does yield bond forces of order 1 nN. An increase in the average bond length of only 0.04 Å yields a large bond force due to the large spring constants of the bond potential. The total energy change per angle is 0.52 kcal/mol. In a similar vein to the bonds, this corresponds to an increase in the average angle of only a few degrees, but that yields angle forces of ∼0.6 nN. These forces and energies are for the full extension, and they sum up to a force per strand of 1.6 nN that is the maximum. Recent data from AFM experiments and ab initio theory for highly stretched oligomers have forces that are in the range 1–2 nN (26). Thus, the magnitude of the forces is in line with this data.
Within the first 1 nm of extension, when the triple helix is uncoiled, and the applied force reaches 1.3 nN. This value is close to recent measurements on collagen.
Gutsmann et al. (8) pulled single collagen molecules from fibrils using AFM. Their maximum pull force is ∼0.9 nN. Bozec and Horton's AFM measurements on single molecules have pulling forces up to 1.6 nN (9), and they wonder whether the triple helix is uncoiled in the curves that have sharp peaks that reach nN values. The results of these simulations show that uncoiling is certainly possible and magnitude of the forces is large enough.
Separating the strands
The force-separation data is much richer in the simulations of separating the helices than for the tensile pull case. The force rises and falls as the residues on the two separating helices come apart (Fig. 4). The basic picture of the dynamics is that each adjoining residue pair in the separating helices is separated sequentially. The sequential nature of the helix separation can be seen in Fig. 5, which shows the separation distance dres between α carbons of each pair of adjoining residues in the triple helix that are being forced apart as a function of d. As it should be, the distance between the two terminal residues that are being forced apart is linear in d (there is an offset between d and dres). We note that residues at the end of the triple helix are not bound together so that dres is initially large compared to the other residue pairs. Subsequent pairs of adjoining residues have an initial flat region with no change in separation and then a linear separation that is in line with the applied strain rate. All of the remaining pairs have the same features with the transition to the linear separation occurring at increasing d for pairs further within the molecule. This data shows that the adjacent residues in the two separating helices are being separated sequentially.
FIGURE 4.

The force of separation as a function of the separation distance.
FIGURE 5.

The distance between all the neighboring residue pairs on the two separating helices is plotted as a function of the separation distance. The top curve is the first residue pair being separated. Subsequent lines are for subsequent pairs in the sequence.
This sequential separation suggests that the rise and fall in the separation force is due to the local forces (e.g., van der Waals, electrostatic, hydrogen bond) resisting separation of the residues pairs. Forces rise as two residues resist separation and then decrease once the barrier has been overcome. We find that the rise in force is due primarily to the reorienting the residues as they are separated from their partners in the other helix. For example, there is a large rise in the force in the separation range of d = 3.2–3.7 nm. Fig. 6 shows three images of the collagen conformation at d = 3, 4, and 6 nm. The images for d = 3 and 4 nm show that the top chains becomes parallel to the separation direction and taut for residues starting from the terminal one and continuing to the last residue to be separated. The applied force is predominantly perpendicular to the orientation of the residues within the helix. Thus, the separation force must reorient the residue. The process of reorienting the residues involves the dihedral and angle bend interactions, which yield the order of magnitude forces in Fig. 4. The calculated magnitude of the forces to separate the residues is an order of magnitude less than the force calculated in the tensile pull. The separation of the strands does not stretch the bonds nor force close contact between atoms. Thus, the applied force is much less in the separation simulations.
FIGURE 6.

Images of the helices of the collagen molecule being separated by pulling on two helices perpendicular to the triple helix axis. Only the backbone atoms are displayed. The separation is applied vertically with respect to the page. Colors: O (red), N (blue), and C (cyan).
Hydrogen bonds between helices are viewed as integral to the stability of the collagen protein (22). With this in mind, we examined the separation of atoms involved in hydrogen bonds. Most of the hydrogen bonding occurs between atoms in backbones within the triple helix structure. There are some waters within the x-ray structure that provide hydrogen bonding between amides and carbonyls. However, these waters are located on the surface of the collagen molecule, not within the interior of the triple helix. As such, they can exchange with bulk waters. There is one notable hydrogen bond involving the side chain atoms; this involves the amine in the side chain of Arg14 and the carbonyl in the backbone of Arg44. At the start of the simulation there are 13 hydrogen bonds using the criterion that the acceptor-donor separation is <3.3 Å and the angle is <30°. At the end of the simulation there are seven hydrogen bonds. The drops in the applied force correspond to the breaking of hydrogen bonds. Table 3 lists most of the hydrogen bonds that break during the simulation. All the entries in Table 3 match drops in the Δf (Fig. 4 a). In the CHARMM force field, breaking a hydrogen bond is an electrostatic interaction of the partial charges that decreases in strength monotonically as the separation increases. Once a hydrogen bond pair begins to break and separate, the force needed to separate the pair is monotonically decreasing. The van der Waals attraction also decreases with separation of the residues. Thus, as a hydrogen bond is stretched the applied force decreases. The sequence of events is as follows. The separating residues are reoriented; this involves unwinding the triple helix. The internal hydrogen bond is exposed to direct separation. The force required to reorient the residues is sufficient to break the hydrogen bond. Further strain requires less force, and the applied force decreases until the next residue begins reorienting.
TABLE 3.
Hydrogen bonds between listed residue pairs that break at the listed separations d
| Residue | Residue | d (nm) |
|---|---|---|
| Gly6 | Pro34 | 1.0 |
| Gly9 | Pro37 | 2.4 |
| Gly12 | Ile40 | 2.9 |
| Hyp8 | Gly66 | 4.0 |
| Ala3 | Gly72 | 5.8 |
| Gly15 | Ala43 | 6.0 |
| Arg14 | Arg44 | 6.9 |
The distance dHB is the separation between the H atom and the O acceptor in two hydrogen bonds. The dynamics of hydrogen bond breaking for two examples is given in Fig. 7. For d < 2.4 nm, there is a hydrogen bond between the amide hydrogen on Gly9 and the carbonyl O in Pro37. The bond breaks at d = 2.4 nm. Most of the hydrogen bonds in Table 3 are between amide hydrogen and carbonyl O atoms and show similar behavior. One exception is the hydrogen bond between the side chain of Arg14 and carbonyl of Arg44. This hydrogen bond is present at the start of the separation simulation. In Fig. 7, only the part for this case for d > 4 is shown to not overlap with the other data. The hydrogen bond breaks finally at d = 6.95 nm. Earlier, at about d = 6.5 nm, there is an exchange with another H atom in the Arg14 side chain in participating in the hydrogen bond, which reverses back to the original H atom at d = 6.8 nm just before the bond completely breaks.
FIGURE 7.

Separation between hydrogen bonded pairs dHB as a function of the separation for the Gly9:H-Pro37:O pair (circles; d < 2.5 nm) and for the Arg14:H-Arg44:O pair (squares; d > 4 nm). Data for the Arg pair at d < 4 nm is not shown for clarity.
There are hydrogen bonds between the third helix and the two other helices, which we are pulling apart. Some of these hydrogen bonds are not involved in the collagen helix separation. However, there are examples of such hydrogen bonds that do break (e.g., one involving Gly66) and play a role in the separation dynamics. Fig. 6 shows that the third helix initially remains bound at the N-terminus to the bottom helix in the figure (d = 3 nm), but at d = 6 nm, the third helix is bound to the top helix. Some of the variation in Δf between d = 5 and 6 nm is due in part to the dynamics of the third helix switching from bound to the free end of one strand to the other. Overall, the processes of residue reorientation and hydrogen bond breaking are the main mechanisms of separation, independent of the individual helices that are separating.
CONCLUSIONS
We have examined the mechanical strength of a collagen molecule by performing tensile pull and strand separation simulations. The magnitude of tensile pull force is consistent with the strong forces of bond stretching and angle bends that are involved in the tensile deformation. Structurally, the consequence of the tensile pull is the unwinding of the triple helix. This corresponds to recent experiments of Bozec and Horton (9). The separation of the helices has a smaller, oscillatory force. The oscillations are due to the sequential separation of the hydrogen-bonded helices. The rise in the force is due to reorienting the residue in the direction of the separation force. The drop in the force is due to the breaking of the hydrogen bonds and resultant slack in the strands.
As this is the initial atomistic simulation of single molecule collagen properties, there remain a variety of questions to be considered. We have treated a specific, somewhat synthetic, residue sequence. It would be interesting to examine sequences that have a different character of hydrogen bonding. Although atomistic simulations of collagen longer than the persistence length are not yet feasible, such simulations will be very interesting; both from the perspective that such molecules are biologically relevant and that most measurements on longer molecules.
Acknowledgments
Sandia National Laboratories is a multi-program laboratory operated by the Sandia Corporation, a Lockheed Martin Company, for the United States Department of Energy's National Nuclear Administration under contract No. DE-AC04-94AL850000.
Editor: Gregory A. Voth.
References
- 1.Lodish, H., D. Baltimore, A. Berk, S. L. Zipursky, P. Matsudaira, and J. Darnell. 2000. Molecular Cell Biology, 4th ed. W. H. Freeman and Co., New York.
- 2.Holmes, D. F., C. J. Gilpin, C. Baldock, U. Ziese, A. J. Koster, and K. E. Kadler. 2001. Corneal collagen fibril structure in three dimensions: structural insights into fibril assembly, mechanical properties, and tissue organization. Proc. Nat. Acad. Sci. 98:7307–7312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fratzl, P. 2003. Cellulose and collagen: from fibers to tissues. Current Op. Coll. Inter. Sci. 8:32–39. [Google Scholar]
- 4.Tinoco, I., Jr. 2004. Force as a useful variable in reactions: unfolding RNA. Annu. Rev. Biophys. Biomol. Struct. 33:363–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bockelmann, U., Ph. Thomen, B. Essevaz-Roulet, V. Viasnoff, and F. Heslot. 2002. Unzipping DNA with optical tweezers: high sequence sensitivity and force flips. Biophys. J. 82:1537–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun, Y. L., Z. P. Luo, and K. N. An. 2001. Stretching short biopolymers using optical tweezers. Biochem. Biophys. Res. Commun. 286:826–830. [DOI] [PubMed] [Google Scholar]
- 7.Gutsmann, T., G. E. Fantner, J. H. Kindt, M. Venturoni, S. Danielsen, and P. K. Hansma. 2004. Force spectroscopy of collagen fibers to investigate their mechanical properties and structural organization. Biophys. J. 86:3186–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gutsmann, T., G. E. Fantner, M. Venturoni, A. Ekani-Nkodo, J. B. Thompson, J. H. Kindt, D. E. Morse, D. K. Fygenson, and P. K. Hansma. 2003. Evidence that collagen fibrils in tendons are inhomogeneously structured in a tubelike manner. Biophys. J. 84:2593–2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bozec, L., and M. Horton. 2005. Topography and mechanical properties of single molecules of type I collagen using atomic force microscopy. Biophys. J. 88:4223–4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wenger, M. P. E., L. Bozec, M. A. Horton, and P. Mesquida. 2007. Mechanical properties of collagen fibrils. Biophys. J. 93:1255–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhuang, X., and M. Rief. 2003. Single-molecule folding. Curr. Opin. Struct. Biol. 13:88–97. [DOI] [PubMed] [Google Scholar]
- 12.Bustamante, C., S. B. Smith, J. Lipardt, and D. Smith. 2000. Single-molecule studies of DNA mechanics. Curr. Op. Struct. Biol. 10:279–285. [DOI] [PubMed] [Google Scholar]
- 13.Mooney, S., P. Kollman, and T. Klein. 2002. Conformational preferences of substituted prolines in the collagen triple helix. Biopolymers 64:63–71. [DOI] [PubMed] [Google Scholar]
- 14.Lorenzo, A., and E. Caffarena. 2005. Elastic properties, Young's modulus determination and structural stability of the tropocollagen molecule: a computational study by steered molecular dynamics. J. Biomech. 38:1527–1533. [DOI] [PubMed] [Google Scholar]
- 15.Buehler, M. J., and S. Y. Wong. 2007. Entropic elasticity controls nanomechanics of single tropocollagen molecules. Biophys. J. 93:37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Konrad, M. W., and J. I. Bolonick. 1996. Molecular dynamics simulation of DNA stretching is consistent with the tension observed for extension and strand separation and predicts a novel ladder structure. J. Am. Chem. Soc. 118:10989–10994. [Google Scholar]
- 17.MacKerell, A. D., Jr., and G. U. Lee. 1999. Structure, force, and energy of a double-stranded DNA oligonucleotide under tensile loads. Eur. Biophys. J. 28:415–426. [DOI] [PubMed] [Google Scholar]
- 18.Lohikoski, R., J. Timonen, and A. Laaksonen, 2005. Molecular dynamics simulation of single DNA stretching reveals a novel structure. Chem. Phys. Lett. 407:23–29. [Google Scholar]
- 19.Harris, S. A., Z. A. Sands, and C. A. Laughton. 2005. Molecular dynamics simulations of duplex stretching reveal the importance of entropy in determining the biomechanical properties of DNA. Biophys. J. 88:1684–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Piana, S. 2005. Structure and energy of a DNA dodecamer under tensile load. Nucleic Acids Res. 33:7029–7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strick, T. R., M.-N. Dessinges, G. Charvin, N. H. Dekker, J. F. Allemand, D. Bensimon, and V. Croquette. 2003. Stretching of macromolecules and proteins. Rep. Prog. Phys. 66:1–45. [Google Scholar]
- 22.Hugel, T., M. Rief, M. Seitz, H. E. Gaub, and R. R. Netz, 2005. Highly stretched single polymers: atomic-force-microscope experiments versus ab initio theory. Phys. Rev. Lett. 94:048301. [DOI] [PubMed] [Google Scholar]
- 23.Kramer, R. Z., J. Bella, P. Mayville, B. Brodsky, and H. M. Berman. 1999. Sequence dependent conformational variations of collagen triple-helical structure. Nat. Struct. Biol. 6:454–457. [DOI] [PubMed] [Google Scholar]
- 24.MacKerell, A. D., Jr., D. Bashford, M. Belott, R. L. Dunbrack, Jr., J. Evanseck, M. J. Field, S. Fisher, J. Gao, H. Guo, S. Ha, D. Joseph, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, I. W. E. Reiher, B. Roux, M. Schlenkrich, J. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin, and M. Karplus. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- 25.Plimpton, S. J. 1995. Fast parallel algorithms for short-range molecular dynamics. J. Comp. Phys. 117:1–19. [Google Scholar]
- 26.in ‘t Veld, P. J., and M. J. Stevens. Translation script for LAMMPS Molecular Dynamics Simulator. Available at http://lammps.sandia.gov/.