

Abstract
The linear peptide gramicidin forms prototypical ion channels specific for monovalent cations and has been used extensively to study the organization, dynamics, and function of membrane-spanning channels. The tryptophan residues in gramicidin channels are crucial for maintaining the structure and function of the channel. We explored the structural basis for the reduction in channel conductance in the case of single-tryptophan analogs of gramicidin with three Trp → hydrophobic substitutions using a combination of fluorescence approaches, which include red edge excitation shift and membrane penetration depth analysis, size-exclusion chromatography, and circular dichroism spectroscopy. We show here that the gramicidin analogs containing single-tryptophan residues adopt a mixture of nonchannel and channel conformations, as evident from analysis of membrane penetration depth, size-exclusion chromatography, and backbone circular dichroism data. These results are potentially useful in analyzing the effect of tryptophan substitution on the functioning of other ion channels and membrane proteins.
INTRODUCTION
The biological membrane provides a unique environment to membrane-spanning proteins and peptides, influencing their structure and function. Membrane-spanning proteins have distinct stretches of hydrophobic amino acids that form the transmembrane domain and are reported to have significantly higher tryptophan content than soluble proteins (1). In addition, it has been observed that tryptophan residues in integral membrane proteins and peptides are not uniformly distributed but rather tend to be localized toward the membrane interface. This is in part because they are involved in hydrogen bonding (2) with the lipid carbonyl or phosphate groups or interfacial water molecules, and in part because of the rigid, aromatic indole ring (3,4). For instance, crystal structures of membrane proteins such as the KcsA potassium channel (5), bacteriorhodopsin (6), maltoporin (7), and others have shown that most tryptophans are located in a saddle-like “aromatic belt” around the membrane interfacial region. Furthermore, for transmembrane peptides and proteins, tryptophan has been found to be an efficient anchor at the membrane interface (1,8) and defines the hydrophobic lengths of transmembrane helices (9).
In molecular terms, tryptophan is a distinctive amino acid since it is capable of both hydrophobic and polar interactions. This is due to the fact that although tryptophan has the polar −NH group which is capable of forming hydrogen bonds, it also has the largest nonpolar accessible surface area among the naturally occurring amino acids (10). Due to its aromaticity, the tryptophan residue is capable of π−π interactions and weakly polar interactions (11). This amphipathic character of tryptophan gives rise to its hydrogen bonding property and ability to function through long-range electrostatic interactions (12). The amphipathic nature of tryptophan also explains its interfacial localization in membranes (13), which is characterized by specialized motional and dielectric characteristics different from both the bulk aqueous phase and the hydrocarbon-like deeper regions of the membrane (14), both of which support more isotropic motions. The experimentally determined interfacial propensity of tryptophan is the highest among the naturally occurring amino acid residues, thereby accounting for its specific interfacial localization in membrane-bound peptides and proteins (13).
Among ion channels, gramicidin is a particularly powerful model for clarifying the importance of tryptophan at the membrane/water interface for channel structure and assembly. In this study, we applied advanced fluorescence-based approaches to characterize single-Trp gramicidins. The linear peptide gramicidin forms prototypical ion channels specific for monovalent cations and has been used extensively to study the organization, dynamics, and function of membrane-spanning channels (15–18). Gramicidin serves as an excellent model for transmembrane channels due to its small size, defined structure, ready availability, and the relative ease with which chemical modifications can be performed. These features make gramicidin unique among small membrane-active peptides and provide the basis for its use to explore the principles that govern the folding and function of membrane-spanning channels in particular and membrane proteins in general (19,20).
The parent peptide gramicidin A (gA) has the sequence shown in Fig. 1 a. The unique sequence of alternating L- and D-chirality renders gramicidin sensitive to the environment in which it is placed (21). Gramicidin therefore adopts a wide range of environment-dependent conformations. Two major folding motifs have been identified for gramicidin in various media: i), the single-stranded helical dimer (the “channel” form), and ii), a family of double-stranded intertwined helices (collectively known as the “nonchannel” form) (22). For native gA, with four tryptophans in each monomer, the preferred (thermodynamically stable) conformation in membranes is the single-stranded β6.3 helical dimer (23). The transmembrane gramicidin channel is formed by the head-to-head dimerization of β6.3 helices (24). The channel interior is lined by the polar carbonyl and amide moieties of the peptide backbone. Although the backbone organization is quite different, it is noteworthy that the selectivity filter of the bacterial KcsA K+ channel is also defined by peptide carbonyl groups (17,21). In the transmembrane conformation, the carboxy terminal ethanolamine is exposed to the membrane/water interface, whereas the amino terminal formyl group is buried within the hydrophobic core of the membrane. This arrangement places the four tryptophan residues in a cluster at the membrane/water interface at the entrance to the channel (24–28). An important aspect of this conformation is the membrane interfacial location of tryptophan residues, a common feature of many transmembrane helices (3,29). On the other hand, double helical nonchannel conformations of gramicidin, incorporated into lipid bilayer membranes using suitable solvents (30), are thermodynamically unstable in membranes and spontaneously convert to the channel conformation (23,28,30). The distribution and depths of the tryptophan residues constitute a major difference between these two forms (28). In the nonchannel form, some of the tryptophan residues are buried within the low dielectric nonpolar region of the membrane, which is an energetically unfavorable location for tryptophan residues (13). Such conformations have been shown to exist in membranes with polyunsaturated lipids (31) and in membranes with increased acyl chain lengths (23,32).
FIGURE 1.

(a) Amino acid sequence and fluorescence emission maxima of gA and the single-tryptophan analogs used (B denotes Ser-t-butyl). Alternating d-amino acid residues are underlined. The excitation wavelength was 280 nm and the data for gA is from Rawat et al. (28). (b) Effect of changing excitation wavelength on the wavelength of maximum emission for W(11,13,15)BgA (•), W(9,13,15)BgA (○), W(9,11,15)BgA (□), and W(9,11,13)BgA (Δ) in POPC membranes. It should be noted that the lines joining the data points are provided merely as viewing guides. (c) The magnitude of REES obtained for the single-tryptophan analogs of gramicidin. The magnitude of REES corresponds to the total shift in emission maximum when the excitation wavelength is changed from 280 to 307 nm, shown in (b). The concentration of POPC was 0.85 mM. The peptide/POPC ratio was 1:50 (mol/mol). See Materials and Methods for other details.
The tryptophan residues in gramicidin channels are crucial for maintaining the structure and function of the channel (25,33,34). The importance of the tryptophans has been demonstrated by the observation that the cation conductivity of the channel decreases upon substitution of one or all of the tryptophan residues by phenylalanine, tyrosine, or naphthylalanine (12,35,36) and also upon ultraviolet (UV) irradiation or chemical modification of the tryptophan side chains (33,37,38). Additionally, gramicidins with Trp → Phe substitutions have greater difficulty in forming membrane-spanning dimeric channels (34,36). In this work, we explored the structural basis for the reduction in channel-forming propensity in the case of gramicidin analogs with three Trp → Ser-t-butyl substitutions (i.e., single-tryptophan analogs, lacking other aromatic rings) using a combination of fluorescence approaches, which include red edge excitation shift (REES), membrane penetration depth analysis, circular dichroism (CD) spectroscopy, and size-exclusion chromatography. The serine-t-butyl side chain was chosen because it is approximately as hydrophobic as Phe yet is not aromatic. Remarkably, the chromatographic and single-channel properties of Trp → Phe and Trp → Ser-t-butyl substituted gramicidins are virtually indistinguishable (R. E. Koeppe, D. V. Greathouse, and O. S. Andersen, unpublished observations). In contrast to Trp, Ser-t-butyl is unable to donate a hydrogen bond. The ether linkage is masked by the large aliphatic t-butyl group, meaning that Ser-t-butyl is expected to be less amphipathic than Trp. The advantage of using single-tryptophan analogs for fluorescence analysis is that ground state heterogeneity in the tryptophan environment (27,39) is avoided, and any information obtained can be correlated to the unique tryptophan residue in the sequence. Moreover, the parallax method used to measure membrane penetration depth (40) works best with single fluorophores in the membrane, a condition satisfied by the single-tryptophan analogs of gramicidin. Our results show that these analogs adopt a predominantly nonchannel conformation in membranes. Furthermore, the small fraction of molecules which fold into single-stranded channels account for the reduced channel-forming potency, whereas the lack of three of the key Trp dipoles could result in reduced ion conductance.
MATERIALS AND METHODS
Materials
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1-palmitoyl-2-(5-doxyl)stearoyl-sn-glycero-3-phosphocholine (5-PC), and 1-palmitoyl-2-(12-doxyl)stearoyl-sn-glycero-3-phosphocholine (12-PC) were obtained from Avanti Polar Lipids (Alabaster, AL). 2-(9-anthroyloxy)stearic acid (2-AS) and 12-(9-anthroyloxy)stearic acid (12-AS) were from Molecular Probes (Eugene, OR). 1,2-Dimyristoyl-sn-glycero-3-phosphocholine (DMPC) was purchased from Sigma Chemical (St. Louis, MO). Ultrapure grade acrylamide was from Invitrogen Life Technologies (Carlsbad, CA). The single-tryptophan analogs of gramicidin were synthesized using methods described by Greathouse et al. (41) with Fmoc-Ser-t-butyl replacing Fmoc-Trp at selected positions in the gA sequence. 10-Doxylnonadecane (10-DN) was a generous gift from Prof. Erwin London, SUNY, Stony Brook. The concentration of each single-Trp gramicidin analog was estimated from a molar extinction coefficient (ɛ) of 5500 M−1cm−1 at 280 nm. The purity of acrylamide was checked from its absorbance using its molar extinction coefficient (ɛ) of 0.23 M−1cm−1 at 295 nm and optical transparency beyond 310 nm (42). The concentration of a stock solution of 10-DN in ethanol was calculated from its molar extinction coefficient (ɛ) of 12 M−1cm−1 at 422 nm (43). Lipids were checked for purity by thin layer chromatography on silica gel precoated plates (Sigma) in chloroform/methanol/water (65:35:5, v/v) and were found to give only one spot in all cases with a phosphate-sensitive spray and subsequent charring (44). The concentration of phospholipids was determined by phosphate assay subsequent to total digestion by perchloric acid (45). DMPC was used as an internal standard to assess lipid digestion. All other chemicals used were of the highest purity available. Solvents used were of spectroscopic grade. Water was purified through a Millipore (Bedford, MA) Milli-Q system and used for all experiments.
Sample preparation
Experiments were performed using small unilamellar vesicles (SUV) of POPC containing 2% (mol/mol) gramicidin analog. In general, 1280 nmol of POPC in chloroform/methanol was mixed with 25.6 nmol of the gramicidin analog in methanol. A few drops of chloroform were added to this solution. The solution was mixed well and dried under a stream of nitrogen while warming gently (∼40°C) and dried further under a high vacuum for at least 12 h. The dried film was swelled in 1.5 ml of 10 mM sodium phosphate, 150 mM sodium chloride, pH 7.2 buffer, and samples were vortexed for 3 min to uniformly disperse the lipids. The samples were sonicated to clarity under argon (∼30 min in short bursts while being cooled in an ice/water mixture) using a Branson model 250 sonifier (Branson Ultrasonics, Dansbury, CT) fitted with a microtip. The sonicated samples were centrifuged at 15,000 rpm for 15 min to remove any titanium particles shed from the microtip during sonication and incubated for 12 h at 65°C with continuous shaking. Samples were incubated in dark at room temperature for 1 h before fluorescence or CD measurements. Background samples were prepared the same way except that the gramicidin analog was omitted. All experiments were done with multiple sets of samples at 25°C.
In experiments involving fluorescence quenching, the total lipid used was reduced (160 nmol) to avoid scattering artifacts. For experiments involving 10-DN, 160 nmol of POPC containing 10 mol % 10-DN was mixed with 3.2 nmol gramicidin analog. The solution was mixed well and dried as described above. The dried lipid film was then carefully sonicated under argon to avoid any damage to the spin label and incubated overnight at 65°C as described above.
Depth measurements using the parallax method
The actual spin (nitroxide) content of the spin-labeled phospholipids (5- and 12-PC) was assayed using fluorescence quenching of anthroyloxy-labeled fatty acids (2- and 12-AS) as described earlier (46). For depth measurements using the parallax method, SUVs were prepared by sonication followed by incubation at 65°C for 12 h as described above. These samples were made by drying 160 nmol of POPC containing 15 mol % spin-labeled phospholipid (5- or 12-PC) and 3.2 nmol of gramicidin under a stream of nitrogen while being warmed gently (35°C) and then under a high vacuum for at least 3 h. Duplicate samples were prepared in each case except for samples lacking the quencher (5- or 12-PC) where triplicates were prepared. Background samples lacking the fluorophore (gramicidin analog) were prepared in all experiments, and their fluorescence intensity was subtracted from the respective sample fluorescence intensity.
Average depths of the individual tryptophan residues in membrane-bound gramicidin analogs were estimated by the parallax method (40) using the equation
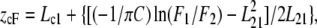 |
(1) |
where zcF = the depth of the fluorophore from the center of the bilayer, Lc1 = the distance of the center of the bilayer from the shallow quencher (5-PC in this case), L21 = the difference in depth between the two quenchers (i.e., the transverse distance between the shallow and the deep quencher), and C = the two-dimensional quencher concentration in the plane of the membrane (molecules/Å2). Here F1/F2 is the ratio of F1/Fo and F2/Fo in which F1 and F2 are fluorescence intensities in the presence of the shallow (5-PC) and deep quencher (12-PC), respectively, both at the same quencher concentration C; Fo is the fluorescence intensity in the absence of any quencher. All the bilayer parameters used were the same as described previously (40).
Steady-state fluorescence measurements
Steady-state fluorescence measurements were performed with a Hitachi F-4010 steady-state spectrofluorometer (Tokyo, Japan) using 1-cm path length quartz cuvettes. Excitation and emission slits with a nominal bandpass of 5 nm were used. Background intensities of samples in which gramicidin was omitted were subtracted from each sample spectrum to cancel out any contribution due to the solvent Raman peak and other scattering artifacts. The spectral shifts obtained with different sets of samples were identical in most cases. In other cases, the values were within ±1 nm of those reported.
Dual quenching measurements
Quenching of gramicidin tryptophan fluorescence was estimated by measurement of fluorescence intensity in the presence and absence of 0.3 M acrylamide (taken from a freshly prepared 4 M stock solution in water) or 10 mol % 10-DN (47). Samples were kept in dark for at least 1 h before measuring fluorescence. For acrylamide quenching, the excitation wavelength was fixed at 295 nm and emission was monitored at 334 nm in all cases. For 10-DN quenching, the excitation wavelength was fixed at 295 nm and emission was monitored at 334 nm except in the case of W(11,13,15)BgA, where emission was monitored at 320 nm. For samples containing acrylamide, corrections for inner filter effect were made using the following equation (48):
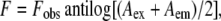 |
(2) |
where F is the corrected fluorescence intensity and Fobs is the background-subtracted fluorescence intensity of the sample. Aex and Aem are the measured absorbances at the excitation and emission wavelengths. The absorbances of the samples were measured using a Hitachi U-2000 UV-visible absorption spectrophotometer. Dual quenching ratios (Q-ratios) were calculated using the equation (43)
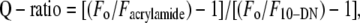 |
(3) |
where Fo is the fluorescence intensity in the absence of any quencher, Facrylamide is the corrected fluorescence intensity in the presence of 0.3 M acrylamide, and F10-DN is the fluorescence intensity in the presence of 10 mol % 10-DN.
Time-resolved fluorescence measurements
Fluorescence lifetimes were calculated from time-resolved fluorescence intensity decays using a Photon Technology International (London, Western Ontario, Canada) LS-100 luminescence spectrophotometer in the time-correlated single-photon counting mode. This machine uses a thyratron-gated nanosecond flash lamp filled with nitrogen as the plasma gas (17 ± 1 inches of mercury vacuum) and is run at 22–25 kHz. Lamp profiles were measured at the excitation wavelength using Ludox (colloidal silica) as the scatterer. To optimize the signal/noise ratio, 5000 photon counts were collected in the peak channel. All experiments were performed using excitation and emission slits with a bandpass of 8 nm or less. The sample and the scatterer were alternated after every 10% acquisition to ensure compensation for shape and timing drifts occurring during the period of data collection. This arrangement also prevents any prolonged exposure of the sample to the excitation beam, thereby avoiding any possible photodamage to the fluorophore. The data stored in a multichannel analyzer were routinely transferred to an IBM PC for analysis. Fluorescence intensity decay curves so obtained were deconvoluted with the instrument response function and analyzed as a sum of exponential terms:
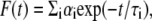 |
(4) |
where F(t) is the fluorescence intensity at time t and αi is a preexponential factor representing the fractional contribution to the time-resolved decay of the component with a lifetime τi such that Σiαi = 1. The decay parameters were recovered using a nonlinear least squares iterative fitting procedure based on the Marquardt algorithm as described earlier (28). A fit was considered acceptable when plots of the weighted residuals and the autocorrelation function showed random deviation about zero with a minimum χ2 value (generally not more than 1.2). Mean (average) lifetimes 〈τ〉 for biexponential decays of fluorescence were calculated from the decay times and preexponential factors using the following equation (48):
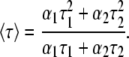 |
(5) |
Circular dichroism measurements
CD measurements were carried out at room temperature (25°C) on a JASCO J-715 spectropolarimeter (Tokyo, Japan), which was calibrated with (+)-10-camphorsulfonic acid. The spectra were scanned in a quartz optical cell with a path length of 0.1 cm. All spectra were recorded in 0.5 nm wavelength increments with a 4 s response and a bandwidth of 1 nm. For monitoring changes in secondary structure, spectra were scanned in the far-UV range from 200 to 280 nm at a scan rate of 100 nm/min. Each spectrum is the average of 12 scans with a full-scale sensitivity of 10 mdeg. All spectra were corrected for background by subtraction of appropriate blanks and were smoothed making sure that the overall shape of the spectrum remained unaltered. Data are represented as mean residue ellipticities and were calculated using the formula
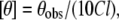 |
(6) |
where θobs is the observed ellipticity in mdeg, l is the path length in cm, and C is the concentration of peptide bonds in mol/L.
Size-exclusion chromatography
Size-exclusion chromatography was performed using an Ultrastyragel 1000 Å column (Waters, Milford, MA) with a tetrahydrofuran mobile phase at a flow rate of 1.0 mL/min, following the methods of Bañó et al. (49).
RESULTS
Fluorescence characteristics and red edge excitation shifts of gramicidin analogs
The sequences of the single-tryptophan gramicidin analogs used are shown in Fig. 1 a. Our objective was to examine the immediate membrane environment experienced by the tryptophan residue in each of these analogs, in the absence of other aromatic rings, when bound to membranes. We have earlier utilized the intrinsic tryptophan fluorescence of gramicidin to effectively distinguish gramicidin conformations in membranes (28,50). In particular, we showed that REES of gramicidin tryptophans is sensitive to the conformation of membrane-bound gramicidin and could be used to distinguish various conformations. A shift in the wavelength of maximum fluorescence emission toward higher wavelengths, caused by a shift in the excitation wavelength toward the red edge of the absorption band, is termed REES (14,51,52). This effect is observed mostly with polar fluorophores in motionally restricted environments, such as viscous solutions or condensed phases where the dipolar relaxation time for the solvent shell around a fluorophore is comparable to or longer than its fluorescence lifetime. Although other fluorescence techniques yield information about the fluorophore itself, REES provides information about the relative rates of solvent relaxation, which is not possible to obtain by other techniques. REES can serve as a powerful tool to monitor gramicidin conformations in membranes and membrane-mimetic environments (27,28,39,53).
The fluorescence emission maximum of gramicidin in the channel conformation in POPC membranes is 333 nm, whereas the nonchannel conformation displays an emission maximum at 335 nm, when excited at 280 nm (28). Fig. 1 a shows the fluorescence emission maxima of the single-tryptophan analogs of gramicidin in POPC membranes. The analog with the sole tryptophan at position 15 displays an emission maximum of 335 nm. When the position of any one of the tryptophans in the sequence is changed, the emission maximum exhibits progressive blue shift. Thus, the gramicidin analogs with tryptophans at positions 13 and 11 show emission maxima at 331 and 328 nm, respectively. Interestingly, the analog with the sole tryptophan at position 9 displays an extremely blue-shifted emission maximum at 318 nm, suggesting that the environment around the tryptophan may be very hydrophobic (see below). The variation in fluorescence emission maximum among the analogs may be indicative of differential localization of the tryptophans along the membrane axis. This is particularly true for the nonchannel conformation of gramicidin where the tryptophan residues are distributed more evenly along the backbone manifold than in the channel conformation (28).
The shifts in the maxima of fluorescence emission of the tryptophan residue of the gramicidin analogs as a function of excitation wavelength are shown in Fig. 1 b (we have used the term “maximum of fluorescence emission” in a somewhat wider sense here. In every case, we monitored the wavelength corresponding to maximum fluorescence intensity, as well as the center of mass of the fluorescence emission. In most cases, both these methods yielded the same wavelength. In cases where minor discrepancies were found, the center of mass of emission has been reported as the fluorescence maximum 0. As the excitation wavelength is changed from 280 to 307 nm, the emission maximum is shifted from 335 to 340 nm in the case of Trp-15, from 331 to 335 nm for Trp-13, and 328 to 335 nm for Trp-11. The shifts correspond to REES of 4–7 nm in these cases (Fig. 1 c). It is possible that there could be further red shift if excitation is carried out beyond 307 nm. We found it difficult to work in this wavelength range due to low signal/noise ratio and artifacts due to the solvent Raman peak that sometimes remains even after background subtraction. Such dependence of the emission maximum on excitation wavelength is characteristic of REES and implies that the individual tryptophans are localized in motionally restricted regions. In contrast, a somewhat surprising enhanced REES of 15 nm is observed for Trp-9, from 318 to 333 nm, when the excitation wavelength is changed from 280 to 307 nm. In addition to the dependence of fluorescence emission maxima on the excitation wavelength, fluorescence polarization also depends on the excitation and emission wavelengths in motionally restricted environments (54). All the single-tryptophan gramicidin analogs display wavelength-dependent fluorescence polarization (not shown). This finding reinforces the motional restriction for the single-tryptophan residues of the analogs.
Fluorescence lifetime serves as a sensitive indicator of the local environment and polarity in which a given fluorophore is placed (55). A typical decay profile of a gramicidin analog with biexponential fitting and statistical parameters is shown in Fig. 2. The fluorescence decays could be fitted well using a biexponential function. The mean fluorescence lifetimes (Table 1), calculated using Eq. 5, for the individual tryptophans in the single-Trp gramicidin analogs are found to be 2.25 (Trp-15), 1.58 (Trp-13), 1.98 (Trp-11), and 1.57 ns in the case of Trp-9. The relatively small variations in mean fluorescence lifetime could possibly be due to different mixtures of conformations (see below) as well as different average Trp positions among the different gramicidin analogs. In addition, fluorescence lifetime is known to depend on the excitation and emission wavelength in motionally restricted environments (54). All the single-tryptophan gramicidin analogs displayed wavelength-dependent fluorescence lifetime (not shown), thereby confirming motional restriction for each of the tryptophan residues.
FIGURE 2.

Representative time-resolved fluorescence intensity decay of W(11,13,15)BgA in POPC membranes. Excitation wavelength was at 297 nm, which corresponds to a peak in the spectral output of the nitrogen lamp. Emission was monitored at 330 nm. The sharp peak on the left is the lamp profile. The relatively broad peak on the right is the decay profile, fitted to a biexponential function. The two lower plots show the weighted residuals and the autocorrelation function of the weighted residuals. All other conditions are as in Fig. 1. See Materials and Methods for other details.
TABLE 1.
Fluorescence lifetimes of single-tryptophan analogs of gramicidin
Gramicidin conformations
Circular dichroism spectroscopy
The variation in fluorescence parameters among the gramicidin analogs may be indicative of differential localization of the tryptophan residues in the membrane. Since the distribution and depths of the tryptophan residues constitute a major difference between the channel and nonchannel conformations of gramicidin (28), we examined the backbone conformation of the gramicidin analogs using CD spectroscopy, which has been used previously to distinguish various conformations of gramicidin (23,28,30). The CD spectra of the gramicidin analogs are shown in Fig. 3, along with the spectrum for gramicidin in the channel conformation for reference. The characteristic single-stranded β6.3-channel conformation has characteristic peaks of positive ellipticity around 218 and 235 nm, a valley around 230 nm, and negative ellipticity below 208 nm.
FIGURE 3.

Far-UV CD spectra of gramicidin (long-dashed line), W(11,13,15)BgA (dotted line), W(9,13,15)BgA (dash-dotted line), W(9,11,15)BgA (short-dashed line), and W(9,11,13)BgA (solid line) in POPC membranes. All conditions are as in Fig. 1. See Materials and Methods for other details.
The CD spectra of the gramicidin analogs are varied (Fig. 3), and each deviates from the spectrum of the channel conformation. Interestingly, the CD spectra of the gramicidin analogs lack the contribution of aromatic residues (i.e., the tryptophan residues that are substituted by ser-t-butyl in the analogs). Although this may be a reason for some of the differences in CD spectra in Fig. 3, a change in backbone conformation of the gramicidin analogs as compared to native gramicidin cannot be ruled out. The spectra of the gramicidin analogs are consistent with spectra expected from mixtures of channel and nonchannel conformations (56), with W(9,11,13)BgA showing the greatest proportion of the nonchannel form. This interpretation of the CD spectra is also consistent with the findings from size-exclusion chromatography.
Size-exclusion chromatography
All the analogs also show substantial amounts of double-stranded conformers when examined by size-exclusion chromatography (Table 2), from ∼10% for W(11,13,15)BgA to ∼50% for W(9,11,13)BgA. There is not a strong lipid dependence for the mixture of conformations, as the results are substantially similar in POPC and DMPC (Table 2). It is therefore clear that the backbone conformation of the single-tryptophan analogs is markedly different from the native gramicidin channel conformation.
TABLE 2.
Nonchannel conformer population for single-tryptophan gramicidin analogs in lipid vesicles based on size-exclusion chromatography
| Gramicidin analog | DMPC | POPC |
|---|---|---|
| W(11,13,15)BgA | 5% | 12% |
| W(9,13,15)BgA | 21% | 32% |
| W(9,11,15)BgA | 37% | 32% |
| W(9,11,13)BgA | 43% | 48% |
The percentage of gramicidin analog present in double-stranded (nonchannel) conformation is listed. The peptide/lipid ratio was 1:50 (mol/mol) in all cases. See Materials and Methods for other details.
Membrane penetration depths of gramicidin analogs
As shown in Fig. 1 a, the tryptophan residues of the gramicidin analogs display a progressive blue shift with the change in position of the residue in the gramicidin sequence. Such a position dependence of the emission maximum indicates different average locations for the various tryptophan residues along the membrane axis. The blue shifts and other data are also consistent, with Trp-9 being 90% single-stranded (Table 2) (yet still relatively buried), Trp-15 being solvent-exposed (in both of its conformations), and Trp-11/Trp-13 being intermediate cases, each within a conformational mixture. The different average locations also would imply that the accessibility of the aqueous environment to the tryptophan residues would also be graded. The location, depth, orientation, and distribution of the tryptophan residues of gramicidin are different in the channel and nonchannel conformations (25–28), although Trp-9 is relatively more buried in both conformations. The distribution of tryptophans along the membrane axis is more pronounced in the nonchannel conformation (57). Analysis of membrane penetration depths of individual tryptophan residues could therefore provide additional information.
Acrylamide quenching of tryptophan fluorescence is widely used to monitor tryptophan environments in proteins (58). However, acrylamide quenching of gramicidin tryptophan fluorescence is relatively insensitive to the distribution of tryptophan residues across the membrane axis (28), which could result from permeation of acrylamide into the membrane bilayer (59). To eliminate such possible complications, we utilized a dual quenching approach (43) to estimate the average location and distribution of the tryptophan residues in the single-Trp gramicidin analogs. This method is based on the differential accessibility of an aqueous and membrane-bound quencher to membrane-bound tryptophan residues. We utilized the aqueous quencher acrylamide and membrane-bound spin label quencher 10-DN to calculate a Q-ratio, which has been found to have an approximate linear relationship with fluorophore depth in the membrane (43). Spin labels are strong quenchers of a wide range of fluorophores, including tryptophans (40,60,61). The use of two quenchers in the Q-ratio should amplify sensitivity by canceling out nondepth related effects on quenching.
Fig. 4 shows the quenching of gramicidin tryptophan fluorescence in the presence of fixed concentrations of acrylamide or 10-DN. Acrylamide quenching does not appear to be sensitive to the different tryptophan residues of the gramicidin analogs. Fluorescence quenching by 10-DN appears to be more sensitive to the gramicidin analogs. The Q-ratios, calculated using Eq. 3, are shown in Fig. 4. The Q-ratio exhibits significant dependence on the position of the tryptophan residues in the sequence of the gramicidin analogs and varies between 0.08 for Trp-9 and 0.93 for Trp-15. The lower Q-ratio for W(11,13,15)BgA is consistent with an (average) tryptophan position deeper in the membrane, whereas a higher Q-ratio for W(9,11,13)BgA is representative of a tryptophan residue at shallow regions of the membrane (43).
FIGURE 4.

Quenching and Q-ratios of tryptophan fluorescence of gramicidin analogs in POPC membranes. Fo is the fluorescence intensity in the absence of quencher (acrylamide or 10-DN), and F is the fluorescence intensity in the presence of quencher (acrylamide or 10-DN). The open bars represent quenching by 0.3 M acrylamide, and the hatched bars represent quenching by 10 mol % 10-DN. Data shown represent mean ± SE of at least three independent measurements. For acrylamide quenching, the excitation wavelength was fixed at 295 nm and emission was monitored at 334 nm in all cases. For 10-DN quenching, the excitation wavelength was fixed at 295 nm and emission was monitored at 334 nm except in the case of W(11,13,15)BgA, where emission was monitored at 320 nm. The filled bars represent Q-ratios calculated according to Eq. 1. The concentration of POPC was 0.11 mM. All other conditions are as in Fig. 1. See Materials and Methods for other details.
Although analysis of membrane depths by the dual quenching approach is useful (62), it should be noted that it does not provide absolute values for fluorophore depth in the membrane bilayer. The parallax method (40) involves determination of the parallax in the apparent locations of fluorophores by comparing fluorescence quenching by phospholipids spin-labeled at two different depths. The average depths of penetration of the individual tryptophan residues in the single-Trp gramicidin analogs, estimated using this method, are shown in Table 3. The average depths of the ensemble of four tryptophans of native gramicidin in the channel and nonchannel conformations are also shown. These results are to be interpreted with some degree of caution due to a number of reasons. Importantly, the depths obtained are influenced by “conformational averaging”. For example, previous work using molecular dynamics simulations have revealed motional flexibility, giving rise to conformational heterogeneity of Trp-9 in gramicidin (63).
TABLE 3.
Membrane penetration depths of the tryptophan residue in single-tryptophan analogs of gramicidin by the parallax method
| Gramicidin analog | Depth from the center of the bilayer zcF (Å) |
|---|---|
| Gramicidin (channel)* | 11.0 |
| Gramicidin (nonchannel)* | 7.3 |
| W(11,13,15)BgA | 1.3 |
| W(9,13,15)BgA | 5.5 |
| W(9,11,15)BgA | 10.7 |
| W(9,11,13)BgA | 12.5 |
Depths were calculated from fluorescence quenchings obtained with samples containing 15 mol % of 5-PC and 12-PC and using Eq. 1. Samples were excited at 280 nm, and emission was monitored at 335 nm. The gramicidin/total lipid ratio was 1:50 (mol/mol). See Materials and Methods for other details.
From Rawat et al. (28).
Since the parallax method is an intensity-based approach, these results would be further complicated by any heterogeneity in fluorescence quantum yields of individual tryptophan residues arising due to environmental sensitivity. In addition, it should be noted that dynamic considerations, in addition to conformational averaging, may also influence the quenching and the (apparent) parallax calculation (Eq. 1). For these reasons, caution should be exercised in interpreting the numbers in Table 3. Importantly, the reported depth for W(11,13,15)BgA might be particularly susceptible to the effects of averaging despite the fact that only a single tryptophan is present in the gramicidin sequence. In the nonchannel conformation, the tryptophans would be placed at a graded series of membrane depths, whereas in the channel conformation all the tryptophans are clustered at the membrane interface. Taken together, the observed data from all the methods support a mixed population of nonchannel and channel conformers for each of the single-Trp gramicidins, possibly involving additional dynamic averaging.
DISCUSSION
Gramicidin serves as an excellent model for transmembrane channels due to its small size, ready availability, and the relative ease with which chemical modifications can be performed. These features make gramicidin unique among small membrane-active peptides and provide the basis for using gramicidin to explore principles that govern the folding and function of membrane-spanning channels in particular and membrane proteins in general. It is interesting to note that gramicidin represents a useful model for realistic determination of conformational preference in a lipid bilayer environment despite the alternating sequence of L-D chirality generally not encountered in naturally occurring peptides and proteins. This is because the dihedral angle combinations generated in the conformation space by various gramicidin conformations are “allowed” according to the Ramachandran plot (64). Gramicidin channels share important structural features, such as membrane interfacial localization of tryptophan residues, a channel interior being made of the peptide backbone, and ion selectivity arising out of backbone interactions, with other naturally occurring channel proteins, including the bacterial KcsA K+ channel (17,21,65).
It should be mentioned here that the anisotropic segmental mobility, typically exhibited by membrane bilayers (14,66), may influence our results. For example, the depth values reported in Table 3 represent averages over the various orientations in the timescale of measurement (nanosecond). Since the deeper regions of the bilayer are characterized by large motions, the depth of Trp-9 in W(11,13,15)BgA would be especially sensitive to such motional variation. These motional variations would be smaller in the case of Trp-15 in W(9,11,13)BgA. Similarly, the observed REES of Trp-9 in W(11,13,15)BgA could correspond to a ground state conformational heterogeneity, which could contribute to a relatively large magnitude of REES.
Tryptophan substitution has been shown to affect channel inactivation and gating in the case of the nicotinic acetylcholine receptor and cardiac sodium channels (67,68). The tryptophan residues in gramicidin channels have been earlier shown to be vital for maintaining the functional conformation of the channel (25,33,34). Indeed, when one subunit contains no tryptophans and the other subunit has reversed chirality, double-stranded channels may result (69). The channel conductance also is lowered irrespective of the manner in which tryptophan residues are perturbed (photolysis, chemical modification, substitution by other amino acids) (12,33,35–38). These findings bring out the crucial importance of the tryptophan residues of gramicidin in its ion channel function.
Interestingly, it has been recently shown that gramicidin tryptophans are also important for proton conduction across the membrane (70). In this work, we show that the gramicidin analogs containing single-tryptophan residues (the others being replaced by Ser-t-butyl) adopt a mixture of channel and nonchannel conformations, as evident from analysis of membrane depth, size-exclusion chromatography, and backbone CD data. The size-exclusion data suggest that the fraction of nonchannel conformer(s) increases as the lone Trp is moved outward from position 9 to 15 (Table 2). The fluorescence quenching analysis indicates that Trp-9 is likely to be accessible to the deep quencher (12-PC) in each of the major conformations. Taken together, the methods used in this study show that the single-Trp gramicidins, as well as gramicidins that lack tryptophans (34,69), fold into mixtures of single-stranded and double-stranded conformations, thereby implying the importance of tryptophan residues in membrane protein folding and function.
Acknowledgments
We gratefully acknowledge Sushmita Mukherjee for preliminary experiments and R. Rukmini for help with the membrane penetration depth experiments. We thank Y.S.S.V. Prasad and G.G. Kingi for technical help and Sandeep Shrivastava for help during the preparation of the manuscript. We thank members of A.C. laboratory for critically reading the manuscript.
This work was supported by research grants from the Council of Scientific and Industrial Research, Government of India (A.C.) and National Institutes of Health grants RR 15569 and GM 70971 to R.E.K. and D.V.G. S.S.R. and D.A.K. thank the Council of Scientific and Industrial Research for the award of Senior Research Fellowships. A.C. is an honorary professor of the Jawaharlal Nehru Centre for Advanced Scientific Research, Bangalore, India.
Satinder S. Rawat's present address is 364 Plantation Street, Room 570R, Lazare Research Building, University of Massachusetts Medical School, Worcester, MA 01605.
Devaki A. Kelkar's present address is Department of Biochemistry and Molecular Biology, Oregon Health and Science University, Portland, OR 97239.
Editor: Kenton J. Swartz.
References
- 1.Schiffer, M., C. H. Chang, and F. J. Stevens. 1992. The functions of tryptophan residues in membrane proteins. Protein Eng. 5:213–214. [DOI] [PubMed] [Google Scholar]
- 2.Ippolito, J. A., R. S. Alexander, and D. W. Christianson. 1990. Hydrogen bond stereochemistry in protein structure and function. J. Mol. Biol. 215:457–471. [DOI] [PubMed] [Google Scholar]
- 3.Yau, W.-M., W. C. Wimley, K. Gawrisch, and S. H. White. 1998. The preference of tryptophan for membrane interfaces. Biochemistry. 37:14713–14718. [DOI] [PubMed] [Google Scholar]
- 4.Koeppe, R. E., H. Sun, P. C. A. van der Wel, E. M. Scherer, P. Pulay, and D. V. Greathouse. 2003. Combined experimental/theoretical refinement of indole ring geometry using deuterium magnetic resonance and ab initio calculations. J. Am. Chem. Soc. 125:12268–12276. [DOI] [PubMed] [Google Scholar]
- 5.Doyle, D. A., J. M. Cabral, R. A. Pfuetzner, A. Kuo, J. M. Gulbis, S. L. Cohen, B. T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- 6.Luecke, H., B. Schobert, H.-T. Richter, J.-P. Cartailler, and J. K. Lanyi. 1999. Structural changes in bacteriorhodopsin during ion transport at 2 angstrom resolution. Science. 286:255–261. [DOI] [PubMed] [Google Scholar]
- 7.Schirmer, T., T. A. Keller, Y. F. Wang, and J. P. Rosenbusch. 1995. Structural basis for sugar translocation through maltoporin channels at 3.1 Å resolution. Science. 267:512–514. [DOI] [PubMed] [Google Scholar]
- 8.de Planque, M. R. R., J.-W. P. Boots, D. T. S. Rijkers, R. M. J. Liskamp, D. V. Greathouse, and J. A. Killian. 2002. The effects of hydrophobic mismatch between phosphatidylcholine bilayers and transmembrane α-helical peptides depend on the nature of interfacially exposed aromatic and charged residues. Biochemistry. 41:8396–8404. [DOI] [PubMed] [Google Scholar]
- 9.Demmers, J. A., E. van Duijn, J. Haverkamp, D. V. Greathouse, R. E. Koeppe, A. J. R. Heck, and J. A. Killian. 2001. Interfacial positioning and stability of transmembrane peptides in lipid bilayers studied by combining hydrogen/deuterium exchange and mass spectrometry. J. Biol. Chem. 276:34501–34508. [DOI] [PubMed] [Google Scholar]
- 10.Chothia, C. 1976. The nature of the accessible and buried surfaces in proteins. J. Mol. Biol. 105:1–14. [DOI] [PubMed] [Google Scholar]
- 11.Burley, S. K., and G. A. Petsko. 1985. Aromatic-aromatic interaction: a mechanism of protein structure stabilization. Science. 229:23–28. [DOI] [PubMed] [Google Scholar]
- 12.Fonseca, V., P. Daumas, L. Ranjalahy-Rasoloarijao, F. Heitz, R. Lazaro, Y. Trudelle, and O. S. Andersen. 1992. Gramicidin channels that have no tryptophan residues. Biochemistry. 31:5340–5350. [DOI] [PubMed] [Google Scholar]
- 13.Wimley, W. C., and S. H. White. 1996. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Biol. 3:842–848. [DOI] [PubMed] [Google Scholar]
- 14.Chattopadhyay, A. 2003. Exploring membrane organization and dynamics by the wavelength-selective fluorescence approach. Chem. Phys. Lipids. 122:3–17. [DOI] [PubMed] [Google Scholar]
- 15.Andersen, O. S., and R. E. Koeppe. 1992. Molecular determinants of channel function. Physiol. Rev. 72:89–158. [DOI] [PubMed] [Google Scholar]
- 16.Killian, J. A. 1992. Gramicidin and gramicidin-lipid interactions. Biochim. Biophys. Acta. 1113:391–425. [DOI] [PubMed] [Google Scholar]
- 17.Wallace, B. A. 2000. Common structural features in gramicidin and other ion channels. Bioessays. 22:227–234. [DOI] [PubMed] [Google Scholar]
- 18.Kelkar, D. A., and A. Chattopadhyay. 2007. The gramicidin ion channel: a model membrane protein. Biochim. Biophys. Acta. 1768:2011–2025. [DOI] [PubMed] [Google Scholar]
- 19.Koeppe II, R. E., and O. S. Andersen. 1996. Engineering the gramicidin channel. Annu. Rev. Biophys. Biomol. Struct. 25:231–258. [DOI] [PubMed] [Google Scholar]
- 20.Andersen, O. S., and R. E. Koeppe II. 2007. Bilayer thickness and membrane protein function: an energetic perspective. Annu. Rev. Biophys. Biomol. Struct. 36:107–130. [DOI] [PubMed] [Google Scholar]
- 21.Chattopadhyay, A., and D. A. Kelkar. 2005. Ion channels and d-amino acids. J. Biosci. 30:147–149. [DOI] [PubMed] [Google Scholar]
- 22.Veatch, W. R., E. T. Fossel, and E. R. Blout. 1974. The conformation of gramicidin A. Biochemistry. 13:5249–5256. [DOI] [PubMed] [Google Scholar]
- 23.Killian, J. A., K. U. Prasad, D. Hains, and D. W. Urry. 1988. The membrane as an environment of minimal interconversion. A circular dichroism study on the solvent dependence of the conformational behavior of gramicidin in diacylphosphatidylcholine model membranes. Biochemistry. 27:4848–4855. [DOI] [PubMed] [Google Scholar]
- 24.O'Connell, A. M., R. E. Koeppe, and O. S. Andersen. 1990. Kinetics of gramicidin channel formation in lipid bilayers: transmembrane monomer association. Science. 250:1256–1259. [DOI] [PubMed] [Google Scholar]
- 25.Hu, W., K.-C. Lee, and T. A. Cross. 1993. Tryptophans in membrane proteins: indole ring orientations and functional implications in the gramicidin channel. Biochemistry. 32:7035–7047. [DOI] [PubMed] [Google Scholar]
- 26.Ketchem, R. R., W. Hu, and T. A. Cross. 1993. High-resolution conformation of gramicidin A in a lipid bilayer by solid-state NMR. Science. 261:1457–1460. [DOI] [PubMed] [Google Scholar]
- 27.Mukherjee, S., and A. Chattopadhyay. 1994. Motionally restricted tryptophan environments at the peptide-lipid interface of gramicidin channels. Biochemistry. 33:5089–5097. [DOI] [PubMed] [Google Scholar]
- 28.Rawat, S. S., D. A. Kelkar, and A. Chattopadhyay. 2004. Monitoring gramicidin conformations in membranes: a fluorescence approach. Biophys. J. 87:831–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reithmeier, R. A. F. 1995. Characterization and modeling of membrane proteins using sequence analysis. Curr. Opin. Struct. Biol. 5:491–500. [DOI] [PubMed] [Google Scholar]
- 30.LoGrasso, P. V., F. Moll, and T. A. Cross. 1988. Solvent history dependence of gramicidin A conformations in hydrated lipid bilayers. Biophys. J. 54:259–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sychev, S. V., L. I. Barsukov, and V. T. Ivanov. 1993. The double ππ5.6 helix of gramicidin A predominates in unsaturated lipid membranes. Eur. Biophys. J. 22:279–288. [DOI] [PubMed] [Google Scholar]
- 32.Zein, M., and R. Winter. 2000. Effect of temperature, pressure and lipid acyl chain length on the structure and phase behaviour of phospholipid-gramicidin bilayers. Phys. Chem. Chem. Phys. 2:4545–4551. [Google Scholar]
- 33.Andersen, O. S., D. V. Greathouse, L. L. Providence, M. D. Becker, and R. E. Koeppe. 1998. Importance of tryptophan dipoles for protein function: 5-fluorination of tryptophans in gramicidin A channels. J. Am. Chem. Soc. 120:5142–5146. [Google Scholar]
- 34.Salom, D., E. Pérez-Payá, J. Pascal, and C. Abad. 1998. Environment- and sequence-dependent modulation of the double-stranded to single-stranded conformational transition of gramicidin A in membranes. Biochemistry. 37:14279–14291. [DOI] [PubMed] [Google Scholar]
- 35.Daumas, P., F. Heitz, L. Ranjalahy-Rasoloarijao, and R. Lazaro. 1989. Gramicidin A analogs: influence of the substitution of the tryptophans by naphthylalanines. Biochimie. 71:77–81. [DOI] [PubMed] [Google Scholar]
- 36.Becker, M. D., D. V. Greathouse, R. E. Koeppe, and O. S. Andersen. 1991. Amino acid sequence modulation of gramicidin channel function: effects of tryptophan-to-phenylalanine substitutions on the single-channel conductance and duration. Biochemistry. 30:8830–8839. [DOI] [PubMed] [Google Scholar]
- 37.Barth, C., and G. Stark. 1991. Radiation inactivation of ion channels formed by gramicidin A. Protection by lipid double bonds and by α-tocopherol. Biochim. Biophys. Acta. 1066:54–58. [DOI] [PubMed] [Google Scholar]
- 38.Sobko, A. A., M. A. Vigasina, T. I. Rokitskaya, E. A. Kotova, S. D. Zakharov, W. A. Cramer, and Y. N. Antonenko. 2004. Chemical and photochemical modification of colicin E1 and gramicidin A in bilayer lipid membranes. J. Membr. Biol. 199:51–62. [DOI] [PubMed] [Google Scholar]
- 39.Kelkar, D. A., and A. Chattopadhyay. 2005. Effect of graded hydration on the dynamics of an ion channel peptide: a fluorescence approach. Biophys. J. 88:1070–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chattopadhyay, A., and E. London. 1987. Parallax method for direct measurement of membrane penetration depth utilizing fluorescence quenching by spin-labeled phospholipids. Biochemistry. 26:39–45. [DOI] [PubMed] [Google Scholar]
- 41.Greathouse, D. V., R. E. Koeppe, L. L. Providence, S. Shobana, and O. S. Andersen. 1999. Design and characterization of gramicidin channels. Methods Enzymol. 294:525–550. [DOI] [PubMed] [Google Scholar]
- 42.Eftink, M. R. 1991. Fluorescence quenching reactions: probing biological macromolecular structure. In Biophysical and Biochemical Aspects of Fluorescence Spectroscopy. T. G. Dewey, editor. Plenum Press, New York. 1–41.
- 43.Caputo, G. A., and E. London. 2003. Using a novel dual fluorescence quenching assay for measurement of tryptophan depth within lipid bilayers to determine hydrophobic α-helix locations within membranes. Biochemistry. 42:3265–3274. [DOI] [PubMed] [Google Scholar]
- 44.Dittmer, J. C., and R. L. Lester. 1964. A simple, specific spray for the detection of phospholipids on thin-layer chromatograms. J. Lipid Res. 5:126–127. [PubMed] [Google Scholar]
- 45.McClare, C. W. F. 1971. An accurate and convenient organic phosphorus assay. Anal. Biochem. 39:527–530. [DOI] [PubMed] [Google Scholar]
- 46.Abrams, F. S., and E. London. 1993. Extension of the parallax analysis of membrane penetration depth to the polar region of model membranes: use of fluorescence quenching by a spin-label attached to the phospholipid polar headgroup. Biochemistry. 32:10826–10831. [DOI] [PubMed] [Google Scholar]
- 47.Kelkar, D. A., and A. Chattopadhyay. 2006. Monitoring ion channel conformations in membranes utilizing a novel dual fluorescence quenching approach. Biochem. Biophys. Res. Commun. 343:483–488. [DOI] [PubMed] [Google Scholar]
- 48.Lakowicz, J. R. 1999. Principles of Fluorescence Spectroscopy. Kluwer-Plenum Press, New York.
- 49.Baño, M. C., L. Braco, and C. Abad. 1988. New high-performance liquid chromatography-based methodology for monitoring the conformational transitions of self-associating hydrophobic peptides, incorporated into liposomes. J. Chromatogr. 458:105–116. [DOI] [PubMed] [Google Scholar]
- 50.Kelkar, D. A., and A. Chattopadhyay. 2007. Modulation of gramicidin channel conformation and organization by hydrophobic mismatch in saturated phosphatidylcholine bilayers. Biochim. Biophys. Acta. 1768:1103–1113. [DOI] [PubMed] [Google Scholar]
- 51.Demchenko, A. P. 2002. The red-edge effects: 30 years of exploration. Luminescence. 17:19–42. [DOI] [PubMed] [Google Scholar]
- 52.Raghuraman, H., D. A. Kelkar, and A. Chattopadhyay. 2005. Novel insights into protein structure and dynamics utilizing the red edge excitation shift approach. In Reviews in Fluorescence 2005, Vol. 2. C. D. Geddes and J. R. Lakowicz, editors. Springer, New York. 199–224.
- 53.Rawat, S. S., D. A. Kelkar, and A. Chattopadhyay. 2005. Effect of structural transition of the host assembly on dynamics of an ion channel peptide: a fluorescence approach. Biophys. J. 89:3049–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mukherjee, S., and A. Chattopadhyay. 1995. Wavelength-selective fluorescence as a novel tool to study organization and dynamics in complex biological systems. J. Fluoresc. 5:237–246. [DOI] [PubMed] [Google Scholar]
- 55.Prendergast, F. G. 1991. Time-resolved fluorescence techniques: methods and applications in biology. Curr. Opin. Struct. Biol. 1:1054–1059. [Google Scholar]
- 56.Greathouse, D. V., J. F. Hinton, K. S. Kim, and R. E. Koeppe II. 1994. Gramicidin A/short-chain phospholipid dispersions: chain length dependence of gramicidin conformation and lipid organization. Biochemistry. 33:4291–4299. [DOI] [PubMed] [Google Scholar]
- 57.Arumugam, S., S. Pascal, C. L. North, W. Hu, K.-C. Lee, M. Cotten, R. R. Ketchem, F. Xu, M. Brenneman, F. Kovacs, F. Tian, A. Wang, S. Huo, and T. A. Cross. 1996. Conformational trapping in a membrane environment: a regulatory mechanism for protein activity? Proc. Natl. Acad. Sci. USA. 93:5872–5876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Eftink, M. R. 1991. Fluorescence quenching: theory and applications. In Topics in Fluorescence Spectroscopy, Vol. 2: Principles. J. R. Lakowicz, editor. Plenum Press, New York. 53–126.
- 59.Moro, F., F. M. Goñi, and M. A. Urbaneja. 1993. Fluorescence quenching at interfaces and the permeation of acrylamide and iodide across phospholipid bilayers. FEBS Lett. 330:129–132. [DOI] [PubMed] [Google Scholar]
- 60.London, E., and G. W. Feigenson. 1981. Fluorescence quenching in model membranes. 1. Characterization of quenching caused by a spin-labeled phospholipid. Biochemistry. 20:1932–1938. [DOI] [PubMed] [Google Scholar]
- 61.Chattopadhyay, A. 1992. Membrane penetration depth analysis using fluorescence quenching: a critical review. In Biomembrane Structure & Function: The State of the Art. B. P. Gaber and K. R. K. Easwaran, editors. Adenine Press, Schenectady, NY. 153–163.
- 62.Musse, A. A., J. Wang, G. P. Deleon, G. A. Prentice, E. London, and A. R. Merrill. 2006. Scanning the membrane-bound conformation of helix 1 in the colicin E1 channel domain by site-directed fluorescence labeling. J. Biol. Chem. 281:885–895. [DOI] [PubMed] [Google Scholar]
- 63.Allen, T. W., O. S. Andersen, and B. Roux. 2003. Structure of gramicidin A in a lipid bilayer environment determined using molecular dynamics simulations and solid-state NMR data. J. Am. Chem. Soc. 125:9868–9877. [DOI] [PubMed] [Google Scholar]
- 64.Andersen, O. S., G. Saberwal, D. V. Greathouse, and R. E. Koeppe. 1996. Gramicidin channels—a solvable membrane “protein” folding problem. Indian J. Biochem. Biophys. 33:331–342. [PubMed] [Google Scholar]
- 65.Kelkar, D. A., and A. Chattopadhyay. 2006. Membrane interfacial localization of aromatic amino acids and membrane protein function. J. Biosci. 31:297–302. [DOI] [PubMed] [Google Scholar]
- 66.Seelig, J. 1977. Deuterium magnetic resonance: theory and application to lipid membranes. Q. Rev. Biophys. 10:353–418. [DOI] [PubMed] [Google Scholar]
- 67.Lasalde, J. A., S. Tamamizu, D. H. Butler, C. R. Vibat, B. Hung, and M. G. McNamee. 1996. Tryptophan substitutions at the lipid-exposed transmembrane segment M4 of Torpedo californica acetylcholine receptor govern channel gating. Biochemistry. 35:14139–14148. [DOI] [PubMed] [Google Scholar]
- 68.Wang, S. Y., C. Russell, and G. K. Wang. 2005. Tryptophan substitution of a putative D4S6 gating hinge alters slow inactivation in cardiac sodium channels. Biophys. J. 88:3991–3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Durkin, J. T., L. L. Providence, R. E. Koeppe, and O. S. Andersen. 1992. Formation of non-β6.3-helical gramicidin channels between sequence-substituted gramicidin analogues. Biophys. J. 62:145–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gowen, J. A., J. C. Markham, S. E. Morrison, T. A. Cross, D. D. Busath, E. J. Mapes, and M. F. Schumaker. 2002. The role of Trp side chains in tuning single proton conduction through gramicidin channels. Biophys. J. 83:880–898. [DOI] [PMC free article] [PubMed] [Google Scholar]