

Abstract
Spin-labeling and multifrequency EPR spectroscopy were used to probe the dynamic local structure of skeletal myosin in the region of force generation. Subfragment 1 (S1) of rabbit skeletal myosin was labeled with an iodoacetamide spin label at C707 (SH1). X- and W-band EPR spectra were recorded for the apo state and in the presence of ADP and nucleotide analogs. EPR spectra were analyzed in terms of spin-label rotational motion within myosin by fitting them with simulated spectra. Two models were considered: rapid-limit oscillation (spectrum-dependent on the orientational distribution only) and slow restricted motion (spectrum-dependent on the rotational correlation time and the orientational distribution). The global analysis of spectra obtained at two microwave frequencies (9.4 GHz and 94 GHz) produced clear support for the second model and enabled detailed determination of rates and amplitudes of rotational motion and resolution of multiple conformational states. The apo biochemical state is well-described by a single structural state of myosin (M) with very restricted slow motion of the spin label. The ADP-bound biochemical state of myosin also reveals a single structural state (M*, shown previously to be the same as the post-powerstroke ATP-bound state), with less restricted slow motion of the spin label. In contrast, the extra resolution available at 94 GHz reveals that the EPR spectrum of the S1.ADP.Vi-bound biochemical state of myosin, which presumably mimics the S1.ADP.Pi state, is resolved clearly into three spectral components (structural states). One state is indistinguishable from that of the ADP-bound state (M*) and is characterized by moderate restriction and slow motion, with a mole fraction of 16%. The remaining 84% (M**) contains two additional components and is characterized by fast rotation about the x axis of the spin label. After analyzing EPR spectra, myosin ATPase activity, and available structural information for myosin II, we conclude that post-powerstroke and pre-powerstroke structural states (M* and M**) coexist in the S1.ADP.Vi biochemical state. We propose that the pre-powerstroke state M** is characterized by two structural states that could reflect flexibility between the converter and N-terminal domains of myosin.
INTRODUCTION
The combination of EPR and site-directed spin labeling is a well-established approach for probing the structure and dynamics of proteins. The anisotropy of the magnetic tensors of a nitroxide spin label produces exceptional sensitivity of the EPR lineshape to nitroxide orientation with respect to the applied magnetic field. Molecular motion in the submicrosecond timescale greatly affects the lineshape of the nitroxide EPR spectrum, making it sensitive to global tumbling of the protein and to local spin-label motion with respect to the protein. Global tumbling usually is considered to be unrestricted rotation, either isotropic or anisotropic; local motion is considered to be restricted rotation. There are several approaches to interpret restricted local motion, such as rapid-limit oscillation (1–3) or slow motion in a restoring potential (4). The first model assumes that the spin-label rotation is fast enough to average the anisotropic magnetic interactions, with a frequency of rotation greater than 2π (gx–gz)βeH0/h = 1.8 ×109 s−1, (βe = Bohr magneton, H0 = 3.3565 T, W-band spectrum center field), τc ≤ 0.09 ns. In the second model, a broad range of the rate of spin label motion is considered. Both models allow characterization of protein local structure via interpretation of EPR spectra in terms of spin-label motion restrictions (angle of oscillation or restoring potential). Fitting of EPR spectra in the model of rapid-limit oscillation provides information about restrictions of spin-label motion directly, due to the assumption of fast motion of the spin label. Fitting of EPR spectra in the slow motion approach produces two parameters: the rotational diffusion coefficient and the equilibrium distribution of orientation probability of the spin label {DR, P0}. The distribution P0 characterizes protein local structure, but it depends on the rate of spin-label motion. In a single-frequency EPR experiment, the simultaneous determination of these two parameters is ambiguous primarily because of the undetermined spin-spin relaxation time T2, which is responsible for spectral broadening. A two-frequency experiment can remove this ambiguity, due to the frequency dependence of the sensitivity to τR (4,5). In a previous study of the membrane protein phospholamban, we have shown that a simultaneous fit of X- and W-band EPR spectra can produce unambiguous results for both rate and amplitude of spin-label motion and can quantitatively resolve two distinct conformational states of the protein (5). In this study, we apply this method to myosin.
We compared two models of spin-label motion in myosin, as described above, by fitting experimental EPR spectra of IASL-labeled myosin subfragment 1 (IASL-S1) in three different biochemical states (apo S1, S1.ADP, and S1.ADP.Vi) that represent three intermediate steps in the myosin ATPase cycle: rigor (M), post-powerstroke (M*), and pre-powerstroke (M**). During its ATPase cycle, myosin exhibits substantial structural transformation of the force-generating domain, reflected in atomic structures (6–9), by intrinsic fluorescence (10,11), fluorescence resonant energy transfer (12), and EPR (13). These data suggest that the structure of the force-generating domain of myosin is determined by the biochemical state that is created by the bound nucleotide ∼4 nm away. However, it remains possible that several structural (conformational) states of the force-generating domain can coexist (and interconvert) within one biochemical state, as defined by the state of the nucleotide at the active site. In this article, we used the high orientational resolution of spin label EPR, augmented by experiments at two microwave frequencies, to resolve multiple conformational states of the force-generating region of spin-labeled myosin. In this manner, we obtained reliable information about the allosteric influence of the nucleotide on the number of conformational states, the relative populations of these states, and both the rate and amplitude of spin-label rotation in each state.
MATERIALS AND METHODS
Chemicals
ATP, ADP, IASL, and Na3VO4 were obtained from Sigma-Aldrich (Milwaukee, WI). All chemicals were of reagent grade.
Proteins
Myosin was prepared from rabbit back and leg muscles. Myosin S1 was obtained by limited α-chymotrypsin digestion (14). Rabbit fast skeletal muscle F-actin was extracted from acetone powder, as described previously (15).
Labeling of S1
S1 was labeled with IASL specifically at Cys-707 (SH1) by the two-step method of Barnett and Thomas (13), except that the label/protein ratio of the first labeling step was 0.9. After labeling, excess unreacted IASL was washed out with size-exclusion spin columns (Pierce, Rockford, IL). IASL-S1 was concentrated to 200 μM with Microcon concentrators (Millipore, Billerica, MA). Labeling buffer contained 50 mM MOPS, 50 mM KCl, 0.1mM EDTA, pH 7.0.
S1 concentration
The concentration of S1 was determined spectrophotometrically at 280 nm using an extinction coefficient of 8.14 × 104 M−1 cm−1. The S1 concentration was 100–150 μM in EPR experiments, 0.9 μM in actin-activated ATPase assays, 0.1–0.9 μM in high-salt ATPase assays, and 2.3 μM and 9 μM in Mg2+-ATPase assays without and with nucleotide analog, respectively.
Preparation of S1-nucleotide complexes
A complex of IASL-S1 with ADP (IASL-S1.ADP) was obtained by incubation of IASL-S1 with 5 mM ADP for 5 min at 25°C. A complex with ADP and vanadate (IASL-S1.ADP.Vi) was obtained from IASL-S1.ADP by addition of 5 mM Na3VO4 for 20 min at 25°C. These complexes were prepared in the same buffer that was used for EPR experiments: 20mM EPPS (pH 8.0), 6 mM MgCl2, and 1 mM EGTA.
Biochemical assays
Actin-activated ATPase activity (T = 25°C in 10 mM Tris, 3 mM MgCl2, 2.5 mM ATP, pH 7.5) was measured from the liberation of inorganic phosphate (16), using a malachite green–enhanced phosphate assay (17). The dependence of S1 activity on actin concentration was fitted to V = Vmax([actin]/([actin] + Km). High-salt ATPase activities were measured at 25°C in buffers containing 50 mM MOPS, 0.6M KCl, pH 7.5, and either 5 mM EDTA (K/EDTA–ATPase) or 10 mM CaCl2 (Ca/K–ATPase). The Mg2+–ATPase activity of IASL-S1.ADP.Vi was measured in 20 mM EPPS, 1 mM EGTA, 6 mM MgCl2, 5 mM ATP, pH 8.0, T = 20°C. The results of biochemical assays are shown in Table 1.
TABLE 1.
S1 ATPase activity
| Actin-activated ATPase
|
High salt ATPase
|
||||
|---|---|---|---|---|---|
| Protein | Vmax | Km | Mg2+− ATPase, V0 | VCa/K | VK/EDTA |
| S1 | 13.1 ± 0.5 s−1 | 13.8 ± 2.3 μM | 0.10 ± 0.02 s−1 | 1.0 ± 0.1 s−1 | 8.5 ± 0.5 s−1 |
| IASL-S1 | 2.4 ± 0.2 s−1 | 7.2 ± 0.3 μM | 0.32 ± 0.13 s−1 | 11.2 ± 1.3 s−1 | 1.4 ± 0.3 s−1 |
| IASL-S1.ADP.Vi | – | – | 0.006 s−1 | – | – |
Mass spectrometry
Mass spectrometry experiments were performed using a quadrupole time-of-flight mass spectrometer (QSTAR; Applied Biosystems, Foster City, CA) with an electrospray ionization source. The protein sample (2 mg/ml S1 or IASL-S1 in 10 mM NH4HCO3 buffer at pH 7.9) was injected into the solvent stream using a 10 μL-injection loop installed in the integrated loop injector. Three to five injections were performed for every sample with 2-min intervals between them. Data were acquired continuously during load buffer infusion and protein infusions over the range 500–2000 m/z. Spectra were analyzed with software (AnalystQS; Applied Biosystems).
Labeling extent and specificity
IASL labeling decreased the rate of K/EDTA ATPase assay to ∼15% and increased the rate of Ca/K–ATPase assay 11-fold, indicating the specific labeling of at least 90% of S1 at Cys-707 (13). The complete labeling of S1 was confirmed by mass spectrometry: the peak of IASL-S1 was shifted by 212.3 Da relative to unmodified S1, matching the molecular weight of the spin label, and no trace of unlabeled S1 was found in the IASL-S1 sample. Control experiments with mixtures showed that S1 and IASL-S1 were detected with similar sensitivity by mass spectrometry, and so the spin-labeled S1 sample contained <5% of unlabeled S1.
IASL-S1 modeling
Apo IASL-S1, IASL-S1.ADP, and IASL-S1.ADP.Vi were modeled using corresponding crystal structures (PDB: 1FMV, 1MMA, and 1VOM) to understand the position of the spin label relative to myosin. CHARMM force field parameters for IASL were taken from previously published values (18). Missing loops of the protein were added and refined using the Homology module of InsightII (Accelrys San Diego, CA). The protein then was energy-minimized, and the spin label was mutated into C707. After mutation, a Monte Carlo search was performed to find the lowest energy conformation for the spin label (18).
EPR
EPR spectra were acquired with an X-band, 9.4-GHz spectrometer (EleXsys E500; Bruker BioSpin, Billerica, MA) and a W-band, 94-GHz spectrometer (EleXsys E680; Bruker BioSpin). At X-band, a super-HQ cavity with a variable temperature accessory (Bruker BioSpin) was used. The 30 μL-sample was contained in a quartz capillary with an outside diameter/inside diameter of 0.84/0.6 mm (VitroCom, Mountain Lakes, NJ) and then sealed (Critoseal; Oxford Labware, St. Louis, MO). The scan width was 120 G; the peak-to-peak modulation amplitude was 1 G; the modulation frequency was 100 kHz. Spectra were recorded below saturation, at H1 = 0.05 G, at 20°C. At the W-band, the standard TE011-mode cylindrical cavity resonator (EN600-1021H; Bruker BioSpin) was used with an ER 4118CF-W variable temperature cryostat. The 0.2-μL sample was contained in a quartz capillary with outside diameter/insider diameter of 0.25/0.15 mm (VitroCom), then flame-sealed and sedimented by low-speed centrifugation. The scan width was 400 G; the peak-to-peak modulation amplitude was 1 G; the modulation frequency was 100 kHz. Spectra were recorded below saturation, at H1 = 0.045 G, at 20°C or 80 K. EPR experiments were performed immediately after S1 labeling and preparation of nucleotide complexes, without freezing. For low-temperature EPR experiments, 30% glycerol (v/v) was added to IASL-S1 as a cryoprotectant. The buffer for EPR contained 20 mM EPPS (pH 8.0), 6 mM MgCl2, and 1 mM EGTA. Before lineshape analysis, the phase of the W-band spectrum was adjusted to correct for a slight admixture of dispersion (19). The quadrature spectrum was calculated from a Hilbert transformation of the experimental spectrum (20), and the weighted quadrature spectrum was subtracted from the experimental spectrum until the integrated spectrum displayed a flat baseline.
IASL-S1: models of motion
Two models of spin-label motion were considered: 1), the model of slow motion in a restoring potential (4), and 2), the model of rapid-limit oscillation about x, y, and z axes of the spin label (1–3). S1 was modeled as a prolate ellipsoid, with axial ratio p = 1 (sphere) or 2.5 (ellipsoid) and a long-axis length of 10.05 nm or 16.25 nm, respectively (21). Theoretical rotational diffusion coefficients in water at 20°C are DRiso= 1.3 × 106 rad2s−1, τc = 128 ns (spheroid, diameter 10.05 nm), and DR⊥ = 1.0 × 106 rad2s−1, τc⊥ = 167 ns, and DR‖ = 2.45 × 106 rad2s−1, τc‖ = 68 ns (ellipsoid) (22).
Spectra simulation and fitting
EPR spectra were simulated according to described models of motion. The four different approaches that were considered are described below.
Approach A: “powder spectrum”
This approach was used to determine the principal values of g- and A-tensor components of spin-labeled myosin. To simulate the EPR spectrum, the resonant magnetic field for a single orientation of a spin label in the applied magnetic field was calculated according to the work of Libertini and Griffith (23) as follows:
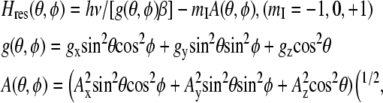 |
(1) |
where Hres is the magnetic field at resonance, ν is the applied frequency, β is the Bohr magneton, and θ and φ define the orientations of the nitroxide relative to the applied magnetic field (24). After subsequent summation of more than 10,000 random orientations of the spin label in the applied magnetic field, convolution with a Lorentzian function, and calculation of the derivative, the simulated EPR spectrum was fitted to the experimental W-band spectrum, which was acquired at low temperature (T = 80 K) to avoid any molecular motion. The g- and A-tensor components (Eq. 1) and the Lorentzian linewidth were the fit variables.
Approach B: rapid-limit oscillation of the spin label
In this approach, it is assumed that the rotational correlation times are all much less than 1 ns, so that the spectrum is determined entirely by the orientational distribution. Under these conditions, the components of magnetic tensors are partially averaged. The apparent values of g and A tensors (g′ and A′) determined from the fit to Eq. 1 of experimental W-band spectra, acquired at T = 20°C, are interpreted in terms of restrictions of spin-label motion (assuming no sensitivity of W-band EPR to slow tumbling of myosin (see below, section “Sensitivity of X- and W-band EPR to S1 tumbling”)). W-band EPR spectra were simulated and fitted as described in Approach A, and restrictions of spin-label motion were derived from the rapid-limit oscillation model (1,3), using Euler transformations as follows:
 |
(2) |
where α is the angle of oscillation and 〈cos2α〉 = (1/2)*(1 + sinαcosα/α).
Approach C: rapid-limit oscillation of the spin label and global tumbling of myosin
In this approach, we assumed rapid-limit oscillation of the spin label within myosin and myosin global tumbling with the diffusion coefficient DRiso = 1.3 × 109 rad2s−1 or DR⊥ = 1.0 × 109 rad2s−1 and DR‖ = 2.45 × 109 rad2s−1. Apparent values of g and A tensors (g′ and A′) were determined from W-band spectra, as described in Approach B, and were used for the NLSL fit (25) of X-band spectra to account for myosin tumbling. The NLSL program uses the microscopic ordering with macroscopic disorder model (26), assuming a random distribution of rotating molecules undergoing unrestricted global rotational motion.
Approach D: slow local motion of the spin label and global tumbling of myosin
This approach was realized with NLSL-SRLS software (27) and so used the slow relaxing local structure (SLRS) model (28), which accounts for two motions: restricted local motion of the spin label and unrestricted tumbling of myosin in solution. The restriction of rotational motion of the spin label is defined in the model by expansion coefficients cLK of spherical harmonics 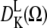 as follows:
as follows:
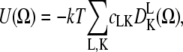 |
(3) |
where U(Ω) is the restoring potential and Ω is a set of angles needed to describe the spin-label orientation relative to the applied magnetic field (25). The equilibrium distribution of orientation probability was defined from the restoring potential as described by Budil et al. (25) as follows:
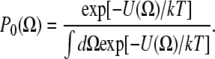 |
(4) |
In this work, we used the first three terms of the series (Eq. 3) to model restrictions of spin-label motion, c20, c22, and c40. The inclusion of higher-order terms (e.g., c42 and c44) did not improve the fit. In the simulation, rates of rotational motion (spin label and S1) are defined by rotational diffusion coefficients DR; we report the rates through the rotational correlation times defined as τc = 1/(6DR). In the case of anisotropic motion, we defined correlation times for rotation about the x, y, and z axes of the spin-label diffusion frame as τci = 1/(6DRi), where i = x, y, z, or τc⊥ = 1/(6DR⊥) and τc‖ = 1/(6DR‖). Spectra were simulated with a fixed rotational correlation time of myosin tumbling, τc = 128 ns or τc⊥ = 167 ns and τc‖ = 68 ns. Only the parameters defining the local (internal) spin-label motion were varied, with initial values for correlation times in the χ range from 0.2 to 200 ns. The basis set was truncated to improve efficiency without significant effects on simulated spectra. The maximum quantum numbers for total angular momentum were Lemax = 12 and Lomax =11; the maximum quantum numbers for the z component of angular momentum in the lab and molecular frames were Kmax = 8 and Mmax = 8, respectively; and the nuclear spin transition index was pl = 2. The same truncation parameters were used to simulate spectra at the X- and W-bands. A Monte Carlo search of initial values for restrictions of spin-label motion and rotational correlation time was used to sample full parameter space (29). All experimental spectra were double-integrated and normalized and the goodness of fit was quantified via the Pearson χ2 test.
RESULTS
Magnetic tensor values
To analyze EPR spectra quantitatively in terms of spin-label orientation and anisotropic rotational motion, it is necessary to accurately determine the values of [gx, gy, gz] and [Ax, Ay, Az]. These values were determined by fitting W-band EPR spectra of apo IASL-S1, IASL-S1.ADP, and IASL-S1.ADP.Vi at T = 80 K (Table 2), assuming a rigid powder (Approach A). Fig. 1 shows the fit for the apo IASL-S1 sample. The values of g and A tensors for all considered myosin samples were the same, reflecting similar polarity of the environment for the spin label (30) in these states of myosin.
TABLE 2.
Magnetic tensor values determined from W-band EPR spectra of frozen samples (Fig. 1)
| gx | gy | gz | Ax (G) | Ay (G) | Az (G) |
|---|---|---|---|---|---|
| 2.0087 ± 0.0001 | 2.0058 ± 0.0001 | 2.0018 ± 0.00005 | 7.1 ± 0.7 | 7.2 ± 0.8 | 36.7 ± 0.2 |
FIGURE 1.

Fit of apo IASL-S1 W-band EPR spectrum, acquired at T = 80 K, to determine g and A magnetic tensor values (Table 2). (Black) experiment; (gray) fit.
Sensitivity of X- and W-band EPR to S1 tumbling
To determine the sensitivity of X- and W- band EPR spectra to global tumbling of S1, spectra were simulated with NLSL software (25) using g and A tensor values from Table 2. Isotropic rotational diffusion was assumed, characterized by a single rotational correlation time τc. The dependence of the apparent hyperfine splitting AZ′ at the X-band (determined from the outer extrema splitting) and at the W-band (determined from the z-component splitting) on τc is shown in Fig. 2. AZ′ shows some sensitivity to τc at the X-band even at 167 ns (the longest correlation time expected for global S1 tumbling at 20°C). This value is virtually constant at the W-band for τc ≥ 68 ns (the shortest correlation time expected for global S1 tumbling). Global S1 tumbling, thus, can be ignored in the analysis of W-band EPR spectra.
FIGURE 2.

Dependence of apparent A′z hyperfine splitting on correlation time of isotropic tumbling, revealed from simulated X- and W-band EPR spectra.
IASL-S1: rapid-limit oscillation of the spin label
W-band EPR spectra were simulated and fitted to experimental spectra according to the model of rapid-limit oscillation of the spin label (Approach B). X-band simulations also included global tumbling of S1 ((Approach C) and were fitted to the experimental spectra (Fig. 3). Apparent values of magnetic tensors determined from these fits are shown in Table 3 with the χ2 values for the fits and the angles of oscillation determined from Eq. 2. The spectra of the apo (IASL-S1) and ADP (IASL-S1.ADP) biochemical states were fitted as single-component spectra; addition of a second component did not improve the fit. The spectra of IASL-S1.ADP.Vi were fitted as two-component spectra, and addition of third component did not improve the fit.
FIGURE 3.

Spectral fits for the model of rapid-limit oscillation. (Black) experiment; (gray) fit. Parameters from the best fits are shown in Table 3.
TABLE 3.
Results for Approach C: rapid-limit oscillation of the spin label within myosin
| Angle of oscillation
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| IASL-S1 | χ2·10−5 | gx′ | gy′ | gz′ | Ax′ | Ay′ | Az′ | α* | β* | γ* |
| Apo | 75.92 | 2.0084 | 2.0057 | 2.0022 | 7.3G | 7.7G | 35.5G | 13° | 24° | 15° |
| ADP | 305.41 | 2.0082 | 2.0055 | 2.0023 | 8.2G | 7.7G | 34.4G | 29° | 25° | 22° |
| ADP Vi component 1, 69% | 34.5 | 2.00814 | 2.0045 | 2.0034 | 8.7G | 17.2G | 27.8G | 64° | 25° | 16° |
| ADP Vi component 2, 31% | 2.0082 | 2.0055 | 2.0023 | 8.2G | 7.7G | 34.4G | 29° | 25° | 22° | |
As shown in Table 3, the angle of oscillation about the y axis of the spin label remains constant in all considered biochemical states of myosin, and the angle of oscillation about the z axis slightly increases in the IASL-S1.ADP state. The major difference in the spin-label dynamics is in oscillation about the x axis: the angle of oscillation changes from 13° in the apo IASL-S1 state to 64° in the IASL-S1.ADP.Vi state.
IASL-S1: slow restricted motion of spin label
X-band and W-band EPR spectra of IASL-S1 were simulated and fitted globally with NLSL-SRLS software (27) in the approach of slow local motion of the spin label and global tumbling of S1 (Approach D). Magnetic tensor values (Table 2) and the rotational correlation time of global S1 tumbling were kept constant. The rotational correlation time and restrictions of spin-label motion within myosin, Lorentzian broadening, and the angle between the magnetic frame and the diffusion frame of the spin label (βd) were variable parameters. We found no significant difference in the quality of spectral fit for isotropic and anisotropic S1 tumbling, and so the results provided are for the isotropic tumbling model. Fig. 4 shows fits of simultaneously simulated X- and W-band EPR spectra to experimental spectra, and fit parameters are shown in Table 4. Fig. 5 shows the probability of the spin-label orientational distribution for each biochemical state of IASL-S1. The spectra of apo IASL-S1 and IASL-S1.ADP were fitted adequately as single-component spectra; addition of an additional spectral component did not improve the fit.
FIGURE 4.

Spectral fits for the model of slow motion. Apo, ADP, and ADP Vi S1; X- and W-band EPR. (Black) experiment; (gray) fit. For S1.ADP.Vi, the quality of the fit is illustrated for 1, 2, and 3 spectral components.
TABLE 4.
Results for model d, slow restricted motion of the spin label within myosin
| IASL-S1 | χ2×10−5 | c20 | c22 | c40 | τcx | τcy | τcz | βd |
|---|---|---|---|---|---|---|---|---|
| Apo | 40.22 | 4.36 | – | – | 9.9 ns | 9.9 ns | 64 ns | 32° |
| ADP | 32.09 | 1.88 | −0.36 | – | 6.8 ns | 6.8 ns | 25.2 ns | 5° |
| ADP Vi component 1, 78% | 7.17 | 4.96 | −4.91 | −1.93 | 1.0 ns | 1.8 ns | >300 ns | – |
| ADP Vi component 2, 16% | 1.88 | −0.36 | – | 6.8 ns | 6.8 ns | 25.2 ns | 5° | |
| ADP Vi component 3, 6% | 6.42 | −6.24 | – | 0.2 ns | 0.1 ns | >300 ns | – |
Parameters are defined in the text.
FIGURE 5.

Orientational distribution of the spin label within myosin in the apo, ADP, and ADP Vi biochemical states, based on the model of slow restricted motion. The distribution of z axis of the spin label is color-coded. X axis in spin-label distribution of the S1.ADP.Vi state (M** structural state) is marked to indicate the symmetry about x axis. (Inset) Position of a spin label within apo S1, after Monte Carlo minimization, showing converter domain (blue), N-terminal domain (gray), and relay helix (yellow). The axes of spin label molecular frame are x, y, and z. The colored mesh indicates myosin atoms located at a distance <0.5 nm relative to the spin label.
In contrast, adequate fits of the IASL-S1.ADP.Vi spectra required three component spectra. Fig. 4 shows the different sensitivity of X- and W-band EPR spectra to the addition of spectral components. The second component of the IASL-S1.ADP.Vi state (Table 4) is better resolved at the X-band than at the W-band, apparently because of different spectral broadening. In contrast, the third component is resolved better at the W-band because of greater spectral resolution than at the X-band. Addition of a fourth component did not improve the fit quality. The determined mole fractions and χ2 of these fits are shown in Table 4. In the global two-frequency fit, the correlation coefficients between rotational correlation time and the orientational parameters (cLK in Eq. 3) were 0.51–0.57. The correlation coefficients among x, y, and z components of the rotational correlation time or among orientational (cLK) parameters were usually higher (0.9–1.0).
DISCUSSION
IASL-S1: models of spin-label motion
For the apo S1 state, both models produce almost the same result for the amplitude of the spin-label motion (at very different rates), apparently because of a high restriction for the spin-label motion. Both models give the angle of spin-label motion as 24° (amplitude of oscillation about the y axis in the rapid-limit oscillation model and half-median angle of the orientational distribution of the spin label in the slow restricted motion model). However, the rates of spin-label motion in the apo S1 state are very model-dependent: τc < 0.09 ns for the model of rapid-limit oscillation and τc ∼10 ns for the model of slow restricted motion. The χ2 value for the model of rapid-limit oscillation is ∼50% greater than the value for the model of slow restricted motion, and so the model of slow restricted motion clearly is preferred.
EPR spectra of the S1.ADP biochemical state show fewer restrictions for the spin-label motion within myosin. In the model of rapid-limit oscillation, the spin label oscillates with an amplitude of 29° about the x axis, whereas the slow motion model reveals a half-median angle of 43° for the spin-label motion about the x axis, with a correlation time of ∼7 ns. The χ2 test shows that the model of slow restricted motion clearly is preferred over the model of rapid-limit oscillation (32.09 × 10−5 vs. 305.41 × 10−5; Tables 3 and 4). This conclusion is clear only with the inclusion of W-band spectra, which provide the extra resolution needed to remove ambiguity.
Both models show that X- and W-band EPR spectra of the S1.ADP.Vi state contain multiple components. The model of slow restricted motion yields three components with mole fractions 16%, 78% and 6%, χ2 = 7.17 × 10−5; the χ2 test for one or two components is worse. The model of rapid-limit oscillation reveals two spectral components with mole fractions 31% and 69%, χ2 = 34.5 × 10−5. In both models, one spectral component is similar to the spectrum of the S1.ADP state, corresponding to highly restricted motion, and the additional component(s) reflect(s) increased angular amplitudes of motion (Tables 3 and 4, Fig. 5). The best model (the model of slow restricted motion; Fig. 5, bottom), shows 78% of the spin label within myosin distributed in two orientations, divided by 90° relative to rotation about the x axis (Fig. 5), with the other mobile component (6%) having a single distribution.
Overall, the model of slow restricted motion produces the best fit, (Table 4), and so we interpret the experimental data in terms of this model.
IASL-S1: structural states
In the apo S1 biochemical state, multifrequency EPR unambiguously detects a single structural state of myosin in the force-generating domain, which presumably corresponds to the M state detected by fluorescence and EPR (13,31). The motion of the spin label is complex; the molecular frame of the spin label is tilted in the diffusion frame (βd = 32°), and all motional parameters (rotational correlation time and restoring potential) are given relative to the diffusion frame. According to our modeling of the structure of Dictyostelium discoideum myosin in the apo S1 state (PDB: 1FMV (32)), the spin label is located between the converter and N-terminal domains of myosin with the relay helix on the side (Fig. 5). The spin label is squeezed between myosin domains, but not buried under the surface, because g and A tensor values do not reflect changes in polarity for different myosin states. In the apo S1 state, the spin label moves slowly on the nanosecond timescale (τcx,y = τc⊥ = 9.9 ns, τcz = 64 ns), and the motion is very restricted. These slow restricted motions probably reflect those of the secondary structural elements in close contact with the labeled site.
In the S1.ADP state, a single structural state is also reported, presumably corresponding to the M* state that is detected by fluorescence and EPR (13,31). In this conformation of the force-generating domain, the spin label moves slightly faster. Its motion is less restricted relative to the apo S1, and the tilt of the molecular frame of the spin label in the diffusion frame is less in this conformation (Table 4, Fig. 5). The structure of Dictyostelium discoideum myosin with bound ADP (PDB: 1MMA (7)) shows a slightly different position of the converter relative to the N-terminal domain compared to that of apo S1, allowing more space for the spin label to move. This is in qualitatively agreement with EPR data: the motion of the spin-labeled side chain becomes less constrained.
A much more complex structural picture emerges in the S1.ADP.Vi complex, which is presumed to correspond to the pre-powerstroke or transition state. The EPR spectrum in this complex shows at least two, and probably three, structural states, although only one structural state is revealed by the crystal structure PDB: 1VOM (9). One spectral component (16%) is similar to that of IASL-S1.ADP (M*), suggesting that the ADP-bound biochemical state is occupied. However, the Mg-ATPase activity of myosin is inhibited completely under the conditions of the EPR experiment, proving that all myosin is in the S1.ADP.Vi biochemical state. There is no significant population in the S1.ADP biochemical state, despite the population of the M* structural state. The structures of Dictyostelium discoideum myosin in ADP and ATP states are very similar (with the root mean-square deviation = 0.3 nm) and, like Mizukura and Maruta (12), we assigned the observed S1.ADP-like spectral component as a post-powerstroke (M* (33)) S1.ATP structural state. The two other spectral components (corresponding to higher mobility, with mole fractions 78% and 6%) have different dynamics of the spin label (Table 4); there is no tilt of the molecular frame relative to the diffusion frame. These spectral components could be assigned as the S1.ADP.Pi (pre-powerstroke M**) state of myosin (13). The spin-label dynamics is complex, but one conclusion is clear—this well-defined biochemical complex trapped by ADP and vanadate is structurally heterogeneous, allowing occupancy of both the post-powerstroke and pre-powerstroke structural states.
According to the structure of the S1.ADP.Vi state, the spin label is located between the converter and N-terminal domains, whereas the relay helix is bent in this structural state and is not likely to affect spin-label motion. The two observed distributions of spin-label orientation in the M** structural state (78% and 6%; Fig. 5) could reflect a slight difference in relative position of the converter and N-terminal domains: rotation of the spin label about the x axis is allowed in one state (M1**, 6%) and stopped in another state (M2**, 78%). This slight difference in relative position of domains within one myosin population (M**) could be interpreted in terms of flexibility between myosin domains in the M** state on the microsecond and slower timescale (34); it also could be treated as a slow exchange between myosin conformations producing two spectral components.
Spin label with flexible linker as a probe for local structure of a protein
The length and flexibility of a spin label's linker plays a substantial role in determining the EPR spectrum (1). The spectrum typically depends less on peptide backbone motion than on restriction of a spin label's motion by adjacent structural elements of a protein (35). Careful interpretation of the EPR spectral lineshape in terms of the spin-label orientational distribution is important for a precise distance determination in DEER (36,37), to find relative orientation of protein domains (38,39) and for a rigorous analysis of multicomponent spectra. In our study, we emphasize the role of multifrequency EPR in spectral analysis, which has allowed us to resolve multiple structural states of the force-generating domain of myosin in S1.ADP.Vi biochemical state. Future studies should combine rigorous spectral interpretation of multifrequency EPR with molecular dynamics simulations (40–42) and transient EPR (31) to define the coupling between the dynamics of the force-generating domain and the active site of myosin during the ATPase cycle.
CONCLUSIONS
We have rigorously analyzed spin-label dynamics within myosin to probe the local structure of the force-generation region. Analysis of multifrequency (X-band and W-band) EPR spectra of spin-labeled myosin unambiguously determines both the rates and amplitudes of restricted spin-label motion within myosin and resolves multiple conformational states. In apo- and ADP-bound states of myosin S1, spin-label dynamics reflects single structural states, which is consistent with the structures of Dictyostelium discoideum myosin II in these states. However, the S1.ADP.Vi biochemical state is resolved into a mixture of three structural states. One state resembles the ADP-bound state (16%), with slow and moderately restricted spin-label motion; two others (78% and 6%) are characterized by fast motion of the spin label distributed symmetrically about its x axis. The presence of the S1.ADP-like structural state in the S1.ADP.Vi biochemical state, with completely inhibited Mg2+-ATPase activity, supports the hypothesis of structural similarity of myosin S1 in ADP- and ATP-bound (postpowerstroke) states (M*). The two other S1.ADP.Vi states, which correspond to higher spin-label mobility, could be assigned to the S1.ADP.Pi (prepowerstroke) structural state (M**), based on similarity of spin-label dynamics. This interpretation supports the conclusion that the post- and prepowerstroke structural states are in a dynamic equilibrium in solution, even with ADP and vanadate tightly bound to the myosin active site. The two populations of spin label detected in the S1.ADP.Pi structural state could be interpreted in terms of domain flexibility within myosin in the S1.ADP.Vi biochemical state on a timescale slower than 1 μs. This hypothesis should be tested further by multifrequency EPR of probes at different positions within the myosin force-generating region.
Acknowledgments
This work was supported by NIH grants AR53562 (Y.E.N.) and AR32961 (D.D.T.). NLSL-SRLS software was kindly provided by Dr. Z. Liang (Cornell University). We appreciate discussions with Dr. D. Budil (Northeastern University), Dr. Z. Liang and Dr. J. H. Freed (Cornell University), and with Jennifer Klein and Jack Surek (University of Minnesota).
Abbreviations used: EPR, electron paramagnetic resonance; IASL, 4-(2-iodoacetamido)-TEMPO; DR, coefficient of rotational diffusion; EPPS, 4-(2-hydroxyethyl)piperazine-1-propanesulfonic acid; MOPS, 3-(N-morpholino) propane sulfonic acid; PDB, Protein Data Bank; τc, rotational correlation time.
Editor: Christopher Lewis Berger.
References
- 1.Van, S. P., G. B. Birrell, and O. H. Griffith. 1974. Rapid anisotropic motion of spin labels. Models for motion averaging of the ESR parameters. J. Magn. Reson. 15:444–459. [Google Scholar]
- 2.Timofeev, V. P., and D. O. Nikolsky. 2003. The role of the fast motion of the spin label in the interpretation of EPR spectra for spin-labeled macromolecules. J. Biomol. Struct. Dyn. 21:367–378. [DOI] [PubMed] [Google Scholar]
- 3.Dzuba, S. A., Y. D. Tsvetkov, and A. G. Maryasov. 1992. Echo-induced EPR spectra of nitroxides in organic glasses: model of orientational molecular motions near equilibrium position. Chem. Phys. Lett. 188:217–222. [Google Scholar]
- 4.Freed, J. H. 1976. Theory of slow tumbling ESR spectra for nitroxides. In Spin Labeling: Theory and Applications. L. J. Berliner, editor. Academic Press, New York. 53–132.
- 5.Nesmelov, Y. E., C. B. Karim, L. Song, P. G. Fajer, and D. D. Thomas. 2007. Rotational dynamics of phospholamban determined by multifrequency electron paramagnetic resonance. Biophys. J. 93:2805–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rayment, I., W. R. Rypniewski, K. Schmidt-Base, R. Smith, D. R. Tomchick, M. M. Benning, D. A. Winkelmann, G. Wesenberg, and H. M. Holden. 1993. Three-dimensional structure of myosin subfragment-1: a molecular motor. Science. 261:50–58. [DOI] [PubMed] [Google Scholar]
- 7.Gulick, A. M., C. B. Bauer, J. B. Thoden, and I. Rayment. 1997. X-ray structures of the MgADP, MgATPgammaS, and MgAMPPNP complexes of the Dictyostelium discoideum myosin motor domain. Biochemistry. 36:11619–11628. [DOI] [PubMed] [Google Scholar]
- 8.Fisher, A. J., C. A. Smith, J. B. Thoden, R. Smith, K. Sutoh, H. M. Holden, and I. Rayment. 1995. X-ray structures of the myosin motor domain of Dictyostelium discoideum complexed with MgADP.BeFx and MgADP.AlF4. Biochemistry. 34:8960–8972. [DOI] [PubMed] [Google Scholar]
- 9.Smith, C. A., and I. Rayment. 1996. X-ray structure of the magnesium(II).ADP.vanadate complex of the Dictyostelium discoideum myosin motor domain to 1.9 Å resolution. Biochemistry. 35:5404–5417. [DOI] [PubMed] [Google Scholar]
- 10.Malnasi-Csizmadia, A., R. J. Woolley, and C. R. Bagshaw. 2000. Resolution of conformational states of Dictyostelium myosin II motor domain using tryptophan (W501) mutants: implications for the open-closed transition identified by crystallography. Biochemistry. 39:16135–16146. [DOI] [PubMed] [Google Scholar]
- 11.Werber, M. M., A. G. Szent-Gyorgyi, and G. D. Fasman. 1972. Fluorescence studies on heavy meromyosin-substrate interaction. Biochemistry. 11:2872–2883. [DOI] [PubMed] [Google Scholar]
- 12.Mizukura, Y., and S. Maruta. 2002. Analysis of the conformational change of myosin during ATP hydrolysis using fluorescence resonance energy transfer. J. Biochem. (Tokyo). 132:471–482. [DOI] [PubMed] [Google Scholar]
- 13.Barnett, V. A., and D. D. Thomas. 1987. Resolution of conformational states of spin-labeled myosin during steady-state ATP hydrolysis. Biochemistry. 26:314–323. [DOI] [PubMed] [Google Scholar]
- 14.Margossian, S. S., and S. Lowey. 1982. Preparation of myosin and its subfragments from rabbit skeletal muscle. Methods Enzymol. 85(Pt B):55–71. [DOI] [PubMed] [Google Scholar]
- 15.Prochniewicz, E., T. F. Walseth, and D. D. Thomas. 2004. Structural dynamics of actin during active interaction with myosin: different effects of weakly and strongly bound myosin heads. Biochemistry. 43:10642–10652. [DOI] [PubMed] [Google Scholar]
- 16.Fiske, C. H., and Y. Subbarow. 1925. The colorimetric determination of phosphorus. J. Biol. Chem. 66:375–400. [Google Scholar]
- 17.Lanzetta, P. A., L. J. Alvarez, P. S. Reinach, and O. A. Candia. 1979. An improved assay for nanomole amounts of inorganic phosphate. Anal. Biochem. 100:95–97. [DOI] [PubMed] [Google Scholar]
- 18.Sale, K., C. Sar, K. A. Sharp, K. Hideg, and P. G. Fajer. 2002. Structural determination of spin label immobilization and orientation: a Monte Carlo minimization approach. J. Magn. Reson. 156:104–112. [DOI] [PubMed] [Google Scholar]
- 19.Earle, K. A., D. E. Budil, and J. H. Freed. 1993. 250-GHz EPR of nitroxides in the slow-motional regime: models of rotational diffusion. J. Phys. Chem. 97:13289–13297. [Google Scholar]
- 20.Ernst, R. R., G. Bodenhausen, and A. Wokaun. 1987. Principles of Nuclear Magnetic Resonance in One and Two Dimensions. Oxford University Press, Oxford.
- 21.Garrigos, M., J. E. Morel, and J. Garcia de la Torre. 1983. Reinvestigation of the shape and state of hydration of the skeletal myosin subfragment 1 monomer in solution. Biochemistry. 22:4961–4969. [DOI] [PubMed] [Google Scholar]
- 22.Tao, T. 1969. Time-dependent fluorescence depolarization and brownian rotational diffusion coefficients of macromolecules. Biopolymers. 8:609–632. [Google Scholar]
- 23.Libertini, L., and O. Griffith. 1970. Orientation dependence of the electron spin resonance spectrum of di-t-butyl nitroxide. J. Chem. Phys. 53:1359–1367. [Google Scholar]
- 24.Fajer, P. G., R. L. H. Bennett, C. F. Polnaszek, E. A. Fajer, and D. D. Thomas. 1990. General method for multiparameter fitting of high-resolution EPR spectra using a simplex algorithm. J. Magn. Reson. 88:111–125. [Google Scholar]
- 25.Budil, D., S. Lee, S. Saxena, and J. Freed. 1996. Nonlinear-least-squares analysis of slow-motion EPR spectra in one and two dimensions using a modified Levenberg-Marquardt algorithm. J. Magn. Reson. A120:155–189. [Google Scholar]
- 26.Schneider, D. J., and J. H. Freed. 1989. Calculating slow motional magnetic resonance spectra: a user's guide. In Biological Magnetic Resonance. L. J. Berliner, editor. Plenum Publishing, New York. 1–76.
- 27.Barnes, J. P., Z. Liang, H. S. McHaourab, J. H. Freed, and W. L. Hubbell. 1999. A multifrequency electron spin resonance study of T4 lysozyme dynamics. Biophys. J. 76:3298–3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Polimeno, A., and J. H. Freed. 1995. Slow motional ESR in complex fluids: the slowly relaxing local structure model of solvent cage effects. J. Phys. Chem. 99:10995–11006. [Google Scholar]
- 29.Khairy, K., D. Budil, and P. Fajer. 2006. Nonlinear-least-squares analysis of slow motional regime EPR spectra. J. Magn. Reson. 183:152–159. [DOI] [PubMed] [Google Scholar]
- 30.Mobius, K., A. Savitsky, C. Wegener, M. Plato, M. Fuchs, A. Schnegg, A. A. Dubinskii, Y. A. Grishin, I. A. Grigor'ev, M. Kuhn, D. Duche, H. Zimmermann, and H. J. Steinhoff. 2005. Combining high-field EPR with site-directed spin-labeling reveals unique information on proteins in action. Magn. Reson. Chem. 43:S4–S19. [DOI] [PubMed] [Google Scholar]
- 31.Ostap, E. M., H. D. White, and D. D. Thomas. 1993. Transient detection of spin-labeled myosin subfragment 1 conformational states during ATP hydrolysis. Biochemistry. 32:6712–6720. [DOI] [PubMed] [Google Scholar]
- 32.Bauer, C. B., H. M. Holden, J. B. Thoden, R. Smith, and I. Rayment. 2000. X-ray structures of the apo and MgATP-bound states of Dictyostelium discoideum myosin motor domain. J. Biol. Chem. 275:38494–38499. [DOI] [PubMed] [Google Scholar]
- 33.Lymn, R. W., and E. W. Taylor. 1970. Transient state phosphate production in the hydrolysis of nucleoside triphosphates by myosin. Biochemistry. 9:2975–2983. [DOI] [PubMed] [Google Scholar]
- 34.Houdusse, A., and H. L. Sweeney. 2001. Myosin motors: missing structures and hidden springs. Curr. Opin. Struct. Biol. 11:182–194. [DOI] [PubMed] [Google Scholar]
- 35.Columbus, L., T. Kalai, J. Jeko, K. Hideg, and W. L. Hubbell. 2001. Molecular motion of spin labeled side chains in α-helices: analysis by variation of side chain structure. Biochemistry. 40:3828–3846. [DOI] [PubMed] [Google Scholar]
- 36.Hustedt, E. J., R. A. Stein, L. Sethaphong, S. Brandon, Z. Zhou, and S. C. Desensi. 2006. Dipolar coupling between nitroxide spin labels: the development and application of a tether-in-a-cone model. Biophys. J. 90:340–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hilger, D., Y. Polyhach, E. Padan, H. Jung, and G. Jeschke. 2007. High-resolution structure of a Na+/H+ antiporter dimer obtained by pulsed electron paramagnetic resonance distance measurements. Biophys. J. 93:3675–3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fajer, P. G., E. A. Fajer, and D. D. Thomas. 1990. Myosin heads have a broad orientational distribution during isometric muscle contraction: time-resolved EPR studies using caged ATP. Proc. Natl. Acad. Sci. USA. 87:5538–5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baumann, B. A., H. Liang, K. Sale, B. D. Hambly, and P. G. Fajer. 2004. Myosin regulatory domain orientation in skeletal muscle fibers: application of novel electron paramagnetic resonance spectral decomposition and molecular modeling methods. Biophys. J. 86:3030–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.LaConte, L. E., V. Voelz, W. Nelson, M. Enz, and D. D. Thomas. 2002. Molecular dynamics simulation of site-directed spin labeling: experimental validation in muscle fibers. Biophys. J. 83:1854–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fajer, M. I., H. Li, W. Yang, and P. G. Fajer. 2007. Mapping electron paramagnetic resonance spin label conformations by the simulated scaling method. J. Am. Chem. Soc. 129:13840–13846. [DOI] [PubMed] [Google Scholar]
- 42.Budil, D. E., K. L. Sale, K. A. Khairy, and P. G. Fajer. 2006. Calculating slow-motional electron paramagnetic resonance spectra from molecular dynamics using a diffusion operator approach. J. Phys. Chem. A. 110:3703–3713. [DOI] [PubMed] [Google Scholar]