

Abstract
Purpose
The goal of this study was to identify mutations in X-chromosomal genes associated with retinitis pigmentosa (RP) in patients from Germany, The Netherlands, Denmark, and Switzerland.
Methods
In addition to all coding exons of RP2, exons 1 through 15, 9a, ORF15, 15a and 15b of RPGR were screened for mutations. PCR products were amplified from genomic DNA extracted from blood samples and analyzed by direct sequencing. In one family with apparently dominant inheritance of RP, linkage analysis identified an interval on the X chromosome containing RPGR, and mutation screening revealed a pathogenic variant in this gene. Patients of this family were examined clinically and by X-inactivation studies.
Results
This study included 141 RP families with possible X-chromosomal inheritance. In total, we identified 46 families with pathogenic sequence alterations in RPGR and RP2, of which 17 mutations have not been described previously. Two of the novel mutations represent the most 3’-terminal pathogenic sequence variants in RPGR and RP2 reported to date. In exon ORF15 of RPGR, we found eight novel and 14 known mutations. All lead to a disruption of open reading frame. Of the families with suggested X-chromosomal inheritance, 35% showed mutations in ORF15. In addition, we found five novel mutations in other exons of RPGR and four in RP2. Deletions in ORF15 of RPGR were identified in three families in which female carriers showed variable manifestation of the phenotype. Furthermore, an ORF15 mutation was found in an RP patient who additionally carries a 6.4 kbp deletion downstream of the coding region of exon ORF15. We did not identify mutations in 39 sporadic male cases from Switzerland.
Conclusions
RPGR mutations were confirmed to be the most frequent cause of RP in families with an X-chromosomal inheritance pattern. We propose a screening strategy to provide molecular diagnostics in these families.
Introduction
Retinitis pigmentosa (RP) is clinically characterized by night blindness, concentric constriction of visual fields, pigment deposits predominantly in the midperiphery of the retina, and attenuation of retinal vessels. It affects one in 3000–4000 individuals and can be caused by mutations in more than 40 genes (Retnet; Retina International). The disease may be inherited as autosomal recessive, autosomal dominant, or X-linked (XL) traits. Approximately 50% of the cases are sporadic [1].
Most mutations lead to a similar phenotype, making genotype-phenotype correlations difficult. Nevertheless, mutations in XLRP genes are associated with a severe phenotype in terms of onset and progression of the disease. The two known RP-associated X-chromosomal genes are designated RPGR (OMIM 312610) and RP2 (OMIM 312600) [2-4]. Additional loci on the X-chromosome have been mapped, but the respective genes have not been identified [5-7]. Indeed, most XLRP cases can be explained by mutations in RPGR and RP2 accounting for up to 80% and 20% of disease alleles, respectively. Moreover, RPGR mutations underlie 10–20% of all familial RP cases, which is higher than most other single RP loci [8,9].
The RPGR gene is composed of 23 exons, including the alternatively spliced exons 9a, ORF15, 15a, and 15b [10-12]. Exon ORF15 is predominantly found in transcripts of the retina and brain [13], whereas mRNAs including exons 1–19 are widely expressed [12]. The N-terminal part of RPGR contains a domain homologous to the regulator of chromosome condensation 1 (RCC1), the guanine-nucleotide-exchange factor for the GTPase Ran that has been shown to be important for the association with binding partners. RPGR mutations are most frequently found in exon ORF15, followed by exons 1–15 [9-12,14,15]. No mutations have been identified in exons 16–19 so far. ORF15 contains a purine-rich domain of approximately 1050 base pairs, which is predicted to encode a repetitive glycine and glutamate region at the C-terminus of the protein. In human, out of frame deletions, duplications, or insertions are frequently found in ORF15, whereas nonsense mutations are rare and disease-relevant missense mutations have not been described. Two canine animal models of ORF15 frame-shift mutations have been published and closely resemble the human phenotype in terms of disease onset and progression [16,17].
RT–PCR studies have suggested that splicing of both, constitutive exons and the ORF15-internal repeat of RPGR, generates numerous transcript variants [11,18]. This indicates that splicing is an important regulator of RPGR function. Recently, we have shown that a novel isoform of RPGR, including exon 9a, is expressed predominantly in cones of the human retina [10], which may explain why mutations in RPGR can also lead to a predominant degeneration of cones starting in the central part of the retina [19-22]. These findings extend the phenotypic spectrum associated with RPGR mutations and suggest a specific role of RPGR isoforms in the survival of cones and rods of the human retina.
RP2 is composed of five coding exons, which are translated to a widely expressed protein of 350 amino acids [4]. The function of RP2 is not completely understood, but it shows homology to cofactor C, a protein involved in the ultimate step of gamma-tubulin folding, and interacts with ADP ribosylation factor-like 3 [23]. Posttranslational acyl modifications at the N-terminus of RP2 mediate its targeting to the plasma membrane, and the disruption of the acylation site leads to RP [24]. The majority of pathogenic sequence alterations found in RP2 represent truncating mutations, whereas missense mutations often locate to the cofactor C-like domain of RP2.
The present study describes the results of mutational screenings in RP families with possible XL inheritance patterns. In addition to known sequence alterations, we found several novel mutations in RPGR and RP2 and characterized an XLRP family with variable disease expression in female carriers. We identified a microdeletion breakpoint in addition to an ORF15 frameshift mutation in a single RP patient. A screening strategy for molecular diagnostic testing in patients or families with presumed X-chromosomal inheritance is discussed.
Methods
Patients
Informed consent was obtained from each patient and healthy control after explanation of the nature and possible consequences of the study. Blood samples were collected to perform routine molecular genetic testing for genes associated with RP or related retinal diseases. We collected a panel of 141 retinal degeneration patients with possible XL inheritance patterns. Among those were 39 patients without history of additional affected family members, six cases with assumed dominant transmission and males preferentially affected, and 90 cases with possible XL-inheritance showing at least two affected male family member and no male-to-male transmission. The latter group also included male sibships. Six simplex cases were clinically diagnosed with cone-rod dystrophy, whereas all others were diagnosed with RP. Clinical evaluations of patients included slit lamp examination, funduscopy, and Ganzfeld-electroretinography (ERG). Ganzfeld-ERG was performed according to standards of the International Society for Clinical Electrophysiology of Vision using a LKC-UTAS 3000 ERG device (LKC Technologies Inc., Gaithersburg, MD) or a Nicolet Spirit examination unit (Nicolet, Madison, WI) [25,26]. For scotopic ERG recordings, we employed a phase of dark-adaptation preceded by single dim white and bright white flashes for stimulation. Bright white flashes were used for single responses and 30 Hz flicker responses in photopic ERG measurements. Goldmann perimetry (Goldmann perimetry module on the Octopus 101 visual field testing device; Haag-Streit, Köniz, Switzerland), fundus photography, and best corrected visual acuity testing were often performed to complement the clinical characterization.
Furthermore, we collected blood samples from 15 individuals of an XLRP family, segregating the disease in three generations (index 25085). In this family, ocular examinations, as approved by the ethics committee of the Institute of Human Genetics of the University Hospital of Cologne, were done in II:3, III:7, and III:12 and included refraction, best corrected visual acuity, intraocular pressure, slit-lamp biomicroscopy, funduscopy, and color discrimination test. ERG, dark adaptation and visual field were also analyzed in these three patients.
Linkage analysis for an XLRP locus with variable heterozygote manifestation
We analyzed DNA samples from 15 individuals of a family that segregates RP in three generations (index patient 25085). PCR fragments of polymorphic microsatellite markers were amplified using fluorescent-labeled oligonucleotides and analyzed on an ABI-377 DNA sequencer (Applied Biosystems, Darmstadt, Germany). Genotypes were determined by GeneScan software (Applied Biosystems). Data provided by the GDB Human Genome Database were used as reference for allele sizes. Segregation analysis was performed by genotyping locus-specific microsatellite markers for the following AD RP genes: NRL (RP27), CRX, RP1, PIM1K (RP9), IMPDH1 (RP10), CA4 (RP17), and FSCN2. Markers were not informative for PRPF31, RDS, and RHO. Therefore, the entire coding regions of the corresponding genes were sequenced in the index patient (III:7). In PRPF3 and PRPF8, mutations have only been described in exons 11 and 42, respectively. These exons were directly sequenced. Primer sequences and PCR conditions are available upon request. The RP31 locus had not yet been described when this study was performed and thus, was not analyzed [27].
To map the disease locus, we performed a genome-wide linkage analysis using the Affymetrix GeneChip Human Mapping 10K Array, version 2.0 (Affymetrix, Santa Clara, CA). Genotypes were called by the GeneChip DNA Analysis Software (GDAS v2.0; Affymetrix). Genders of samples were verified by counting heterozygous single nucleotide polymorphisms (SNPs) on the X chromosome. Relationship errors were evaluated with the help of the program Graphical Relationship Representation [28]. The program PedCheck was applied to detect Mendelian errors [29], and data for SNPs with such errors were removed from the data set. Non-Mendelian errors were identified by using the program Merlin [30] and unlikely genotypes for related samples were deleted. Nonparametric linkage analysis using all genotypes of a chromosome simultaneously was performed with Merlin. Parametric linkage analysis was performed by a modified version of the program Genehunter 2.1 [31,32] through stepwise use of a sliding window with sets of 100 SNPs and by the program Allegro [33] assuming autosomal dominant inheritance with reduced penetrance and a disease allele frequency of 0.0001. Haplotypes were reconstructed with Allegro or Merlin and presented graphically with HaploPainter [34]. All data handling was performed using the graphical interface Alohomora [35].
Mutation screening
The genomic DNA of the index patient panel was screened for disease-associated sequence alterations in exon ORF15 of RPGR by direct sequencing of PCR products. A single PCR fragment, spanning the entire coding region of exon ORF15, was amplified from 100 ng genomic DNA using primers ORF15-F3 and ORF15-R6 (Appendix 1). As described elsewhere, patients’ genomic DNA was either extracted by a salting out method or by affinity purification mediated by magnetic beads [36,37]. PCR was performed with either HotfireTaq (Solis Biodyne, Tartu, Estonia) or HotstarTaq Polymerase (Qiagen AG, Hombrechtikon, Switzerland) as recommended by the manufacturers. Q-solution and 3µM MgCl2 were added to the reaction mixture. Cycling conditions of the PCR were conducted for 45 cycles as follows: initial denaturation 95 °C for 15 min, denaturation 95 °C for 45 s, annealing 60 °C for 1 min, elongation 72 °C for 3.5 min. Subsequently, the PCR fragments were sequenced (ABI 3100; Applied Biosystems, Rotkreuz, Switzerland) with primers ORF15-R7b, -R8b, -R9, -R5, -R4, and -F10 (Appendix 1). The sequenced PCR profiles were compared to the RPGR reference database entry NM_001034853 (NCBI database). If genomic DNA were available only from female members of the family, sequence analysis of ORF15 was occasionally not complete, since polymorphic heterozygote deletions or duplications may resulted in overlapping sequence spectrums. If no mutations in RPGR exon ORF15 were detected, we subsequently analyzed all coding exons of RP2 and the RPGR exons 1 through 15, 9a, 15a, and 15b, including flanking intronic sequences. RP2 mutational screening was performed using either single strand conformation polymorphism analysis or direct sequencing [4] (Appendix 1). Long-range PCR was conducted as recommended by the manufacturers (Expand long range PCR system; Roche, Mannheim, Germany) using the primer ORF15-F10 and ORF15–15a-R10 (annealing temperature of 60 °C; elongation time of 10 min). Potential mutations of RP2 or RPGR were verified by sequence analysis of the respective position in either 100 or 300 control alleles. The database entry BC043348 was used as reference sequence for RP2 mutation analysis. The identified nucleotide substitutions and protein consequences are described as recommended (for guidelines see the Human Genome Variation Society) and, to be comparable to previously published data, as suggested by Sharon et al. [15], or Bader et al. [9].
X-chromosome inactivation studies
A nonrandom X-inactivation may explain variable phenotypic expression in females that carry a single RPGR mutation. To test this possibility, we performed X-chromosome inactivation studies and examined the methylation patterns at the androgen receptor (AR) locus as described before [38]. Briefly, for each proband, we prepared two reactions. In the first, 400 ng genomic DNA were digested with HpaII (8 U) in a total volume of 10 μl for 30 min at 37 °C. In the second reaction, 150 ng genomic DNA were incubated without enzyme. Subsequent PCR amplification was done using oligonucleotide primers AR-A (5′-CTT TCC AGA ATC TGT TCC AG-3′) and AR-B (5′-AAG GTT GCC TGT TCC TCA TC-3′) in a PTC-thermal cycler (MJ Research, BioRad, München, Germany) at 95 °C for 1 min, 55 °C for 1 min, and 72 °C for 1 min for 35 cycles. About 2–10 μl of the product was mixed with 2–6 μl loading buffer and run at 70 W for 3–3.5 h on an 8% nondenaturing polyacrylamide gel. After electrophoresis, the gel was silverstained for evaluation. DNA samples of a male control as well as two female controls with previously confirmed unilateral X-inactivation were included into the analysis.
Results
Identification of mutations
We analyzed DNAs from 141 patients with retinal degeneration for mutations in X-chromosomal RP-associated genes. DNA was collected from various geographical regions, including Germany, Switzerland, Denmark, and The Netherlands and contained 90 samples from families indicative for an XL-inheritance pattern. Within the subgroup of 90 index patients, we identified 31 (35%) mutations in exon ORF15 of RPGR. Among these ORF15 mutations, 14 sequence alterations had been reported previously, whereas eight were novel mutations (Figure 1A, Table 1). In addition to two nonsense mutations leading to a premature stop codon, we identified three different duplications, 16 distinct deletions, and one insertion. All cause a frameshift and were predicted to result in altered amino acids followed by a premature stop codon (Table 1). We excluded that additional nucleotide variations in RP2 and RPGR may explain the phenotype of RP in patients with ORF15 mutations and performed mutation screenings in exons 1 through 15, 9a, 15a, and 15b of RPGR and all coding exons of RP2. In addition to pathogenic mutations, we identified several polymorphic sequence alterations (Table 2 and Table 3).
Figure 1.
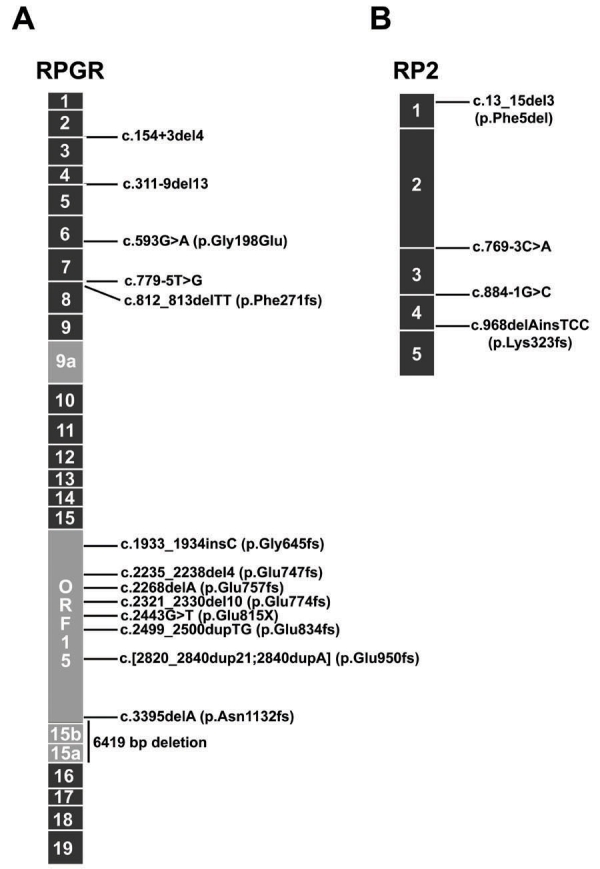
Location of novel RPGR and RP2 mutations identified in this study. The schematic drawing shows novel mutations and their approximate positions in RPGR (A) and RP2 (B). Alternatively spliced exons are shown in light gray, whereas other exons are depicted in black.
Table 1. Mutations found in RPGR and RP2 in X-linked retinitis pigmentosa families.
| Patient ID | Gene (exon/intron) | ORF15 mutation (1) | ORF15 protein change (1) | Mutation (2) | Protein change (2) | Mutation first reported by |
| XRP51 | RP2 (exon 1) | - | - | c.13_15del3 | p.Phe5del | novel mutation |
| XRP6 | RP2 (intron 2) | - | - | c.769-3C>A | - | novel mutation |
| XRP21***** | RP2 (exon 2) | - | - | c.358C>T | p.Arg120X | Hardcastle et al., 1999 [51] |
| XRP26 | RP2 (intron 3) | - | - | c.884-1G>C | - | novel mutation |
| 24748 | RP2 (exon 4) | - | - | c.968delAinsTCC | p.Lys323fs | novel mutation |
| XRP11 | RPGR (intron 2) | - | - | c.154+3del4 | - | novel mutation |
| XRP19 | RPGR (intron 4) | - | - | c.311-9del13 | - | novel mutation |
| 24750 | RPGR (exon 6) | - | - | c.593G>A | p.Gly198Glu | novel mutation |
| XRP16 | RPGR (exon 7) | - | - | c.644G>T | p.Gly215Val | Vervoort et al., 2002 [45] |
| 24739 | RPGR (intron 7) | - | - | c.779-5T>G | - | novel mutation |
| 24746 | RPGR (exon 8) | - | - | c.812_813delTT | p.Phe271fs | novel mutation |
| XRP32 | RPGR (ORF15) | g.ORF15+180_181insC | ORF15Gly60fs | c.1933_1934insC | p.Gly645fs | novel mutation |
| XRP70 | RPGR (ORF15) | g.ORF15+482_485delAGAA | ORF15Glu162fs | c.2235_2238delAGAA | p.Glu747fs | novel mutation |
| 2460***** | RPGR (ORF15) | g.ORF15+483_484delGA | ORF15Glu161fs | c.2236_2237delGA | p.Glu746fs | Vervoort et al., 2000 [11] |
| 24520 | RPGR (ORF15) | g.ORF15+515delA | ORF15Glu172fs | c.2268delA | p.Glu757fs | novel mutation |
| 2554 | RPGR (ORF15) | g.ORF15+517_518delAG | ORF15Glu172fs | c.2270_2271delAG | p.Glu757fs | Sharon et al., 2003 [15] |
| XRP72 | RPGR (ORF15) | g.ORF15+568_577delAGAGGAAAAA | ORF15Glu189fs | c.2321_2330delAGAGGAAAAA | p.Glu774fs | novel mutation |
| XRP48 | RPGR (ORF15) | g.ORF15+587_588delAG | ORF15Arg195fs | c.2340_2341delAG | p.Arg780fs | Bader et al., 2003 [9] |
| 25085° **** | RPGR (ORF15) | g.ORF15+652_653delAG | ORF15Glu217fs | c.2405_2406delAG | p.Glu802fs | Vervoort et al., 2000 [11] |
| XRP13 | RPGR (ORF15) | g.ORF15+673_674delAG | ORF15Glu224fs | c.2426_2427delAG | p.Glu809fs | Vervoort et al., 2000 [11] |
| 24751 | RPGR (ORF15) | g.ORF15+690G>T(stop) | ORF15Glu230Ter | c.2443G>T | p.Glu815X | novel mutation |
| XRP27 | RPGR (ORF15) | g.ORF15+714A>T (stop) | ORF15Lys238Ter | c.2467A>T | p.Lys823fs | Pelletier et al., 2006 [14] |
| 2249 | RPGR (ORF15) | g.ORF15+747_748insTG | ORF15Glu249fs | c.2499_2500dupTG | p.Glu834fs | novel mutation |
| 24745 | RPGR (ORF15) | g.ORF15+748delA | ORF15Glu249fs | c.2501delA | p.Glu834fs | Bader et al., 2003 [9] |
| 2604 | RPGR (ORF15) | g.ORF15+753delG | ORF15Glu251fs | c.2506delG | p.Glu836fs | Pelletier et al., 2006 [14] |
| 10005° | RPGR (ORF15) | g.ORF15+795delG | ORF15Glu265fs | c.2548delG | p.Glu850fs | Prokisch et al., 2007 [52] |
| 24731° | RPGR (ORF15) | g.ORF15+795delG | ORF15Glu265fs | c.2548delG | p.Glu850fs | Prokisch et al., 2007 [52] |
| 2678 | RPGR (ORF15) | g.ORF15+801_802insG | ORF15Glu267fs | c.2554dupG | p.Glu852fs | Bader et al., 2003 [9] |
| 25333 | RPGR (ORF15) | g.ORF15+833_834delGG | ORF15Glu278fs | c.2586_2587delGG | p.Glu863fs | Yokoyama et al., 2001 [53] |
| 2865 | RPGR (ORF15) | g.ORF15+875_876delGG | ORF15Glu292fs | c.2628_2629delGG | p.Glu877fs | Push et al., 2002 [54] |
| 2814 | RPGR (ORF15) | g.ORF15+1087_1088ins22 | ORF15Glu365fs | c.[2820_2840dup21; 2840dupA] | p.Glu950fs | novel mutation |
| XRP23 | RPGR (ORF15) | g.ORF15+1254_1257delGGAG | ORF15Gly418fs | c.3007_3010delGGAG | p.Gly1003fs | Sharon et al., 2003 [15] |
| 24747** | RPGR (ORF15) | g.ORF15+1339delA | ORF15Glu446fs | c.3092delA | p.Glu1031fs | Bader et al., 2003 [9] |
| 2557°° | RPGR (ORF15) | g.ORF15+1642delA | ORF15Asn547fs | c.3395delA | p.Asn1132fs | novel mutation |
The table shows families with at least two affected males and no male to male transmission. A degree symbol (°) denotes cases with assumed dominant inheritance pattern. Two degree symbols (°°) show that in this patient an additional deletion was identified previously (Roepman et al., 1996) [39]. As shown in this study, the additional deletion does not remove parts of the coding region of exon ORF15 and deletes 6419 bp including exon 15a and 15b. Two asterisks (**) indicate that the mutation of this patient ID was identified two times. Four asterisks (****) and five asterisks (*****) show that the mutation was identified four and five times, respectively. The mutations are either presented in approved nomenclature according to Human Genome Variation Society (2) or as recommended by Bader et al., 2003 [9] and Sharon et al., 2003 [15] (1) (BC043348 and NM_001034853 were used as reference sequence for, respectively, RP2 and RPGR mutation analysis).
Table 2. Polymorphic sequence variations in exon ORF15 of RPGR found in X-linked retinitis pigmentosa patients.
| Patient ID | Deletions or duplications found in exon ORF15 (1) | Deletions or duplications found in exon ORF15 (2) | Nucleotide substitutions found in exon ORF15 (1) | Nucleotide substitutions found in exon ORF15 (2) | Protein change resulting from nucleotide substitutions (1)/(2) |
| 24731 | g.ORF15+1060_1080dup21 | c.2813_2833dup21 | g.ORF15+1643C>T | c.3396C>T | p.N547N / p.N1132N |
| 24731 | - | - | g.ORF15+1677G>A | c.3430G>A | p.V559I / p.V1144I |
| 24747 | - | - | g.ORF15+1643C>T | c.3396C>T | p.N547N / p.N1132N |
| 24747 | - | - | g.ORF15+1677G>A | c.3430G>A | p.V559I / p.V1144I |
| 2678 | - | - | g.ORF15+1813A>C | c.3566A>C | 3'UTR |
| 2814 | - | - | g.ORF15+1643C>T | c.3396C>T | p.N547N / p.N1132N |
| 2814 | - | - | g.ORF15+1677G>A | c.3430G>A | p.V559I / p.V1144I |
| 2814 | - | - | g.ORF15+1813A>C | c.3566A>C | 3'UTR |
| 2865 | g.ORF15+1307_1318delAGTGGAAGGGGA | c.3060_3071del12 | - | - | - |
| 3044 | g.ORF15+914_916delGGA | c.2667_2669del3 | g.ORF15+588G>A | c.2341G>A | p.A196T / p.A781T |
| 25085 | g.ORF15+1307_1318delAGTGGAAGGGGA | c.3060_3071del12 | - | - | - |
The sequence alterations are either presented in approved nomenclature according to Human Genome Variation Society (2) or as recommended by Bader et al., 2003 [9] and Sharon et al., 2003 [15] (1) (BC043348 and NM_001034853 were used as reference sequence for, respectively, RP2 and RPGR mutation analysis).
Table 3. Polymorphic sequence variations in exons 1 to 15 of RPGR found in X-linked retinitis pigmentosa patients.
|
Patient ID |
Position in RPGR |
Nucleotide substitutions found in RPGR exons 1 to 15 |
SNP ID |
Heterozygosity frequency |
Protein change |
| 24731 | intron 1 | c.29-15G>A | rs6651585 | 0.474 +/- 0.110 | - |
| 24745 | exon 10 | c.1164G>A | rs1801686 | 0.189 +/- 0.242 | p.A388A |
| 24747 | intron 1 | c.29-15G>A | rs6651585 | 0.474 +/- 0.110 | - |
| 24747 | exon 9 | c.1033A>G | - | 0.007** | p.N345D |
| 2249 | exon 11 | c.1367A>G | - | - | p.Q456R |
| 2604 | exon 10 | c.1164G>A | rs1801686 | 0.189 +/- 0.242 | p.A388A |
| 2814 | exon 9 | c.1033A>G | - | 0.007** | p.N345D |
| 2814 | intron 12 | c.1507-101A>T | rs5918520 | - | - |
| 2557 | intron 12 | c.1507-101A>T | rs5918520 | - | - |
| 24520 | intron 12 | c.1507-101A>T | rs5918520 | - | - |
| 25428 | intron 1 | c.29-15G>A | rs6651585 | 0.474 +/- 0.110 | - |
| 25428 | exon 9 | c.1033A>G | - | 0.007** | p.N345D |
| 25428 | intron 12 | c.1507-101A>T | rs5918520 | - | - |
| 24748* | intron 4 | c.310+10T>C | - | - | - |
The sequence alterations are presented in approved nomenclature according to Human Genome Variation Society (NM_001034853 was used as reference sequence for mutation analysis). The asterisk (*) indicates that this sequence variant was not detected in over 300 control alleles. The two asterisks (**) indicate that this value was published by Sharon et al., 2000 [15].
The deletion c.2548delG in exon ORF15 of RPGR was detected in two independent families. In one of them (index patient 24731), the mother as well as three of her sons were affected by RP, whereas one son and one daughter were unaffected. Since the mother was affected, this suggests that female carriers of this mutation can be affected by RP. The second family, in which we found the mutation c.2548delG in the index patient 10005, showed variable disease manifestation in female carriers. The finding that two apparently unrelated families with the same RPGR mutation showed disease expression in female carriers, suggests a genotype-phenotype correlation. Further families with this mutation need to be analyzed to verify this observation.
In patient 2557, we identified the single bp deletion c.3395delA at the 3’ end of exon ORF15 (Table 1). Remarkably, the same patient had been shown to have an approximately 6.4 kb deletion in the RPGR gene [39]. We now have determined the breakpoints in the genomic DNA to verify whether the 6.4 kb deletion, in addition to c.3395delA, affects the exon ORF15 coding region. We performed long-range PCR and sequencing and found that the 6.4 kb deletion started 27 bp after the stop codon of exon ORF15 and ended 72 bp downstream of exon 15a (Figure 2A,B,C). Thus, the larger deletion of patient 2557 includes 6419 bp and removes exon 15a and 15b of RPGR, but does not affect the coding region of exon ORF15. In contrast, the mutation c.3395delA removes an adenine 62 bp upstream of the ORF15 stop codon. Consequently, the mutation was predicted to result in a frameshift at the 3’-terminal end of ORF15, which affects only the last 21 amino acids and results in a premature stop codon after inclusion of 19 RPGR-unrelated amino acids. The two deletions found in the male index patient 2557 locate to one allele at a distance of only 90 bp within RPGR.
Figure 2.
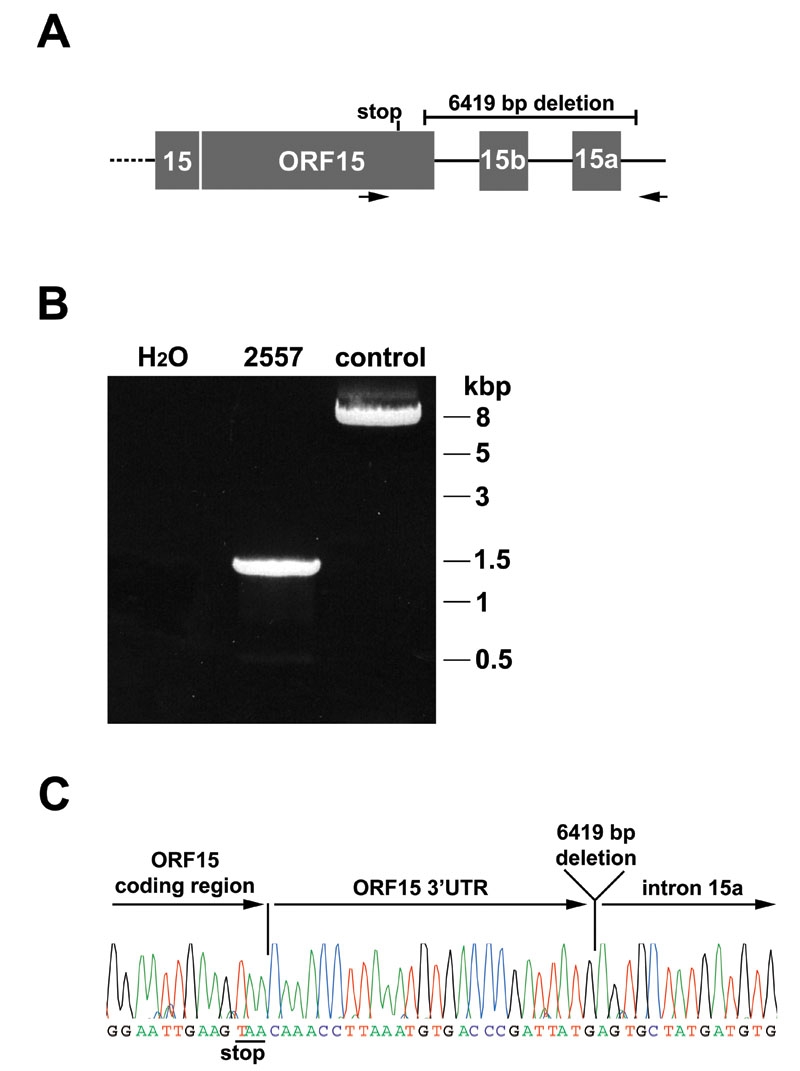
Characterization of the deletion breakpoint in patient 2557. A: Schematic drawing of RPGR exons ORF15, 15a, and 15b. Primers used for long-range PCR are marked by arrows and flank the 6419 bp deletion. B: Long-range PCR for patient 2557 (male) and a control sample (male). The difference of the PCR products represents the 6.4 kb deletion in patient 2557. C: The sequence profile shows the breakpoint of the deletion in patient 2557. The open reading frame of exon ORF15 was not affected by the deletion. In total, 6419 bp were deleted, starting 27 bp after the stop codon in ORF15 and ending 72 bp after exon 15a. Note that exon 15a is downstream of 15b. Thus, the deletion affected both exons 15a and 15b of RPGR.
In XLRP patients without an ORF15 mutation, we screened the five exons of RP2 by direct sequencing and identified five pathogenic sequence variations, which either lead to a frameshift, an altered amino acid composition, or a presumed splice defect of RP2 (Figure 1B, Table 1).
DNA from index patients who did not show mutations in ORF15 and RP2 were analyzed for sequence variations in exons 1 through 15, 9a, 15a, and 15b of RPGR. We were able to identify pathogenic sequence alterations in six additional XLRP cases. One mutation, found in exon 8, deletes two bp of codon 271, introducing a frameshift to the transcript. Three mutations affect intronic sequences close to RPGR exons and presumably result in splice defects. Furthermore, two mutations alter a glycine in either exon 6 or 7 of RPGR (Figure 1A, Table 1). None of these sequence alterations were identified in 100 to 300 control alleles. Sequence alterations summarized in Table 3 likely represent polymorphisms.
RPGR ORF15 mutations were neither detected in 39 simplex male cases with RP, nor in six simplex male cases affected by cone-rod dominated degenerations.
Analyses of a retinitis pigmentosa family with an assumed dominant inheritance pattern
Among the six families with dominant inheritance pattern, we found three deletions in exon ORF15 of RPGR. In one of these six families (index patient 25085, Figure 3A) linkage analysis by microsatellite genotyping was performed and excluded seven known ADRP loci. Sequencing of PRPF31, RDS, RHO, HPRP3, and PRPF8 did not reveal causative mutations. Microarray-based linkage analysis suggested an 11.5 Mb region on the X-chromosome comprising the RPGR gene.
Figure 3.
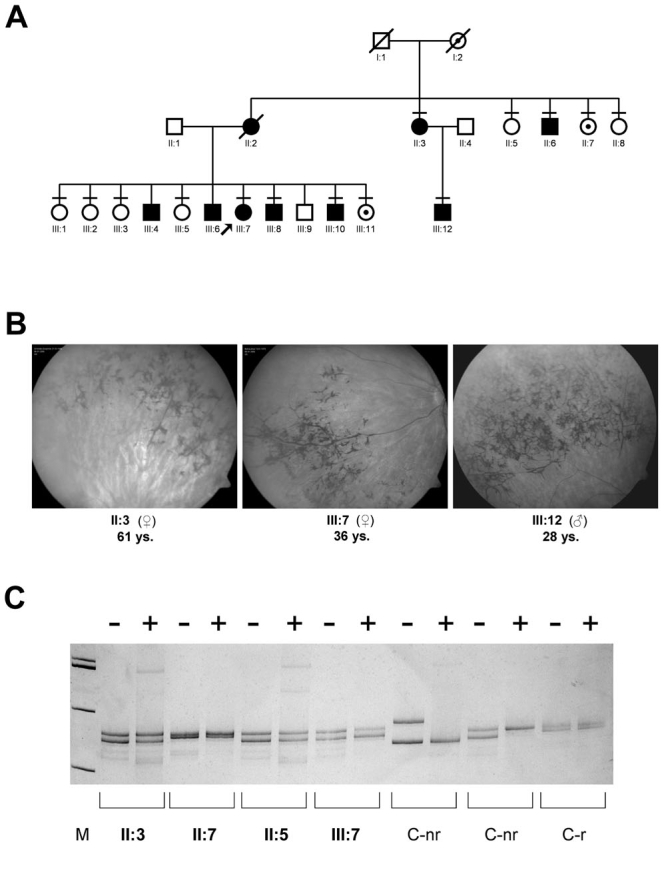
Characterization of a family with X-linked retinitis pigmentosa and variable expressivity in mutation carriers. A: Pedigree with three generations. Circles represent females and squares represent males. Slashed symbols indicate deceased family members. Filled black symbols denote family members with retinitis pigmentosa (RP), and circles with a dot indicate female mutation carriers who had no history of visual complaints. Horizontal bars designate family members whose genotype was determined by molecular genetic testing. Arrow marks the index patient 25085. The mutation c.2405_2406delAG in exon ORF15 of RPGR segregates with the disease in males, and shows variable heterozygote manifestation in females. B: Fundus pictures of three affected family members show typical pigmentations found in the peripheral retina of patients with RP. Patient age and gender are provided below each fundus photograph. C: Pattern of X-chromosome inactivation of selected female family members. None showed a unilateral X-inactivation at the AR-locus. The following abbreviations and symbols are used: control nonrandom X-inactivation (C-nr), control random X-inactivation (C-r), HpaII digestion (+), and no HpaII digestion (-).
Subsequent analysis of RPGR identified a 2 bp deletion within the alternatively spliced exon ORF15 (c.2405_2406delAG, Table 1). This deletion was predicted to result in a frameshift, an altered amino acid composition, and a premature stop codon after 97 bp truncating the full length protein by 320 amino acids. In addition, an in-frame 12 bp deletion, previously described as a polymorphism [9], was found in each patient with the 2 bp deletion. This suggested that both deletions were located on the same allele. The 12 bp deletion was further downstream of the 2 bp deletion and affected positions c.3060_3071del12. No additional sequence alteration was identified in RP2 and RPGR. The segregation of the deletions was in accordance with linkage data.
The mutation was identified in four males and four females of the family, whereas six healthy female relatives did not carry the mutation (Figure 3A). All male mutation carriers showed the RP phenotype, whereas only two of the four female carriers were affected (Figure 3A). In this family, the ORF15 mutation caused a variable heterozygote manifestation of the phenotype in females, although the majority of previously reported RPGR mutations are recessive.
Clinical evaluation and interviews with the patients showed that the course of the disease, with respect to age-of-onset and disease progression, varied both in affected males and females. Males reported awareness of visual impairment between seven and 24 years of age, and disease progression varied from slow to rapid. Females first noticed visual impairment at 24 years (II:2), 28 years (III:7), and 32 years (II:3). Individual II:3 (mother of III:12; 61 years old) rapidly lost vision within a few years, a disease course similar to the male family member III:4, who reported normal vision until age 24, followed by rapid visual loss. In contrast to the severe course of the disease in female II:3, kinetic perimetry, scotopic and photopic ERG, and dark adaptation indicated mild expression of RP in III:7 eight years after the diagnosis was made. However, she had pigmentary deposits in the midperiphery of her fundus (Figure 3B).
The phenotypic variability within female carriers of the family might be caused by an altered X-chromosomal inactivation. We analyzed the X-inactivation pattern in different family members, but did not find skewed signal intensities in any of the symptomatic female carriers (Figure 3C).
Discussion
We identified 17 novel and 16 known mutations in RPGR and RP2 by screening of 141 DNA samples from RP patients. In the subgroup of 90 possible XLRP families, including also male sibships, we identified five different RP2 mutations and 28 distinct pathogenic sequence alterations in RPGR. Approximately 35% of the XLRP families described herein showed mutations in exon ORF15 of RPGR. This detection rate for ORF15 mutations was within the range found in other studies [8,14]. RPGR exon ORF15 mutations have been reported to occur either in 30% or 52% in North American families with XLRP [15,40], whereas 43% were reported for Spanish cohorts [41]. Furthermore, detection rates of 49% have been described for families from France and 60% for families from the UK/Ireland [11,14].
Of all published RPGR mutations, 55% have been reported to reside in exon ORF15 [1]. We detected a total of 37 RPGR mutations, including 31 located to exon ORF15. Thus, we identified over 80% of the RPGR mutations in ORF15, a proportion higher than described recently [1]. These variations in detection rates might be due to different ethnical backgrounds of the patient cohorts.
Up to 28% of sporadic XLRP cases from France were reported to carry mutations in exon ORF15 of RPGR [14]. In contrast, we did not find ORF15 mutations in 39 sporadic cases from Switzerland. This difference again indicates that the ethnical background of the cohorts influences the detection rate. Nevertheless, it might also be necessary to pre-select sporadic cases for early-onset RP phenotypes, as done by Pelletier et al. [14], to increase the chance to identify ORF15 mutations in sporadic cases.
In one XLRP family, we found the mutation c.968delAinsTCC in RP2. This pathogenic sequence alteration deletes a single nucleotide and inserts three novel nucleotides at the same position. It leads to a frameshift starting in exon 4 and was predicted to result in a premature stop in exon 5. Moreover, the mutation inserts three nucleotides in the penultimate position in exon 4 of RP2 and thus alters the consensus sequence of the splice donor site. This might lead to mis-splicing with increased exon 4 skipping from RP2 transcripts. The position of this mutation is, to the best of our knowledge, the most 3’-terminal disease-associated variant in RP2 identified so far. This also holds true for the mutation c.3395delA (patient 2557) in exon ORF15 of RPGR. In the same patient, an additional deletion of approximately 6.4 kb removing RPGR exons 15a and 15b was identified, a rearrangement that helped to identify the gene in 1996 [39]. In this study, we determined the breakpoint of this 6.4 kb microdeletion and found that the coding region of ORF15 was not affected. In contrast, the mutation c.3395delA (p.Asn1132fs) of patient 2557 leads to a frameshift in the 3’-terminal part of ORF15 and alters the composition of the last 21 amino acids of RPGR, which are not part of the repetitive and highly charged region in ORF15. Interestingly, 3’-terminal mutations in RPGR exon ORF15 are occasionally associated with cone-rod degenerations rather than classic RP [19-21,42]. However, in the case described herein, a typical RP phenotype was observed. It is not clear whether or not the microdeletion of over 6.4 kb influenced the phenotype in patient 2557.
Disease expression in males with RPGR mutations is severe. It has been shown previously that the phenotype of female carriers of pathogenic RPGR mutations is highly variable. Some exhibit severe symptoms, while others are asymptomatic. We have identified three pathogenic sequence alterations in the mutational hot spot exon ORF15 that lead to disease expression also in female carriers. The mutation c.2548delG was found in a family in which the mother, in addition to three of her sons, was affected. The same mutation occurred in a second family with reduced disease expression in female carriers (family 10005). The finding that a mutation leads to a phenotype only in a fraction of female carriers could be due to genetic modifiers. We identified additional in-frame variations in ORF15 that might influence the clinical picture in female carriers. Nevertheless, in-frame deletions and duplications were frequently described as polymorphisms of exon ORF15, which makes it unlikely to be the only factor causing the differences found in carriers. Other modifiers such as age, environmental factors, additional changes in the second allele of the carrier females, or genetic variability in other genes may influence the disease expression. Further investigations will identify and characterize the possible modifiers that determine the clinical outcome of RPGR mutations in female carriers. In contrast, no RP2 mutations have been described that result into heterozygote carrier manifestations.
The mutation c.2405_2406delAG, found in one of the families with assumed dominant inheritance of this study, has been described previously [43,44]. The family characterized here showed severe retinal degeneration in males and mild (III:7) to severe (II:3) ocular symptoms in two female carriers. Moreover, we identified two female mutation carriers who had no history of visual complaints: II:7 (51 years old) and III:11 (29 years old). In accordance with Rozet et al. [43], we conclude that this mutation leads to variable heterozygote manifestation of the phenotype. With the assumption that the families are unrelated, this observation suggests a genotype-phenotype correlation associated with the RPGR mutation c.2405_2406delAG. We also identified an additional in-frame (12 bp) deletion in all carriers of the pathogenic 2 bp deletion. It is likely that the 12 bp deletion represents a polymorphism as described by Bader et al. [9], although an additional effect on the phenotype cannot be excluded. Skewed X chromosome inactivation as an explanation of the largely variable disease manifestations in females was not supported by our data. Full expression of disease symptoms in females with an RPGR mutation may be misleading in molecular analyses and genetics counseling. Thus, ORF15 mutations in RPGR should be excluded in families that seem to fit an AD pattern of RP, but lack male-to-male transmission.
We propose a screening strategy for routine molecular genetics testing of possible XLRP cases by direct sequencing. Between 30% and 80% of mutations are found in exon ORF15 of RPGR [40,45], followed by mutation frequencies that are similar for RP2 and RPGR exons 1 through 15. In total, approximately 70%–80% of XLRP cases carry mutations in RPGR and up to 20% in RP2. Based on these results and to keep costs low, we recommend beginning with the screening of an XLRP male patient by ORF15 mutation analysis. Our approach to amplify the complete exon ORF15 in a single PCR reaction reduces costs and effort. After identification of an ORF15 mutation, the molecular diagnosis might be considered as confirmed, since patients with an additional mutation in exons 1 through 15 of RPGR and RP2 have not been reported. Nevertheless, this cannot be excluded, and a complete screening of known XLRP genes should be offered. In addition, the segregation of the ORF15 mutation within the family might be verified. If no ORF15 mutation is identified, we recommend to continue the screen for mutations with RP2 rather than RPGR. This recommendation is based on the fact that RP2 is composed of five exons only, whereas 15 exons need to be screened to complete the sequence analysis of RPGR without any clear benefit in terms of likelihood to detect the causative mutation.
So far, no RP2 mutation has been identified in families with likely dominant inheritance pattern, except for a single case, where the only affected female showed a structural chromosome aberration and a unilateral X-inactivation due to a balanced X-autosome translocation [14]. In contrast, dominant inheritance was reported for a couple of RPGR mutations [43,46]. Furthermore, ORF15 mutations have also been associated with phenotypes of the central part of the retina, including cone-rod or cone dystrophies and atrophic macular degeneration [19-21,42]. Consequently, in XLRP cases with manifestations of the phenotype in females or in patients with X-linked cone-dominated retinopathies, we recommend screening exon ORF15 first, followed by the other exons of RPGR.
In addition to the phenotype of RP, mutations in exons 6, 8, and 10 of RPGR have been associated with features of primary ciliary dyskinesia, hearing dysfunction, sinusitis, and recurrent infections [47-50]. Thus, in XLRP cases with additional phenotypes, the best candidate region of RPGR seems to be exons 1 through 15.
Acknowledgments
We thank the following for financial support: Velux Foundation (Switzerland), Olga Mayenfish Foundation (Switzerland), Novartis Foundation (Switzerland), ‘Schweizerischer Fonds zur Verhütung und Bekämpfung der Blindheit’ (Switzerland), Foundation Fighting Blindness, British Retinitis Pigmentosa Society, Deutsche Forschungsgemeinschaft (DFG LO 457/3 and LO 457/5), Maria-Pesch-Stiftung, the CEC (EVI-GENORET LSHG-CT-2005–512036), and De Nederlandse Vereniging ter Voorkoming van Blindheid (ANVVB). We also thank E.M. Sankila, E. van Nouhuys, M.J. van Shooneveld, C.W.R.J. Cremers, C.B. Hoyng, and L.I. van den Born for help in counseling patients. We are grateful to Birgit Langer and Ursula Biendl (ERG), Jaya Balakrishnan for technical assistance, and we thank Gabor Matyas for sequencing support. We are indebted to the families who participated in this study.
Appendix 1: Primer combinations used to amplify and sequence RPGR or RP2 exons including flanking intronic regions.
To access the data, click or select the words “Appendix 1”. This will initiate the download of a PDF file.
References
- 1.Shu X, Black GC, Rice JM, Hart-Holden N, Jones A, O'Grady A, Ramsden S, Wright AF. RPGR mutation analysis and disease: an update. Hum Mutat. 2007;28:322–8. doi: 10.1002/humu.20461. [DOI] [PubMed] [Google Scholar]
- 2.Meindl A, Dry K, Herrmann K, Manson F, Ciccodicola A, Edgar A, Carvalho MR, Achatz H, Hellebrand H, Lennon A, Migliaccio C, Porter K, Zrenner E, Bird A, Jay M, Lorenz B, Wittwer B, D'Urso M, Meitinger T, Wright A. A gene (RPGR) with homology to the RCC1 guanine nucleotide exchange factor is mutated in X-linked retinitis pigmentosa (RP3). Nat Genet. 1996;13:35–42. doi: 10.1038/ng0596-35. [DOI] [PubMed] [Google Scholar]
- 3.Roepman R, van Duijnhoven G, Rosenberg T, Pinckers AJ, Bleeker-Wagemakers LM, Bergen AAB, Post J, Beck A, Reinhardt R, Ropers HH, Cremers FPM, Berger W. Positional cloning of the gene for X-linked retinitis pigmentosa 3: homology with the guanine-nucleotide-exchange factor RCC1. Hum Mol Genet. 1996;5:1035–41. doi: 10.1093/hmg/5.7.1035. [DOI] [PubMed] [Google Scholar]
- 4.Schwahn U, Lenzner S, Dong J, Feil S, Hinzmann B, van Duijnhoven G, Kirschner R, Hemberger M, Bergen AAB, Rosenberg T, Pinckers AJ, Fundele R, Rosenthal A, Cremers FPM, Ropers HH, Berger W. Positional cloning of the gene for X-linked retinitis pigmentosa 2. Nat Genet. 1998;19:327–32. doi: 10.1038/1214. [DOI] [PubMed] [Google Scholar]
- 5.Melamud A, Shen GQ, Chung D, Xi Q, Simpson E, Li L, Peachey NS, Zegarra H, Hagstrom SA, Wang QK, Traboulsi EI. Mapping a new genetic locus for X linked retinitis pigmentosa to Xq28. J Med Genet. 2006;43:e27. doi: 10.1136/jmg.2005.031518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hardcastle AJ, Thiselton DL, Zito I, Ebenezer N, Mah TS, Gorin MB, Bhattacharya SS. Evidence for a new locus for X-linked retinitis pigmentosa (RP23). Invest Ophthalmol Vis Sci. 2000;41:2080–6. [PubMed] [Google Scholar]
- 7.Gieser L, Fujita R, Goring HH, Ott J, Hoffman DR, Cideciyan AV, Birch DG, Jacobson SG, Swaroop A. A novel locus (RP24) for X-linked retinitis pigmentosa maps to Xq26–27. Am J Hum Genet. 1998;63:1439–47. doi: 10.1086/302121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferreira PA. Insights into X-linked retinitis pigmentosa type 3, allied diseases and underlying pathomechanisms. Hum Mol Genet. 2005;14:R259–67. doi: 10.1093/hmg/ddi272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bader I, Brandau O, Achatz H, Apfelstedt-Sylla E, Hergersberg M, Lorenz B, Wissinger B, Wittwer B, Rudolph G, Meindl A, Meitinger T. X-linked retinitis pigmentosa: RPGR mutations in most families with definite X linkage and clustering of mutations in a short sequence stretch of exon ORF15. Invest Ophthalmol Vis Sci. 2003;44:1458–63. doi: 10.1167/iovs.02-0605. [DOI] [PubMed] [Google Scholar]
- 10.Neidhardt J, Glaus E, Barthelmes D, Zeitz C, Fleischhauer J, Berger W. Identification and characterization of a novel RPGR isoform in human retina. Hum Mutat. 2007;28:797–807. doi: 10.1002/humu.20521. [DOI] [PubMed] [Google Scholar]
- 11.Vervoort R, Lennon A, Bird AC, Tulloch B, Axton R, Miano MG, Meindl A, Meitinger T, Ciccodicola A, Wright AF. Mutational hot spot within a new RPGR exon in X-linked retinitis pigmentosa. Nat Genet. 2000;25:462–6. doi: 10.1038/78182. [DOI] [PubMed] [Google Scholar]
- 12.Kirschner R, Rosenberg T, Schultz-Heienbrok R, Lenzner S, Feil S, Roepman R, Cremers FPM, Ropers HH, Berger W. RPGR transcription studies in mouse and human tissues reveal a retina-specific isoform that is disrupted in a patient with X-linked retinitis pigmentosa. Hum Mol Genet. 1999;8:1571–8. doi: 10.1093/hmg/8.8.1571. [DOI] [PubMed] [Google Scholar]
- 13.Kirschner R, Erturk D, Zeitz C, Sahin S, Ramser J, Cremers FPM, Ropers HH, Berger W. DNA sequence comparison of human and mouse retinitis pigmentosa GTPase regulator (RPGR) identifies tissue-specific exons and putative regulatory elements. Hum Genet. 2001;109:271–8. doi: 10.1007/s004390100572. [DOI] [PubMed] [Google Scholar]
- 14.Pelletier V, Jambou M, Delphin N, Zinovieva E, Stum M, Gigarel N, Dollfus H, Hamel C, Toutain A, Dufier JL, Roche O, Munnich A, Bonnefont JP, Kaplan J, Rozet JM. Comprehensive survey of mutations in RP2 and RPGR in patients affected with distinct retinal dystrophies: genotype-phenotype correlations and impact on genetic counseling. Hum Mutat. 2007;28:81–91. doi: 10.1002/humu.20417. [DOI] [PubMed] [Google Scholar]
- 15.Sharon D, Sandberg MA, Rabe VW, Stillberger M, Dryja TP, Berson EL. RP2 and RPGR Mutations and Clinical Correlations in Patients with X–Linked Retinitis Pigmentosa. Am J Hum Genet. 2003;73:1131–46. doi: 10.1086/379379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beltran WA, Hammond P, Acland GM, Aguirre GD. A frameshift mutation in RPGR exon ORF15 causes photoreceptor degeneration and inner retina remodeling in a model of X-linked retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2006;47:1669–81. doi: 10.1167/iovs.05-0845. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Q, Acland GM, Wu WX, Johnson JL, Pearce-Kelling S, Tulloch B, Vervoort R, Wright AF, Aguirre GD. Different RPGR exon ORF15 mutations in Canids provide insights into photoreceptor cell degeneration. Hum Mol Genet. 2002;11:993–1003. doi: 10.1093/hmg/11.9.993. [DOI] [PubMed] [Google Scholar]
- 18.Hong DH, Li T. Complex expression pattern of RPGR reveals a role for purine-rich exonic splicing enhancers. Invest Ophthalmol Vis Sci. 2002;43:3373–82. [PubMed] [Google Scholar]
- 19.Ayyagari R, Demirci FY, Liu J, Bingham EL, Stringham H, Kakuk LE, Boehnke M, Gorin MB, Richards JE, Sieving PA. X-linked recessive atrophic macular degeneration from RPGR mutation. Genomics. 2002;80:166–71. doi: 10.1006/geno.2002.6815. [DOI] [PubMed] [Google Scholar]
- 20.Demirci FY, Rigatti BW, Wen G, Radak AL, Mah TS, Baic CL, Traboulsi EI, Alitalo T, Ramser J, Gorin MB. X-linked cone-rod dystrophy (locus COD1): identification of mutations in RPGR exon ORF15. Am J Hum Genet. 2002;70:1049–53. doi: 10.1086/339620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang Z, Peachey NS, Moshfeghi DM, Thirumalaichary S, Chorich L, Shugart YY, Fan K, Zhang K. Mutations in the RPGR gene cause X-linked cone dystrophy. Hum Mol Genet. 2002;11:605–11. doi: 10.1093/hmg/11.5.605. [DOI] [PubMed] [Google Scholar]
- 22.Ebenezer ND, Michaelides M, Jenkins SA, Audo I, Webster AR, Cheetham ME, Stockman A, Maher ER, Ainsworth JR, Yates JR, Bradshaw K, Holder GE, Moore AT, Hardcastle AJ. Identification of novel RPGR ORF15 mutations in X-linked progressive cone-rod dystrophy (XLCORD) families. Invest Ophthalmol Vis Sci. 2005;46:1891–8. doi: 10.1167/iovs.04-1482. [DOI] [PubMed] [Google Scholar]
- 23.Evans RJ, Hardcastle AJ, Cheetham ME. Focus on molecules: X-linked Retinitis Pigmentosa 2 protein, RP2. Exp Eye Res. 2006;82:543–4. doi: 10.1016/j.exer.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 24.Schwahn U, Paland N, Techritz S, Lenzner S, Berger W. Mutations in the X-linked RP2 gene cause intracellular misrouting and loss of the protein. Hum Mol Genet. 2001;10:1177–83. doi: 10.1093/hmg/10.11.1177. [DOI] [PubMed] [Google Scholar]
- 25.Gerber DM, Munier FL, Niemeyer G. Cross-sectional study of visual acuity and electroretinogram in two types of dominant drusen. Invest Ophthalmol Vis Sci. 2003;44:493–6. doi: 10.1167/iovs.01-0787. [DOI] [PubMed] [Google Scholar]
- 26.Marmor MF, Zrenner E. Standard for clinical electroretinography (1994 update). Doc Ophthalmol. 1995;89:199–210. doi: 10.1007/BF01203373. [DOI] [PubMed] [Google Scholar]
- 27.Papaioannou M, Chakarova CF, Prescott DC, Waseem N, Theis T, Lopez I, Gill B, Koenekoop RK, Bhattacharya SS. A new locus (RP31) for autosomal dominant retinitis pigmentosa maps to chromosome 9p. Hum Genet. 2005;118:501–3. doi: 10.1007/s00439-005-0063-3. [DOI] [PubMed] [Google Scholar]
- 28.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. GRR: graphical representation of relationship errors. Bioinformatics. 2001;17:742–3. doi: 10.1093/bioinformatics/17.8.742. [DOI] [PubMed] [Google Scholar]
- 29.O'Connell JR, Weeks DE. PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet. 1998;63:259–66. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 31.Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES. Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet. 1996;58:1347–63. [PMC free article] [PubMed] [Google Scholar]
- 32.Strauch K, Fimmers R, Kurz T, Deichmann KA, Wienker TF, Baur MP. Parametric and nonparametric multipoint linkage analysis with imprinting and two-locus-trait models: application to mite sensitization. Am J Hum Genet. 2000;66:1945–57. doi: 10.1086/302911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gudbjartsson DF, Jonasson K, Frigge ML, Kong A. Allegro, a new computer program for multipoint linkage analysis. Nat Genet. 2000;25:12–3. doi: 10.1038/75514. [DOI] [PubMed] [Google Scholar]
- 34.Thiele H, Nurnberg P. HaploPainter: a tool for drawing pedigrees with complex haplotypes. Bioinformatics. 2005;21:1730–2. doi: 10.1093/bioinformatics/bth488. [DOI] [PubMed] [Google Scholar]
- 35.Ruschendorf F, Nurnberg P. ALOHOMORA: a tool for linkage analysis using 10K SNP array data. Bioinformatics. 2005;21:2123–5. doi: 10.1093/bioinformatics/bti264. [DOI] [PubMed] [Google Scholar]
- 36.Neidhardt J, Barthelmes D, Farahmand F, Fleischhauer JC, Berger W. Different amino acid substitutions at the same position in rhodopsin lead to distinct phenotypes. Invest Ophthalmol Vis Sci. 2006;47:1630–5. doi: 10.1167/iovs.05-1317. [DOI] [PubMed] [Google Scholar]
- 37.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Allen RC, Zoghbi HY, Moseley AB, Rosenblatt HM, Belmont JW. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet. 1992;51:1229–39. [PMC free article] [PubMed] [Google Scholar]
- 39.Roepman R, Bauer D, Rosenberg T, van Duijnhoven G, van de Vosse E, Platzer M, Rosenthal A, Ropers HH, Cremers FPM, Berger W. Identification of a gene disrupted by a microdeletion in a patient with X-linked retinitis pigmentosa (XLRP). Hum Mol Genet. 1996;5:827–33. doi: 10.1093/hmg/5.6.827. [DOI] [PubMed] [Google Scholar]
- 40.Breuer DK, Yashar BM, Filippova E, Hiriyanna S, Lyons RH, Mears AJ, Asaye B, Acar C, Vervoort R, Wright AF, Musarella MA, Wheeler P, MacDonald I, Iannaccone A, Birch D, Hoffman DR, Fishman GA, Heckenlively JR, Jacobson SG, Sieving PA, Swaroop A. A comprehensive mutation analysis of RP2 and RPGR in a North American cohort of families with X-linked retinitis pigmentosa. Am J Hum Genet. 2002;70:1545–54. doi: 10.1086/340848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garcia-Hoyos M, Garcia-Sandoval B, Cantalapiedra D, Riveiro R, Lorda-Sánchez I, Trujillo-Tiebas MJ, Rodriguez de Alba M, Millan JM, Baiget M, Ramos C, Ayuso C. Mutational screening of the RP2 and RPGR genes in Spanish families with X-linked retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2006;47:3777–82. doi: 10.1167/iovs.06-0323. [DOI] [PubMed] [Google Scholar]
- 42.Mears AJ, Hiriyanna S, Vervoort R, Yashar B, Gieser L, Fahrner S, Daiger SP, Heckenlively JR, Sieving PA, Wright AF, Swaroop A. Remapping of the RP15 locus for X-linked cone-rod degeneration to Xp11.4-p21.1, and identification of a de novo insertion in the RPGR exon ORF15. Am J Hum Genet. 2000;67:1000–3. doi: 10.1086/303091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rozet JM, Perrault I, Gigarel N, Souied E, Ghazi I, Gerber S, Dufier JL, Munnich A, Kaplan J. Dominant X linked retinitis pigmentosa is frequently accounted for by truncating mutations in exon ORF15 of the RPGR gene. J Med Genet. 2002;39:284–5. doi: 10.1136/jmg.39.4.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jin ZB, Liu XQ, Hayakawa M, Murakami A, Nao-i N. Mutational analysis of RPGR and RP2 genes in Japanese patients with retinitis pigmentosa: identification of four mutations. Mol Vis. 2006;12:1167–74. [PubMed] [Google Scholar]
- 45.Vervoort R, Wright AF. Mutations of RPGR in X-linked retinitis pigmentosa (RP3). Hum Mutat. 2002;19:486–500. doi: 10.1002/humu.10057. [DOI] [PubMed] [Google Scholar]
- 46.Banin E, Mizrahi-Meissonnier L, Neis R, Silverstein S, Magyar I, Abeliovich D, Roepman R, Berger W, Rosenberg T, Sharon D. A non-ancestral RPGR missense mutation in families with either recessive or semi-dominant X-linked retinitis pigmentosa. Am J Med Genet A. 2007;143:1150–8. doi: 10.1002/ajmg.a.31642. [DOI] [PubMed] [Google Scholar]
- 47.Moore A, Escudier E, Roger G, Tamalet A, Pelosse B, Marlin S, Clément A, Geremek M, Delaisi B, Bridoux AM, Coste A, Witt M, Duriez B, Amselem S. RPGR is mutated in patients with a complex X linked phenotype combining primary ciliary dyskinesia and retinitis pigmentosa. J Med Genet. 2006;43:326–33. doi: 10.1136/jmg.2005.034868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Iannaccone A, Breuer DK, Wang XF, Kuo SF, Normando EM, Filippova E, Baldi A, Hiriyanna S, MacDonald CB, Baldi F, Cosgrove D, Morton CC, Swaroop A, Jablonski MM. Clinical and immunohistochemical evidence for an X linked retinitis pigmentosa syndrome with recurrent infections and hearing loss in association with an RPGR mutation. J Med Genet. 2003;40:e118. doi: 10.1136/jmg.40.11.e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koenekoop RK, Loyer M, Hand CK, Al Mahdi H, Dembinska O, Beneish R, Racine J, Rouleau GA. Novel RPGR mutations with distinct retinitis pigmentosa phenotypes in French-Canadian families. Am J Ophthalmol. 2003;136:678–87. doi: 10.1016/s0002-9394(03)00331-3. [DOI] [PubMed] [Google Scholar]
- 50.Zito I, Downes SM, Patel RJ, Cheetham ME, Ebenezer ND, Jenkins SA, Bhattacharya SS, Webster AR, Holder GE, Bird AC, Bamiou DE, Hardcastle AJ. RPGR mutation associated with retinitis pigmentosa, impaired hearing, and sinorespiratory infections. J Med Genet. 2003;40:609–15. doi: 10.1136/jmg.40.8.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hardcastle AJ, Thiselton DL, Van Maldergem L, Saha BK, Jay M, Plant C, Taylor R, Bird AC, Bhattacharya S. Mutations in the RP2 gene cause disease in 10% of families with familial X-linked retinitis pigmentosa assessed in this study. Am J Hum Genet. 1999;64:1210–5. doi: 10.1086/302325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Prokisch H, Hartig M, Hellinger R, Meitinger T, Rosenberg T. A population-based epidemiological and genetic study of X-linked retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2007;48:4012–8. doi: 10.1167/iovs.07-0071. [DOI] [PubMed] [Google Scholar]
- 53.Yokoyama A, Maruiwa F, Hayakawa M, Kanai A, Vervoort R, Wright AF, Yamada K, Niikawa N, Naōi N. Three novel mutations of the RPGR gene exon ORF15 in three Japanese families with X-linked retinitis pigmentosa. Am J Med Genet. 2001;104:232–8. [PubMed] [Google Scholar]
- 54.Pusch CM, Broghammer M, Jurklies B, Besch D, Jacobi FK. Ten novel ORF15 mutations confirm mutational hot spot in the RPGR gene in European patients with X-linked retinitis pigmentosa. Hum Mutat. 2002;20:405. doi: 10.1002/humu.9072. [DOI] [PubMed] [Google Scholar]