

Abstract
L1, a highly conserved transmembrane glycoprotein member of the immunoglobulin superfamily of cell adhesion molecules, mediates many developmental processes in the nervous system. Here we present the biophysical characterization and the binding properties of the least structurally defined part of this receptor: its cytoplasmic tail (CT). We have shown by analytical ultracentrifugation and dynamic light scattering experiments that it is mostly monomeric and unstructured in aqueous solution. We have defined by nuclear magnetic resonance the molecular details of L1-CT binding to two major targets: a membrane-cytoskeletal linker (MCL), ezrin, and an endocytosis mediator, AP2. Surprisingly, in addition to the two previously identified ezrin binding motifs, the juxtamembrane and the 1176YRSLE regions, we have discovered a third one, a part of which has been previously associated with binding to another MCL, ankyrin. For the L1 interaction with AP2 we have determined the precise interaction region surrounding the 1176YRSLE binding site and that this overlaps with the second ezrin binding site. In addition, we have shown that the juxtamembrane region of L1-CT has some binding affinity to AP2-μ2, although the specificity of this interaction needs further investigation. These data indicate that L1-CT belongs to the class of intrinsically disordered proteins. Endogenous flexibility of L1-CT might play an important role in dynamic regulation of intracellular signaling: the ability of cytoplasmic tails to accommodate different targets has the potential to fine-tune signal transduction via cell surface receptors.
A transmembrane glycoprotein member of the immunoglobulin superfamily of cell adhesion molecules (IgCAM),1 L1 is essential for many developmental processes. Mutation of the single gene encoding L1 in humans results in a number of devastating neurological abnormalities and mental retardation syndromes (1). L1 involvement in the metastatic progression has been well documented, and it is now considered an important target for treating specific tumor types, including ovarian carcinomas (2).
The importance of understanding L1-CT oligomeric state comes from the notion that L1-mediated cell adhesion is associated with the activation of the MAP-kinase signaling pathway (3), the initial stages of which are characterized by L1 clustering. There are many classic examples, such as avidity modulation in integrins (4, 5), showing that receptor oligomerization is an important initial step during activation. Any part of the receptor may be involved in this process. Recent studies indicate that the third fibronectin type III domain of the L1 extracellular domain spontaneously homomultimerizes, leading to the formation of trimeric L1 and the concomitant recruitment of integrins (6). The transmembrane domain of human L1 contains a potential homooligomerization motif, GXXXG (7). However its role in L1 clustering has yet to be investigated. In this work we present evidence that the cytoplasmic domain of L1 is mostly monomeric in aqueous solution.
In the nervous system L1 mediates cell migration, axon extension, branching, fasciculation, guidance, and interactions with glia (8, 9). Neuronal L1-CT (sequence presented in Figure 1A) contains four additional amino acids (1177RSLE) compared to L1-CT expressed in nonneuronal cells or in a variety of human tumors (10, 11). These are coded for by the alternatively spliced exon 27. Although several L1-associated cytosolic molecules participating in the axon growth and branching have been identified (12–14), the structural basis for L1-mediated intracellular signaling and cell remodeling still remains a mystery. Here we present biophysical and biochemical data characterizing free L1-CT in aqueous solution and in complexes with two of its binding partners, the ezrin FERM domain and the AP2-μ2 chain.
Figure 1.
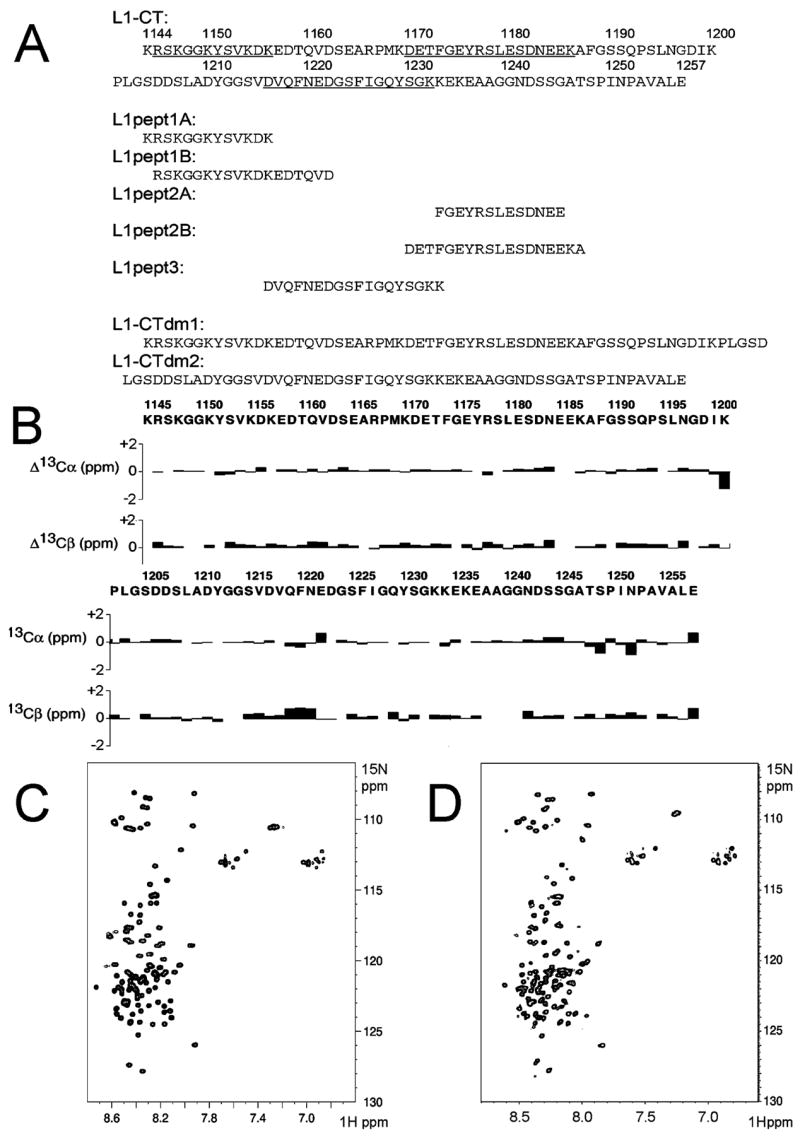
(A) Sequences of L1-CT and the peptides derived from it. Three major regions, which are involved in ezrin binding, are underscored. (B) Differences between 13Cα/13Cβ shifts of each residue from the free L1-CT in aqueous solution and the corresponding random coil values. Very small deviations indicate the mostly unstructured nature of L1-CT. (C) 1H–15N-HSQC spectrum of L1-CT in aqueous solution (50 mM potassium phosphate, 300 mM NaCl, pH 6.07, 14 ° C, 0.05 mM). (D) 1H–15N-HSQC spectrum of L1-CT in detergent (100 mM deuterated DPC, pH 5.4, 25 °C, 0.5 mM)
The involvement of L1 in regulation of the axonal outgrowth and the neuronal migration requires coordination with the actin cytoskeleton. IgCAMs, such as L1, do not bind directly to the actin cytoskeleton; their interactions are regulated by MCLs. Ezrin, radixin, and moesin (ERM) form a family of highly homologous proteins that are ideal candidates for coordinating these interactions between L1 and the actin cytoskeleton during the axonal outgrowth and migration. ERM proteins have a C-terminal actin binding site (15) and an N-terminal FERM (f our point one, ezrin, radixin, and moesin, common to all members of the band 4.1 superfamily (16)) domain that can bind to the transmembrane molecules. Direct interactions between L1 and ezrin have been identified in vitro and confirmed in primary hippocampal neurons and nerve growth factor-treated PC12 cells (14). The L1-CT interaction with the ezrin FERM domain plays a crucial role in neurite branching (9) and involves two major sites: the juxtaproximal 1147KGGKYS-VKDK homologue of the nonpolar RxxTYxVxxA motif of ICAM2 (17) and the 1176YRSLE region (14). In this study we have confirmed these findings by NMR. Moreover, we have identified an additional region, which has previously been associated with L1 binding to another MCL, ankyrin (18).
L1-controlled endocytosis is implicated in the motility of the nerve growth cones (19). The neuronal 1176YRSLE motif coordinates L1 endocytosis via clathrin-coated pits (13) via a tyrosine-based sorting signal (YxxΦ, where Φ is a residue with a bulky hydrophobic side chain) found in many other proteins (20): the 1176YRSL sequence of L1 serves as a docking site for the μ2 chain of the clathrin-associated AP2 complex (21). Along with this motif, an additional hydrophobic residue located three amino acids upstream (1176Y-3; 1173F in L1) is anticipated to be involved in this complex formation, as has been shown for the cytoplasmic domain of human P-selectin (22). In this work we have confirmed by NMR that the 1173FxxYxxL motif of L1 is indeed a major AP2-μ2 binding site. Our data, however, suggest that the L1-CT and AP2-μ2 interaction requires a longer motif, 1170D–1182D, which encompasses the above-mentioned residues.
MATERIALS AND METHODS
Expression and Purification
Human L1-CT (K1144–E1257) and its shorter peptides, L1-CTdm1 (K1144–D1205) and L1CTdm2 (L1202–E1257), as well as the mouse ezrin FERM domain (V (7)–T299) and the AP2-μ2 chain (T156–C435) were subcloned into the pET15b (Novagen, Inc.) vector containing an N-terminal His tag. We used NdeI forward/BamHI reverse cutting sites for L1-CT and its shorter peptides, and NdeI forward/XhoI reverse cutting sites for ezrin and AP2-μ2. L1-CT and ezrin constructs were expressed in BL21(DE3) cells; AP2-μ2 was expressed in the BL21(DE3)/pLys cell line to improve the expression levels. Lowering the temperature of the cell cultures to 30 °C before (preconditioning) and after the induction with IPTG considerably improved the solubility of both the ezrin FERM domain and the AP2-μ2 chain. Purification of all of the proteins was completed under native conditions according to the manufacturer’s instructions (Novagen, Inc.), followed by gel filtration on a HiLoad 16/ 60 Superdex 75 column in 50 mM potassium phosphate (300 mM NaCl, pH 7.0) buffer: although L1-CT solubility was not dependent upon salt concentration, both ezrin and AP2 required a high salt content to prevent aggregation. Millipore Amicon centrifugal units with different cutoff filters (5K for L1-CT and 10K for ezrin and AP2-μ2) were used to concentrate the proteins. Isotopically labeled samples were produced by growing cultures in M9 minimal medium containing [15N]ammonium chloride (1.1 g/L) and/or [13C]glucose (3 g/L) as the sole nitrogen and carbon sources, respectively. The short peptides (L1pept1A, L1pept1B, L1pept2A, L1pept2B, and L1pept3) were synthesized chemically (Biosynthesis Inc.).
NMR Spectroscopy
HSQC (heteronuclear single-quantum correlation) titration experiments were performed in 50 mM potassium phosphate (300 mM NaCl, pH 6.07) buffer at 14 °C. Peak volume ratios were compared to peak intensity ratios in several experiments, and no significant differences were observed. Thus we present all of the differential line-broadening data in the form of peak intensity ratio. Sequence-specific assignments of the double-labeled L1-CT were made using the standard triple resonance experiments at 14 °C at pH 6.07 as described in Clore and Gronenborn (23) on a Bruker Avance 500 MHz spectrometer with a TXI probe. Briefly, HNCACB and HNCA experiments were used for the backbone and H(CC)(CO)HN, (H)CC(CO)HN, and HCCH-TOCSY experiments for the side chain assignments. All of the spectra were processed with nmrPipe (24) or TopSpin (Bruker, Inc.) and visualized with PIPP (25) or AUREMOL (Bruker Inc.). Transferred NOESY (nuclear Overhauser effect spectroscopy) experiments for the different peptides were performed on a Varian Inova 600 MHz spectrometer equipped with an inverse triple-resonance cryoprobe at 14 °C (and in several cases at 25 °C) in the same buffer (50 mM potassium phosphate, 300 mM NaCl, pH 6.07). Different ratios of the peptides to the binding partner were investigated to find the optimal range of NOE transfer for each particular analysis. The resonance assignments of unlabeled peptides were made using the conventional 2D-1H TOCSY (total correlation spectroscopy) and NOESY spectra (26).
Analytical Ultracentrifugation
Sedimentation velocity analysis was conducted at 20 °C and 55000 rpm using interference optics with a Beckman-Coulter XL-I analytical ultracentrifuge. Double sector synthetic boundary cells equipped with sapphire windows were used to match the sample and reference menisci. Molecular mass, partial specific volume (v¯), and hydrodynamic parameters were calculated using Sednterp (27). Model-independent analysis was performed using Sedfit (28) to obtain c(s) distributions. Data were globally fit to a hybrid continuous-discrete model using Sedphat (29).
Dynamic Light Scattering
The study was conducted on Malvern Instruments’ Zetasizer Nano (Worcestershire, U.K.) at 25 °C. Prior to the measurements L1-CT solution was passed through a 0.1 μm Millipore (Billerica, MA) Millex-W syringe filter and centrifuged on a Fisher Scientific Eppendorf minicentrifuge (Fair Lawn, NJ) at 5600g for 10 min. Molecular masses were determined from the mean hydrodynamic radius using the Malvern DTS software.
RESULTS
L1-CT in Aqueous Solution
To define the conformation and oligomeric state of L1-CT, we have employed a number of different methods. We started with NMR, a particularly powerful high-resolution technique, to examine proteins at near physiological conditions. On the basis of 13Cα and 13Cβ chemical shifts, which do not significantly deviate from the random coil values (Figure 1B), and the narrow distribution of the amide peaks in the 15N-HSQC spectrum (Figure 1C), we conclude that L1-CT is predominantly unstructured in aqueous solution. Although according to the secondary structure prediction (Neural Network Prediction, http://npsa-pbil.ibcp.fr/) three regions of L1-CT, 1147K–1156K, 1177R–1182D, and 1208L–1239G, demonstrate some tendency (not very high; data not shown) to form short -strands, we do not observe any by NMR: the 1214S–1220N motif is the only potential nucleation point for a β-strand in our data. This is consistent with the circular dichroism data for the neurofascin (an L1-CAM family member) cytoplasmic domain, which was found to be mostly random with some tendency to form β-structures at higher temperatures (30). In the case of L1-CT, raising the temperature causes differential line broadening and a consequent disappearance above 37 °C of a number of resonances in the 15N-HSQC spectrum (data not shown). The disappearance of specific peaks has previously been observed in dynamic regions of globular proteins (31), when intermediate conformational exchange results in significant broadening of the spectra, often beyond the experimental detection. In addition to conformational exchange, temperature increase in partially disordered systems (32) stimulates exchange between non-protected amides and water protons, resulting in further broadening of the spectra. We have embedded L1-CT in dodecylphosphocholine (DPC) micelles to mimic the interactions with membrane surface, which could enforce and stabilize the structural rearrangements in otherwise unstructured cytoplasmic tails of integral membrane proteins (33). Apparently, the 15N-HSQC pattern (Figure 1D) is not significantly perturbed and is very similar to the one in aqueous solution (Figure 1C), which demonstrates small, if any, changes in L1-CT structure upon addition of the detergent.
To further characterize L1-CT shape and to determine its oligomerization state, we have used sedimentation velocity analytical ultracentrifugation. The normalized c(s) plots for two concentrations of L1-CT, 0.02 and 0.06 mM, are presented in Figure 2A. A major peak is observed near s = 1.7 S with minor features near 3 and 4 S. The sedimentation coefficient of the major peak and the relative amplitudes of the higher S features are independent of concentration, indicating that L1-CT does not undergo a reversible self-association over this concentration range. Global analysis of the two data sets using a hybrid continuous-discrete model yields a best fit molecular mass of 18.2 kDa for the major species, with a corrected sedimentation coefficient of s20,w = 1.63 S and a frictional ratio of f/f0 = 1.59. These data indicate that L1-CT exists as a monomer at this concentration range. However, the deduced molecular mass is somewhat higher than the monomer value of 14.4 kDa. This discrepancy may be due to the incorrect value for the partial specific volume (&vmacr;) calculated from the amino acid composition, as has been observed in other systems (34). The frictional ratio is much higher than the value of 1.1–1.3 typically observed for globular proteins and is consistent with the NMR data indicating an unstructured extended conformation.
Figure 2.
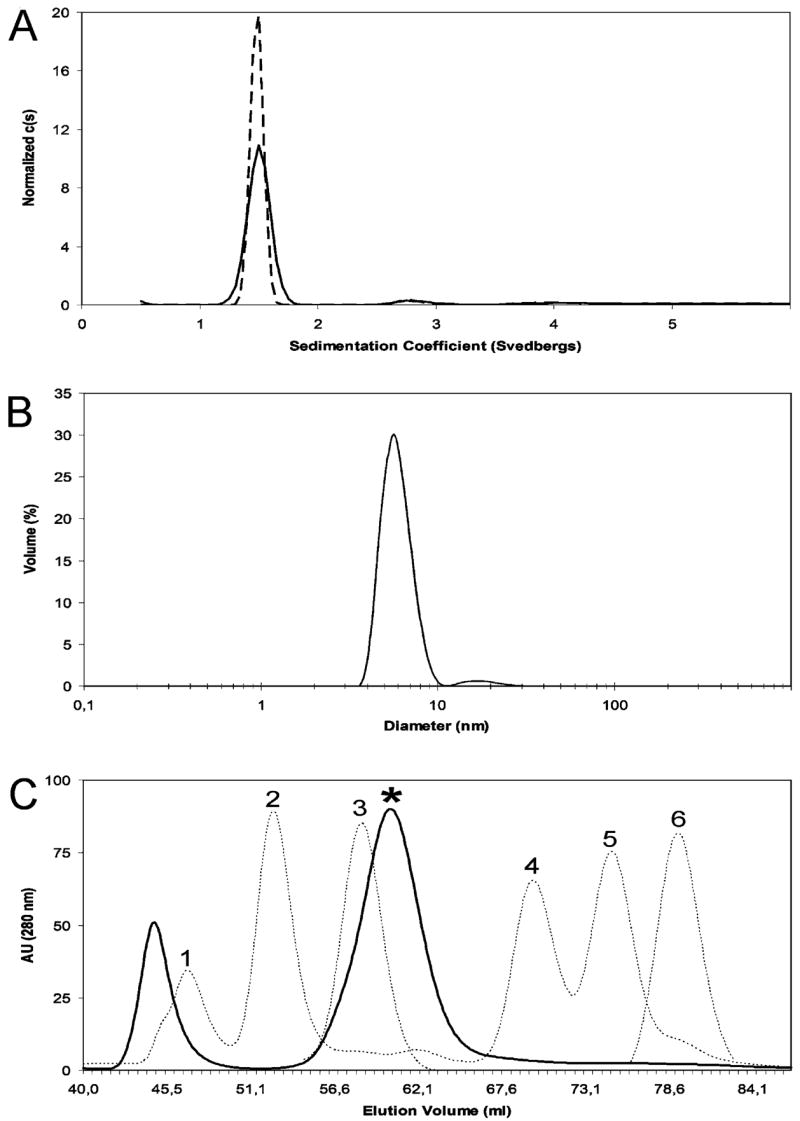
(A) The normalized distribution of L1-CT sedimenting species is shown for two concentrations: 0.06 mM (dashed line) and 0.02 mM (solid line). Global analysis of the two data sets using a hybrid continuous-discrete model yields a best fit molecular mass of 18.2 kDa for the major species, with a corrected sedimentation coefficient of s20,w = 1.63 S and a frictional ratio of f/f0 = 1.59. (B) The normalized volume distribution for the L1-CT particles in aqueous solution (at 0.03 mM, pH 8.0, IS 300 mM). The major peak is observed around a diameter of 5.93 nm with the minor features near 15 nm for the higher order aggregates. (C) Size-exclusion chromatography analysis. The L1-CT elution profile is shown by a solid curve with the major peak corresponding to the monomeric fraction indicated by a star. Dotted curves represent commercial molecular mass markers (MPBio Inc.): 1, γ-globulin, 160 kDa; 2, bovine albumin, 67 kDa; 3, ovalbumin, 45 kDa; 4, chymotrypsinogen, 24 kDa; 5, myoglobin, 18 kDa; 6, cytochrome c, 13 kDa.
Dynamic light scattering experiments provided us with similar results. The normalized volume distribution for the L1-CT particles in aqueous solution (at 0.03 mM, pH 8.0, IS 300 mM) is presented in Figure 2B. The major peak is observed around a diameter of 5.93 nm with minor features near 15 nm for the higher order aggregates. The resulting hydrodynamic radius (2.96 nm) represents an approximately 40 kDa globular (spherical) protein (Malvern DTS software). According to our NMR data, however, L1-CT is not a globular protein, and it exists mostly as a random coil in solution, i.e., as a linear polymer. For a linear polypeptide the estimated MW is 15.6 kDa (Malvern DTS software), which correlates nicely with 14.4 kDa, the theoretically calculated value for the L1-CT monomer.
Size-exclusion chromatography confirms this conclusion. The elution position of L1-CT from the Superdex-75 column is in between 45 and 24 kDa globular proteins [Figure 2C, solid curve, peak marked with the star; dotted curves represent molecular mass markers (peak 3, ovalbumin, 45 kDa; peak 4, chymotrypsinogen, 24 kDa)]. This elution position, however, does not correspond to a 14.4 kDa globular monomeric protein [Figure 2C (peak 5, myoglobin, 18 kDa; peak 6, cytochrome c, 13 kDa, shown for comparison)], which we believe is due to the disordered nature of the L1-CT monomer characterized by a significantly increased hydrodynamic radius. We also observe another major peak at a position of high molecular mass aggregates (around an elution volume of 44 mL). When the contents of the fractions, corresponding to each major peak, have been concentrated separately (beyond 0.5 mM) and then injected onto the column during consequent runs, we observed the reappearance of both peaks in each resulting graph (data not shown). However, the distributions of the peak intensities were different from the original pattern. This observation allows us to speculate that some sort of reversible exchange between oligomeric and monomeric forms of L1-CT might exist at a higher concentration range not accessible for AU or DLS techniques due to a number of optical artifacts.
To conclude, we found that L1-CT is predominantly monomeric and unstructured in aqueous solution. Knowing the molecular details of its binding patterns, described in the following section, we can suggest that L1-CT belongs to a very interesting class of natively unfolded or intrinsically unstructured proteins, commonly referred to as IDPs. Below we present the detailed characterization of L1-CT interactions with two different target proteins as determined by NMR.
L1-CT Interactions with Ezrin and AP2-μ2
We have completed L1-CT backbone assignments using modern triple-resonance NMR experiments as described in the Materials and Methods section. Because of the enormous degree of degeneracy in chemical shifts, mainly due to the disordered nature of L1-CT, this task was not straight forward. We were, however, able to assign the majority of the 114 (with 11 exceptions) backbone resonance combinations (1Hα, 13Cα, 13Cβ, 13CO, 15N). Titrating the unlabeled proteins, the ezrin FERM domain and the AP2-μ2 chain, into the solution of isotopically labeled L1-CT and monitoring associated perturbations in the 15N-HSQC spectra allowed us to map the binding surface of L1-CT in both complexes. We further confirmed our findings for the L1 ezrin interaction by monitoring the perturbations in 3D 15N/13C-HNCO spectra of L1-CT upon addition of the ezrin FERM domain. In all of these cases we observed a differential line broadening and disappearance of the peaks rather than shifts in resonance frequencies. This phenomenon is due to the large molecular mass of the target proteins (37 kDa for the ezrin FERM domain and 34 kDa for the AP2-μ2 chain) and intermediate exchange between L1 conformations in free and bound states (35). Figure 3 shows a plot of the ratios of the peak intensities for the free and bound forms of L1-CT as a function of residue number. Despite some similarities we do see significant differences in L1 binding patterns to these two different targets.
Figure 3.
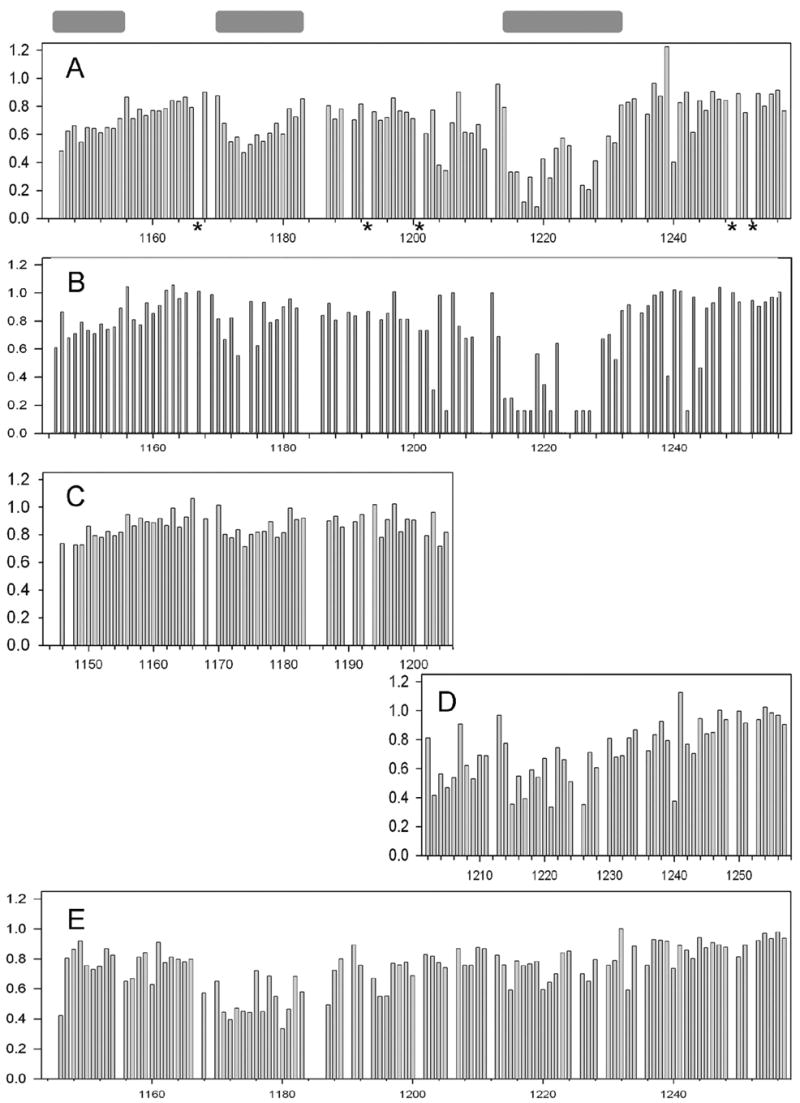
Summary of the observed changes in peak intensities indicating L1 interaction with ezrin and AP2-μ2. All experiments were performed in 50 mM potassium phosphate (pH 6.07, 300 mM NaCl) buffer at 14 °C. Normalized intensity ratios of the peaks corresponding to free L1-CT (and its smaller peptides in panels D and E, 0.05 mM) in aqueous solution versus bound to the target proteins are plotted as a function of the residue number: (A) perturbations of L1-CT amides upon titration of the ezrin FERM domain (0.1 mM); (B) perturbations of L1-CT (0.05 mM) carbonyl carbons upon titration of FERM (0.1 mM); for the peaks broadened beyond detection in the complex a ratio of 0.15 is arbitrarily assigned; (C) perturbations of L1-CTdm1 amides upon titration of the FERM domain (0.15 mM); (D) perturbations of L1-CTdm2 amides upon titration of the FERM domain (0.15 mM); (E) perturbations of L1-CT amides upon titration of AP2-μ2 (0.2 mM). Unassigned or seriously overlapping residues are represented by gaps. Prolines of the L1-CT sequence are marked by stars in panel A. Proposed ezrin binding motifs are shown as gray boxes positioned over the corresponding amino acids in the sequence of L1-CT.
The L1-CT binding interface with the ezrin FERM domain is much broader than with the AP2-μ2 chain. Surprisingly, in addition to the two ERM binding sites identified in previous studies, the 1176YRSLE region and the juxtamembrane region, we identified a significant line broadening in a third one, composed of residues from 1213G to 1232K (Figure 3A,B). This site overlaps with the classical ankyrin binding motif, 1220NEDGS-FIGQY. Yeast two-hybrid analysis (14), however, indicated that deletion of the L1-CT C-terminal to the RSLE, including this third binding site, did not affect the interaction between L1 and ezrin. In order to determine the importance of this motif, we performed additional experiments described below.
In the case of AP2-μ2, as expected, the1176YRSL motif of L1-CT is in the center of the cluster of resonances with reduced peak intensities (Figure 3E). Thus it is, indeed, the major binding determinant. The residue three amino acids upstream of 1176Y, 1173F, is also involved in the L1-AP2-μ2 complex. Contrary to the L1 interaction with ezrin, the C-terminus of L1-CT is not involved in the interaction with AP2-μ2. Surprisingly enough, we do see additional perturbations in the juxtamembrane region of L1-CT. However, the affected residues (mostly 1146S, although first two residues in L1-CT we were not able to assign unambiguously) do not form a well-defined cluster and are different from the ones affected by interaction with ezrin. To find out whether this is a real binding site, rather than the influence of the fused His tag in the L1-CT construct, we performed additional experiments described below.
Transferred NOE experiments (36) were used to confirm the binding loci for AP2-μ2 and ezrin. This method is limited to systems where the free and bound forms are in fast exchange. On the basis of the NMR titration data in Figure 3, we have designed and tested several short peptides, which correspond to the different binding motifs of L1-CT (Figure 1A).
It appears that 1145RSKGGKYSVKDK (L1pept1A) is sufficient to define the ezrin juxtamembrane binding site, with major hydrophobic interactions around the 1151YSV triplet (Figure 4A). Even though we see several residues on the right side of the L1-CT juxtamembrane region also being perturbed in the titration data above (Figure 3A,B), the extension of L1pept1A by an additional six residues to L1pept1B has no significant effect on its interaction with ezrin. Neither is the conformation of the 1151YSV triplet in the bound state perturbed, as being judged from the comparison of NOE patterns (Figure 4A,B), nor is the binding affinity profoundly altered (the most pronounced trNOE effects are seen at the ratios of 10:1 and 15:1 for these two peptides, respectively). Next, despite the fact that it contains all of the defining hydrophobic core residues, 1173FGEYRSLESDNEE (L1pept2A) does not show any trNOE effects under conditions we tried (data not shown). Inclusion of the three additional residues on the left side in L1pept2B and the optimization of the peptide/ezrin ratio to 100:1 allowed us to see additional peaks in the NOESY spectrum (Figure 4D). Unexpectedly, the third peptide, 1216DVQFNEDGSFIGQYSGKK (L1pept3), which was designed to mimic the third ezrin binding region of L1-CT, does not show any additional peaks in trNOE experiments under all of the conditions we tried (data not shown). This may be, as in previous case, because the construct is lacking some crucial residues, such as 1213GSV or even 1203GSD/ 1208LADY, which are absolutely vital for binding to the FERM domain. Alternatively, the binding affinity of this construct could be very low and cooperative in nature, and thus it cannot be measured separately even by this extremely sensitive NMR approach.
Figure 4.
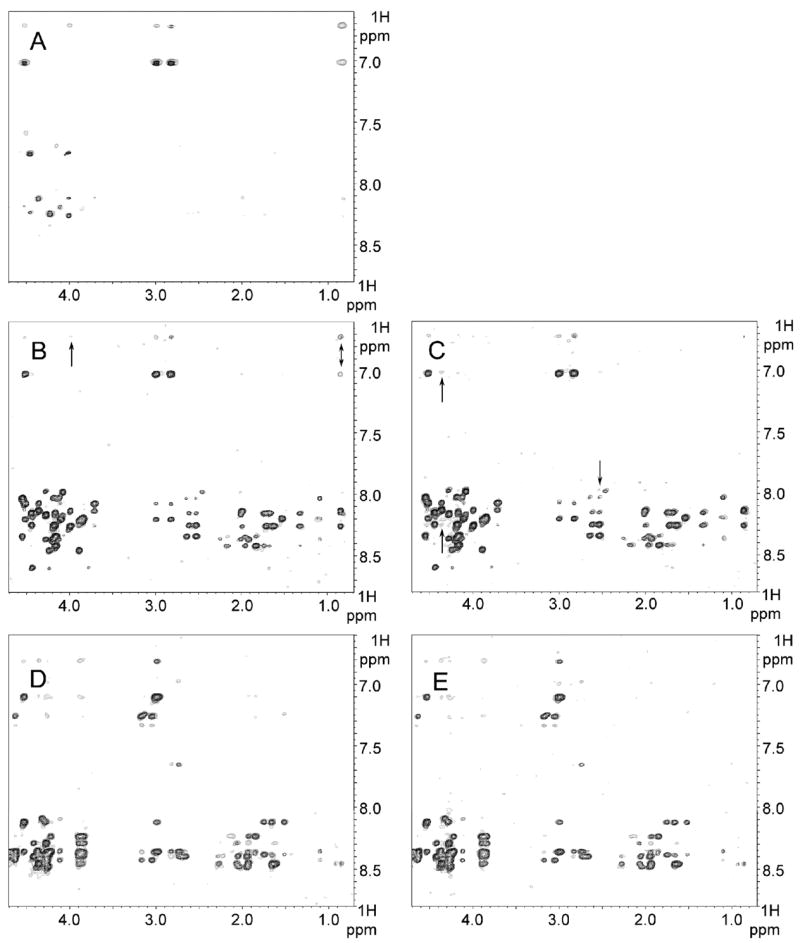
Transferred NOE evidence for L1-CT motifs interacting with ezrin and AP2-μ2. All shown trNOE experiments were performed at 14 °C (400 ms mixing time) in 50 mM potassium phosphate (300 mM NaCl, pH 6.07) buffer. Superposition of NOESY spectra are presented as follows (peptide alone is shown in black; peptide in complex, in gray): (A) L1pept1A alone and in complex with the ezrin FERM domain at the ratio of 10:1; (B) L1pept1B alone and in complex with the ezrin FERM domain at the ratio of 15:1 with peaks originated from the major triad 151YSV marked by arrows; (C) L1pept1B alone and in complex with the AP2-μ2 chain at the ratio of 100:1 with new peaks originated from residues different from the 151YSV triad marked by arrows; (D) L1pept2B alone and in complex with the ezrin FERM domain at the ratio of 100:1; (E) L1pept2B alone and in complex with the AP2-μ2 chain at the ratio of 100:1.
To distinguish between these possibilities, we have designed two long L1-CT peptides with four overlapping residues (Figure 1A): the first one, N-terminus starting from 1144K and ending at 1205D (L1-CTdm1) and the second one, C-terminus starting from 1202L up to the final 1257E (L1-CTdm2). We analyzed these two peptides using the same type of 15N-HSQC titration experiments as on full L1-CT (Figure 3A). Upon ezrin binding both peptides show line-broadening patterns similar to the corresponding regions of full L1-CT (Figure 3C,D). The only difference is that the intensity drops in the affected regions are slightly less pronounced, while the 203GSDD stretch of L1-CTdm2 (Figure 3D) is slightly more perturbed (probably due to the influence of the N-terminally fused His tag). Theses data allow us to conclude that ezrin binding to the third motif of L1-CT is independent of the other two sites at least in vitro. The same is true for the combination of the juxtamembrane and the 1176YRSLE regions and explains why deletion of the L1-CT C-terminus did not eliminate L1-ezrin interaction (14): these two motifs are sufficient on their own for the stable L1-CT/ ezrin FERM domain complex formation.
The similarities of the L1 interaction with AP2-μ2, compared to the L1-ezrin interaction defined by the trNOE technique, come from the 1176YRSLE region. While L1pept2A mixed with AP2-μ2 does not show any additional peaks in NOESY spectra (data not shown), L1pept2B does (Figure 4E). Furthermore, the patterns of trNOE peaks for L1pep2B mixed with both, ezrin and AP2-μ2, are remarkably similar (Figure 4D,E). This finding confirms the presence of a common binding site in L1-CT: the region encompassing 1176YRSLE is involved in interactions with both the ezrin FERM domain and the AP2-μ2 chain, and it accommodates homologous conformations in both complexes. The picture is very different for the juxtamembrane region. Even though L1pept1B (but not L1pept1A; data not shown) shows additional (very limited) peaks in NOESY spectra upon interaction with AP2-μ2 (Figure 4C), the pattern is different from the one present due to interaction with ezrin: the major triad 1151YSV peaks (marked by arrows in Figure 4B) do not appear in this case. A few new very weak peaks (marked by arrows in Figure 4C) are associated with the C-terminus of the peptide and possibly 1152S in a totally different conformation from that seen with ezrin.
The number of additional peaks observed in L1-CT plus ezrin or AP2-μ2 is limited compared to L1-CT alone, which precluded total structure determination of the bound peptides.
DISCUSSION
It is common for many cell surface receptors to undergo avidity modulation upon an activation stimulus, resulting in receptor clustering (4). L1-CAMs follow this paradigm in mediating cell adhesion associated with activation of the MAP-kinase signaling pathway (3). Our original gel filtration data indicated the possibility for L1-CTs playing a role in L1-CAM clustering at high, beyond 0.5 mM, concentration levels. Nevertheless, according to our AU and DLS data, below 0.1 mM in aqueous solution L1-CT exists as a stable monomer, and we have used this concentration range to map its binding surface to two different adaptor proteins, ezrin and AP2-μ2. However, the membrane environment may significantly increase the local “planar concentration” of L1-CT. Combined with the polarizing effect of the transmembrane domain in prealigning L1-CT, this could stimulate oligomerization of the intact L1 via its cytoplasmic tails. Further investigation is required to find out if the interactions within transmembrane domains of L1 may provide a necessary additional force to stimulate/stabilize the oligomerization in question and to define the role of specific lipids, such as sphingolipids, in this process.
In this study, we have performed detailed NMR experiments not only to confirm the direct interaction of L1-CT with the ezrin FERM domain and the AP1-μ2 chain but also to pinpoint the specific regions involved. In addition to two suggested in previous studies of ezrin binding motifs, the juxtamembrane and the 1176YRSLE surrounding regions, we have shown for the first time the presence of a third one, previously associated with binding to another MCL, ankyrin. In the case of AP2-μ2 we have confirmed the extended 1176YRSLE surrounding region, which overlaps with the second ezrin binding motif, as a major binding site. We have also shown that the membrane proximal region of L1-CT has some binding affinity to AP2-μ2, even though the specificity of this binding needs to be further investigated. It is worth noting that L1-CT binding to both target proteins is very weak indeed.
Furthermore, there are few methods which allow in vitro studies such as ours describing the behavior of a mostly unstructured protein of over 100 residues in aqueous solution. This is why the differential line broadening in HSQC titration series and limited trNOEs we have observed are valuable indications of specific interactions. Moreover, the biological significance of low-affinity binding should not be underestimated. Cell surface receptors, especially cell adhesion molecules, form clusters upon activation, so the local concentration of L1-CTs and their binding partners will be very high at least at certain stages of neuronal adhesion.
To conclude, we have demonstrated that the cytoplasmic domain of L1 belongs to a very interesting class of intrinsically disordered proteins (IDPs), which establish stable conformations only upon binding to their respective partners. A large fraction of eukaryotic proteins is thought to be composed of IDPs (37). There are two prevalent theories regarding the evolutional advantage of IDP existence in the cell. According to the first one, the lack of structure and the disordered nature of IDPs allow them to interact with different ligands, giving them a unique multifaceted functional ability. The second one takes into account overall cell size and crowding: in order to have a large binding interface, as observed in many IDPs, a globular protein would have to be 2–3 times larger, potentially increasing the overall size of the cell (38). Our data, in fact, support both of these hypotheses. L1-CT can be classified as an IDP, and it shows the functional flexibility which is a hallmark of this class. It also presents a significant binding interface during its interaction with ezrin. Thus both hypotheses are equally applicable.
Recent findings (39) indicate that, despite having a much lower aggregation propensity than globular proteins, the IDPs do have a potential for amyloidosis. Some IDPs (such as human prion protein, synuclein, and Tau protein) are clearly involved in formation of insoluble plaques or aggregates, which ultimately leads to protein conformational disorders such as Alzheimer’s syndrome, Creutzfeld–Jacob syndrome, Parkinson’s disease, Huntington disease, Gerstmann–Straussler–Scheinker syndrome, and fatal familial insomnia. Thus further investigation of systems like L1-CT is essential to unravel the mechanism of coupling binding to folding, which might ultimately provide the strategies to fight these diseases in the future.
Acknowledgments
We thank Frank Delaglio and Dan Garrett for NMR software.
Footnotes
This work was supported in parts by grants from the American Heart Association (0335075N) and the University of Connecticut Research Foundation to O.V.
Abbreviations: Ig, immunoglobulin superfamily; CAM, cell adhesion molecule; CT, cytoplasmic tail; MW, molecular weight; DLS, dynamic light scattering; AU, analytical ultracentrifugation; NMR, nuclear magnetic resonance; MCL, membrane-cytoskeletal linker; HSQC, heteronuclear single-quantum correlation; trNOE, transferred nuclear Overhauser effect; NOESY, nuclear Overhauser effect spectroscopy; TOCSY, total correlation spectroscopy; DPC, dodecylphosphocholines; IPTG, isopropyl β-D-1-thiogalactopyranoside; IS, ionic strength; FERM, four point one, ezrin, radixin, and moesin; IDP, intrinsically disordered protein; Kd, dissociation constant.
References
- 1.Yamasaki M, Thompson P, Lemmon V. CRASH syndrome: mutations in L1CAM correlate with severity of the disease. Neuropediatrics. 1997;28:175–178. doi: 10.1055/s-2007-973696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gutwein P, et al. Cleavage of L1 in exosomes and apoptotic membrane vesicles released from ovarian carcinoma cells. Clin Cancer Res. 2005;11:2493–2501. doi: 10.1158/1078-0432.CCR-04-1688. [DOI] [PubMed] [Google Scholar]
- 3.Schaefer AW, et al. Activation of the MAPK signal cascade by the neural cell adhesion molecule L1 requires L1 internalization. J Biol Chem. 1999;274:37965–37973. doi: 10.1074/jbc.274.53.37965. [DOI] [PubMed] [Google Scholar]
- 4.Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 5.Qin J, Vinogradova O, Plow EF. Integrin bidirectional signaling: a molecular view. PLoS Biol. 2004;2:726–729. doi: 10.1371/journal.pbio.0020169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Silletti S, et al. Plasmin-sensitive dibasic sequences in the third fibronectin-like domain of L1-cell adhesion molecule (CAM) facilitate homomultimerization and concomitant integrin recruitment. J Cell Biol. 2000;149:1485–1502. doi: 10.1083/jcb.149.7.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Russ WP, Engelman DM. The GxxxG motif: a framework for transmembrane helix-helix association. J Mol Biol. 2000;296:911–919. doi: 10.1006/jmbi.1999.3489. [DOI] [PubMed] [Google Scholar]
- 8.Hortsch M. The L1 family of neural cell adhesion molecules: old proteins performing new tricks. Neuron. 1996;17:587–593. doi: 10.1016/s0896-6273(00)80192-0. [DOI] [PubMed] [Google Scholar]
- 9.Cheng L, Itoh K, Lemmon V. L1-mediated branching is regulated by two ezrin-radixin-moesin (ERM)-binding sites, the RSLE region and a novel juxtamembrane ERM-binding region. J Neurosci. 2005;25:395–403. doi: 10.1523/JNEUROSCI.4097-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miura M, et al. Molecular cloning of cDNA encoding the rat neural cell adhesion molecule L1. Two L1 isoforms in the cytoplasmic region are produced by differential splicing. FEBS Lett. 1991;289:91–95. doi: 10.1016/0014-5793(91)80915-p. [DOI] [PubMed] [Google Scholar]
- 11.Linnemann D, Raz A, Bock E. Differential expression of cell adhesion molecules in variants of K1735 melanoma cells differing in metastatic capacity. Int J Cancer. 1989;43:709–712. doi: 10.1002/ijc.2910430428. [DOI] [PubMed] [Google Scholar]
- 12.Davis JQ, Bennett V. Ankyrin-binding activity of nervous system cell adhesion molecules expressed in adult brain. J Cell Sci, Suppl. 1993;17:109–117. doi: 10.1242/jcs.1993.supplement_17.16. [DOI] [PubMed] [Google Scholar]
- 13.Kamiguchi H, et al. The neural cell adhesion molecule L1 interacts with the AP-2 adaptor and is endocytosed via the clathrin-mediated pathway. J Neurosci. 1998;18:5311–5321. doi: 10.1523/JNEUROSCI.18-14-05311.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dickson TC. Functional binding interaction identified between the axonal CAM L1 and members of the ERM family. J Cell Biol. 2002;157:1105–1112. doi: 10.1083/jcb.200111076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Turunen O, Wahlstrom T, Vaheri A. Ezrin has a COOH-terminal Actin-binding site that is conserved in the ezrin protein family. J Cell Biol. 1994;126:1445–1453. doi: 10.1083/jcb.126.6.1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chishti AH, et al. The FERM domain: a unique module involved in the linkage of cytoplasmic proteins to the membrane. Trends Biochem Sci. 1998;23:281–282. doi: 10.1016/s0968-0004(98)01237-7. [DOI] [PubMed] [Google Scholar]
- 17.Hamada K, et al. Structural basis of adhesion-molecule recognition by ERM proteins revealed by the crystal structure of the radixin-ICAM-2 complex. EMBO J. 2003;22:502–514. doi: 10.1093/emboj/cdg039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davis JQ, Bennett V. Ankyrin binding activity shared by the neurofascin/L1/NrCAM family of nervous system cell adhesion molecules. J Biol Chem. 1994;269:27163–27166. [PubMed] [Google Scholar]
- 19.Kamiguchi H, Yoshihara F. The role of endocytic l1 trafficking in polarized adhesion and migration of nerve growth cones. J Neurosci. 2001;21:9194–9203. doi: 10.1523/JNEUROSCI.21-23-09194.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ohno H, et al. Structural determinants of interaction of tyrosine-based sorting signals with the adaptor medium chains. J Biol Chem. 1996;271:29009–29015. doi: 10.1074/jbc.271.46.29009. [DOI] [PubMed] [Google Scholar]
- 21.Schaefer AW, et al. L1 endocytosis is controlled by a phosphorylation-dephosphorylation cycle stimulated by outsidein signaling by L1. J Cell Biol. 2002;157:1223–1232. doi: 10.1083/jcb.200203024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Owen DJ, et al. A third specificity-determining site in mu 2 adaptin for sequences upstream of Yxx phi sorting motifs. Traffic. 2001;2:105–110. doi: 10.1034/j.1600-0854.2001.020205.x. [DOI] [PubMed] [Google Scholar]
- 23.Clore GM, Gronenborn AM. NMR structure determination of proteins and protein complexes larger than 20 kDa. Curr Opin Chem Biol. 1998;2:564–570. doi: 10.1016/s1367-5931(98)80084-7. [DOI] [PubMed] [Google Scholar]
- 24.Delaglio F, et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 25.Garrett DS, et al. A common sense approach to peak picking in two- three- and four-dimensional spectra using automatic computer analysis of contour diagrams. J Magn Reson. 1991;95:214–220. doi: 10.1016/j.jmr.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 26.Wuthrich K. NMR of proteins and nucleic acids. John Wiley & Sons; New York: 1986. [Google Scholar]
- 27.Laue TM, et al. Computer-aided interpretation of analytical sedimentation data for proteins. In: Harding S, Rowe A, Horton J, editors. Analytical Ultracentrifugation in Biochemistry and Polymer Science. Royal Society of Chemistry; Cambridge, U.K.: 1992. pp. 90–125. [Google Scholar]
- 28.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schuck P. On the analysis of protein self-association by sedimentation velocity analytical ultracentrifugation. Anal Biochem. 2003;320:104–124. doi: 10.1016/s0003-2697(03)00289-6. [DOI] [PubMed] [Google Scholar]
- 30.Zhang X, et al. Structural requirements for association of neurofascin with ankyrin. J Biol Chem. 1998;273:30785–30794. doi: 10.1074/jbc.273.46.30785. [DOI] [PubMed] [Google Scholar]
- 31.Odaert B, et al. Solution NMR study of the monomeric form of p13suc1 protein sheds light on the hinge region determining the affinity for a phosphorylated substrate. J Biol Chem. 2002;277:12375–12381. doi: 10.1074/jbc.M111741200. [DOI] [PubMed] [Google Scholar]
- 32.Vinogradova O, et al. A membrane setting for the sorting motifs present in the adenovirus E3–13.7 protein which down-regulates the epidermal growth factor receptor. J Biol Chem. 1998;273:17343–17350. doi: 10.1074/jbc.273.28.17343. [DOI] [PubMed] [Google Scholar]
- 33.Vinogradova O, et al. A structural basis for integrin activation by the cytoplasmic tail of the alpha IIb-subunit. Proc Natl Acad Sci USA. 2000;97:1450–1445. doi: 10.1073/pnas.040548197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Philo JS, Yang TH, LaBarre M. Re-examining the oligomerization state of macrophage migration inhibitory factor (MIF) in solution. Biophys Chem. 2004;108:77–87. doi: 10.1016/j.bpc.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 35.Matsuo H, et al. Identification by NMR spectroscopy of residues at contact surfaces in large, slowly exchanging macro-molecular complexes. J Am Chem Soc. 1999;121:9903–9904. [Google Scholar]
- 36.Clore GM, Gronenborn AM. The two-dimensional transferred nuclear Overhauser effect. J Magn Reson. 1982;48:402–417. [Google Scholar]
- 37.Fink AL. Natively unfolded proteins. Curr Opin Struct Biol. 2005;15:35–41. doi: 10.1016/j.sbi.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 38.Gunasekaran K, et al. Extended disordered proteins: targeting function with less scaffold. Trends Biochem Sci. 2003;28:81–85. doi: 10.1016/S0968-0004(03)00003-3. [DOI] [PubMed] [Google Scholar]
- 39.Linding R, et al. A comparative study of the relationship between protein structure and beta-aggregation in globular and intrinsically disordered proteins. J Mol Biol. 2004;342:345–353. doi: 10.1016/j.jmb.2004.06.088. [DOI] [PubMed] [Google Scholar]