

Abstract
Epithelial tumorigenesis has been linked to AKT up-regulation. Human papillomaviruses (HPV) cause anogenital cancers and anogenital HPV infection up-regulates AKT activity. Mounting evidence points to a role for cutaneous HPVs as etiologic factors in skin tumorigenesis. High-risk cutaneous β HPVs have been linked to carcinogenesis in immunosuppressed patients, and high-risk cutaneous HPV8 genes enhance tumorigenesis in transgenic mice. We find that, in contrast to anogenital HPVs, cutaneous HPV8 early genes down-regulate epidermal AKT activity by down-regulating AKT1 isoform levels. This down-regulation occurs before papilloma formation or tumorigenesis and leads to cutaneous differentiation changes that may weaken the epidermal squame for viral release. We find that, in viral warts (papillomas) and HPV gene-induced epidermal tumors, AKT activitycan be activated focally by up-regulation and phosphorylation of the AKT2 isoform. In squamous cell carcinomas (SCC), AKT1 down-regulation is also common, consistent with a viral influence, whereas AKT2 up-regulation is widespread. Activation of up-regulated AKT2 by serine phosphorylation associates with high-grade tumors. Our data suggest that AKT2 up-regulation is characteristic of SCC and that coincident AKT2 activation through serine phosphorylation correlates with malignancy. These findings highlight differences between the effects of anogenital and cutaneous HPV on epithelial AKT activity and furthermore show that AKT isoforms can behave differently during epidermal tumorigenesis. These findings also suggest AKT2 as a possible therapeutic tumor target in SCC.
Introduction
Human papillomaviruses (HPV) are small dsDNA viruses. Anogenital HPVs are responsible for almost all cervical neoplasia. The role of β cutaneous HPVs in the etiology of nonmelanoma skin cancer is more contentious. High-risk β cutaneous HPVs are thought to be cofactors with UVB radiation in causing tumors, particularly in immunosuppressed individuals (e.g., renal transplant recipients) and in sufferers of the genetic disorder epidermodysplasia verruciformis (reviewed in ref. 1). It was recently shown that high-risk cutaneous HPV8 early genes enhance tumorigenesis rates in transgenic mice (2), adding considerable weight to the proposal that β cutaneous HPVs can be tumorigenic.
Anogenital HPV gene expression has been linked to increased AKT activity (3, 4). AKT is a serine/threonine kinase and downstream mediator of the phosphatidylinositol 3-kinase pathway, which regulates cell survival, differentiation, and growth factor responsiveness. AKT1 and AKT2 isoforms are widely expressed, whereas AKT3 has restricted expression (reviewed in ref. 5). AKT isoforms have redundant activities (6) but also isoform-specific activities during normal physiology (7-9) and carcinogenesis (10-12). Dysregulated AKT activity is associated with cancer in many cell types, including keratinocytes (13, 14).
AKT1 and AKT2 isoforms are active in epidermal keratinocytes (15) where AKT activity has been linked with differentiation (15-18). Although neither the AKT1-null nor AKT2-null mice display an overt epidermal defect (7, 8), the AKT1 and AKT2 double-null mice lack stratum corneum and die neonatally, probably from a skin barrier defect (6). Thus, AKT1 and AKT2 can behave redundantly within epidermis. However, there is also strong evidence that in normal skin AKT1 and AKT2 have specific or preferred targets. AKT1 isoform associates prominently with keratinocytes undergoing late terminal differentiation (granular layer keratinocytes), which are forming stratum corneum (18). AKT1 is important for stratum corneum formation (15, 16, 18). AKT2, in contrast, is expressed ubiquitously at low levels in epidermis and AKT2 activity is detected mainly in fetal epidermis, associated with keratinocytes leaving the basal proliferative layer (18).
HPV virions are assembled intracellularly and do not lyse their host cell. Therefore, to accomplish the release of virus particles from the terminally differentiated keratinocytes of stratum corneum, HPV has to escape from the keratinocyte cornified envelope, a protein-lipid–linked cell envelope designed to provide a biological barrier (19). Infection of mucosal keratinocytes by anogenital HPVs induces cornified envelope fragility by a variety of means (20, 21). The mechanism used by cutaneous HPV viruses for disruption of the tough epidermal cornified envelope is unknown, and because epidermal AKT activity is linked to stratum corneum formation (18), we speculated that HPV may achieve squame disruption by modulating AKT activity.
In this work, we asked if cutaneous HPVs modulate epidermal AKT activity in a manner analogous to anogenital HPVs. Because AKT activity is associated with stratum corneum function (18), we asked if cutaneous HPVs also modulate stratum corneum properties acting through AKT1 or AKT2. Because both cutaneous HPVs and AKT have been linked to squamous cell carcinoma (SCC), we explored the relationship between HPV gene expression, AKT isoform expression, and carcinogenesis. We find that, indeed, high-risk cutaneous HPV8 early region genes down-regulate AKT1 and that this is sufficient to induce differentiation and cornified envelope changes. AKT down-regulation was unexpected, given that AKT up-regulation has been linked to SCC. However, we find that, in human epidermis and experimentally induced mouse skin tumors, AKT2 isoform activity up-regulates with tumor progression and malignancy. Our work highlights major differences between the consequences of cutaneous and anogenital HPV infection and suggests novel markers for cutaneous SCC.
Materials and Methods
Explant culture of embryonic mouse skin and inhibitor treatment
Explant culture of murine fetal skin was similar to Komuves et al. (22). Skin (epidermis plus dermis) was dissected from the torso region of E15.5 CD1 mice, rinsed in sterile PBS, and transferred, dermis side down, to sterile 0.25-μm Millipore HA filter discs, which had been prewetted with culture medium (below). The skin explants on the filter disc were cultured at the air-liquid interface on metal grids so that the dermis was in contact with medium and the epidermis was exposed to air. Culture was for 48 h, covering the period of barrier formation. Explants were cultured in Williams' medium E supplemented with 10 μg/mL insulin, 10 ng/mL hydrocortisone, 2 mmol/L glutamine, 100 IU/L penicillin, 100 mg/L streptomycin, and 25 μg/mL amphotericin (all reagents from Life Technologies). For the inhibitor experiments, LY294002 (150 μmol/L; Cell Signaling Technology) or wortmannin (200 nmol/L and 2 μmol/L; Sigma-Aldrich) in DMSO vehicle were added to the medium before culture.
Immunohistochemical analysis and tissue
Tissue for immunohistochemistry was harvested from adult mice, fetal mice, or fetal skin explants (ages as specified), human clinical tissue, or human skin cancer arrays (Biomax). Adult mice used in immunolocalization of AKT1 and AKT2 were young adults of 6 to 20 weeks. However, from necessity, the age of transgenic mice and their wild-type (WT) controls yielding spontaneously occurring papillomas, dysplasia, and tumors were older (14–20 months). No changes in AKT staining were found in older WT mice. Clinical material was obtained with informed written consent from patients attending dermatology clinics at Barts and The London NHS Trust. Ethical approval was granted by the East London and City local research ethics committee. Immunochemistry on Bouin's or paraformaldehyde-fixed paraffin-embedded sections or frozen sections was by standard techniques (as described in ref. 18), with the addition that antigen retrieval was routinely carried out on paraffin-embedded sections by boiling for 10 min in 0.01 mol/L citric acid (pH 6.0). Antigen was detected by Elite avidin-biotin complex system and 3,3′-diaminobenzidine (Vector Laboratories). Primary antibodies were as follows: anti–phosphorylated AKT substrate and phosphorylated AKT (Ser473; 1:50), anti-AKT1 (1:50), and anti-AKT2 (1:10; all from Cell Signaling Technology); antimouse involucrin (1:500; Covance); and antimouse filaggrin (1:500; Zymed). Human skin cancer tissue arrays were treated as above but baked for 2 h at 60°C before dehydration down an ethanol gradient and antibody incubation.
Western blot analysis
Epidermis was removed from the dermis of embryonic skin explants by incubation in 5 mmol/L EDTA for 3 to 5 min and then boiling the epidermis in 10 mmol/L Tris-HCl (pH 7.5), 5% SDS, and 20% β-mercaptoethanol for 5 min. Protein lysates were prepared from adult mouse skin by boiling minced skin (epidermis plus dermis) in the same buffer for 10 min. Primary antibodies were as follows: anti–phosphorylated AKT (Ser473; 1:2,000), anti-AKT substrate (1:2,000), total AKT1 (1:1,000), and AKT2 (1:500; all from Cell Signaling Technology); antimouse filaggrin (1:1,000); mouse anti-actin (clone AC13; 1:2,000; Sigma-Aldrich); and antimouse keratin 14 (K14) and keratin 10 (1:4,000; Covance). Western blot analysis was by standard technique using secondary antibodies, goat anti-rabbit horseradish peroxidase (1:5,000; Jackson ImmunoResearch), and rabbit anti-mouse (1:2,000; DakoCytomation).
Retroviral infection of skin equivalent culture models
The construction of retroviral vectors pLXSN-8-E6 and pLXSN-8-E7 is described elsewhere (23, 24). The HPV8-E2 open reading frame was amplified by PCR using the primers 5′-AAGCTTGAATTCGTGATCAAGAAGACGAGGGCG-3′ and 5′-AAGCTTGGATCCTGTGTTAGTAGCAAGGCAGCG-3′ containing EcoRI and BamHI restriction endonuclease sites at their 5′ ends, respectively. The amplimers were cloned into the EcoRI and BamHI sites of pLXSN, thus obtaining pLXSN-8-E2. Amphotrophic recombinant retroviruses were generated by transfecting pLXSN-derived plasmid DNA into the packaging cell line PT67. Human keratinocytes were isolated from discarded abdominal skin after plastic surgery as described (23) and used at passage 1. Keratinocytes were seeded out in defined keratinocyte serum-free medium (0.07 mmol/L calcium; Invitrogen) at a cell density of 9 × 104 cells/cm2 in 6-cm dishes. Retroviral supernatants were mixed with an equal volume of DMEM in the presence of 5 μg/mL of hexadimethrine bromide (polybrene; Sigma-Aldrich) and added to the keratinocytes. Spin infection was made by centrifugation for 1 h at 300 × g, and the cells were washed with PBS and then cultured in defined keratinocyte serum-free medium. After 2 days, cells were selected with G418 (500 μg/mL) for 3 days after which time only infected keratinocytes survived. The cultures were trypsinized before reaching confluency and used immediately in skin equivalent (organotypic) cultures as described (20).
Cornified envelope analysis
Cornified envelopes were extracted by initially boiling whole skin (epidermis and dermis) for 10 min in extraction buffer [50 mmol/L Tris-HCl (pH 7.5), 2% SDS, 5 mmol/L EDTA] and then collected by centrifugation (13,000 × g, 10 min, room temperature). Pellets were washed in 10 mmol/L Tris-HCl and 0.1% SDS before mounting in Aquamount (BDH) containing 0.01% Nile red (Sigma-Aldrich) and visualized on a Nikon Eclipse E600 fluorescent microscope.
Results
HPV8 earlygene expression alters keratinocyte late terminal differentiation and down-regulates AKT activity by specifically down-regulating AKT1
When the early region genes from the high-risk cutaneous HPV8 are expressed in murine epidermal basal keratinocytes under the control of the K14 promoter, mice spontaneously develop papillomas with moderate and severe dysplasia, and carcinomas, without any further treatment with physical or chemical carcinogens (2). We compared nonlesional transgenic skin to WT skin to investigate the effects of HPV8 early gene expression without the added complication of tumorigenesis (Fig. 1; Supplementary Fig. S1).
Figure 1.
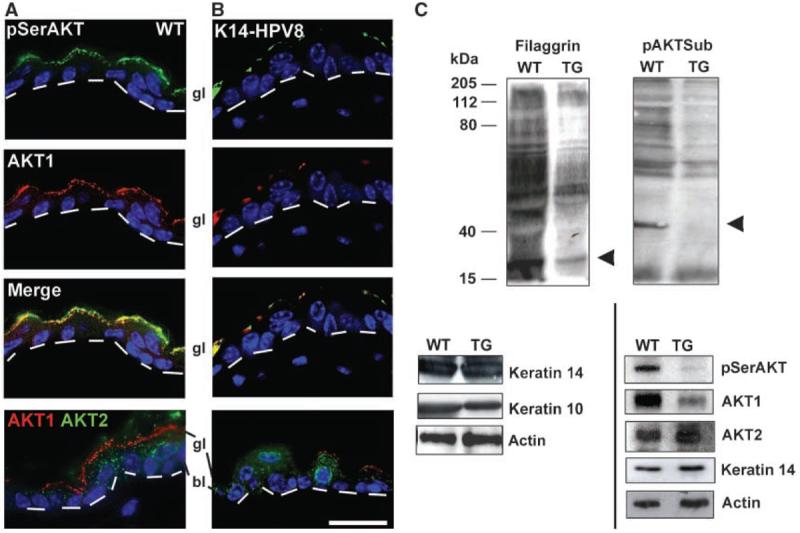
Expression of HPV8 early genes reduces expression of both pSerAKT and AKT1. AKT1 and pSerAKT were coexpressed in the granular layer (gl) of both WT epidermis (A) and K14-HPV8 epidermis (B), although expression of AKT1 and pSerAKT was markedly reduced in the K14-HPV8 epidermis (B). AKT1 is specifically reduced in expression in the K14-HPV8 epidermis (B), whereas the expression of AKT2 was unchanged. Dotted lines, dermoepidermal boundary. Bar, 20 μm (A and B). C, Western blot analysis confirmed the reduction in filaggrin, AKT1, and pSerAKT and that AKT2 levels were unaffected. Arrowhead, mature filaggrin subunit. AKT activity, indicated by an antibody that detects phosphorylated substrates of AKT (RXRXX(pS/pT) (pAKTSub), was reduced in the K14-HPV8 transgenic. Arrowhead, a particular AKT substrate that is ablated by HPV8 early genes (see text). TG, transgenic.
Anogenital HPV infection produces late terminal differentiation changes and changes to the cornified envelope of mucosal epithelium (20, 21). We show that cutaneous HPV8 early genes down-regulate filaggrin, a key late terminal differentiation marker that is processed through multiple steps to produce a small molecular weight form crucial for terminal differentiation [Fig. 1C, arrowhead (mature filaggrin); Supplementary Fig. S1]. Involucrin and keratin 10, markers for early terminal differentiation, seem unchanged by HPV8 early genes (Supplementary Fig. S1). This suggests that cutaneous HPVs, like anogenital HPVs, affect terminal differentiation. Cutaneous HPVs specifically affect epidermal late differentiation, and so we investigated whether late terminal differentiation changes are via upper granular layer AKT1 modulation.
In WT, normal adult epidermis, most AKT1 is in the upper granular layer subjacent to the stratum corneum, whereas AKT2 is expressed in all strata (Fig. 1A; ref. 18). AKT3 is undetectable by PCR in normal adult epidermis, although readily detected in cultured keratinocytes (data not shown; ref. 17). Ser473-phosphorylated AKT (pSerAKT) expression, an indicator of AKT activity in epidermis (17), is coincident with AKT1 in the granular layer (Fig. 1A). HPV8 early gene expression in transgenic nonlesional epidermis severely reduces pSerAKT and AKT1 isoform levels, although AKT2 levels are unchanged (Fig. 1B and C). Analysis of epidermal AKT substrates using an antibody to the phosphorylated AKT substrate motif (Fig. 1C; ref. 25) confirmed that AKT activity (i.e., levels of the phosphorylated substrates) was decreased in epidermis from HPV8 early region transgenics. Most putative AKT substrates were down-regulated (Fig. 1C). However, a specific 40-kDa protein band was ablated (Fig. 1C, arrowhead), suggesting that as well reducing AKT activity the HPV8 viral early genes are specifically down-regulating a particular protein, either directly or as a consequence of AKT-mediated changes. The identity of this protein is the subject of further investigation. We conclude that HPV8 early region genes are down-regulating epidermal AKT activity by specifically down-regulating the AKT1 isoform.
AKT down-regulation is sufficient to induce cornified envelope fragility and epidermal differentiation changes caused by HPV8 early gene expression
AKT1-null epidermis appears phenotypically similar to WT (8). To find whether the differentiation changes caused by HPV8 early gene expression are mediated via AKT1 down-regulation, we compared HPV8 transgenic and AKT1-null epidermis. We extracted cornified envelopes and analyzed mechanical strength by a standard sonication assay (26). Both HPV transgenic cornified envelopes and AKT1-null (AKT1−/−) cornified envelopes were significantly more labile than control WT cornified envelopes (K14-HPV8, P = 0.036, unpaired t test, n = 5; AKT1−/−, P = 0.008, unpaired t test, n = 5; Fig. 2A). These data show that there is a defect in cornified envelope strength in HPV8 early region transgenic epidermis and that AKT1 down-regulation is sufficient to confer this change.
Figure 2.

Inhibition of AKT and reduction of AKT1 expression cause differentiation changes and cornified envelope fragility. A, sonication assay shows that cornified envelopes (CEs) from the HPV8 early region transgenic mouse (K14-HPV8) and the AKT1-null mouse are more fragile. The graph shows significant differences in the percentage resistant cornified envelopes after 30-s sonication in the K14-HPV8 mouse and AKT1−/− mouse compared with WT littermates (K14-HPV8, P = 0.036, unpaired t test, n = 5; AKT1−/−, P = 0.008, unpaired t test, n = 5). Bars, SD. B, immunofluorescent analysis of pSerAKT in explants treated with LY294002 or wortmannin (Wort) or DMSO (vehicle) for 48 h shows that complete loss of pSerAKT by LY294002 treatment leads to failure to form stratum corneum. At higher wortmannin concentrations, pSerAKT is down-regulated. Bar, 50 μm. C, Western blot analysis of lysates from the same epidermal explants confirms the effects of LY294002 or wortmannin on pSerAKT levels and shows corresponding changes to filaggrin levels. β-Actin is the loading control. Arrowhead, mature form of filaggrin. D, the relative percentages of intact cornified envelopes in the treated explants show that proportion of intact cornified envelopes are decreased on LY294002 treatment and at higher wortmannin levels (significant differences between DMSO and LY294002 (*, P = 0; n = 8) and DMSO and 2 μmol/L wortmannin (**, P = 0.015, unpaired t test; n = 8). Bars, SD.
The relationship between pSerAKT levels in epidermis and epidermal differentiation and cornified envelope production was further explored in a developing fetal explant model that recapitulates in vivo development and forms a bona fide differentiated epidermis with stratum corneum (Fig. 2B, DMSO control). This experimental system was used to modulate AKT activity using well-characterized inhibitors of phosphatidylinositol 3-kinase function, wortmannin, and LY294002. Explants treated with LY294002 had markedly reduced pSerAKT (Fig. 2B and C), whereas those treated with wortmannin has a less marked reduction compared with the vehicle (DMSO)-treated control (Fig. 2B and C). The different effects of these inhibitors on AKT activity in developing explant skin are consistent and may reflect a difference in efficiency of delivery of inhibitors to target cells in this system. Strong reduction of pSerAKT via LY294002 caused substantial reduction in filaggrin and produced fragile cornified envelopes, as occurs after HPV8 early region gene expression (Fig. 2B-D). Wortmannin induced minor reduction in pSerAKT, leading to slightly reduced mature filaggrin levels at the highest wortmannin concentration (Fig. 2C, arrowhead) and more fragile cornified envelopes at high wortmannin levels (Fig. 2D). These data confirm that reduction in AKT activity (pSerAKT levels) can produce the differentiation changes and cornified envelope fragility conferred by HPV8 early genes.
HPV8 E2 gene activity is responsible for the reduction in pSerAKT and AKT1
The K14 HPV8 transgenic mouse has been shown to express E2, E6, and E7 gene products in epidermis (2). To confirm that AKT1 reduction was caused by HPV8 gene products and to find the gene product responsible for AKT1 down-regulation, constitutive expression of individual HPV8 early genes E2, E6, and E7 was analyzed in a human skin equivalent culture model (Fig. 3). Compared with controls, only the E2-transfected culture had reduced levels of pSerAKT and AKT1, in line with the HPV8 transgenic data and showing that the E2 transcription factor regulates AKT1 levels in skin organotypic culture.
Figure 3.

E2 is responsible for the reduction in pSerAKT and AKT1 in skin equivalent cultures expressing HPV8 early genes. Immunohistochemical analysis of the expression of pSerAKT and AKT1 in human keratinocyte skin equivalent cultures expressing the HPV8 early genes E2, E6, and E7 under the retroviral long terminal repeat promoter. Note the reduction in expression of both pSerAKT and AKT1 in the E2-expressing skin equivalent culture compared with the control. pSerAKT and AKT1 are associated with the upper layers as in epidermis in control, E2, and E7 cultures. However, both AKT1 and pSerAKT are expressed in all strata of the E6-expressing skin equivalent culture, which may reflect differentiation changes when this oncoprotein is expressed in all layers under the long terminal repeat promoter. Dotted lines, dermoepidermal boundary. Bar, 20 μm.
It is also noteworthy that, in the E7 cultures, pSerAKT and AKT1 levels were comparable with controls. In contrast, anogenital HPV E7 expression has been reported to up-regulate AKT (4). It is also apparent that, in the E6-transfected culture, the distribution of pSerAKT and AKT1 is altered (Fig. 3), which may be secondary to differentiation changes induced by more widespread expression of oncogenic E6 under the viral long terminal repeat promoter in culture compared with basal E6 expression in the transgenic model.
A novel AKT2-associated pSerAKT activity correlates with tumor progression in the K14-HPV8 mouse
AKT up-regulation has been reported in skin tumors, yet we report cutaneous HPV-mediated AKT down-regulation. Therefore, we examined the status of AKT during tumor progression in the K14-HPV8 mouse (Fig. 4). Significantly, papillomas did not express pSerAKT; however, expression was reinitiated in dysplasia and still apparent in SCC [Fig. 4A, asterisk (dysplasia) and arrowhead (carcinoma)]. Analysis of adjacent tissue sections shows that pSerAKT up-regulation in dysplastic epidermis and carcinoma corresponds to up-regulation of the AKT2 isoform (compare Fig. 4A and B). Indeed, AKT2 first seems up-regulated in papillomas (Fig. 4B), preceding pSerAKT up-regulation (Fig. 4A), consistent with the idea that AKT2 up-regulation precedes its activation by serine phosphorylation. AKT1 is down-regulated in nonlesional transgenic epidermis as expected (Fig. 4C). AKT1 down-regulation is maintained during dysplasia and carcinoma (Fig. 4C, note that the inflammatory dermal infiltrate stains positively for AKT1). Coimmunofluorescence analysis confirms that AKT2 and pSerAKT activity occur in the same cells in carcinoma (Fig. 4D). Taken together, these data show that AKT2 is responsible for the up-regulation of AKT activity in HPV8-induced tumors.
Figure 4.
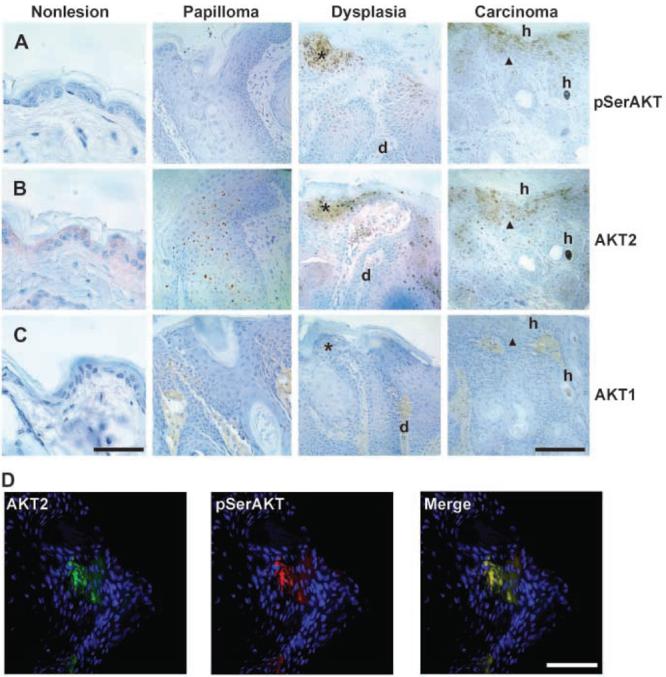
AKT2-associated pSerAKT activity correlates with tumor progression in the K14-HPV8 mouse. A to C, immunohistochemical analysis of pSerAKT (A), AKT2 (B), and AKT1 (C) during tumor progression (papilloma to dysplasia to carcinoma) in the K14-HPV8 mouse. AKT1 is undetectable (C), AKT2 is up-regulated in papilloma, dysplasia, and carcinoma (B), whereas pSerAKT is up-regulated in dysplasia and carcinoma (A). Asterisks and arrowheads point to regions of co–up-regulation of pSerAKT and AKT2 in dysplasia and carcinoma, respectively (A and B). C, in contrast, AKT1 expression was not detected in these regions of dysplasia or carcinoma (asterisk and arrowhead, respectively). d and h, equivalent points in dermis and hair follicle for orientation in adjacent tissue sections. D, coimmunofluorescence showing that the same cells express AKT2 and pSerAKT in SCC tissue from the K14-HPV8 mouse. Bars, 100 μm (A–C) and 50 μm (D).
AKT1 is lost in most human viral warts, whereas up-regulated serine-phosphorylated AKT2 occurs in samples from immunosuppressed patients with poor prognosis
To show that AKT1 down-regulation detected after expression of HPV8 early genes also occurs in a valid disease setting, we examined human cutaneous warts (i.e., productive viral lesions undergoing viral replication and early gene expression) from immunosuppressed (renal transplant recipients) and immunocompetent patients. In normal human scalp epidermis, pSerAKT and AKT1 colocalize in the granular layer as expected and AKT2 is expressed in all strata (Fig. 5A-C; compare with mouse epidermis in Fig. 1A; note that pSerAKT and AKT2 antibodies have reduced sensitivity on archival, formalin-fixed patient samples). pSerAKT and AKT1 are down-regulated (Fig. 5A and B) beginning in the wart margin, and AKT1 usually remains down-regulated within the wart body. pSerAKT-expressing cells can reappear in more dysplastic-like regions of warts (Fig. 5A), consistent with the initiation of pSerAKT in tumors in the K14-HPV8 mouse. Nests of strong AKT2 expression can also reappear (Fig. 5C), most commonly within papillomas from immunosuppressed, renal transplant recipients (Fig. 5C and D). Coimmunofluorescence on serial sections of a dysplastic region of a renal transplant recipient wart was used to show that a subset of the cells that express AKT2 also express pSerAKT and that these AKT2-expressing cells specifically lack detectable AKT1 (Fig. 5D). This is consistent with the proposal that AKT2 up-regulation precedes its phosphorylation and shows that the serine-phosphorylated isoform is predominantly or completely AKT2.
Figure 5.
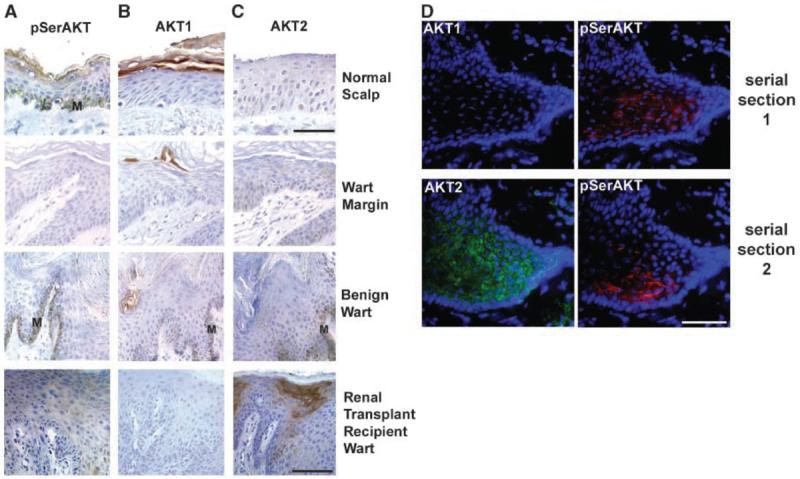
The majority of human viral warts display loss of AKT1 and pSerAKT, whereas a subset of renal transplant recipient warts show co–up-regulation of pSerAKT and AKT2. A to C, representative immunohistochemical analysis of pSerAKT (A), AKT1 (B), and AKT2 (C) in viral warts. Immunocompetent individuals (benign wart), renal transplant recipients, and wart margins were compared with normal scalp epidermis as a control. Although pSerAKT and AKT1 are down-regulated in wart margins and wart bodies compared with the control (A and B), there is focal up-regulation of pSerAKT and AKT2 in a wart from a renal transplant recipient. M, melanin. D, serial sections of a frozen papilloma from a renal transplant recipient. Whereas AKT1 expression was not coincident with pSerAKT expression in the first section, AKT2 was coincident with pSerAKT in the second serial section, although there is clearly much AKT2 that is not activated by phosphorylation (see text). Bars, 50 μm (A–C; normal scalp), 100 μm (A–C; wart margin, benign wart, and renal transplant recipient wart), and 50 μm (D).
Analysis of the frequency of AKT1 loss shows it occurs at a similar frequency in immunosuppressed and immunocompetent patients (Supplementary Table S1), consistent with the proposal that HPVs down-regulate AKT1. AKT2 up-regulation is detected more commonly in warts from immunosuppressed renal transplant recipients (P < 0.05, Fisher's exact test). However, coexpressed pSerAKT and AKT2 are confined to warts from renal transplant recipients (Supplementary Table S1), and the two patients where this occurred both developed skin cancers, one dying from metastatic SCC. These data confirm our previous finding that HPV genes down-regulate epidermal AKT1 and that this down-regulation occurs in human viral warts. Although patient numbers are low, these data also suggest that AKT2 and pSerAKT coexpression could be used as an indicator of poor prognosis. This latter proposal was tested in a range of SCCs.
SCCs have a characteristic AKT profile that changes with malignancy; AKT2 up-regulation is characteristic of SCC, whereas serine-phosphorylated AKT2 correlates with malignancy
To find if the profile of AKT isoforms and activity correlated with tumor type and malignancy, a panel of SCCs, basal cell carcinomas, and malignant melanomas was screened (Fig. 6; Supplementary Fig. S2).
Figure 6.
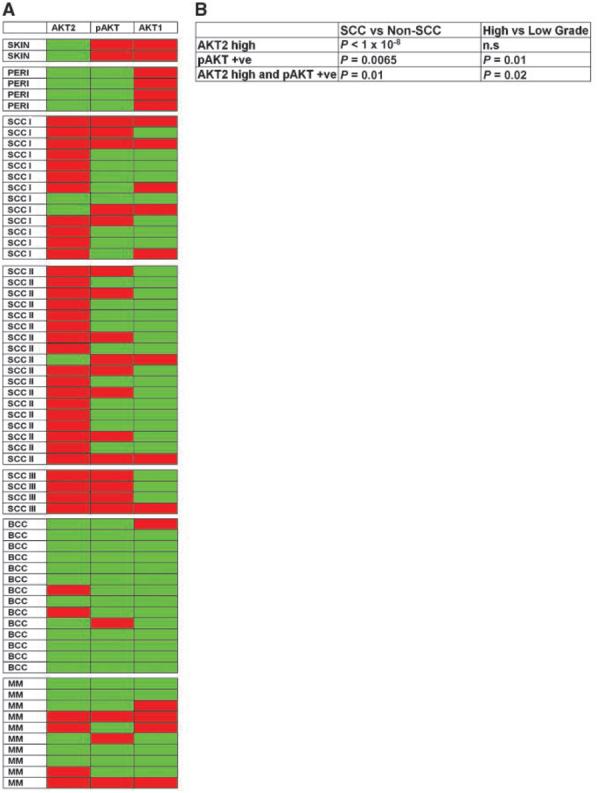
Coexpression of AKT2 and pSerAKT is a common feature of high-grade SCC. A, tissue array comparison of AKT2, pSerAKT (pAKT), and AKT1 expression in normal skin, perilesional skin, low-grade (I and II, differentiated) SCC (SCC I and II), high-grade (III, poorly differentiated) SCC (SCC III), basal cell carcinoma (BCC), and malignant melanoma (MM). All samples were analyzed on serial tissue arrays for AKT2 level (red, up-regulated; green, equivalent to controls) and the presence or absence of pSerAKT or AKT1 (red, present; green, absent). See Supplementary Fig. S2 for representative immunohistochemical outcomes. The analysis shows common up-regulation of AKT2 in SCC, whereas serine phosphorylation of AKT and AKT2 up-regulation correlates with poorly differentiated SCC (SCC III). In contrast, basal cell carcinoma and malignant melanoma lack consistent AKT2 up-regulation and show markedly different expression profiles. B, tests of association of AKT2 up-regulation, pSerAKT positivity, and AKT2 up-regulation/pSerAKT coexpression with SCCs versus non-SCCs and high-grade (SCC III) versus low-grade SCCs (SCC I and II). Significance was tested with Fisher's exact test (n.s, not significant).
AKT1 down-regulation is common in SCCs (Fig. 6A; Supplementary Fig. S2), consistent with its association with HPV activity and the hypothesis that cutaneous HPVs are linked to skin tumorigenesis. The apparent absence of AKT1 in basal cell carcinomas is because tumor nodules and not adjacent stratum corneum are screened (Supplementary Fig. S2).
SCCs show a distinct profile compared with basal cell carcinomas and malignant melanomas (Fig. 6A; Supplementary Fig. S2) with almost all SCCs showing increased AKT2 expression (P < 1 × 10−8, Fisher's exact test; Fig. 6B). This suggests that high focal AKT2 expression could be a SCC marker.
pSerAKT is down-regulated at tumor borders. However, within SCCs, pSerAKT is often focally up-regulated. Coincident high AKT2 and up-regulated pSerAKT occurred in all poorly differentiated SCCs examined (grade III; Fig. 6A; Supplementary Table S1) and can discriminate between better-differentiated grades (grades I and II) and poorly differentiated grade III SCCs (P > 0.02; Fig. 6B). This adds weight to the proposal that up-regulation of Ser473-phosphorylated AKT2 is a marker of malignancy.
Discussion
In this work, we show that cutaneous HPVs, in contrast to anogenital HPVs, down-regulate AKT activity. AKT1 is specifically down-regulated leading to changes in late terminal differentiation and the epidermal cornified envelope. AKT2, which is present in all keratinocytes at low levels, is unaffected. Later in tumor progression, AKT2 is specifically up-regulated and activated by Ser473 phosphorylation in papillomas from immunosuppressed patients, in HPV-induced tumors, and in SCCs. We show that up-regulated AKT2 is characteristic of SCC and that AKT2 activation by serine phosphorylation correlates with tumor grade.
We find that epidermal AKT1 regulates late terminal differentiation and cornified envelope integrity. This is consistent with previous findings that epidermal AKT1 phosphorylates heat shock protein 27, altering its interaction with keratinocyte terminal differentiation proteins, such as filaggrin (18). We speculate that AKT1 activity is targeted by cutaneous HPVs to render the cornified envelope “fragile” to effect escape. Anogenital HPV-associated change to cornified envelopes is well documented and also achieved through changes to late terminal differentiation proteins (20, 21) and, possibly, by expression of the HPV E1∧E4 early gene, which binds keratin and causes keratin collapse (27). However, cutaneous papillomaviruses express an E1∧E4 protein that is unable to induce intermediate filament collapse (28). It is possible that anogenital and cutaneous viruses are using variant strategies to achieve the same end—to modify squames for escape.
Down-regulation of AKT activity by the cutaneous HPV E2 transcription factor shows a fundamental difference between cutaneous and anogenital HPV biology, as anogenital HPV gene expression has been linked to increased AKT activity. High-risk anogenital HPV16 oncoprotein E7 sequesters PP2A subunits, preventing dephosphorylation of AKT and increasing activation (3), although it was recently reported that HPV16 E7 down-regulates Rb protein, causing increased AKT activity, an increase also shown during anogenital viral carcinogenesis (4). The structure of E2 transcription factors differs between anogenital and cutaneous viruses. Cutaneous E2 contains an extended “hinge” domain, absent in anogenital E2 (29), which may underlie the functional difference, although further investigations of molecular differences are required.
The E2 transcription factor regulates HPV E6 and E7 genes as well as a variety of endogenous keratinocyte genes (30). It is not yet determined whether HPV8 E2 directly regulates the AKT1 promoter, although we consider it improbable because E2 is expressed lower in the epidermal strata than AKT (31), suggesting that regulation is indirect.
AKT1 is down-regulated on HPV8 early gene expression and in human viral warts. Because cutaneous HPVs are ubiquitously carried in the outer layers of epidermis (32), it has been difficult to distinguish viral infection from carriage, complicating efforts to relate cutaneous HPV presence to carcinogenesis (33, 34). Therefore, AKT1 status could be used as a marker to distinguish cutaneous HPV infection from carriage.
We show AKT isoform-specific behavior during tumorigenesis; that is, we report AKT1 down-regulation via viral activity and AKT2 up-regulation and activation during tumorigenesis. It has recently been reported that down-regulation of AKT1, but not AKT2, causes invasion and metastasis in breast epithelial cells (10, 35), suggesting that AKT1 can act as a tumor suppressor. On the other hand, it has also been reported that epidermal AKT1 constitutive expression can cause epithelial-mesenchymal transition and increased invasiveness in SCC cell lines (36). It is not known whether HPV down-regulation of AKT1 shown here predisposes to or changes the progression of skin tumorigenesis. However, the AKT1-null mouse does not develop spontaneous skin tumors (8), suggesting that AKT1 is not acting as a primary tumor suppressor in epidermis.
We propose that AKT2 is a marker for SCC and its activation by phosphorylation corresponds with malignancy. Although there are reports of increased AKT1 levels in tumors (9, 37, 38), reports of AKT amplification and the majority of reports of increased AKT gene expression in tumors involve AKT2 [e.g., pancreatic cancer (39, 40), ovarian cancer (41, 42), breast carcinoma (35, 42-44), colorectal cancer (45, 46), and hepatocellular carcinoma (47)]. This has led to the proposal that AKT2 may be the most important member of the AKT family in promoting tumorigenesis (48, 49). Additionally, activation of AKT isoforms has been linked with tumor aggressiveness or poor clinical outcome (refs. 38, 42; reviewed in ref. 48). Hence, our finding that AKT2 is a marker for SCC and its activation by phosphorylation corresponds with malignancy fits well with findings in additional tissues. We propose that in epidermis active AKT2 is a tumor promoter and suggest it as a possible target for therapy.
Acknowledgments
Grant support: Medical Research Council UK grant CEG G9803920 (C. Byrne), Fellowship from St. Bartholomew's and The London Charitable Fund RAB 04/F1 (R.F.L. O'Shaughnessy), Cancer Research UK (C. Byrne, C.A. Harwood, and A. Storey), Center for Molecular Medicine Cologne (H. Pfister), and Mildred-Scheel-Stiftung für Krebsforschung/Deutsche Krebshilfe (B. Akgũl).
We thank Dr. James Cooke and Jon Welti for experimental assistance, Stuart Brown for critical comment, and Dr. Morris Birnbaum and Bob Monks for AKT1-null tissue.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
References
- 1.Akgũl B, Cooke JC, Storey A. HPV-associated skin disease. J Pathol. 2006;208:165–75. doi: 10.1002/path.1893. [DOI] [PubMed] [Google Scholar]
- 2.Schaper ID, Marcuzzi GP, Weissenborn SJ, et al. Development of skin tumors in mice transgenic for early genes of human papillomavirus type 8. Cancer Res. 2005;65:1394–400. doi: 10.1158/0008-5472.CAN-04-3263. [DOI] [PubMed] [Google Scholar]
- 3.Pim D, Massimi P, Dilworth SM, Banks L. Activation of the protein kinase B pathway by the HPV-16 E7 oncoprotein occurs through a mechanism involving interaction with PP2A. Oncogene. 2005;24:7830–8. doi: 10.1038/sj.onc.1208935. [DOI] [PubMed] [Google Scholar]
- 4.Menges CW, Baglia LA, Lapoint R, McCance DJ. Human papillomavirus type 16 E7 up-regulates AKT activity through the retinoblastoma protein. Cancer Res. 2006;66:5555–9. doi: 10.1158/0008-5472.CAN-06-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Woodgett JR. Recent advances in the protein kinase B signaling pathway. Curr Opin Cell Biol. 2005;17:150–7. doi: 10.1016/j.ceb.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 6.Peng XD, Xu PZ, Chen ML, et al. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev. 2003;17:1352–65. doi: 10.1101/gad.1089403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cho H, Mu J, Kim JK, et al. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKBβ) Science. 2001;292:1728–31. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- 8.Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ. Akt1/PKBα is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem. 2001;276:38349–52. doi: 10.1074/jbc.C100462200. [DOI] [PubMed] [Google Scholar]
- 9.Heron-Milhavet L, Franckhauser C, Rana V, et al. Only Akt1 is required for proliferation, while Akt2 promotes cell cycle exit through p21 binding. Mol Cell Biol. 2006;26:8267–80. doi: 10.1128/MCB.00201-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Irie HY, Pearline RV, Grueneberg D, et al. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. J Cell Biol. 2005;171:1023–34. doi: 10.1083/jcb.200505087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Altomare DA, Tanno S, De Rienzo A, et al. Frequent activation of AKT2 kinase in human pancreatic carcinomas. J Cell Biochem. 2003;88:470–6. doi: 10.1002/jcb.10287. [DOI] [PubMed] [Google Scholar]
- 12.Toker A, Yoeli-Lerner M. Akt signaling and cancer: surviving but not moving on. Cancer Res. 2006;66:3963–6. doi: 10.1158/0008-5472.CAN-06-0743. [DOI] [PubMed] [Google Scholar]
- 13.Segrelles C, Ruiz S, Perez P, et al. Functional roles of Akt signaling in mouse skin tumorigenesis. Oncogene. 2002;21:53–64. doi: 10.1038/sj.onc.1205032. [DOI] [PubMed] [Google Scholar]
- 14.Mao JH, To MD, Perez-Losada J, Wu D, Del Rosario R, Balmain A. Mutually exclusive mutations of the Pten and ras pathways in skin tumor progression. Genes Dev. 2004;18:1800–5. doi: 10.1101/gad.1213804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thrash BR, Menges CW, Pierce RH, McCance DJ. AKT1 provides an essential survival signal required for differentiation and stratification of primary human keratinocytes. J Biol Chem. 2006;281:12155–62. doi: 10.1074/jbc.M512116200. [DOI] [PubMed] [Google Scholar]
- 16.Janes SM, Ofstad TA, Campbell DH, Watt FM, Prowse DM. Transient activation of FOXN1 in keratinocytes induces a transcriptional programme that promotes terminal differentiation: contrasting roles of FOXN1 and Akt. J Cell Sci. 2004;117:4157–68. doi: 10.1242/jcs.01302. [DOI] [PubMed] [Google Scholar]
- 17.Calautti E, Li J, Saoncella S, Brissette JL, Goetinck PF. Phosphoinositide 3-kinase signaling to Akt promotes keratinocyte differentiation versus death. J Biol Chem. 2005;280:32856–65. doi: 10.1074/jbc.M506119200. [DOI] [PubMed] [Google Scholar]
- 18.O'Shaughnessy RF, Welti JC, Cooke JC, et al. AKT-dependent HspB1 (Hsp27) activity in epidermal development. J Biol Chem. 2007;282:17297–305. doi: 10.1074/jbc.M610386200. [DOI] [PubMed] [Google Scholar]
- 19.Candi E, Schmidt R, Melino G. The cornified envelope: a model of cell death in the skin. Nat Rev Mol Cell Biol. 2005;6:328–40. doi: 10.1038/nrm1619. [DOI] [PubMed] [Google Scholar]
- 20.Lehr E, Jarnik M, Brown DR. Human papillomavirus type 11 alters the transcription and expression of loricrin, the major cell envelope protein. Virology. 2002;298:240–7. doi: 10.1006/viro.2002.1445. [DOI] [PubMed] [Google Scholar]
- 21.Lehr E, Brown DR. Infection with the oncogenic human papillomavirus type 59 alters protein components of the cornified cell envelope. Virology. 2003;309:53–60. doi: 10.1016/s0042-6822(02)00100-9. [DOI] [PubMed] [Google Scholar]
- 22.Komuves LG, Hanley K, Jiang Y, Elias PM, Williams ML, Feingold KR. Ligands and activators of nuclear hormone receptors regulate epidermal differentiation during fetal rat skin development. J Invest Dermatol. 1998;111:429–33. doi: 10.1046/j.1523-1747.1998.00296.x. [DOI] [PubMed] [Google Scholar]
- 23.Akgũl B, Garcia-Escudero R, Ghali L, et al. The E7 protein of cutaneous human papillomavirus type 8 causes invasion of human keratinocytes into the dermis in organotypic cultures of skin. Cancer Res. 2005;65:2216–23. doi: 10.1158/0008-5472.CAN-04-1952. [DOI] [PubMed] [Google Scholar]
- 24.Leverrier S, Bergamaschi D, Ghali L, et al. Role of HPV E6 proteins in preventing UVB-induced release of pro-apoptotic factors from the mitochondria. Apoptosis. 2007;12:549–60. doi: 10.1007/s10495-006-0004-1. [DOI] [PubMed] [Google Scholar]
- 25.Zhang H, Zha X, Tan Y, et al. Phosphoprotein analysis using antibodies broadly reactive against phosphorylated motifs. J Biol Chem. 2002;277:39379–87. doi: 10.1074/jbc.M206399200. [DOI] [PubMed] [Google Scholar]
- 26.Hewett DR, Simons AL, Mangan NE, et al. Lethal, neonatal ichthyosis with increased proteolytic processing of filaggrin in a mouse model of Netherton syndrome. Hum Mol Genet. 2005;14:335–46. doi: 10.1093/hmg/ddi030. [DOI] [PubMed] [Google Scholar]
- 27.Wang Q, Griffin H, Southern S, et al. Functional analysis of the human papillomavirus type 16 E1∧E4 protein provides a mechanism for in vivo and in vitro keratin filament reorganization. J Virol. 2004;78:821–33. doi: 10.1128/JVI.78.2.821-833.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roberts S, Ashmole I, Johnson GD, Kreider JW, Gallimore PH. Cutaneous and mucosal human papillomavirus E4 proteins form intermediate filament-like structures in epithelial cells. Virology. 1993;197:176–87. doi: 10.1006/viro.1993.1578. [DOI] [PubMed] [Google Scholar]
- 29.Steger G, Schnabel C, Schmidt HM. The hinge region of the human papillomavirus type 8 E2 protein activates the human p21(WAF1/CIP1) promoter via interaction with Sp1. J Gen Virol. 2002;83:503–10. doi: 10.1099/0022-1317-83-3-503. [DOI] [PubMed] [Google Scholar]
- 30.Hadaschik D, Hinterkeuser K, Oldak M, Pfister HJ, Smola-Hess S. The papillomavirus E2 protein binds to and synergizes with C/EBP factors involved in keratinocyte differentiation. J Virol. 2003;77:5253–65. doi: 10.1128/JVI.77.9.5253-5265.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haller K, Stubenrauch F, Pfister H. Differentiation-dependent transcription of the epidermodysplasia verruciformis-associated human papillomavirus type 5 in benign lesions. Virology. 1995;214:245–55. doi: 10.1006/viro.1995.0028. [DOI] [PubMed] [Google Scholar]
- 32.Forslund O, Lindelof B, Hradil E, et al. High prevalence of cutaneous human papillomavirus DNA on the top of skin tumors but not in “stripped” biopsies from the same tumors. J Invest Dermatol. 2004;123:388–94. doi: 10.1111/j.0022-202X.2004.23205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Elder JT. Warts and skin cancer: only skin deep? J Invest Dermatol. 2004;123i:vi. [Google Scholar]
- 34.Orth G. Human papillomaviruses and the skin: more to be learned. J Invest Dermatol. 2004;123:XI–XIII. doi: 10.1111/j.0022-202X.2004.23243.x. [DOI] [PubMed] [Google Scholar]
- 35.Yoeli-Lerner M, Yiu GK, Rabinovitz I, Erhardt P, Jauliac S, Toker A. Akt blocks breast cancer cell motility and invasion through the transcription factor NFAT. Mol Cell. 2005;20:539–50. doi: 10.1016/j.molcel.2005.10.033. [DOI] [PubMed] [Google Scholar]
- 36.Grille SJ, Bellacosa A, Upson J, et al. The protein kinase Akt induces epithelial mesenchymal transition and promotes enhanced motility and invasiveness of squamous cell carcinoma lines. Cancer Res. 2003;63:2172–8. [PubMed] [Google Scholar]
- 37.Staal SP. Molecular cloning of the akt oncogene and its human homologues AKT1 and AKT2: amplification of AKT1 in a primary human gastric adenocarcinoma. Proc Natl Acad Sci U S A. 1987;84:5034–7. doi: 10.1073/pnas.84.14.5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun M, Wang G, Paciga JE, et al. AKT1/PKBα kinase is frequently elevated in human cancers and its constitutive activation is required for oncogenic transformation in NIH3T3 cells. Am J Pathol. 2001;159:431–7. doi: 10.1016/s0002-9440(10)61714-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miwa W, Yasuda J, Murakami Y, et al. Isolation of DNA sequences amplified at chromosome 19q13.1-q13.2 including the AKT2 locus in human pancreatic cancer. Biochem Biophys Res Commun. 1996;225:968–74. doi: 10.1006/bbrc.1996.1280. [DOI] [PubMed] [Google Scholar]
- 40.Ruggeri BA, Huang L, Wood M, Cheng JQ, Testa JR. Amplification and overexpression of the AKT2 oncogene in a subset of human pancreatic ductal adenocarcinomas. Mol Carcinog. 1998;21:81–6. [PubMed] [Google Scholar]
- 41.Yuan ZQ, Sun M, Feldman RI, et al. Frequent activation of AKT2 and induction of apoptosis by inhibition of phosphoinositide-3-OH kinase/Akt pathway in human ovarian cancer. Oncogene. 2000;19:2324–30. doi: 10.1038/sj.onc.1203598. [DOI] [PubMed] [Google Scholar]
- 42.Bellacosa A, de Feo D, Godwin AK, et al. Molecular alterations of the AKT2 oncogene in ovarian and breast carcinomas. Int J Cancer. 1995;64:280–5. doi: 10.1002/ijc.2910640412. [DOI] [PubMed] [Google Scholar]
- 43.Bacus SS, Altomare DA, Lyass L, et al. AKT2 is frequently upregulated in HER-2/neu-positive breast cancers and may contribute to tumor aggressiveness by enhancing cell survival. Oncogene. 2002;21:3532–40. doi: 10.1038/sj.onc.1205438. [DOI] [PubMed] [Google Scholar]
- 44.Cheng GZ, Chan J, Wang Q, Zhang W, Sun CD, Wang LH. Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Res. 2007;67:1979–87. doi: 10.1158/0008-5472.CAN-06-1479. [DOI] [PubMed] [Google Scholar]
- 45.Roy HK, Olusola BF, Clemens DL, et al. AKT protooncogene overexpression is an early event during sporadic colon carcinogenesis. Carcinogenesis. 2002;23:201–5. doi: 10.1093/carcin/23.1.201. [DOI] [PubMed] [Google Scholar]
- 46.Parsons DW, Wang TL, Samuels Y, et al. Colorectal cancer: mutations in a signalling pathway. Nature. 2005;436:792. doi: 10.1038/436792a. [DOI] [PubMed] [Google Scholar]
- 47.Xu X, Sakon M, Nagano H, et al. Akt2 expression correlates with prognosis of human hepatocellular carcinoma. Oncol Rep. 2004;11:25–32. [PubMed] [Google Scholar]
- 48.Altomare DA, Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene. 2005;24:7455–64. doi: 10.1038/sj.onc.1209085. [DOI] [PubMed] [Google Scholar]
- 49.Cheng JQ, Lindsley CW, Cheng GZ, Yang H, Nicosia SV. The Akt/PKB pathway: molecular target for cancer drug discovery. Oncogene. 2005;24:7482–92. doi: 10.1038/sj.onc.1209088. [DOI] [PubMed] [Google Scholar]