

Abstract
Nanoparticles of ~10 nm in diameter made with chitosan or lactic acid-grafted chitosan were developed for high drug loading and prolonged drug release. A drug encapsulation efficiency of 92% and a release rate of 28% from chitosan nanoparticles over a 4-week period were demonstrated with bovine serum protein. To further increase drug encapsulation, prolong drug release, and increase chitosan solubility in solution of neutral pH, chitosan was modified with lactic acid by grafting D,L-lactic acid onto amino groups in chitosan without using a catalyst. The lactic acid-grafted chitosan nanoparticles demonstrated a drug encapsulation efficiency of 96% and a protein release rate of 15% over 4 weeks. With increased protein concentration, the drug encapsulation efficiency decreased and drug release rate increased. Unlike chitosan, which is generally soluble only in acid solution, the chitosan modified with lactic acid can be prepared from solutions of neutral pH, offering an additional advantage of allowing proteins or drugs to be uniformly incorporated in the matrix structure with minimal or no denaturization.
Keywords: chitosan, lactic acid-g-chitosan, drug delivery, nanoparticles
Introduction
A number of polymeric nanoparticles have been synthesized and studied in the past few years as promising drug delivery systems to improve delivery efficiency and reduce side-effects of drug toxicity (Uhrich et al 1999; Ranney 2000; Soppimath et al 2001). Nanoscale drug systems can circumvent the rapid recognition by the immune system and deliver drugs to cells with high efficiency compared with microparticle-based system (LaVan et al 2003). Among those investigated, chitosan-based materials have drawn considerable attention in view of chitosan’s excellent biocompatibility, biodegradability, and reactive surface functional groups for easy surface modification (Dodane and Vilivadam 1998; Paul and Sharma. 2000; Janes et al 2001; Drury and Mooney 2003). The positively charged amino groups of chitosan tend to adhere to the negatively charged cell surfaces, facilitating the penetration of chitosan nanoparticles across the cell membrane (Artursson et al 1994; Borchard et al 1996; Janes et al 2001).
Chitosan–drug nanoparticles have been prepared by a number of methods as reviewed by Janes et al (2001). 5-fluorouracil drug was incorporated in chitosan nanoparticles by chemical crosslinking with glutaraldehyde, targeted at anticancer drug delivery (Ohya et al 1994). Alternative methods were sought after the discovery of the negative effect of glutaraldehyde crosslinking on cell viability and the degraded integrity of the incorporated drug. Bodmeier et al (1989) and Calvo et al (1997) used an ionic gelation method to prepare chitosan particles with sizes ranging from micron to submicron. In this method, an anionic cross-linking agent, sodium tripolyphosphate (TPP), was introduced into an aqueous solution of chitosan in acetic acid (Calvo et al 1997; Majeti 2000; Janes et al 2001; Kumar et al 2004; Zhang et al 2004). The major advantage of this method is the ease in manipulation of particle size by changing pH values. Berthold et al (1996), Calvo et al (1997), and Janes et al (2001) synthesized chitosan nanoparticles using desolvating agents (eg, sodium sulfate) through the interaction of chitosan with sulfate. Though many advances have been made on the synthesis of chitosan nanoparticles and their use in short-term (eg, weeks) drug delivery (Calvo et al 1997; Janes and Alonso 2003; Xu and Du 2003; Zhang et al 2004), long-term (eg, months) drug release by nanoparticles remains a major challenge.
In the present study, chitosan-based nanoparticles with a high degree of size uniformity were prepared by grafting D,L-lactic acid on chitosan to serve as a drug carrier for prolonged drug release. The lactic acid-grafted chitosan (LA-g-chitosan) was prepared by dehydrating the solvent cast thin film of chitosan containing lactic acids. The LA-g-chitosan nanoparticles were fabricated via a co-precipitation process by LA-g-chitosan in ammonium hydroxide to form coacervate drops. The structure of nanoparticles was investigated by transmission electron microscopy (TEM). The chemical structure and bonding were studied by nuclear magnetic resonance (NMR) and Fourier transformed infrared (FTIR) spectroscopy. Bovine serum albumin (BSA) was used as a model protein to examine the drug absorption and release characteristics of both chitosan and LA-g-chitosan nanoparticles in phosphate buffer saline (PBS) at pH 7.4.
Experimental procedure
Materials
Chitosan from crab shells with 88% of deacetylation (weight average molar mass MW 190 kDa and Brookfield viscosity 200–800 cps in 1% solution with 1% acetic acid), BSA (MW 68 kDa), and D,L-lactic acid (90% aqueous solution) were purchased from Sigma-Aldrich (St Louis, MO, USA). Coomassie Blue G 250 was purchased from Bio-Rad Laboratories (Hercules, CA, USA).
Preparation of LA-g-chitosan copolymer
To prepare LA-g-chitosan copolymer, chitosan powder was first mixed with an aqueous solution of lactic acid, and the mixture was stirred overnight using a magnet stirrer to create a final solution at a chitosan concentration of 2 wt.%. The solution was placed in polystyrene petri dishes and maintained at 70°C for 5 hours for film formation. The as-produced film (~0.08 mm in thickness) was heated at 80–90°C under high vacuum for 5 hours, and the grafted copolymer was formed as a result of the dehydration of the chitosan lactate salts and the formation of the corresponding amide linkages. The unreacted lactic acid and oligo(lactic acid) (OLLA) were removed from the grafted chitosan polymer using a Soxhlet apparatus by washing with chloroform and methanol solvents for 48 hours each.
Characterization of LA-g-chitosan copolymer
NMR spectroscopy and polarized Fourier transformed infrared (FTIR) spectroscopy were used to characterize the chemical structure and bonding characteristics of LAg-chitosan copolymer, respectively. For NMR, samples of 10–20 mg each were prepared by dissolving LA-g-chitosan in 0.7 mL of D2O containing 0.5 M DCl/D2O. 1H-NMR spectra were acquired with a Bruker AV-301 spectrometer.
For FTIR spectroscopy, a dried 5-mg sample was mixed with 300 mg dry KBr and pressed into a pellet using a macro KBr die kit. The solid pellet was placed in a magnetic holder and the system was purged with air before testing. FTIR spectra of 200 scans at 4 cm−1 resolution were acquired using a Nicolet 5DX spectrometer equipped with a deuterated triglycine sulfate (DTGS) detector and a solid transmission sample compartment. Spectrum analysis and display were performed using standard Nicolet and Microcal Origin software.
The ninhydrin assay was used to determine the percentile decrease of free amino groups in LA-g-chitosan versus pure chitosan, which was defined as the percentage of free amino groups in chitosan that had reacted with lactic acid. Both 0.1 mg/mL solutions of chitosan and LA-g-chitosan were prepared in a mixture of CH3COOH (3% w/v) and HCl (1% w/v) under constant stirring at 20°C for 24 hours. Acetate buffer (0.5 mL, 4 M, pH 5.5) was then added to the resulting polymer solutions (0.1–0.5 mL, corresponding to 10–50 μg of chitosan or LA-g-chitosan). All the sample solutions were prepared in 5-mL glass tubes capped with a rubber septum. Ninhydrin reagent (2 mL, Sigma) was added to the sample solutions prepared above, and the tubes containing solution mixtures were maintained in a boiling water bath for 30 minutes. The solutions were then cooled down to room temperature, and UV absorbance of the solutions was acquired at 570 nm. A standard absorbance curve was generated using D-glucosamine (Sigma) solutions (100% free amino groups).
Preparation of chitosan and LA-g-chitosan nanoparticles
Chitosan nanoparticles were prepared by dissolving chitosan powder in 0.2 M acetic acid to produce a 2 wt.% solution, followed by addition of the solution in ammonium hydroxide solution at pH 8.5–9.0. A constant solution flow rate of 0.1 mL/min was maintained using a mini-pump. As the acetic chitosan solution was introduced into the basic solution, the opalescent suspension was formed under vigorous magnetic stirring at room temperature. The particles were separated from the solution by centrifugation. After separation, the particles were washed with deionized water until a pH of 7.4 was reached. LA-g-chitosan nanoparticles were prepared by dissolving LA-g-chitosan film in 0.2 M acetic solution and then following the same procedure as for chitosan nanoparticles.
BSA absorption on nanoparticles
BSA absorption on nanoparticles was carried out in PBS at pH 7.4 at room temperature. 200 mg of nanoparticles were dispersed in 6 mL of PBS solution under sonication, and BSA was added at varying concentrations of 0, 1.0, 2.0, 3.0, 4.0, or 5.0 mg/mL. The nanoparticle suspension was then left for 24 hours in a rotating agitator. After protein adsorption, the nanoparticles were separated by centrifugation, and the amount of free protein in the supernatant was measured. The supernatant in 96-well plates was analyzed using a modified Coomassie blue protein assay and by UV spectroscopy at 590 nm. A calibration curve was generated at each time interval using nanoparticles without BSA loaded to correct for the intrinsic absorbance of chitosan. The BSA encapsulation efficiency (AE) of the process was calculated as follows:
where T is the total amount of BSA added in PBS and F is the amount of free BSA left in supernatant after the nanoparticles were removed from the solution. For each BSA concentration, the samples were examined in triplicate, and results were presented as mean value ± standard deviation.
Morphology and structure characterization of nanoparticles
The morphology and size distribution of nanoparticles were examined by TEM (CM 100 TEM) at an accelerating voltage of 40 kV. Samples were prepared by depositing a drop of chitosan nanoparticle suspension on a copper grid fitted with a carbon support film and dried under vacuum.
In vitro release of BSA from nanoparticles
BSA loaded nanoparticles were placed in a test tube containing 6 mL of PBS at pH 7.4 and incubated at 37°C for 24 hours. At specified time intervals, 1 mL out of 6 mL solution was removed to a siliconized 1.5-mL microcentrifuge tube, and the medium in the test tube was replenished with 1 mL of fresh PBS. The amount of BSA released from the nanoparticles was evaluated by the modified Coomassie blue protein assay method as noted above. All release tests were run in triplicate, and the results were reported as mean value ± standard deviation.
Results and discussion
Characterization of LA-g-chitosan polymer
Two types of LA-g-chitosan copolymer films were obtained depending on the amount of lactic acid used. Copolymer films obtained with a lactic acid/chitosan mole ratio lower than 1.5 (Table 1) were transparent and readily soluble in solutions with pH up to 7.5, while those with a lactic acid/chitosan ratio higher than 1.5 were light brown and yielded swollen hydrogels under the same condition. Compositions and other material properties of these LA-g-chitosan copolymers are shown in Table 1. During the dehydrating process, the formation of amide and polycondensation of lactic acid took place at the same time. The unreacted lactic acid and oligomeric lactic acid in the reaction mixture were washed out as impurities using excess chloroform and methanol.
Table 1.
LA-g-chitosan copolymers with different compositions
| No. | Mole ratio (acid/amine group)a | % degree of substitution or percentage of decrease of amino groupsb | Water solubility |
|---|---|---|---|
| Chitosan | – | 13.2c | Soluble in pH <6 |
| C1 | 1.5 | 35.8 | Soluble in pH <7.5 |
| C2 | 2.9 | – | Slightly soluble at pH 7.5 |
| C3 | 5.9 | – | Insoluble at pH 7.5 |
| C4 | 11.6 | – | Insoluble at pH 7.5 |
Different ratios of reactants were maintained by adding different amounts of lactic acid in a fixed amount of chitosan (0.5 g). Degree of substitution was not calculated for the LA-g-chitosan with a higher fraction of lactic acid because these samples were not soluble in water.
Estimated by ninhydrin assay.
Degree of substitution in unmodified chitosan, ie, 13.2 corresponds to the degree of deacetylation.
Figure 1 shows FTIR spectra of chitosan and LA-g-chitosan with two different ratios of lactic acid to chitosan. The peaks at 1655 and 1325 cm−1 in the IR spectrum of chitosan (a) correspond to amide I and amide III, respectively. The peak at 1585 cm−1 is the free amino band of chitosan. The two peaks at 902 and 1157 cm−1 are the result of the saccharide structure of chitosan. The peak at 1377 cm−1 is the characteristic band of CH3 symmetrical deformation mode. The LA-g-chitosan spectrum (b) has a broad band around 1591 cm−1, owing to the overlapping of the peaks from the free amino band of chitosan and the amide that couples chitosan and lactic acid oligomers. The peak at 1736 cm−1 is attributed to carbonyl of ester or carboxylic groups on the lactic acid side chains. When LA-g-chitosan was prepared with a high content of lactic acid (c), the broad band around 1591 cm−1 was absent and multiple peaks appeared corresponding to the N-acetylated and free amino groups of chitosan. The increased intensity of the amide I peak (1655 cm−1) indicated an increase in amidation resulting from the reaction of chitosan with increased amount of lactic acid. The peak of the amino groups shifted slightly from 1591 to 1598 cm−1, while the intensity of the corresponding peak of the carbonyl group at 1740 cm−1 increased. Compared with the reported IR spectrum of lactic acid and their oligomers (Kister et al 1998), the well-resolved peaks at 1452 and 1315 cm−1 were attributed to -CH3 and -CH groups in LA-g-chitosan.
Figure 1.
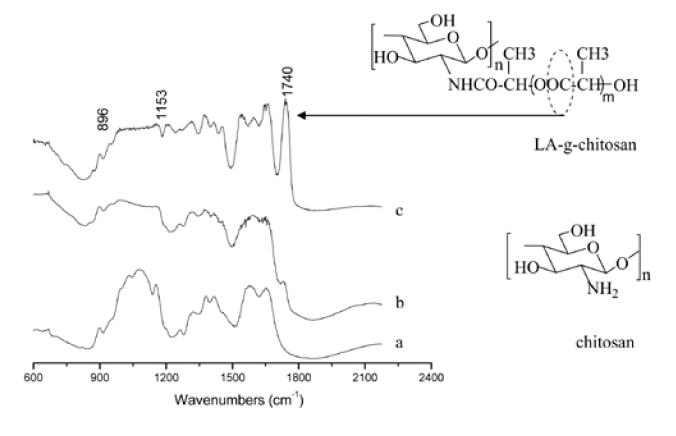
Infrared spectra of (a) chitosan, (b) LA-g-chitosan with acid/amine ratio=1.5 (sample C1, Table 1), and (c) LA-g-chitosan with acid/amine ratio=11.6 (sample C4, Table 1).
Figure 2 shows 1H-NMR spectra of chitosan and LAg-chitosan. The characteristic peaks from LA-g-chitosan associated with chitosan and lactic acid segment were assigned as follows according to the data reported in the literature on pure chitosan and lactide oligomer (Rashkov et al 1996; Schwach et al 1997; Sashiwa et al 2003). Chemical shifts for chitosan: δ4.9–5.2 (br, H-1), 3.45–3.90 (br, H-3, H-4, H-5, and H-6), 2.95 (0.85H, br s, H-2), 1.85 ppm (br s, NHAc). Chemical shifts for LA-g-chitosan: δ 5–5.15 (br, H-1 of GlcN), 4.90–5.00 (q, -CH of lactyl unit), 4.25 (q, -CH of hydroxylated lactyl unit), 3.50–4.0 (m, H-3, H-4, H-5, and H-6), 2.90–3.10 (br s, H-2), 2.05 ppm (br s, NHAc), 1.40–1.50 (d, -CH3 of lactyl units), 1.30–1.40 (d, -CH3 of hydroxylated lactyl units). NMR spectra shown in Figure 2 were taken at 20°C. The appearance of a new H-2 proton signal at 3.22 ppm from LA-g-chitosan corresponding to N-alkylation of chitosan (Kumar et al 2004) confirms the bonding between chitosan and lactic acid. Since two peaks corresponding to lactyl units connected to the chitosan and the hydroxylated lactyl units are well separated, the 1H-NMR spectra can be used to measure the degree of polymerization of LA (number of lactyl units) side chain grafted onto the chitosan. For example, the number of lactyl units for sample C1 shown in Table 1 was evaluated to be 1.52. When the number of methyl proton in hydroxylated lactyl unit is set to 3, the number of lactyl units multiplied by the repeat unit weight of 72 g mole−1 yields the length of LA side chain 110 g mole−1.
Figure 2.
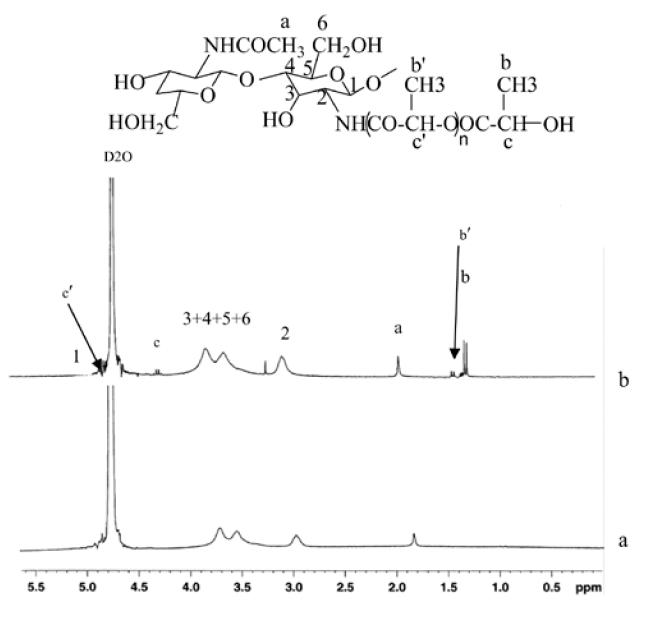
1H-NMR (nuclear magnetic resonance) spectra of (a) chitosan and (b) LA-g-chitosan (sample C1)
Characterization of nanoparticles
Figure 3 shows an exemplary TEM image of LA-g-chitosan nanoparticles at two magnifications. TEM images of pure chitosan nanoparticles were not presented since they appeared similar to the LA-g-chitosan nanoparticles. From TEM images, both chitosan and LA-g-chitosan nanoparticles were spherical and have an average diameter of ~10 nm and a fairly uniform size distribution. Advantages of using small-size particles as a carrier system include high cellular uptake, good suspensibility, and easy penetration into arterial walls (Labhasetwar et al 1997). Furthermore, the particles with smaller sizes have larger surface area to volume ratios and thus may have a high drug-loading capacity and a slow drug-diffusion rate.
Figure 3.

Transmission electron micrograph of LA-g-chitosan nanoparticles.
Protein encapsulation and interaction with chitosan
Drug AE of chitosan and LA-g-chitosan nanoparticles in BSA solutions of different concentrations was evaluated following the procedure outlined above. The AEs of both chitosan and LA-g-chitosan nanoparticles in BSA solution with an initial BSA concentration of 1 mg/mL were measured to be 92% and 96%, respectively. The high AEs of chitosan and LA-g-chitosan may be attributed to the small size of particles that have a high surface to volume ratio and high electrostatic interaction between the negatively charged moieties on BSA and the positively charged amine groups on chitosan. The fact that LA-g-chitosan nanoparticles had a higher AE than chitosan nanoparticles may be attributed to additional hydrophobic interactions of the LA-g-chitosan particles with BSA. In the present work, no linker molecules were used to bind BSA with the polymer and thus only the adsorption owing to the electrostatic attractions between BSA proteins and nanoparticles and the direct entrapment of protein into the polymer matrix would contribute to the drug loading. This helps retain protein integrity and biofunctionality. Our preliminary results on polyacrylamide gel electrophoresis showed that the integrity of the BSA released from the nanoparticles was retained throughout the release period (data not shown).
Protein release study
Figure 4 shows the percent cumulative release rate of BSA from chitosan and LA-g-chitosan nanoparticles loaded with BSA at different concentrations, over a 4-week period. For both types of nanoparticles, the protein release rates increased with increased BSA loading concentrations. The release of BSA from nanoparticles is mainly driven by the protein concentration gradient. The encapsulation of high concentration produced a greater concentration gradient between the polymer and release medium leading to a higher diffusion rate (Miyazaki et al 1988; Janes et al 2001; Xu and Du 2003). Consequently, these results show that it is possible to modulate the protein release rate by adjusting the initial protein concentration. All the drug-release profiles exhibit an initial burst release, presumably from the particle surface, followed by a sustained release driven by diffusion of the protein through the polymer wall and polymer erosion. The release profiles of chitosan and LA-g-chitosan particles are similar, but the BSA release rate from chitosan nanoparticles is significantly higher than from LA-g-chitosan nanoparticles. For example, the burst releases from chitosan and LA-g-chitosan loaded with BSA at a concentration of 1 mg/mL were about 15% and 10%, respectively. The BSA releases from chitosan and LA-g-chitosan over a 4-week period at BSA concentration of 1 mg/mL were 28% and 15%, respectively. The sustained BSA releases from both chitosan and LA-g-chitosan nanoparticles might be attributed to strong intermolecular interactions, including hydrogen bonding and dipole–dipole interactions between chitosan and BSA molecules. Previous studies (Miyazaki et al 1988; Janes et al 2001; Xu and Du 2003) suggested that the mechanism of association of proteins with chitosan is, at least, partially mediated by the ionic interaction between chitosan and protein macromolecules. It is known that lactic acid oligomers are hydrophobic, and introduction of this hydrophobic moiety to chitosan could substantially alter the physicochemical properties of the chitosan nanoparticles for BSA absorption. Thus, the slower drug release from LAg-chitosan than chitosan might be attributed to additional intermolecular forces, including hydrophobic interactions and hydrogen bonding. More in-depth research is needed to reveal and confirm the underlying mechanism and their roles in association of BSA with LA-g-chitosan.
Figure 4.

Bovine serum albumin (BSA) release profiles of (a) chitosan and (b) LA-g-chitosan nanoparticles at different BSA loading concentrations. Data shown are the mean ± standard deviation (n=3).
Summary
Spherical and uniformly dispersed chitosan and lactic acid-modified chitosan (LA-g-chitosan) nanoparticles with a mean diameter of ~10 nm were prepared. Albumin encapsulation efficiency as high as 92% and 96% was attained for chitosan and LA-g-chitosan nanoparticles, respectively. The BSA release profiles of both chitosan and LA-g-chitosan nanoparticles exhibit an initial burst release, followed by a sustained quasi-linear release. The chitosan nanoparticles have a protein release rate of 28% over a 4-week period and predicated complete protein release up to 3 months, while LA-g-chitosan nanoparticles had a protein release rate of 15% at the same protein concentration over a 4-week period and predicated complete release up to 6 months. By incorporating the lactyl segment into the chitosan backbone, the resulting nanoparticle reduces the burst release, but the release pattern was similar to that of the pure chitosan nanoparticles. Since most proteins and cell membranes are negatively charged, these nanoparticles are also expected to be potential vehicles to associate more easily with other proteins and subsequently internalized by the target cells than negatively charged nanoparticles.
References
- Artursson P, Lindmark T, Davis S, et al. Pharm Res. Vol. 11. 1994. Effect of chitosan on the permeability of monolayers of intestinal epithelial-cells (Caco-2) pp. 1358–61. [DOI] [PubMed] [Google Scholar]
- Berthold A, Cremer K, Kreuter J. J Control Release. Vol. 39. 1996. Preparation and characterization of chitosan microspheres as drug carrier for prednisolone sodium phosphate as model for antiinflammatory drugs; pp. 17–25. [Google Scholar]
- Bodmeier R, Chen HG, Paeratakul O. Pharm Res. Vol. 6. 1989. A novel approach to the oral delivery of micro and nanoparticles; pp. 413–7. [DOI] [PubMed] [Google Scholar]
- Borchard G, Luessen HL, de Boer AG, et al. J Control Release. Vol. 39. 1996. The potential of mucoadhesive polymers in enhancing intestinal peptide drug absorption.3. Effects of chitosan-glutamate and carbomer on epithelial tight junctions in vitro; pp. 131–8. [Google Scholar]
- Calvo P, RemunanLopez C, VilaJato JL, et al. J Appl Polm Sci. Vol. 63. 1997. Novel hydrophilic chitosan-polyethylene oxide nanoparticles as protein carriers; pp. 125–32. [Google Scholar]
- Dodane V, Vilivalam VD. Pharm Sci Technol Today. Vol. 1. 1998. Pharmaceutical applications of chitosan; pp. 246–53. [Google Scholar]
- Drury JL, Mooney DJ. Biomaterials. Vol. 24. 2003. Hydrogels for tissue engineering:scaffold design variables and applications; pp. 4337–51. [DOI] [PubMed] [Google Scholar]
- Janes KA, Alonso MJ. J Appl Polym Sci. Vol. 88. 2003. Depolymerized chitosan nanoparticles for protein delivery:Preparation and characterization; pp. 2769–76. [Google Scholar]
- Janes KA, Calvo P, Alonso MJ, et al. Adv Drug Deliv Rev. Vol. 47. 2001. Polysaccharide colloidal particles as delivery systems for macromolecules; pp. 83–97. [DOI] [PubMed] [Google Scholar]
- Janes PC, Alonso MJ. Adv Drug Deliv Rev. Vol. 47. 2001. Polysaccharide colloidal particles as delivery systems for macromolecules; pp. 83–97. [DOI] [PubMed] [Google Scholar]
- Kister G, Cassanas G, Vert M, et al. Polymer. Vol. 39. 1998. Effects of morphology, conformation and configuration on the IR and Raman spectra of various poly(lactic acid)s; pp. 267–73. [Google Scholar]
- Kumar MNVR, Muzzarelli RAA, Muzzarelli C, et al. Chem Rev. Vol. 104. 2004. Chitosan chemistry and pharmaceutical perspectives; pp. 6017–84. [DOI] [PubMed] [Google Scholar]
- LaVan DA, McGuire T, Langer R. Nature Biotechnol. Vol. 21. 2003. Small scale systems for in vivo drug delivery; pp. 1184–91. [DOI] [PubMed] [Google Scholar]
- Labhasetwar V, Song C, Levy RJ, et al. Adv Drug Deliv Rev. Vol. 24. 1997. Nanoparticle drug delivery system for restenosis; pp. 63–85. [Google Scholar]
- Majeti NV, RK . React Funct Polym. Vol. 46. 2000. A review of chitin and chitosan applications; pp. 1–27. [Google Scholar]
- Miyazaki S, Yamaguchi H, Yokouchi C, et al. J Pharm Pharmacol. Vol. 40. 1988. Sustained-release of indomethacin from chitosan granules in beagle dogs; pp. 642–3. [DOI] [PubMed] [Google Scholar]
- Ohya Y, Shiratani M, Kobayashi H, et al. J Macromol Sci Pure Appl Chem. A31. 1994. Release behavior of 5-fluorouracil from chitosan-gel nanospheres immobilizing 5-fluorouracil coated with polysaccharides and their cell-specific cytotoxicity; pp. 629–42. [Google Scholar]
- Paul W, Sharma CP. Stp Pharma Sci. Vol. 10. 2000. Chitosan, a drug carrier for the 21st century: a review; pp. 5–22. [Google Scholar]
- Ranney DF. Biochem Pharmacol. Vol. 59. 2000. Biomimetic transport and rational drug delivery; pp. 105–14. [DOI] [PubMed] [Google Scholar]
- Rashkov I, Manolova N, Li SM, et al. Macromolecules. Vol. 29. 1996. Synthesis, characterization, and hydrolytic degradation of PLA/PEO/PLA triblock copolymers with short poly(L-lactic acid) chains; pp. 50–6. [Google Scholar]
- Sashiwa H, Yajima H, Aiba S, et al. Biomacromolecules. Vol. 4. 2003. Synthesis of a chitosan-dendrimer hybrid and its biodegradation; pp. 1244–9. [DOI] [PubMed] [Google Scholar]
- Schwach G, Coudane J, Engel R, et al. J Macromol Sci Pure Appl Chem. Vol. 35. 1997. More about the polymerization of lactides in the presence of stannous octoate; pp. 3431–40. [Google Scholar]
- Soppimath KS, Aminabhavi TM, Kulkarni AR, et al. J Control Release. Vol. 70. 2001. Biodegradable polymeric nanoparticles as drug delivery devices; pp. 1–20. [DOI] [PubMed] [Google Scholar]
- Uhrich KE, Cannizzaro SM, Langer RS, et al. Chem Rev. Vol. 99. 1999. Polymeric systems for controlled drug release; pp. 3181–98. [DOI] [PubMed] [Google Scholar]
- Xu Y, Du Y. Int J Pharm. Vol. 250. 2003. Effect of molecular structure of chitosan on protein delivery properties of chitosan nanoparticles; pp. 215–26. [DOI] [PubMed] [Google Scholar]
- Zhang H, Oh M, Allen C, et al. Biomacromolecules. Vol. 5. 2004. Monodisperse chitosan nanoparticles for mucosal drug delivery; pp. 2461–8. [DOI] [PubMed] [Google Scholar]