

Abstract
This work combines expertise in stem cell biology and bioengineering to define the system for geometric control of proliferation and differentiation of myogenic progenitor cells. We have created an artificial niche of myogenic progenitor cells, namely, modified extracellular matrix (ECM) substrates with spatially embedded growth or differentiation factors (GF, DF) that predictably direct muscle cell fate in a geometric pattern. Embedded GF and DF signal progenitor cells from specifically defined areas on the ECM successfully competed against culture media for myogenic cell fate determination at a clearly defined boundary. Differentiation of myoblasts into myotubes is induced in growth-promoting medium, myotube formation is delayed in differentiation-promoting medium, and myogenic cells, at different stages of proliferation and differentiation, can be induced to coexist adjacently in identical culture media. This method can be used to identify molecular interactions between cells in different stages of myogenic differentiation, which are likely to be important determinants of tissue repair. The designed ECM niches can be further developed into a vehicle for transplantation of myogenic progenitor cells maintaining their regenerative potential. Additionally, this work may also serve as a general model to engineer synthetic cellular niches to harness the regenerative potential of organ stem cells.
Keywords: satellite cell, myoblast, myotube, ECM, myogenic cell fate
Introduction
Skeletal muscle is maintained and repaired by endogenous stem cells, called satellite cells, which constitute all the regenerative potential in this organ (Zammit et al 2002; Sherwood et al 2004). Satellite cells reside in direct contact with the differentiated, multinucleated muscle cells (myofibers or myotubes), under the basal lamina. Two to five percent of all muscle nuclei are in satellite cells in the adult muscle (Morgan and Partridge 2003). In resting adult muscle, 99.9% of satellite cells are mitotically quiescent until muscle injury activates satellite cells to proliferate and differentiate along myogenic lineage into the myogenic progenitor cells, which progress to become fusion-competent myoblasts (Morgan and Partridge 2003).
Myoblasts are still capable of division but can also fuse to form new multinucleated myofibers. This coordinated cell-fate determination, which consists of cell expansion followed by differentiation, serves to repair or replace the damaged muscle (Morgan and Partridge 2003).
Muscle regeneration is a complex process of tissue remodeling that involves myogenesis, formation of new neuro-muscular junctions, and re-vascularization and is regulated by an intricate network of biochemical pathways, including those initiated by inflammatory cytokines, growth factors, integrins, and the evolutionarily conserved Notch, Wnt, and Shh signaling pathways (Husmann et al 1996; Yang et al 1996; Taverna et al 1998; Conboy and Rando 2002; Pola et al 2003; Seale et al 2003; Tidball 2005). Injury promotes the release of growth factors that bind to extracellular matrix (ECM) proteins, such as proteoheparan sulfates (Husmann et al 1996). During later stages of regeneration, interactions between the remodeled ECM and cell-surface integrin receptors play a key role in the adhesion and spreading of newly generated myoblasts, and thus the organization of the regenerated muscle fibers (Disatnik and Rando 1999; Disatnik et al 2002; Zaidel-Bar et al 2004). Among the best-characterized growth factors, the most important in muscle repair are FGF-2, IGF-1, TGF-β, and GDF-8 (myostatin). FGF-2 promotes the proliferation of myogenic progenitor cells and delays their differentiation, in part by inhibiting the expression of myogenic regulatory factors (Maley et al 1994; Miller et al 2000). IGF-1 promotes myogenic differentiation (Florini et al 1996) and is a key determinant of muscle mass, as it enhances protein synthesis in differentiated myofibers (Bodine et al 2001; Latres et al 2005). At the same time, IGF-1 down-regulates protein degradation in muscle cells (Sandri et al 2004; Stitt et al. 2004) and has anti-apoptotic effects (Lawlor and Rotwein 2000; Downward 2004). Also, IGF-1 reduces age-related muscle atrophy and attenuates experimentally induced muscle wasting (Chakravarthy et al 2001; Shavlakadze et al 2005). TGF-β and the muscle-specific TGF-β family member, myostatin, are on the opposite end of the proliferative spectrum. These factors inhibit proliferation of myogenic progenitor cells during both embryonic development and adult muscle regeneration (McPherron et al 1997; Thomas et al 2000; Moustakas et al 2002; Zimmers et al 2002; McCroskery et al 2003). Myostatin mRNA has been shown in vivo to accumulate progressively during muscle repair (Armand et al 2003), while the mRNA levels of its inhibitor, follistatin, has been shown to be present in the mono-nucleated muscle cells located near the injury site and in newly formed myofibers (Armand et al 2003). This well-studied interplay of growth factors in regenerating muscle serves to restore cellular homeostasis during injury repair (Husmann et al 1996).
In stark contrast to young animals, aged organisms produce very few myoblasts in response to muscle injury, and thus not enough cells are available to form new myofibers (Schultz and Lipton 1982; Bockhold et al 1998; Conboy et al 2003). Decline in the generation of myoblasts in aged muscle has been proven not to be caused by a physical loss of satellite cells related to aging (Conboy et al 2003), but rather by a failure in their ability to become activated and proliferate in response to injury. Remarkably, the intrinsic satellite cell regenerative potential is not irreversibly lost with age, but rather is simply not triggered in old muscle due to extrinsic systemic factors (Conboy et al 2005).
It is of great interest to the scientific community to be able to control regeneration in chronically degenerating or aged organs either by in situ activation of endogenous stem cells or by stem cell transplantation. Satellite cells have often been viewed as a promising source of regenerative reserves in transplantation studies. These cells are numerous in adults, readily available, and relatively easily harvested; they rapidly expand in culture and their progeny myogenic progenitor cells also proliferate and are able to differentiate into new muscle tissue in vivo and in vitro (Morgan and Partridge 2003). However, despite the decades of attempts using electroporation and other techniques, there is no known cell transplantation-based cure for repair of aged or pathologically degenerating muscle (Partridge 2004). Notably, taking into account the dependence of the satellite cells’ regenerative potential on the extrinsic environment described above, the ability of transplanted cells to repair muscle efficiently is likely to be inhibited in the aged environment of a degenerating organ, even if the transplantation itself were successful.
Therefore, several key requirements appear to be necessary for improving both the success of transplantation and the regenerative outcome. We hypothesized that creating an optimized micro-environment for myogenic progenitor cells would provide these requirements by allowing deliberate control of their expansion and differentiation, thereby overcoming the negative effects of the endogenous physiologic niche.
This study focuses on creating biologically active artificial niches for myogenic progenitor cells – ECM substrates containing spatially embedded growth factors (GF) and differentiation factors (DF) – which predictably direct muscle cell fate in a defined geometric pattern. The very first steps for a successful design of such an artificial stem cell niche are to test specific combinations of GF and DF that are able to direct myogenic cell fate in vitro, as well as to create a reliable method of spatial response of myogenic progenitor cells adherent to ECM modified with GF and DF. These goals have been accomplished in the present work.
The results presented here firstly demonstrate that it is possible to create modified ECM adhesion substrates with embedded GF or DF in a geometrically defined area. Secondly, such modified ECM substrates override external conditions imposed by culture media and promote specific cell fate determination of myogenic progenitor cells. Finally, our data directly show that cells with different cell fates determined by their exposure to the geometrically embedded GF or DF can coexist adjacent to each other on the same plate sharing identical culture media.
Methods
Animal strain and primary myoblast cultures
C57BL/6 mice were obtained from Jackson Laboratories and housed at UC Berkeley Animal Care Facility. Both muscle injury and acquisition of muscle progenitor cells from myofiber fragments were performed as previously published (Conboy and Rando 2002). Briefly, 3 days after muscle injury, hind leg muscle was dissected and dissociated into myofibers, which were cultured overnight, during which time activated satellite cells gave rise to colonies of myogenic progenitor cells. These cells, called primary myoblasts, were then expanded and used in this work. Myofibers as well as myoblasts were cultured on ECM-coated plates (1:500 ECM-phosphate buffer solution [PBS]) in growth-promoting medium (GM).
Reagents
Antibodies to bromodeoxyuridine (BrdU) and to embryonic myosin heavy chain (eMHC), and nuclear stain Hoechst were obtained from Abcam Inc. (Cambridge, MA, USA), Vector Laboratories (Burlingame, CA, USA), and Sigma (St Louis, MO, USA), respectively. Secondary antibodies were obtained from Molecular Probes (Eugene, OR, USA). TGF-β, GDF-8, b-FGF, α-TGF-β, and IGF-1 were all obtained from R&D Labs (Minneapolis, MN, USA). Follistatin was obtained from Sigma (St Louis, MO, USA). Ham’s F10, DMEM medium, and p/s were obtained from Mediatech Inc. (Herndon, VA, USA) and OptiMEM medium and FBS were from Invitrogen Corp. (Carlsbad, CA, USA). Horse serum (HS) was also obtained from Mediatech Inc. (Herndon, VA, USA). PBS was obtained from Fisher Scientific (Fairlawn, NJ, USA) and ECM gel from Engelbreth Holm-Swarm (EHS) mouse sarcoma from Sigma (St Louis, MO, USA). This ECM gel contains collagens, non-collagenous glycoproteins, and proteoglycans. Precisely, its major component is laminin, and it also contains collagen type IV, heparan sulfate proteoglycan, entactin, and other minor components.
Slides and cylinders
Two- and 4-chamber culture slides were obtained from BD Biosciences (Bedford, MA, USA) and cloning cylinders were obtained from VWR International (West Chester, PA, USA).
Slide preparation
Two-chamber slides were pre-coated with ECM gel 1 day prior to experiments. To create a separate environment, cloning cylinders were placed into the middle of each chamber during pre-coating. The pre-coated slide with a cylinder was allowed to congeal overnight at room temperature. A seal between the interior and the exterior of the cloning cylinder was formed during the gelation of the ECM as the cylinder penetrated the ECM due to gravity. This seal is vital in preventing exchange of GF or DF between the interior and the exterior environment of the cylinder. To facilitate uniform cell adhesion and proliferation, the seal must also be such that it does not damage the ECM underneath it when the cylinder is removed prior to cellular seeding. To achieve these parameters, different concentrations of ECM were tested and a concentration of 40 μg/ml was finally selected.
For control experiments, 4-chamber slides were used and pre-coated in similar fashion without the cylinders.
Growth and differentiation factor preparation and placement
GF: b-FGF (0.05 μg/ml), follistatin (0.5 μg/ml), and α-TGF-β (10 μg/ml); and DF: GDF-8 (0.1 μg/ml), IGF-1 (0.5 μg/ml), and TGF-β (0.02 μg/ml) were prepared separately in a ECM–PBS solution (12 μg/ml) and placed inside different cylinders of each chamber. In order to maintain the seal formed, pressure equilibrium between the outside and inside environments of the cylinders was maintained while adding factors. This allows us to confine the factors to a specific area on the slide determined by the geometry and size of the inner diameter of the cloning cylinders. Different cloning cylinders were also tested and those with the best surface finishing of the cross section (Scienceware cloning cylinders from VWR International) produced the best seal and thus the best geometric boundary.
This preparation was kept overnight (~24 hours) at 4°C to allow integration of factors into the ECM layer.
Media preparation and cell placement
GM consisted of Ham’s F10 + 20% FBS + FGF-2 (5 ng/ml) + 1% p/s, differentiation-promoting medium (DM) consisted of DMEM + 2% HS + 1% p/s, and neutral medium (NM) consisted of OptiMEM + 5% FBS + 1% p/s. Prior to seeding cells into each chamber, all residual liquid inside chambers and cylinders was aspirated and afterwards cloning cylinders were removed from the chambers, leaving behind areas of modified ECM with embedded GF and DF. Myoblasts were re-suspended into desired media (GM, DM, or NM), seeded under each experimental condition into corresponding chambers, and cultured for 48 hours.
Immunofluorescence analysis
In order to measure cell proliferation or differentiation by immunofluorescence as previously described (Conboy and Rando 2002), myoblasts were fixed with 70% EtOH in PBS after 36 or 48 hours of specific culture conditions. BrdU was added to cultured media 2 hours prior to cell fixation in order to label replicating cells. After fixing cells, they were washed with staining buffer (PBS + 1% FBS + 0.5% Na azide) and permeabilized in staining buffer containing 0.25% Triton X-100. Afterwards, cells were incubated with antibodies specific for both proliferation (BrdU) and differentiation (eMHC) for 1 hour at room temperature. Hoechst stain was added during secondary antibody incubation. For BrdU detection, cells were incubated with 4M HCl at room temperature prior to permeabilization, to denature DNA. α-eMHC was used at 1:25 hybridoma supernatant dilution and α-BrdU at 2.5 μg/ml. Secondary antibodies and Hoechst were used at 1:500 hybridoma supernatant dilution.
Quantification and statistics
Cells were counted from triplicate experiments with at least 300 cells per experiment. Cells expressing BrdU proliferation marker were counted as proliferating cells. Cells expressing eMHC and containing 2 or less nuclei per fiber were counted as early-differentiating cells D1 and those with more than 2 nuclei per fiber were D2. Statistical significance confidence intervals were analyzed with p-value test (Anova: Single Factor) and error bars.
Results
The main goal of this study was to direct myogenic progenitor cells to either proliferation or differentiation when these cells coexist on the same dish but are exposed to different areas of their adhesion substrate. We have used as the adhesion substrate ECM gels with geometrically embedded factors that differ from each other in their effects on cell fate (see references in Table 1). In this way, cells are subjected to the same culture conditions imposed by the media but to different culture conditions imposed by the adhesion substrates. Specifically, we set out to promote differentiation of myogenic progenitor cells into myotubes in GM, growth of progenitor cells in DM, and more efficient proliferation or robust differentiation in NM.
Table 1.
Growth- and differentiation-promoting factors (GF, DF) used (see Methods section for more detailed information about these reagents; references about their effects on myogenesis are given here)
| Factor | Effects on myogenesis | GF combinations | DF combinations | References |
|---|---|---|---|---|
| FGF-2 | Enhanced proliferation of myogenic progenitor cells | + | (Maley et al 1994; Miller et al 2000) | |
| GDF-8(myostatin) | Inhibition of proliferation of myogenic progenitor cells | + | (McPherron et al 1997; Thomas et al 2000; Zimmers et al 2002; McCroskery et al 2003) | |
| Follistatin | Inhibition of myostatin | + | (Armand et al 2003) | |
| IGF-1 | Enhanced differentiation and increased myofiber size–mass | + | (Florini et al 1996; Lawlor and Rotwein 2000; Bodine et al 2001; Heszele and Price 2004; Sandri et al 2004; Shavlakadze et al 2005) | |
| TGF-β | Inhibitor of cell cycle progression, promotes differentiation and wound healing | + | (Derynck and Zhang 2003; Jakubowiak et al. 2000; Massague and Chen 2000; Massague et al 2000) | |
| α-TGF-β | Neutralization of TGF-β activity | + |
To do this, ECM gels have been modified with a combination of GF and DF which had been previously shown to direct myogenic proliferation or differentiation when added to culture media (Husmann et al 1996; Conboy and Rando 2002; Pola et al 2003; Tidball 2005; Wagers and Conboy 2005). The factors used in this study and their main effects on myogenesis have been summarized in Table 1.
Primary myogenic progenitor cell cultures have been generated from satellite cells activated by muscle injury, as previously described (Conboy and Rando 2002; Wagers and Conboy 2005). These myogenic cells or primary myoblasts have been cultured in 3 specific media conditions: GM, DM, and NM (see Methods section for media composition details).
We embedded geometrically combinations of GF and DF (Table 1) into ECM gel, which is frequently used as a substrate for cell growth in vitro and is similar to physiological substrate of myogenic progenitor cells in vivo. We aimed to examine whether these factors would be able to override the cell fate imposed by the aforementioned media and, at the same time, whether a clearly defined boundary between cells with alternative myogenic cell fates could be created by their adhesion to modified ECM substrates which contain either GF or DF.
To achieve these goals, we have developed the experimental technique described in detail in the Methods section and summarized in Scheme 1. Briefly, ECM gels were divided into different geometric areas using cloning cylinders and mixtures of either GF or DF were placed inside them, creating modified areas of ECM which contained factors, vs the unmodified ECM areas. Afterwards cells were uniformly seeded onto the whole ECM substrate, so that after approximately 1 hour cells adhered to both unmodified and modified ECM areas and shared the same media. Adherence of cells to unmodified and modified ECM was simultaneous and there was no difference in cell survival (data not shown). Experiments with GF or DF uniformly embedded into the whole ECM area have also been carried out as positive controls.
Scheme 1.
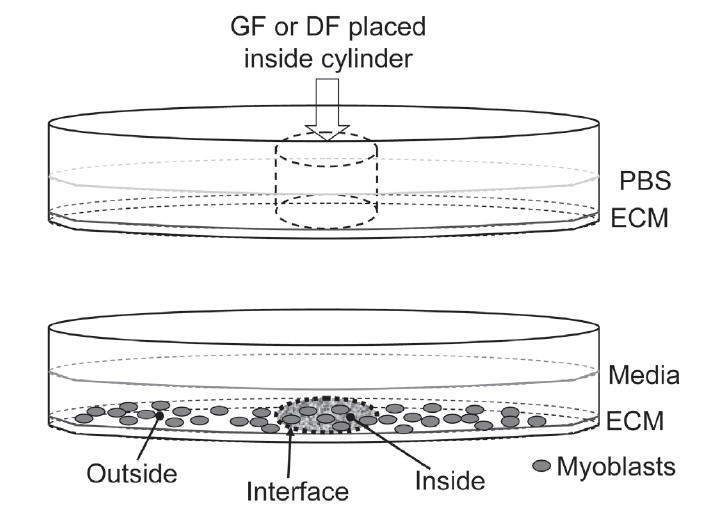
1. ECM:PBS coating (1:100) + cylinder placement. Overnight @RT
2. GF/DF placement inside cylinder. Overnight @4°C
3. Removal of cylinders
4. Media and cells placement
5. Addition of BrdU 2 hours prior to fixation
6. Cells fixing after 48 hours
7. Indirect immunofluorescence (BrdU for proliferation, eMHC for differentiation, nuclear dye Hoechst)
Manipulating cell fate in GM
Firstly, we have examined the behavior of cells in GM without any factors embedded in ECM. Consistent with previously published results (Morgan and Partridge 2003; Conboy and Rando 2005), cells rapidly proliferate and do not differentiate in GM, ie, they incorporate BrdU and only less than 1% express the marker of differentiated myotubes, eMHC (Figure 1a-GM, quantified in Figure 1b-GM). In contrast, embedded DF in ECM successfully promote myogenic differentiation of primary myoblasts (Figure 1a-GM + DF), as shown by their reduced proliferation and enhanced expression of eMHC. Interestingly, such directed differentiation occurs even in the presence of highly mitogenic GM, which contains 20% FBS and FGF-2 (Figure 1a-GM + DF, quantified in Figure 1b-GM + DF). Figure 1b demonstrates quantification of multiple experiments and statistically shows that the effects caused by DF-modified ECM significantly promote the differentiation of myogenic cells exposed to mitogenic media. Specifically, cells attached to areas of DF-modified ECM show higher expression of eMHC+ (number of cells at early stage of differentiation, D1, significantly rises from 0.5% to 11.8%: p = 0.003). In addition, even though cells exposed to DF-modified ECM continue to proliferate, the rate was slower (Figure 1b: Number of proliferating cells P drops from 20.9% in GM to 9.6% in GM + DF; p = 0.077) and a higher percentage of these cells expresses the differentiation marker, eMHC (Figure 1b-GM + DF: D1 = 11.8%). Thus, these findings reveal that it is possible to force the differentiation of cells under proliferative media conditions through DF-modified ECM substrates.
Figure 1.
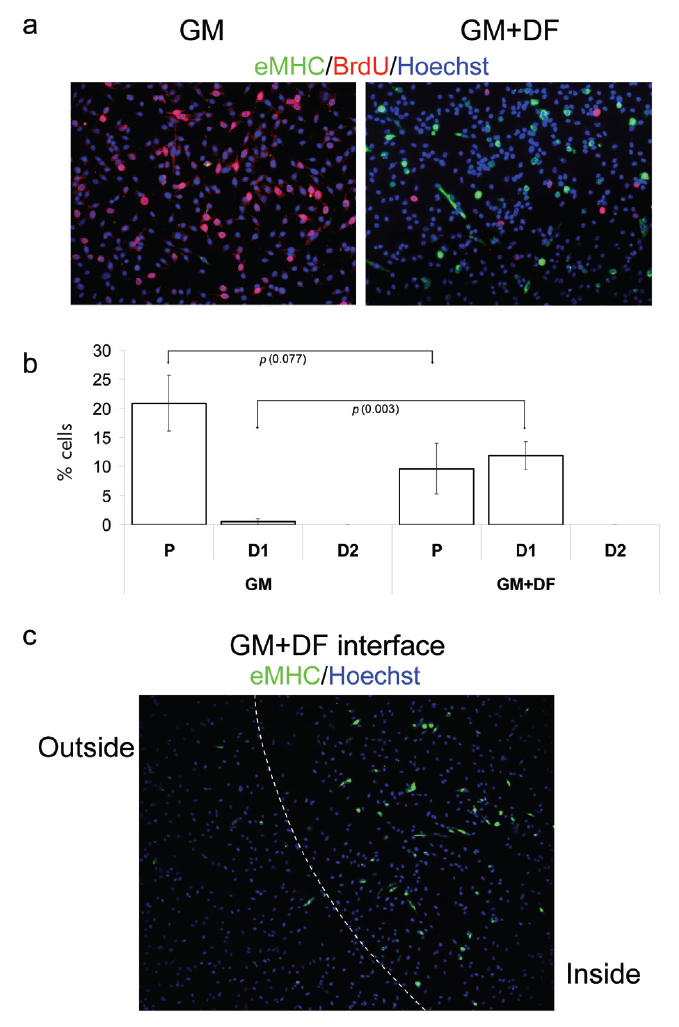
Geometric control of terminal myogenic differentiation in GM. Myogenic progenitor cells have been plated at 50% confluency in chamber slides in GM.
(a) GM with unmodified ECM (GM) and GM with DF-modified ECM (GM + DF). (b) Quantification of P, differentiated cells with less than 2 nuclei (D1 = early stage of differentiation), and differentiated cells with more than 2 nuclei (D2 = later stage of differentiation). On unmodified ECM, there is a significantly higher percentage of proliferative cells vs differentiated cells. Looking at cells grown on DF-modified ECM, we see higher numbers of differentiated cells, but most cells do not form multinucleated myotubes. (c) The boundary between unmodified ECM substrate (outside) and DF-modified ECM (inside). Geometric boundary between DF-modified and unmodified ECM substrate was created as described in Methods. Cells were uniformly seeded throughout the ECM area, cultured for 48 hours and fixed. Immunofluorescence was performed with the indicated antibodies: α-BrdU (red), α-eMHC (green), and Hoechst (blue) was used to label all nuclei. Proliferation (incorporation of BrdU) is observed in GM with unmodified ECM and inhibited proliferation and differentiation (expression of eMHC) is observed in GM with DF-modified ECM. Similar results have been obtained in at least 3 independent experiments. Magnification: (a) 20x; (c) 10x.
Abbreviations for Figures 1-3, Scheme 1, SOM 1: D, differentiated cells; DF, differentiation factors; DM, differentiation-promoting medium; ECM, extracellular matrix; GM, growth-promoting medium; GF, growth factors; NM, neutral medium; P, proliferating cells; PBS, phosphate buffered saline.
Moreover, we achieved geometric control of cell fate determination, as clearly shown in Figure 1c, where an obvious interface between eMHC+ and eMHC− myogenic progenitor cells cultured under identical media conditions was created by exposing these cells to the different regions of ECM substrate (with vs without DF).
Manipulating cell fate in DM
To confirm and extrapolate these findings, we tested whether the reciprocal cell fate determination could also be achieved within our experimental system. Specifically, we cultured cells in DM with unmodified vs GF-modified ECM. Unsurprisingly, fusion competent myoblasts terminally differentiate and form eMHC+ myotubes when cultured in DM (Conboy and Rando 2002; Morgan and Partridge 2003; Conboy and Rando 2005) (Figure 2a-DM, quantified in Figure 2b-DM). In contrast, Figure 2a-DM + GF shows that much fewer eMHC+ myotubes are formed in the area where cells are exposed to GF, compared with the area devoid of GF. Additionally, a significant fraction of cells incorporate BrdU (Figure 2a-DM + GF), thus overcoming the effect imposed by highly differentiating media when attached to GF-modified ECM substrates.
Figure 2.
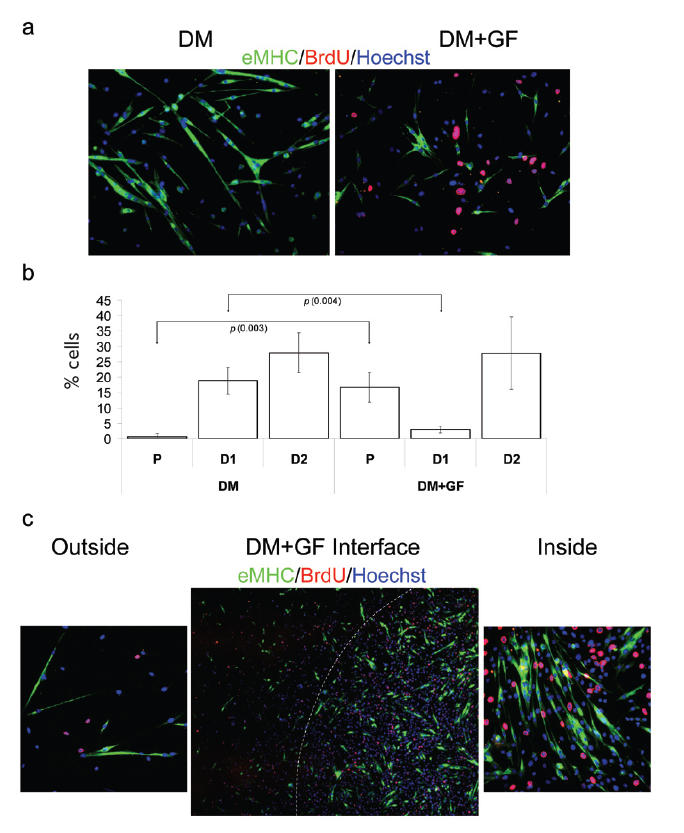
Geometric delay of myotube formation in DM by locally embedded growth factors. Myogenic progenitor cells have been plated at 50% confluency in chamber slides in DM. Cells were cultured for 48 hours and fixed. Immunofluorescence was performed with the indicated antibodies: α-BrdU (red), α-eMHC (green), and Hoechst (blue) was used to label all nuclei.
(a) DM with unmodified ECM (DM) and DM with GF-modified ECM (DM+GF). There is a clear difference in the fate of cells cultured in DM on unmodified ECM (terminally differentiated, multinucleated myotubes) vs on GF-modified ECM (higher numbers of proliferative cells and smaller myotubes). (b) Quantification of P, early stage-differentiated cells with less than 2 nuclei (D1) and later stage-differentiated cells with more than 2 nuclei (D2). Cells cultured on unmodified ECM in DM show low percentage of proliferating cells and high percentage of differentiated cells. Alternately, when cultured on GF-modified ECM, cells show higher percentage of proliferating cells and lower numbers of differentiated cells. (c) The boundary between unmodified ECM substrate and DF-modified ECM is shown (DM + GF interface). Magnified photographs (20x) of cells seeded on unmodified ECM (outside) and on GF-modified ECM (inside) areas of the culture plate are also shown. Cells were originally seeded at uniform confluency; however, as expected, cells adherent to the GF-modified ECM proliferated at a higher rate, resulting in a higher number of cells compared with those adherent to control ECM. Similar results have been obtained in at least 3 independent experiments. Magnification: (a) 20x; (c) “Outside”, “Inside” 20x; (c) “Interface” 10x.
Both the robustness and reproducibility of the aforementioned regulation of cell fate by GF-signaling from specific areas of ECM were confirmed by the quantification of at least 3 replicated experiments, as illustrated in Figure 2b-DM + DF. Proliferation (P) dramatically increases from 0.6% in DM to 16.7% in DM + GF (p = 0.003) and the number of cells at early stage of differentiation significantly drops from 18.9% to 2.9% (p = 0.004).
Notably, similar to the data shown in Figure 1c, a clearly defined interface was created between the region of modified ECM, which contained embedded GF, and the unmodified control ECM area, thus allowing cells with different fates to coexist in the same culture medium (Figure 2c). This interface is discernable not only because some cells incorporate BrdU and some instead form myotubes, but also because of different cell densities. Specifically, there are approximately 4 times more cells in the area of ECM embedded with GF. Since cells were uniformly seeded on the plate at uniform density, the higher number of cells can only be attributed to a predictably higher rate of cell proliferation in regions of GF-modified ECM.
It is well known that plating myoblasts at high density will lead to exit of cell cycle and differentiation, even in the presence of GM (Conboy and Rando 2002; Morgan and Partridge 2003). Thus, we decided to test whether we can inhibit differentiation under that specific condition. Even under high cell density (80% confluency), myogenic differentiation is delayed by GF-modified ECM, although not completely avoided (Supplemental Online Material, SOM 1). Remarkably, when plated at high density, cells attached to GF-modified ECM area show higher levels of both proliferation and differentiation (SOM 1). Therefore, as cell numbers increase, differentiation seems inescapable, despite initial placement of GF into ECM substrate.
SOM 1.
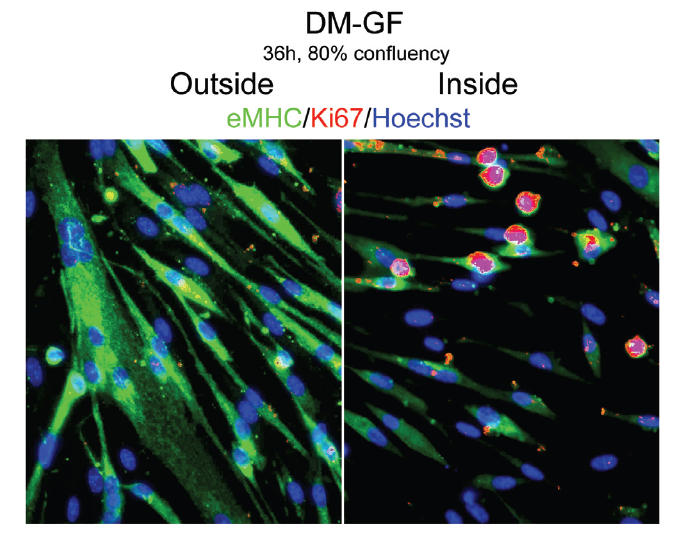
Delay of myogenic differentiation by locally embedded growth factors into ECM under high cell density (80% confluency). Myogenic progenitor cells have been plated at 80% confluency in chamber slides in DM for 36 hours. Immunofluorescence was performed after fixation with the indicated antibodies: α-Ki67 (red), α-eMHC (green), and Hoechst (blue) was used to label all nuclei. As specified in Table 1, GF were embedded into ECM in geometric fashion, as shown in Scheme 1. Consistent with the control DM shown in Figure 2a-DM, cells outside the geometric boundary (outside) form eMHC-positive robust myotubes and do not proliferate. Myogenic cell differentiation is diminished, as indicated by the lower number of nuclei per myotubes, and some Ki67+ proliferating cells persist inside the geometric boundary (inside). Thus, GF embedded in the ECM are capable of diminishing differentiation even when cell numbers increase, but differentiation seems inescapable despite initial placement of GF into ECM. Magnification: 10x.
Manipulating cell fate in NM
After characterizing the effect of modified ECM substrates on cell fate in strongly differentiating or mitogenic media, we tested our GF- and DF-modified ECM substrates in NM conditions. As expected, in NM condition myoblasts slowly proliferate and infrequently produce eMHC+ terminally differentiated cells, which usually have no more than 1 or 2 nuclei (Figure 3a-NM and quantified in Figure 3b-NM: fraction of proliferation cells P = 9.8% and cells at early stage of differentiation D1 = 9.1%). This verifies that NM does not impose any strong determination of cell fate, thus potentially allowing a more discernable effect of our selected factors on the myogenic proliferation and differentiation. It has not yet been determined whether the physiological environment in regenerating muscle is either mitogenic (proliferating), or differentiating or both–neutral. Since proliferation and differentiation overlap during tissue repair, neutral media conditions might mimic the environment of regenerating muscle more accurately than GM or DM alone. As shown in Figure 3a-NM + GF and quantified in Figure 3b-NM + GF, cells cultured on modified ECM with embedded GF robustly proliferate (fraction of proliferating cells P = 32.2%) and do not significantly differentiate (fraction of cells at early stage of differentiation D1 = 2.3%). This is confirmed by the robust incorporation of BrdU and absence of eMHC staining. In parallel, myoblasts attached to modified ECM containing embedded DF efficiently differentiate and lack proliferation (Figure 3a, NM + DF, quantified in Figure 3b, NM + DF; fraction of proliferating cells P = 5.2%; fraction of cells at early stage of differentiation D1 = 13.7%; fraction of cells at late stage of differentiation D2 = 12.4%). As above, quantification of at least 3 replicated experiments demonstrated high reproducibility of this geometric regulation of myogenic cell fate determination (Figure 3b).
Figure 3.
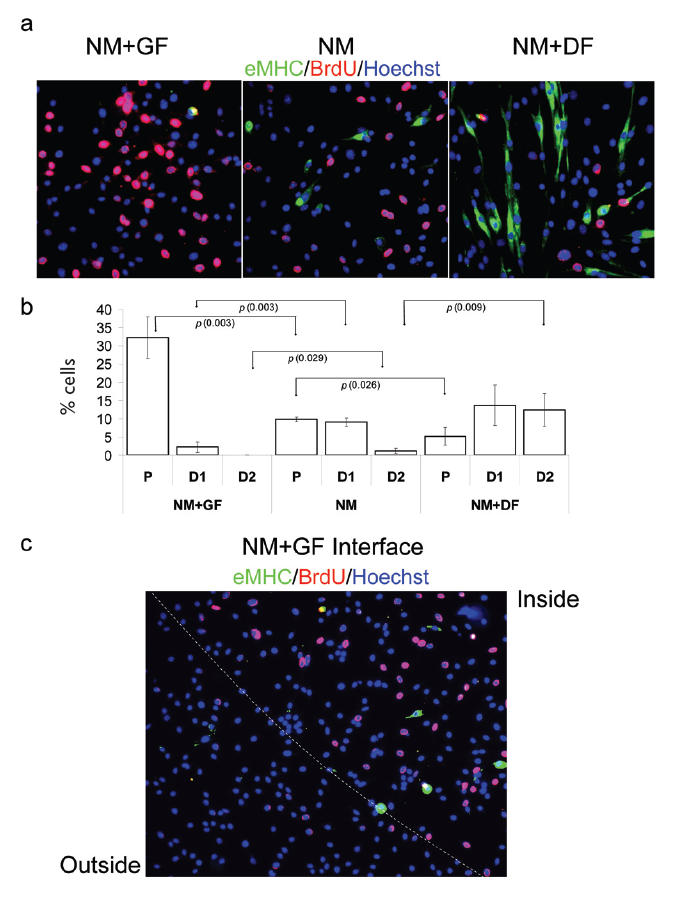
Geometric control of proliferation or terminal differentiation in NM. Myogenic progenitor cells have been plated at 50% confluence in chamber slides in NM for 48 hours. Immunofluorescence was performed with the indicated antibodies: α-BrdU (red), α-eMHC (green), and Hoechst (blue) was used to label all nuclei.
(a) Cells cultured in NM on unmodified ECM substrate (NM) show no distinct tendency towards proliferation or differentiation. When cultured on GF-modified ECM (NM + GF) cells proliferate (BrdU incorporation) without any tendency to form myotubes and differentiate, ie, eMHC+. Conversely, when exposed to DF-modified ECM (NM + DF), cells terminally differentiate (eMHC+) and form myotubes while proliferation is reduced. (b) Quantification of P, early stage-differentiated cells with less than 2 nuclei (D1), and later stage-differentiated cells with more than 2 nuclei (D2). When cultured in NM on unmodified ECM, cells infrequently differentiate and have slow proliferation rate. However, when cells are plated on GF-modified ECM (NM + GF) there is a much larger percentage of proliferating cells; and when they are plated on DF-modified ECM (NM + DF) there are higher percentages of not only eMHC+ differentiated cells but also yield higher percentages of multinucleated myotubes (D2). (c) Boundary between GF-modified ECM and unmodified ECM. There are higher numbers of proliferating cells on GF-modified ECM (inside) than on the unmodified ECM (outside). Similar results have been obtained in at least 3 independent experiments. Magnification: (a) 20x; (c) 10x.
Consistently with data shown above, Figure 3c demonstrates a clearly defined interface between cells with different rates of myogenic proliferation cultured in identical media (NM), which was created by the exposure of cells to the geometrically embedded GF into ECM.
In summary, data presented in this work demonstrate that GF and DF geometrically placed in adhesion substrates of myogenic progenitor cells compete against culture media for myogenic cell fate determination. As expected, embedding GF and DF into ECM substrate yields uniform and opposite effects on the proliferation and differentiation of myogenic progenitor cells. Importantly, the magnitude of the effects on either proliferation or differentiation is identical between uniformly modified ECM (Figures 1a, 2a, 3a) and the spatially modified areas of ECM (Figures 1c, 2c, 3c). This strongly suggests that geometrically embedded factors do not significantly diffuse throughout the ECM and that their initial concentrations are not diluted. Similar effects on proliferation and differentiation of myoblasts have been observed when these GF and DF (listed in Table 1) were added directly to culture media (data not shown). Thus, the biological activity of these factors remains the same whether they are embedded in ECM or added to culture media. However, unlike GF- or DF-modified ECM substrates, directly applied factors are, of course, not capable of creating a geometric boundary between cells with different fates coexisting in the same culture dish. There is no doubt that these factors added to media signal via their specific receptors on cells (Husmann et al 1996; Wagers and Conboy 2005); thus, identical regulation of cell fate from ECM-embedded factors shown here strongly suggests that these factors also signal to cells attached to specific areas of modified ECM.
Discussion
This work demonstrates that myogenic cells at different stages of proliferation and differentiation can deliberately be orchestrated to coexist adjacent to each other on the same plate with identical culture media by their attachment to modified ECM substrates. Thus, cells with different fates and at different stages of the cell cycle can interact, and their direct interactions can be studied in the experimental system developed. During embryonic organogenesis, as well as in regenerating adult tissues, rapidly proliferating and terminally differentiated cells coexist and signal via both multiple cell–cell contacts and soluble molecules. Therefore, our developed technique can be used to identify the important molecular cross-talk regulating cell fate determination in embryonic development and in adult tissue repair.
Yet another useful outcome of our work is the potential to develop a better microenvironment for cell transplantation studies. No existing method allows successful transplantation of myogenic progenitor cells. In this study, we have defined conditions that could improve the regenerative potential of transplanted cells, by allowing local control of their proliferation and terminal myogenic differentiation. In future applications, it would also be interesting to test whether negative effects of the aged or pathologic environments might be overcome, and therefore muscle regenerative potential could be controlled efficiently when myogenic progenitor cells are transplanted in the context of the modified ECM tested in this work. If necessary, the concentration and specific combinations of growth-promoting and differentiation-promoting factors in the ECM could be attenuated to produce maximum myogenic potential in the presence of aged or pathologic environments.
Our data strongly suggest that differentiation of myogenic progenitor cells always prevails over their growth, even when GF are embedded in ECM (Figures 2a, c and SOM 1). Specifically, control cells cultured in DM on unmodified ECM fuse into multinucleated myotubes (Figure 2b-DM; fraction of cells at late stage of differentiation, D2 = 27.9%), while cells cultured on DF-modified ECM remain mono-nucleated in GM (Figure 1b-GM + DF; fraction of cells at early stage of differentiation, D1 = 11.8%, and no multinucleated cells have been found), which signifies a lower degree of terminal myogenic differentiation (Morgan and Partridge 2003). This suggests that DF-modified ECM have a differentiating effect, which is not completely dominant over the growth-promoting media (Figure 1b-GM + DF vs Figure 2b-DM). Therefore, there is no danger of continuous cell proliferation or oncogenic phenotype from the exposure of myogenic progenitor cells to exogenous GF in the described method.
In contrast, we show that, once cells expand, new myotubes are likely to be formed more efficiently and robustly in the presence of GF-modified ECM (SOM 1). Such data are physiologically significant, since the loss of muscle strength and mass, known as muscle atrophy, often accompanies old age and muscle dystrophies. Thus, cell transplantation under conditions that are known to result in muscle hypertrophy could be especially beneficial for aged or pathologic organs.
In summary, the approach presented in this work combines expertise in stem cell biology and bioengineering to create ECM adhesion substrates for deliberate geometric control of proliferation and differentiation of myogenic progenitor cells, which overrides the cell fate determination imposed by the culture media. Such modified ECM substrates could be used as a supportive microenvironment able to promote satellite cell expansion and differentiation in normal, aged, and disease-afflicted muscle. Additionally, this work may also serve as a model to engineer synthetic cellular niches to control and harness optimally the regenerative potential of stem cells in general.
Acknowledgments
This work was supported by Ellison’s Medical Foundation and NIH KO1 AG 025652 grants to I.M.C.; and by the Torres Quevedo Program from the Spanish Ministry of Education and Science, by the European Social Fund and by CEIT and Technum University of Navarra to E.M.J.P.
References
- Armand AS, Della GB, Launay T, et al. Expression and neural control of follistatin vs myostatin genes during regeneration of mouse soleus. Dev Dyn. 2003;227:256–65. doi: 10.1002/dvdy.10306. [DOI] [PubMed] [Google Scholar]
- Bockhold KJ, Rosenblatt JD, Partridge TA. Aging normal and dystrophic mouse muscle: analysis of myogenicity in cultures of living single fibers. Muscle Nerve. 1998;21:173–83. doi: 10.1002/(sici)1097-4598(199802)21:2<173::aid-mus4>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Bodine SC, Stitt TN, Gonzalez M, et al. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol. 2001;3:1014–19. doi: 10.1038/ncb1101-1014. [DOI] [PubMed] [Google Scholar]
- Chakravarthy MV, Fiorotto ML, Schwartz RJ, et al. Long-term insulin-like growth factor-I expression in skeletal muscles attenuates the enhanced in vitro proliferation ability of the resident satellite cells in transgenic mice. Mech Ageing Dev. 2001;122:1303–20. doi: 10.1016/s0047-6374(01)00263-9. [DOI] [PubMed] [Google Scholar]
- Conboy IM, Conboy MJ, Smythe GM, et al. Notch-mediated restoration of regenerative potential to aged muscle. Science. 2003;302:1575–7. doi: 10.1126/science.1087573. [DOI] [PubMed] [Google Scholar]
- Conboy IM, Conboy MJ, Wagers AJ, et al. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature. 2005;433:760–4. doi: 10.1038/nature03260. [DOI] [PubMed] [Google Scholar]
- Conboy IM, Rando TA. The regulation of Notch signaling controls satellite cell activation and cell fate determination in postnatal myogenesis. Dev Cell. 2002;3:397–409. doi: 10.1016/s1534-5807(02)00254-x. [DOI] [PubMed] [Google Scholar]
- Conboy IM, Rando TA. Aging, stem cells and tissue regeneration: lessons from muscle. Cell Cycle. 2005;4:407–10. doi: 10.4161/cc.4.3.1518. [DOI] [PubMed] [Google Scholar]
- Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–84. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- Disatnik MH, Boutet SC, Lee CH, et al. Sequential activation of individual PKC isozymes in integrin-mediated muscle cell spreading: a role for MARCKS in an integrin signaling pathway. J Cell Sci. 2002;115:2151–63. doi: 10.1242/jcs.115.10.2151. [DOI] [PubMed] [Google Scholar]
- Disatnik MH, Rando TA. Integrin-mediated muscle cell spreading. The role of protein kinase c in outside-in and inside-out signaling and evidence of integrin cross-talk. J Biol Chem. 1999;274:32486–92. doi: 10.1074/jbc.274.45.32486. [DOI] [PubMed] [Google Scholar]
- Downward J. PI 3-kinase, Akt and cell survival. Semin Cell Dev Biol. 2004;15:177–82. doi: 10.1016/j.semcdb.2004.01.002. [DOI] [PubMed] [Google Scholar]
- Florini JR, Ewton DZ, Coolican SA. Growth hormone and the insulin-like growth factor system in myogenesis. Endocr Rev. 1996;17:481–517. doi: 10.1210/edrv-17-5-481. [DOI] [PubMed] [Google Scholar]
- Heszele MF, Price SR. Insulin-like growth factor I: the yin and yang of muscle atrophy. Endocrinology. 2004;145:4803–5. doi: 10.1210/en.2004-1037. [DOI] [PubMed] [Google Scholar]
- Husmann I, Soulet L, Gautron J, et al. Growth factors in skeletal muscle regeneration. Cytokine Growth Factor Rev. 1996;7:249–58. doi: 10.1016/s1359-6101(96)00029-9. [DOI] [PubMed] [Google Scholar]
- Jakubowiak A, Pouponnot C, Berguido F, et al. Inhibition of the transforming growth factor beta 1 signaling pathway by the AML1/ETO leukemia-associated fusion protein. J Biol Chem. 2000;275:40282–7. doi: 10.1074/jbc.C000485200. [DOI] [PubMed] [Google Scholar]
- Latres E, Amini AR, Amini AA, et al. Insulin-like growth factor-1 (IGF-1) inversely regulates atrophy-induced genes via the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR) pathway. J Biol Chem. 2005;280:2737–44. doi: 10.1074/jbc.M407517200. [DOI] [PubMed] [Google Scholar]
- Lawlor MA, Rotwein P. Coordinate control of muscle cell survival by distinct insulin-like growth factor activated signaling pathways. J Cell Biol. 2000;151:1131–40. doi: 10.1083/jcb.151.6.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maley MA, Fan Y, Beilharz MW, et al. Intrinsic differences in MyoD and myogenin expression between primary cultures of SJL/J and BALB/C skeletal muscle. Exp Cell Res. 1994;211:99–107. doi: 10.1006/excr.1994.1064. [DOI] [PubMed] [Google Scholar]
- Massague J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000;103:295–309. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- Massague J, Chen YG. Controlling TGF-beta signaling. Genes Dev. 2000;14:627–44. [PubMed] [Google Scholar]
- McCroskery S, Thomas M, Maxwell L, et al. Myostatin negatively regulates satellite cell activation and self-renewal. J Cell Biol. 2003;162:1135–47. doi: 10.1083/jcb.200207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997;387:83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- Miller KJ, Thaloor D, Matteson S, et al. Hepatocyte growth factor affects satellite cell activation and differentiation in regenerating skeletal muscle. Am J Physiol Cell Physiol. 2000;278:C174–181. doi: 10.1152/ajpcell.2000.278.1.C174. [DOI] [PubMed] [Google Scholar]
- Morgan JE, Partridge TA. Muscle satellite cells. Int J Biochem Cell Biol. 2003;35:1151–6. doi: 10.1016/s1357-2725(03)00042-6. [DOI] [PubMed] [Google Scholar]
- Moustakas A, Pardali K, Gaal A, et al. Mechanisms of TGF-beta signaling in regulation of cell growth and differentiation. Immunol Lett. 2002;82:85–91. doi: 10.1016/s0165-2478(02)00023-8. [DOI] [PubMed] [Google Scholar]
- Partridge TA. Stem cell therapies for neuromuscular diseases. Acta Neurol Belg. 2004;104:141–7. [PubMed] [Google Scholar]
- Pola R, Ling LE, Aprahamian TR, et al. Postnatal recapitulation of embryonic hedgehog pathway in response to skeletal muscle ischemia. Circulation. 2003;108:479–85. doi: 10.1161/01.CIR.0000080338.60981.FA. [DOI] [PubMed] [Google Scholar]
- Sandri M, Sandri C, Gilbert A, et al. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117:399–412. doi: 10.1016/s0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz E, Lipton BH. Skeletal muscle satellite cells: changes in proliferation potential as a function of age. Mech Ageing Dev. 1982;20:377–83. doi: 10.1016/0047-6374(82)90105-1. [DOI] [PubMed] [Google Scholar]
- Seale P, Polesskaya A, Rudnicki MA. Adult stem cell specification by Wnt signaling in muscle regeneration. Cell Cycle. 2003;2:418–19. [PubMed] [Google Scholar]
- Shavlakadze T, White JD, Davies M, et al. Insulin-like growth factor I slows the rate of denervation induced skeletal muscle atrophy. Neuromuscul Disord. 2005;15:139–46. doi: 10.1016/j.nmd.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Sherwood RI, Christensen JL, Conboy IM, et al. Isolation of adult mouse myogenic progenitors: functional heterogeneity of cells within and engrafting skeletal muscle. Cell. 2004;119:543–54. doi: 10.1016/j.cell.2004.10.021. [DOI] [PubMed] [Google Scholar]
- Stitt TN, Drujan D, Clarke BA, et al. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell. 2004;14:395–403. doi: 10.1016/s1097-2765(04)00211-4. [DOI] [PubMed] [Google Scholar]
- Taverna D, Disatnik MH, Rayburn H, et al. Dystrophic muscle in mice chimeric for expression of alpha5 integrin. J Cell Biol. 1998;143:849–59. doi: 10.1083/jcb.143.3.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas M, Langley B, Berry C, et al. Myostatin, a negative regulator of muscle growth, functions by inhibiting myoblast proliferation. J Biol Chem. 2000;275:40235–43. doi: 10.1074/jbc.M004356200. [DOI] [PubMed] [Google Scholar]
- Tidball JG. Inflammatory processes in muscle injury and repair. Am J Physiol Regul Integr Comp Physiol. 2005;288:345–53. doi: 10.1152/ajpregu.00454.2004. [DOI] [PubMed] [Google Scholar]
- Wagers AJ, Conboy IM. Cellular and molecular signatures of muscle regeneration: current concepts and controversies in adult myogenesis. Cell. 2005;122:659–67. doi: 10.1016/j.cell.2005.08.021. [DOI] [PubMed] [Google Scholar]
- Yang JT, Rando TA, Mohler WA, et al. Genetic analysis of alpha 4 integrin functions in the development of mouse skeletal muscle. J Cell Biol. 1996;135:829–35. doi: 10.1083/jcb.135.3.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidel-Bar R, Cohen M, Addadi L, et al. Hierarchical assembly of cell-matrix adhesion complexes. Biochem Soc Trans. 2004;32:416–20. doi: 10.1042/BST0320416. [DOI] [PubMed] [Google Scholar]
- Zammit PS, Heslop L, Hudon V, et al. Kinetics of myoblast proliferation show that resident satellite cells are competent to fully regenerate skeletal muscle fibers. Exp Cell Res. 2002;281:39–49. doi: 10.1006/excr.2002.5653. [DOI] [PubMed] [Google Scholar]
- Zimmers TA, Davies MV, Koniaris LGJ, et al. Induction of cachexia in mice by systemically administered myostatin. Science. 2002;296:1486–8. doi: 10.1126/science.1069525. [DOI] [PubMed] [Google Scholar]