

Abstract
Among several promising new drug-delivery systems, liposomes represent an advanced technology to deliver active molecules to the site of action, and at present several formulations are in clinical use. Research on liposome technology has progressed from conventional vesicles (“first-generation liposomes”) to “second-generation liposomes”, in which long-circulating liposomes are obtained by modulating the lipid composition, size, and charge of the vesicle. Liposomes with modified surfaces have also been developed using several molecules, such as glycolipids or sialic acid. A significant step in the development of long-circulating liposomes came with inclusion of the synthetic polymer poly-(ethylene glycol) (PEG) in liposome composition. The presence of PEG on the surface of the liposomal carrier has been shown to extend blood-circulation time while reducing mononuclear phagocyte system uptake (stealth liposomes). This technology has resulted in a large number of liposome formulations encapsulating active molecules, with high target efficiency and activity. Further, by synthetic modification of the terminal PEG molecule, stealth liposomes can be actively targeted with monoclonal antibodies or ligands. This review focuses on stealth technology and summarizes pre-clinical and clinical data relating to the principal liposome formulations; it also discusses emerging trends of this promising technology.
Keywords: liposomes, stealth liposomes, targeted liposomes, immunoliposomes
Introduction
Clinical medicine possesses an extremely broad range of drug molecules currently in use, and new drugs are added to the list every year. One of the main goals of any treatment employing xenobiotics is to increase the therapeutic index of the drug while minimizing its side-effects. The clinical utility of most conventional chemotherapeutics is limited either by the inability to deliver therapeutic drug concentrations to the target tissues or by severe and harmful toxic effects on normal organs and tissues. Different approaches have been attempted to overcome these problems by providing “selective” delivery to the affected area; the ideal solution would be to target the drug only to those organs, tissues, or cells affected by the disease. Selected carriers, such as molecular conjugates and colloidal particulates, can be suitable for this purpose. Colloidal particulates result from physical incorporation of the drug into a particulate colloidal system such as liposomes, niosomes, micro- and nano-spheres, erythrocytes, and polymeric and reverse micelles. Among these carriers, liposomes have been most studied. Their attraction lies in their composition, which makes them biocompatible and biodegradable. They consist of an aqueous core entrapped by one or more bilayers composed of natural or synthetic lipids. Liposomes composed of natural phospholipids are biologically inert and weakly immunogenic, and they possess low intrinsic toxicity. Further, drugs with different lipophilicities can be encapsulated into liposomes: strongly lipophilic drugs are entrapped almost completely in the lipid bilayer, strongly hydrophilic drugs are located exclusively in the aqueous compartment, and drugs with intermediate logP easily partition between the lipid and aqueous phases, both in the bilayer and in the aqueous core (Gulati et al 1998).
Liposomes can be classified according to their lamellarity (uni-, oligo-, and multi-lamellar vesicles), size (small, intermediate, or large) and preparation method (such as reverse phase evaporation vesicles, VETs). Unilamellar vesicles comprise one lipid bilayer and generally have diameters of 50–250 nm. They contain a large aqueous core and are preferentially used to encapsulate water-soluble drugs. Multilamellar vesicles comprise several concentric lipid bilayers in an onion-skin arrangement and have diameters of 1–5 μm. The high lipid content allows these multilamellar vesicles to passively entrap lipid-soluble drugs.
Since liposomes were first developed (around 1980) the related technology has made considerable progress, and several important formulations for the treatment of different diseases are now available commercially or in advanced clinical trials. As shown in Figure 1, the interest in liposome technology and clinical applications remains high, and almost 800 papers and 50 reviews were published in 2005 alone.
Figure 1.
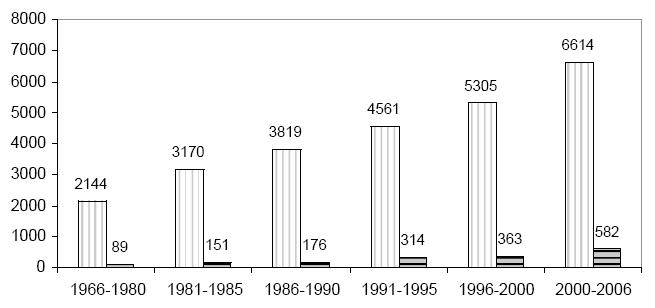
Increase in scientific research on liposomes: papers (vertical line) and reviews (horizontal line) published (total numbers on vertical axis). Data obtained from Ovid-Medline search keyword “liposomes”.
The present review will briefly outline the characteristics of liposomes and look at the related problems and solutions proposed, with a focus on advanced liposome formulations. In particular, we revisit the literature relating to high-stability, long-circulating liposomes (stealth liposomes), their field of application, and future trends concerning novel stealth formulations.
Conventional liposomes
Liposomal formulations of several active molecules are currently in pre-clinical and clinical trials in different fields, with promising results. Two of the key problems in drug therapy (biodistribution throughout the body and targeting to specific receptors) can be overcome by using liposomal formulations: liposomes protect encapsulated molecules from degradation and can passively target tissues or organs that have a discontinuous endothelium, such as the liver, spleen, and bone marrow. On intravenous administration, liposomes are rapidly captured by the mononuclear phagocyte system (MPS) and removed from the blood circulation (Scherphof 1985). This behavior has been exploited for efficient delivery of antiparasitic and antimicrobial drugs to treat infections localized in the mononuclear phagocytic system (eg, antimonial drugs against leishmaniasis) (Alving 1978; Agrawal and Gupta 2000; Basu and Lala 2004), or in order to encapsulate immunomodulators in activated macrophages in cancer models, to produce tumoricidal agents.
However, when the target site is beyond the MPS, efficient liposome uptake by the macrophages, and their consequent removal from circulation, is one of the main disadvantages for possible use of liposomes as drug delivery systems.
Binding of selected serum proteins (opsonins) is the first signal for removal of liposomes: the MPS does not recognize the liposomes themselves but, rather, recognizes opsonins, which are bound to the surface of the liposomes. A limited number of possible opsonizing proteins that affect the fate of liposomes have been identified, eg, immunoglobulins (Patel 1992), fibronectin (Falcone 1986; Patel 1992), beta 2-glycoprotein (Chonn et al 1995), C-reactive protein (CRP) (Volanakis and Narkates 1981), and beta 2-macroglobulin (Murai et al 1995).
Complement components (Patel 1992; Devine et al 1994; Harashima et al 1994) comprise another important system able to recognize liposomes, which evolved as an immediate host defense against invading pathogens. This system acts through initiating membrane lysis and enhancing uptake by the MPS cells (neutrophils, monocytes, macrophages). In particular, the assembly of C5b-9 complexes (membrane attack complex: MAC) of the complement system is able to produces lytic pores, which induce cell lysis or, in the case of liposomes, the release of their contents. The complement-dependent release of liposomal contents appears to be one of the dominant factors in determining the biological fate of liposomes. However, serum components that inhibit the phagocytosis of pathogens or particles, referred to as dysopsonins, have also been identified. Human serum albumin and IgA possess dysopsonic properties and their presence on particle surfaces has been shown to reduce recognition and phagocytosis. A balance between blood opsonic proteins and suppressive proteins has been found to regulate the rate of liposome clearance (Ishida et al 2002).
The instability of liposomes in plasma due to their interaction with high (HDL) and low density (LDL) lipoproteins is another limitation, since this interaction results in the rapid release of the encapsulated drug into the plasma.
The physicochemical properties of liposomes, such as net surface charge, hydrophobicity, size, fluidity, and packing of the lipid bilayers, influence their stability and the type of proteins that bind to them (Chonn et al 1992; Oja et al 1996). One of the first attempts to overcome these problems was focused on manipulation of lipid membrane components in order to modify bilayer fluidity. Damen et al (2005) demonstrated that incorporation of cholesterol (CHOL), by causing increased packing of phospholipids in the lipid bilayer, reduces transfer of phospholipids to HDL; Senior (1982) demonstrated that liposomes obtained from phosphatidylcholine (PC) with saturated fatty acyl chains (with a high liquid crystalline transition temperature) or from sphingomyelin (SM) are more stable in the blood than liposomes prepared from PC with unsaturated fatty acyl chains.
Several approaches have also involved modulating liposome size and charge, so as to reduce MPS uptake. In general, larger liposomes are eliminated from the blood circulation more rapidly than smaller ones (Senior 1982). Small unilamellar vesicles (SUVs) have a half-life longer than that of multilamellar liposomes (MLVs) (500–5000 nm). This suggests that phagocytes can distinguish between the sizes of foreign particles.
Based on these observations, it is evident that the binding of opsonins to liposomes depends on the size of the liposomes, and that in consequence the enhanced MPS uptake of liposomes by the liver is likewise size-dependent (Harashima et al 1994).
Negatively charged liposomes have a shorter half-life in the blood than do neutral liposomes, although the contrary has also been found (Nishikawa 1990; Funato 1992); positively charged liposomes are toxic and thus quickly removed from circulation (Senior 1987).
The complement system has been reported to be activated by both negatively charged and positively charged liposomes in man (Chonn et al 1991; Marjan et al 1994; Bradley et al 1998; Price et al 2001). Chonn et al (1991) report that, for both human and guinea-pig serum, surface charge is a key determinant in complement-system activation by liposomes: negatively charged liposomes activate the complement system via the classical pathway, while positively charged liposomes activate it via the alternative pathway.
Clinical applications of conventional liposomes
Based on these studies, “conventional” liposomes composed of neutral and/or negatively charged lipids plus CHOL have been prepared; some of these formulations have reached the market (Table 1) or are now entering clinical trials. Ambisome® (Gilead Sciences, Foster City, CA, USA) in which the encapsulated drug is the antifungal amphotericin B (Veerareddy and Vobalaboina 2004), Myocet® (Elan Pharmaceuticals Inc., Princeton, NJ, USA) encapsulating the anticancer agent doxorubicin (Alberts et al 2004), and Daunoxome® (Gilead Sciences), in which the drug incorporated is daunorubicin (Allen and Martin 2004), are the principal examples of such formulations. To obtain stable formulations incorporating a constant amount of drug, various mechanisms are exploited. Ambisome is lyophilized; Myocet is supplied in three separate vials, one containing doxorubicin as dry powder, one a solution of empty liposomes in citric buffer, and the third a solution of sodium carbonate. In this case drug entrapment must be achieved immediately prior to administration. Daunoxome is at present the only pure-lipid MPS-avoiding liposomal formulation; it is available as a stable ready-to-inject liposomal formulation.
Table 1.
Approved and emerging liposome formulations
| Active agent (product name) | Composition | Stealth | Company, year of product marketing | Application | Trial phase |
|---|---|---|---|---|---|
| DaunoXome® (daunorubicin) | DSPC/CHOL | no | Nexstar Pharmaceuticals, 1995 | Kaposi’s sarcoma | Approved |
| DOXIL®/Caelyx® (doxorubicin) | SoyHPC/CHOL/DSPE-PEG | yes | Sequus Pharmaceuticals, 1997 | Kaposi’s sarcoma | Approved |
| Myocet®/Evacet® (doxorubicin) | EPC/CHOL | no | Elan Pharma, 2000 | Metastatic breast cancer | Approved |
| SPI-077 (cisplatin) | SoyHPC/CHOL/DSPE-PEG | yes | Sequus Pharmaceuticals | Head and neck cancer, Lung cancer | Phase I/II |
| Lipoplatin™ (cisplatin) | SoyPC/DPPG/CHOL | yes | Regulon Inc. | Several cancer type | Phase II/III |
| S-CKD602 (camptothecin analog) | — | yes | Alza Co. | Several cancer type | Phase I |
| Aroplatin (oxaliplatin analog) | DMPC/DMPG | no | Antigenics Inc | Colorectal cancer | Phase II |
| Depocyt | DOPC/DPPG/CHOL/triolein | no | SkyePharma 1999 | Lymphomatous meningitis | Approved |
| LEP-ETU (paclitaxel) | DOPE/CHOL/cardiolipin | no | NeoPharm Inc | ovarian, breast, and lung cancer | Phase I |
| LEM-ETU (mitoxantrone) | DOPE/CHOL/cardiolipin | no | NeoPharm Inc | leukemia, breast, stomach, liver, ovarian cancers | Phase I |
| LE-SN38 (irinotecan) | DOPE/CHOL/cardiolipin | no | NeoPharm Inc | advanced cancer | Phase I |
| MBT-0206 (paclitaxel) | DOPE/DO- trimethylammoniumpropane | no | MediGene AG | Anti-angiogenic proprieties Breast cancer | Phase I |
| OSI-211 (lurtotecan) | SoyHPC/CHOL | no | Enzon Co. | Ovarian cancer Head and neck cancer | Phase II |
| Marqibo® (vincristine) | DSPPC/CHOL/sphingosine | no | Inex Pharm | Non-Hodgkin’s lymphoma | Phase II/III |
| Atragen® (t-retinoic acid) | DMPC, and soybean oil | no | Aronex Pharm | advanced renal cell ca, acute pro-myelocytic leucemia | Phase I/II |
| INX-0125 (vinorelbine) | DSPPC/CHOL/sphingosine | no | Inex Pharm | breast, colon and lung cancer | Preclinical Phase I |
| INX-0076 (topotecan) | DSPPC/CHOL/sphingosine | no | Inex Pharm | advanced cancer | Preclinical |
| Liposomal-Annamycin® | DSPC/DSPG/Tween | no | MD Anderson CC | breast cancer | Phase II |
| Ambisome® (amphotericin) | SoyHPC/DSPPC/CHOL | no | Fujisawa USA Inc. and Nexstar Pharm 1997 | Fungal infections in immuno-compromised patients | Approved |
| Nyotran® (nistatin) | DMPC/DMPG/CHOL | no | Aronex Pharm | Fungal infections in immuno-compromised patients | Phase II/III |
By addition of sphingomyelin and saturated fatty acid chain lipids to the lipidic formulation, two commercial liposome formulations have been produced. A novel liposomal formulation of vincristine (Marqibo®, formerly Onco TCS; Inex Pharmaceuticals Co., Vancouver, BC, Canada and Enzon Pharmaceuticals Inc., Bridgewater, NJ, USA), based on sphingomyelin-cholesterol uni-lamellar vesicles, has recently been shown to be efficacious in the treatment of relapsed non-Hodgkin’s lymphoma (Anonymous 2004). Inex Pharmaceuticals Co. has in development two further candidate formulations: INX-0125™ (liposomal vinorelbine) (Semple et al 2005) and INX-0076™ (liposomal topotecan) (Tardi et al 2000), also based on the use of sphingomyelin–CHOL mixture.
Another liposomal formulation composed of hydrogenated soy phosphatidylcholine (HSPC) and CHOL and containing lurtotecan has been developed and named OSI-211™ (OSI Pharmaceuticals, Inc., Melville, NY, USA). Clinical results have shown that incorporation of OSI-211 in the acid aqueous core of the vesicle, in addition to providing the known therapeutic advantages of a liposomal carrier, can also favor maintenance of lactone ring closure of lurtotecan (the active form), which increases stability of the active compound, consequently improving its tumor toxicity (Seiden et al 2004; Dark et al 2005).
Two other liposome formulations employing saturated phospholipids have been launched for clinical development: Nyotran® (Aronex Pharmaceuticals, The Woodlands, TX, USA) and Aroplatin® (Antigenics Inc., Lexington, MA, USA). These are multilamellar liposomal formulations consisting of dimyristoyl phosphatidylcholine (DMPC) and dimyristoyl phosphatidylglycerol (DMPG). Nyotran contains nystatin A1, a membrane-active polyene antifungal antibiotic that is structurally related to amphotericin B (Anonymous 1999; Arikan and Rex 2001). Aroplatin is a multilamellar liposomal formulation of cis-bis-neodecanoato-trans-R,R-1,2-diaminocyclohexane platinum (II), a hydrophobic structural analog of oxaliplatin. Aroplatin is in clinical trials for a wide range of tumors and more recently for advanced solid tumors and B-cell lymphoma (Han et al 1993; Verschraegen et al 2003; Lu et al 2005).
Aronex Pharmaceuticals has also developed a liposomal-all/trans/retinoic acid formulation (Atragen®) containing tretinoin, dimyristoyl phosphatidylcholine (DMPC), and soybean oil. The formulation has shown efficacy in the treatment of acute promyelocytic leukemia and other retinoid-responsive cancers (Ozpolat et al 2003).
Several interesting liposome preparations have been developed by NeoPharm (NeoPharm Inc., Waukegan, IL, USA including LEM-ETU™, LEP-ETU™, and LE-SN38™). In LEM-ETU, mitoxantrone (Ugwu et al 2005) is encapsulated in multilamellar liposomes (composed of dioleoyphosphatidyl choline [DOPC] and CHOL) after charge interaction with cardiolipin. LEP-ETU (Zhang et al 2005), a liposome-entrapped “easy to use” paclitaxel formulation, recently demonstrated bio-equivalence with Taxol® (Bristol-Myers Squibb, New York, NY, USA) and interesting activity in Phase I trials. This formulation, composed of DOPC, CHOL, and cardiolipin, is capable of carrying paclitaxel in the liposome bilayer at a maximum mole percent of about 3.5%. This is a higher loading capacity than those obtained in our previous studies with paclitaxel (Crosasso et al 2000) and docetaxel (Immordino et al 2003). In LE-SN38™, the active metabolite of irinotecan (7-ethyl-10-hydroxycamptothecin) is encapsulated in PC, CHOL, and cardiolipin liposomes (Zhang et al 2004a). LE-SN38 was in Phase I trials in 2004 (Kraut et al 2004).
Paclitaxel has also been encapsulated in other modern formulations of cationic lipid complexes (MBT-0206) that have been shown to be bounded and internalized selectively by angiogenic tumoral endothelial cells after intravenous injection. A Phase II clinical trial is in progress for candidate drug EndoTAG™-1 (MediGene A.G., Martinsried, Germany) in the treatment of advanced pancreatic cancer (Eichhorn et al 2006).
Cytarabine is an anti-metabolite, anti-neoplastic agent used in clinical applications for acute lymphoid leukemia, myeloid leukemia and meningeal leukemia. DepoCyt® (SkyePharma PLC, London, UK) is a slow-release formulation of cytarabine designed for intrathecal administration in the treatment of neoplastic meningitis due to breast cancer (Jaeckle et al 2001; Bomgaars et al 2004). In DepoCyt, cytarabine is encapsulated in the aqueous compartment of a spherical 20-μm matrix comprised of lipids biochemically similar to normal human cell membranes (phospholipids, triglycerides and CHOL) (also called DepoFoam™) (Mantripragada 2002).
Annamycin (3’-deamino-4’-epi-3’-hydroxy-2’-iodo-4-demethoxy doxorubicin) is a “second-generation” highly lipophilic anthracycline developed by the M.D. Anderson Cancer Center. Its molecular design will be able to avoid the multi-drug-resistance mechanism of tumor cells, and its lipophilicity produces increased encapsulation in liposomes (Priebe and Perez-Soler 1993). Liposomal annamycin (composed of DMPC, DMPG, and Tween) (Andreff and Estey 2004) was in Phase II trials in late 2005 in patients with refractory or relapsed acute lymphocytic leukemia.
Long-circulating liposomes
The use of saturated phospholipids and cholesterol in the formulation of liposome delivery systems cannot fully overcome their binding with serum components, and the consequently decreased MPS uptake of the vesicles: saturation of the MPS with a previous administration of “empty” liposomes may be necessary. Moreover, SUVs possess the disadvantage of low aqueous entrapment volume, and the use of charged liposomes can be toxic. Several different strategies have been developed to overcome these difficulties by coating the surface of the liposomes with inert molecules to form a spatial barrier.
The first strategy studied was the preparation of liposomes mimicking the erythrocyte membrane; in this case the liposome surface was modified with gangliosides and sialic acid derivatives, such as monosialoganglioside (GM1) (Gabizon and Papahadjopoulos 1988; Allen et al 1989). The subsequent step was to increase the hydrophilicity of the liposomal surface by using hydrophilic polymers.
The mechanism whereby steric stabilization of liposomes increases their longevity in circulation has been extensively discussed (Drummond et al 1999). The basic concept is that a hydrophilic polymer or a glycolipid, such as PEG or GM1, possessing a flexible chain that occupies the space immediately adjacent to the liposome surface (“periliposomal layer”), tends to exclude other macromolecules from this space. Consequently, access and binding of blood plasma opsonins to the liposome surface are hindered, and thus interactions of MPS macrophages with such liposomes are inhibited.
By reducing MPS uptake, long-circulating liposomes can passively accumulate inside other tissues or organs. This phenomenon, called passive targeting, is especially evident in solid tumors undergoing angiogenesis: the presence of a discontinuous endothelial lining in the tumor vasculature during angiogenesis facilitates extravasation of liposomal formulations into the interstitial space, where they accumulate due to the lack of efficient lymphatic drainage of the tumor, and function as a sustained drug-release system. This causes the preferential accumulation of liposomes in the tumor area (a process known as enhanced permeation and retention effect or EPR) (Maeda et al 2001). Liposome formulations do not extravasate from the bloodstream into normal tissues that have tight junctions between capillary endothelial cells. These mechanisms appear to be responsible for the improved therapeutic effects of liposomal anticancer drugs versus free drugs. However, the processes involved in delivery of these carriers and release of active agent, the variability of such processes, and the degree to which the active agent is released into the tumor’s extracellular fluid or into tumor cells, are still unknown.
GM1 and glucoronide liposomes
Several glycolipids have been tested in studies of MPS uptake of liposomes after iv injection: the glycolipid GM1 (a brain-tissue-derived monosialoganglioside) significantly decreased MPS uptake when incorporated on the liposome surface, and the formulation remained in blood circulation for several hours. GM1 grafted liposomes with a diameter in the 90–200 nm range have longer blood retention, with consequent accumulation in tumor tissues, than those out of this size range (Liu et al 1992). Large liposomes with a diameter of >300 nm preferentially accumulate in the spleen, whereas those with a diameter of <40 nm probably penetrate the interstitial spaces of the liver.
Unfortunately, the effect of GM1 liposomes in prolonging half-life in bloodstream has been observed only in mouse models. Among different gangliosides, only GM3-grafted liposomes showed increased circulation time in rats, whereas sialic-acid-modified liposomes exhibit prolonged blood retention in both mice and rats.
The degree of macrophage uptake depends on the concentration of GM1 in PC liposomes: a concentration of 10 mol% decreased MPS uptake by 90%. This MPS-avoiding effect was reversed by the removal of the sialic acid moiety, demonstrating the important role played by this molecule in MPS-trapping avoidance (Yamauchi et al 1994).
On the basis of the studies reported above, two possibilities have been proposed to explain the mechanism of MPS avoidance by ganglioside liposomes. (1) Specific moieties (ie, GM1, but not other gangliosides in mice) grafted to the liposome surface reduce opsonization. (2) These moieties bind dysopsonins, reducing recognition by the MPS. It has also been suggested that the topology of carboxyl groups on the liposome surface plays an important role in liposome circulation time. One problem for the extensive use of sialic acid is its cost, a fact which stimulated the investigation of other derivatives, including glucuronic acid (Park et al 1992). Oku and Namba (2005) recently summarized their research on glucuronate-modified long circulating liposomes. Utilizing palmityl-D-glucuronide, they obtained reduced MPS recognition in vitro, reduced hepatic accumulation in vivo, and consequently increased tumor accumulation and thus therapeutic efficacy of adriamicin encapsulated in long circulating liposomes.
Other recent interesting applications of GM-coated liposomes involve their use for oral administration and delivery to the brain. In particular, Taira et al (2004) suggest that among liposomal formulations used as oral drug carriers, those containing GM1 and GM type III have better possibilities of surviving through the gastrointestinal tract. Mora et al (2002) observed higher brain-tracer uptake for GM1 liposomes than for control liposomes in the cortex, basal ganglia, and mesencephalon of both hemispheres; conversely, no significant changes were observed in liver uptake or blood concentration of the tracer.
PEG-coated liposomes (stealth liposomes)
Among the different polymers investigated in the attempt to improve the blood circulation time of liposomes, poly-(ethylene glycol) (PEG) has been widely used as polymeric steric stabilizer. It can be incorporated on the liposomal surface in different ways, but the most widely used method at present is to anchor the polymer in the liposomal membrane via a cross-linked lipid (ie, PEG- distearoylphosphatidylethanolamine [DSPE] as schematized in Figure 2) (Allen et al 1991, 2002).
Figure 2.
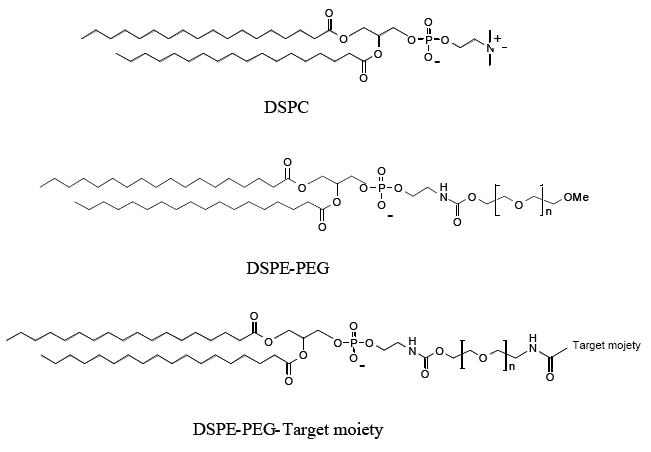
Chemical structures of distearoylphophatidylcholine (DSPC), distearoylphophatidylethanolamine after conjugation with poly-(ethylene glycol) (PEG) (DSPE-PEG) and DSPE-PEG linked with a targeting moiety.
PEG (CAS number 25322-68-3) is a linear polyether diol with many useful properties, such as biocompatibility (Powell 1980), solubility in aqueous and organic media (Powell 1980), lack of toxicity, very low immunogenicity and antigenicity (Dreborg and Akerblom 1990), and good excretion kinetics (Yamaoka et al 1994). These properties allow its use in a variety of applications (Harris 1992), including the biomedical field.
Moreover, unlike GM1, molecular weight and structure of PEG molecules can be freely modulated for specific purposes, and it is easier and cheaper to conjugate the polymer with the lipid.
Poly-ethylene glycols have been used to derivatize therapeutic proteins and peptides, increasing drug stability and solubility, lowering toxicity, increasing half-life (Caliceti and Veronese 2003), decreasing clearance and immunogenicity. These benefits have been particularly observed using branched PEG in the derivatization (Monfardini and Veronese 1998). For the most part, reaction with PEG derivatives does not alter the mechanism of action of a therapeutic protein; rather it enhances its therapeutic effect by altering its pharmacokinetics. PEG-ademase (utilized to treat immunodeficiency), PEG-visomant (human growth hormone), PEG-aspargase (for leukemias), PEG-interferon-alpha (for chronic hepatitis C), PEG-aldesleukin (PEG-IL-2) (an anticancer agent), and PEG-filgrastim (for chemotherapy-induced transferase neutropenia) are the principal PEGylated proteins in clinical use (Mahmood and Green 2005).
Surface modification of liposomes with PEG can be achieved in several ways: by physically adsorbing the polymer onto the surface of the vesicles, by incorporating the PEG-lipid conjugate during liposome preparation, or by covalently attaching reactive groups onto the surface of preformed liposomes.
Grafting PEG onto liposomes has demonstrated several biological and technological advantages. The most significant properties of PEGylated vesicles are their strongly reduced MPS uptake and their prolonged blood circulation and thus improved distribution in perfused tissues. Moreover, the PEG chains on the liposome surface avoid the vesicle aggregation, improving stability of formulations.
Needham et al (1992) demonstrated that the presence of PEG on the liposome surface provides a strong interbilayer repulsion that can overcome the attractive Van der Waals forces, thus stabilizing liposome preparations by avoiding aggregation. In particular, from X-ray analysis of bilayers incorporating PEG1900-lipid, their research showed that the grafted polymer moiety extends about 50Å from the lipid surface and gives rise to strong inter-membrane repulsive forces.
Regarding MPS uptake, Blume et al have found that the increased blood circulation time is due to a reduced interaction with plasma proteins and cell-surface proteins (Blume 1993; Vert and Domurado 2000), although other studies have found no direct evidence of this reduced interaction with plasma components (Johnstone et al 2001). One possible explanation for the reduced interaction is the steric hindrance effect, which is generated by the surface-grafted methoxy-PEG molecules. Complement fixation on PEG-bearing liposomes thus appears to occur in a cryptic location inaccessible to ligation to complement receptors. Another possible contributor to the stealth behavior of such vesicles is competition for CR3 between surface-bound and free-complement proteins iC3b. Furthermore, degradation of surface-bound C3b to fragments inhibiting recognition by phagocytic complement receptors might also explain the anti-phagocytic effect. Studies with freshly isolated macrophages have also indicated the presence of unidentified serum factors (called dysopsonins) that act synergistically with the steric barrier of long circulating particles, thereby further suppressing particle recognition by phagocytic cells (Moghimi et al 1993; Johnstone et al 2001).
A number of reports have indicated that PEG does not completely avoid cumulative uptake by cells of the MPS, and an interesting review updates progress in this area. Moghimi and Szebeni (2003) critically examine the supposed mechanisms that contribute to prolonged circulation times of sterically protected liposomes. They point out that PEGylated liposomes are not completely biologically inert and that there is some evidence the polymer can still induce activation of complement systems: a PEGylated liposomal doxorubicin (PLD) (DOXIL® in the US, Caelyx® in Europe, Schering-Plough, Kenilworth, NJ, USA) (Gabizon and Muggia 1998), is a strong activator of the human complement system, with activation taking place within minutes.
The behavior of PEGylated liposomes depends on the characteristics and properties of the specific PEG linked to the surface. Figure 3 represents the regimens proposed by deGennes, when polymers are attached to the liposome surface, depending on the graft density of the polymer (de Gennes 1980). The molecular mass of the polymer, as well as the graft density, determine the degree of surface coverage and the distance between graft sites.
Figure 3.
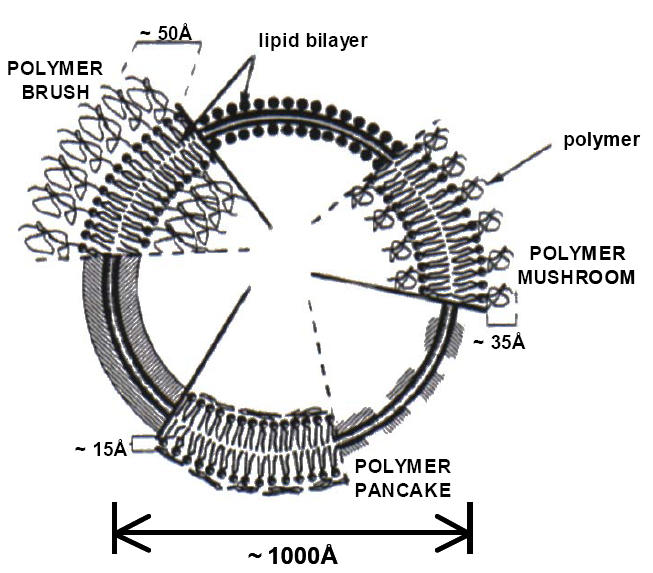
Schematic diagrams of poly-(ethylene glycol) (PEG) configurations regimes (mushroom, brush and pancake) for polymer grafted to the surface of liposome bilayer.
The most evident characteristic of PEG-grafted liposomes (PEGylated-liposomes) is their circulation longevity, regardless of surface charge or the inclusion of stabilizing agent such as cholesterol. Figure 4 represents the degree of longevity as determined by a pharmacokinetic evaluation of PEGylated liposomes containing docetaxel (Immordino et al 2003).
Figure 4.
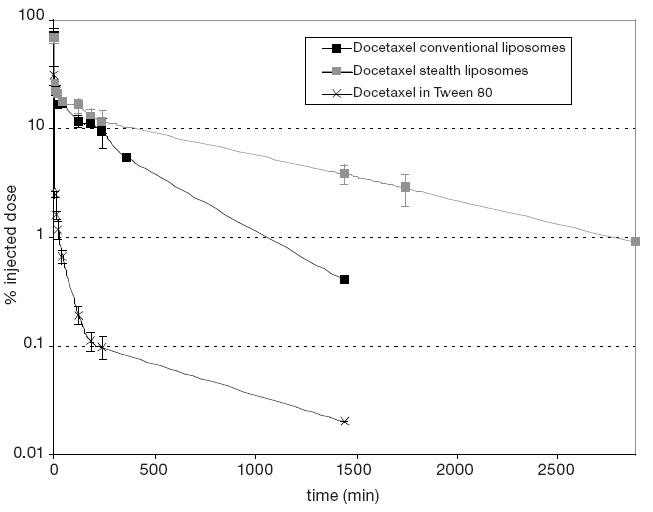
Plasma values obtained from mice iv injected determined by 14C radioactivity associated to docetaxel in Tween 80 (crosses), docetaxel in conventional (black squares), and in PEGylated (gray squares) liposomes.
The ability of the hydrophilic shell of PEG to avoid aggregation between liposomal particles and to decrease the extent of particle-protein interaction in biological fluids is due not only to the molecular mass of the bound polymer and its uniformity (“molecular cloud”) but also to its considerable conformational flexibility (Torchilin et al 1994a). A more rigid polymer, like dextran, grafted to liposomes and used in comparable quantities, does not cause an analogous decrease in liposome-protein interactions.
In liposomes composed of phospholipids and cholesterol, the ability of PEG to increase the circulation lifetime of the vehicles has been found to depend on both the amount of grafted PEG and the length or molecular weight of the polymer (Allen et al 1991). In most cases, the longer-chain PEGs have produced the greatest improvements in blood residence time. For example, Allen et al (1991) reported that blood levels were higher for SM/PC/CHOL/DSPE-PEG liposomes with longer molecular weight PEG (ie, PEG 1900 and PEG 5000) than for liposomes containing PEG-lipid with a shorter chain PEG (ie, PEG 750 and PEG 120). The presence of PEG 2000 doubled the amount of lipid remaining in the plasma compared to formulations containing PEG 350 to 750.
The area of protection of PEG molecules of different molecular weights may be calculated: the calculation is based on a simple approach that was developed by Torchilin and Papisov (Torchilin and Papisov 1994).
A conclusive link has not yet been established between chemical and physical properties of PEG and its ability to extend circulation lifetime. Although the accepted opinion is that PEG increases circulation longevity of drug carriers by reducing or preventing protein binding and/or by inhibiting cell binding/uptake, there is sufficient conflicting data to warrant a reassessment of the mechanism(s) by which surface grafted PEGs improve liposome properties (Allen et al 2002).
Subcutaneous administration of PEGylated liposomes also appears interesting; this administration route could become very important, especially for targeting to the lymph nodes and to achieve sustained drug release in vivo. Targeting liposomes to the lymph nodes for administration of antitumor, antibacterial, and antiviral drugs is of particular interest, and the pharmacokinetics and biodistribution of PEG-DSPE liposomes have been examined after subcutaneous administration (Allen et al 1993). Furthermore, the use of subcutaneously administered liposomes in the field of vaccination and rheumatism, with the aim of prolonging release of antigens or forming a local drug depot, is also in the focus of interest (Babai et al 1999; Corvo et al 1999). PEG-coating on liposomal surface may have a negative effect on lymph node uptake (reduced adsorption by phagocytosis). However, as hypothesized above, lymphatic absorption from the s.c. injection site may increase as a results of the steric stabilization effect and, therefore, the net amount of liposomes taken up by the lymph nodes may be slightly higher despite the stealth effect. Indeed Oussoren and Storm (1997) compared liposome formulations composed of ePC/CHOL and DPPC/CHOL with or without PEG 2000 or 5000 (5 mol%) with diameter 70 or 150 nm. After s.c. administration, the highest blood peak was in all cases that of the PEGylated liposomes; the authors concluded that “the results did not indicate that steric stabilization of liposome has profound effects on lymphatic uptake”.
Although these favorable characteristics have extended clinical applications of PEGylated liposomes, the evidence suggests some caution is needed, and that investigation of some aspects in greater depth, such as multiple dose administration or biodistribution in tumor tissues, is indicated.
Ishida et al (2005) and Laverman et al (2001) recently reported that intravenous injection in rats of PEG-grafted liposomes may significantly alter the pharmacokinetic behavior of a second dose when this second dose is administered after an interval of several days. This phenomenon, called “accelerated blood clearance” (ABC), appears to be inversely related to the PEG content of liposomes. By the same token, an inverse relationship has been observed between dose and magnitude of the ABC effect.
Stealth liposomes are important in cancer treatment for their passive targeting effect, which may lead to preferential accumulation in tumor tissue, but this phenomenon is not fully understood: Stealth liposomes are able to lodge in the interstitial spaces among tumor cells but, once in the tumor area, they locate in the extracellular fluid surrounding the tumor cell without entering it. Thus, to deliver the active form of an anticancer agent, such as doxorubicin or cisplatin, the drug must be released from the liposomes into the tumor extracellular fluid and then diffuse into the cell (Harrington et al 2000a, 2000b). As a result, the ability of liposomes to carry the anticancer agent to the tumor (which depends on their stability) and to release it into the tumor extracellular fluid (depending on membrane composition and fluidity) are equally important factors in determining the anti-tumor effect of liposome-encapsulated anticancer agents. Unfortunately, little is known of the kinetics of drug release from liposomes into the interstitial space: only the free drug can penetrate into the solid tumor, and it is difficult to determinate the ratio between free/liposome-encapsulated drug in the tumor extracellular fluid (Zamboni et al 2004).
Current research on PEG liposomes has concentrated on attaching PEG to the liposome surface in a removable fashion, in order to facilitate subsequent liposome capture by the cells. After PEG liposomes accumulate at the target site through the EPR effect, the PEG coating is detached under the action of local pathological conditions (decreased pH in tumors). New, detachable PEG conjugates have been described (Zalipsky et al. 1999) in which the PEG release process is based on mild thiolysis of the dithiobenzylurethane linkage between PEG and an amino-containing substrate (such as PE).
Clinical applications of stealth liposomes
PEGylated liposomal doxorubicin (PLD) (DOXIL/ Caelyx) was the first and is still the only stealth liposome formulation to be approved in both the USA and Europe for treatment of Kaposi’s sarcoma (Krown et al 2004) and recurrent ovarian cancer (Rose 2005). DOXIL/Caelyx is now undergoing trials for treatment of other malignancies such as multiple myeloma (Hussein and Anderson 2004), breast cancer (Robert et al 2004), and recurrent high-grade glioma (Hau et al. 2004). Several studies are under way to investigate the anticancer activities of PLD in combination with other therapeutics, including the taxanes (paclitaxel or docetaxel) (Briasoulis et al 2004; Vorobiof et al 2004), temozolomide (Temodal® Schering-Plough, Kenilworth, NJ, USA) (Awada et al 2004), and vinorelbine (Katsaros et al 2005).
The rigid bilayer of PLD is composed of HSPC, CHOL, and mPEG-DSPE (molecular weight 2000) at a molar ratio of 55:40:5. Liposomes with a mean diameter of 85 nm are able to incorporate doxorubicin at a concentration of 2 mg/mL. The pharmacokinetics is very slow: plasma elimination follows a biexponential curve, with half-lives of 1.5 and 45 hours (median values); in comparison, plasma half-lives are 0.2 hours for free drug, 2–3 hours for Myocet and 5 hours for Daunoxome. Nearly 100% of the drug detected in the plasma after PLD injection was in liposome-encapsulated form; plasma clearance is clearly slow (0.1 L/hour) and the distribution volume small (4 L).
Due to its pharmacokinetic behavior, cardiotoxicity, myelosuppression, alopecia and nausea are significantly decreased with PLD compared with an equi-effective dose of conventional doxorubicin. These bio-distribution characteristics also make skin treatment of localized cancers such as Kaposi’s sarcoma possible; on the other hand, due to its reduced clearance, the palmar-plantar skin reaction and stomatitis/mucositis are the chief dose-related toxicities of PLD (Lyass et al 2000).
Another stealth liposome formulation is SPI-077™ (Alza Corporation, Mountain View, CA, USA), in which cisplatin is encapsulated in the aqueous core of sterically stabilized liposomes (fully hydrogenated soy HSPC, CHOL, and DSPE-PEG). The stealth behavior of these compounds is evident from their apparent half-life of approximately 60–100 hours. Phase I/II clinical trials have been run to treat head and neck cancer and lung cancer (Kim et al 2001). Although the toxicity profile was promising, the therapeutic efficacy requires improvement (Harrington et al 2001). To obtain the desired balance between encapsulation and release of cisplatin from liposomes, another formulation was evaluated by Alza Corporation (SPI-077 B103); they chose B103, in which HSPC is replaced by unsaturated phospholipids, because of its greater theoretical propensity to release cisplatin (Alza Corporation, data on file). However, Zamboni et al (2004) were not able to detect released cisplatin in in vitro systems, plasma, or tumor extracellular fluid after administration of either stealth formulation of liposomal cisplatin.
Recently, S-CKD602 (Alza Corporation), a PEGylated stealth liposomal formulation of CKD-602, which is a semi-synthetic analog of camptothecin, was submitted for a Phase I trial. After administration of S-CKD602 at doses of 0.5 mg/m2, the plasma AUC was 50-fold that of non-liposomal CKD-602; S-CKD602 showed minimal toxicity and interesting activity (Zamboni et al 2005).
Lipoplatin™ (Regulon Inc. Mountain View, CA, USA) is another liposomal cisplatin formulation composed of dipalmitoyl phosphatidyl glycerol (DPPG), soy PC, CHOL, and mPEG2000-DSPE. Its reported half-life is 60–117 hours, depending on the dose (Boulikas et al 2005; Stathopoulos et al 2005). The study found that Lipoplatin has no nephrotoxicity up to a dose of 125 mg/m2 every 14 days without the serious side effects of cisplatin.
Mitoxantrone (Novantrone®, Wyeth Lederle, Madison, NJ, USA) is a drug used for the treatment of acute myeloid leukemia, multiple sclerosis, and prostate cancer. Despite the promising early results of a PEGylated mitoxantrone formulation (dlakha-Hutcheon et al 1999) the only currently existing formulations with lipids and cardiolipine are in clinical trials (as described above).
PEG alternatives
Despite the well-developed chemistry of PEG coupled to pharmaceuticals, the search for alternative polymers is ongoing; this might be explained both by the patent limitations on PEG and its derivatives, and by the hope of achieving even better control over the properties of modified drugs and drug carriers. Suggested theoretical models for modified protein behavior, and the experimental data available, enable some general requirements for polymers to confer steric protection to drug carriers to be formulated. These polymers should be soluble, hydrophilic, have highly flexible main chain, and high biocompatibility. Synthetic polymers, such as poly(vinyl pyrrolidone) (PVP) and poly(acryl amide) (PAA), are the most prominent examples of other potentially protective polymers (Torchilin et al 1994b, 1994c, 1995). Liposomes containing DSPE covalently linked to poly(2-methyl-2-oxazoline) or to poly(2-ethyl-2-oxazoline) also exhibit extended blood circulation time and decreased uptake by the liver and spleen (Woodle et al 1994). Similar observations have been reported for phosphatidyl polyglycerols (Maruyama et al 1994).
More recent papers describe long circulating liposomes prepared using poly[N-(2-hydroxypropyl) methacrylamide] (Whiteman et al 2001), amphiphilic poly-N-vinylpyrrolidones (Torchilin et al 2001), L-amino-acid-based biodegradable polymer-lipid conjugates (Metselaar et al 2003), and polyvinyl alcohol (Takeuchi et al 2001). All groups of polymer-coated liposomes reported have been found to extend blood circulation time, while liver capture was diminished. These results are comparable with those for PEG-liposomes; the efficacy of the steric effect quite naturally depends on the quantity of incorporated polymer. The prolonged circulation time of polyvinyl alcohol-(molecular weight: 20000) coated liposomes (1.3 mol% coating) was comparable with that of a stealth liposome prepared with 8 mol% of DSPE–PEG2000.
Also, L-amino-acid-based polymers also showed prolonged circulation time and reduced uptake by the MPS, to the same extent as DSPE–PEG2000. Furthermore, these polymers appear to be attractive alternatives for designing long-circulation liposomes, because they have the advantage of being biodegradable.
Targeted liposomes
To increase liposomal drug accumulation in the desired tissues, producing higher and more selective therapeutic activity, the use of targeted liposomes has been suggested. This involves the coupling of targeting moieties capable of recognizing target cells, binding to them, and inducing the internalization of liposomes or encapsulated drugs. Targeting moieties include monoclonal antibodies (MAb) or fragments, peptides, growth factors, glycoproteins, carbohydrates, or receptor ligands (Lopes De Menezes et al 1999; Sapra and Allen 2003; Medina et al 2004). Targeted liposomes offer various advantages over individual drugs targeted by means of polymers or antibodies. One of the most compelling advantages is the dramatic increase in drug amount that can be delivered to the target. Furthermore, the number of ligand molecules exposed on the liposome surface can be increased, improving ligand avidity and degree of uptake. Immunoliposomes also provide a “bystander killing” effect, because the drug molecules can diffuse into adjoining tumor cells.
In order to behave in this fashion, immunoliposomes are built so as to be long-circulating and non-immunogenic, using the stealth technique. Briefly, a PEG (MW 3400) derivative of PE or DSPE containing a maleimide group at the end of the PEG chain is mixed into the liposome formulation (Figure 2). After liposome preparation, reduced thiol groups of Fab or scFv fragments are joined via surface linkage to the maleimide group of the aforementioned PEG-liposome, obtaining a stable thioether bond. This method for preparing immunoliposomes starting from preformed liposomes is the most widely used. Alternatively, commercially available doxorubicin-loaded long-circulating liposomes (DOXIL) have been modified by post-insertion of a monoclonal antibody. MAb, modified with DSPE-PEG conjugate, in which the free PEG terminus is activated with the p-nitrophenylcarbonyl group, has been inserted into preformed liposomes (Lukyanov et al 2004). Therapeutic targets and immunoliposomal compositions are reported in Table 2.
Table 2.
Targeted liposomes in advanced phase of trial
| Targeted with | Encapsulated drug | Disease | References |
|---|---|---|---|
| Anti-HER2 (trastuzumab) | DOXIL® | breast, ovarian cancer | (Park et al 2002) |
| Anti-EGF | doxorubicin, vinorelbine, methotrexate, DNA | solid tumors | (Mamot et al 2003) |
| Anti CD19 | vincristine | lymphoma | (Tseng et al 1999) |
| Anti CD22 | doxorubicin | anti-B-cell lymphoma | (Lundberg et al 2000) |
| Anti CD19 | imatinib | ALL | (Harata et al 2004) |
| Anti-beta1 integrin | doxorubicin | several cancers | (Sugano et al 2000) |
| Anti GD2 | doxorubicin | neuroblastoma | (Pastorino et al 2003) |
| GAH MAb | doxorubicin | gastric, colon and breast cancer | (Hamaguchi et al 2004) |
| Anti-EGF receptor | RNA | brain cancer | (Zhang et al 2004b) |
At present, the only immunoliposome formulation undergoing clinical trials is the PEGylated liposome DXR targeted with the F(ab’)2 fragment of the human MAb GAH, which is able to recognize gastric, colon, and breast cancer cells (Matsumura et al 2004). Another important target antigen is the p185 HER2/neu glycoprotein (HER2), a member of the epidermal growth factor receptor family. This antigen is suitable for targeting because, although HER2 is expressed in healthy tissue, overexpression is unique to tumors, specifically some human breast cancers (25%–30% of cases), gastric, colon, ovarian, and non-small-cell lung carcinoma (Baselga and Metselaar 2000).
The monoclonal antibody anti-HER2 trastuzumab (Herceptin®, Genentech Inc., Vacaville, CA, USA) was the first humanized Mab approved by the FDA and in the EU as monotherapy for metastatic breast cancer. This antibody is widely used in breast cancer, both alone and in combination with chemotherapy agents, and two recent papers describe the GMP- compliant process for producing the F5 single-chain Fv antibody fragment-PEG-lipid conjugate; therapeutic assemblies are made by post-insertion into preformed doxorubicin-encapsulating liposomes (Nellis et al 2005a, 2005b).
In the last few years, antibody-based therapeutics have emerged as important components of therapies for an increasing number of human malignancies (Adams and Weiner 2005) and it is expected that several immunoliposomes will be in trials in the near future.
The main therapeutic area for immunoliposome application is against cancer, but interesting papers describe other applications, such as reducing hemorrhagic transformation after thrombolytic therapy with tissue plasminogen activator in cerebral ischemia (Asahi et al 2003).
Other ligands have been used to specifically target stealth liposomes to receptors over-expressed in cancer cells. Folic acid, which has been used for liposome-specific targeting of DXR, daunorubicin, cisplatin, and other drugs, is one of the most extensively studied (for a recent review (Stephenson et al 2004)) and it has been proposed for boron neutron capture therapy (Stephenson et al 2003). Transferrin is a popular ligand for specific delivery of anticancer drugs, proteins and genes to malignant cells (Ishida et al 2001) as well as for boron neutron capture therapy (Maruyama et al 2004).
Sigma receptors (subtype of opioid receptor) have recently been proposed as an interesting target for different malignancies (breast, melanoma, prostate). An anisamide-derivatized stealth liposomal formulation (DXR) showed high specific toxicity and superior therapeutic effect versus untargeted liposomes (Banerjee et al 2004) and recently haloperidol-associated stealth liposomes can efficiently target genes to sigma receptor overexpressing breast cancer cells (Mukherjee et al 2005).
Further, peptides involved in cell-to-cell interactions have been used as targeting agents for liposomes (now in preclinical trials). Targeting with L-peptide increased liposomal DXR toxicity on nasopharyngeal cells (Lee et al 2004), while vasoactive intestinal peptide 28-mer conjugated with (99mTc) loaded liposome increased the imaging of breast cancer cells (Dagar et al 2003). In addition, stealth liposomes (DXR or 5-FU) have been targeted through RGD-sequence peptides to the integrin of tumor vasculature, demonstrating interesting activity on neuroblastoma, melanoma, and colon in vivo models. Stealth liposomes conjugated with sequences of an angiogenic homing peptide (for instance GPLPLR, APRPG) were able to suppress tumor neovasculature in colon, melanoma and sarcoma cells growing in mice (Kondo et al 2004; Asai and Oku 2005).
Liposome applications
Cationic liposomes for gene delivery
Among various synthetic carriers currently in use in gene therapy, cationic liposomes are the most suitable transfecting vectors. Gene encapsulation in liposomal vesicles allows condensation of DNA plasmid into a highly organized structure, and protects DNA against degradation during storage and in the systemic circulation of the gene encoding a therapeutic protein. Moreover, structural organization of the gene-delivery system must bypass the cell membrane and facilitate endosomal escape, avoiding DNA degradation in the lysosomal compartment (Figure 5).
Figure 5.
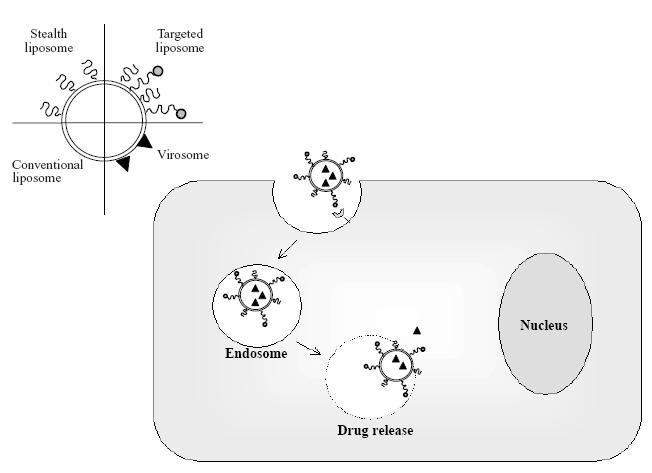
Schematic representation of conventional, stealth, targeted liposomes, and virosomes. Among different mechanism of intracellular uptake of liposomes, endocytosis of targeted liposomes is exemplified.
Numerous cationic lipids have been tested in the formulation of liposomes for gene delivery (the structural formulas of some of them are presented in Figure 6). Transfection efficiency is strongly affected by the presence of three components in the structure of these lipids: a positively charged head-group that interacts with negatively charged DNA, a linker group (which determines the lipid’s chemical stability and biodegradability), and a hydrophobic region to anchor the cationic lipid into the bilayer. Among these, the most often used are N-[1-dioleyloxy)propyl]-N,N,N-trimethylammonium (DOTMA) and dioleoylphophatidylethanolamine (DOPE) in a 1:1 phospholipid mixture (Lipofectin®, Invitrogen Corporation, Carlsbad, CA, USA). Other commercially-available lipids are 2,3-dioleyloxy-N-[2(sperminecarboxamido)ethyl]-N,N-dimethyl-1-propanammonium trifluoroacetate (DOSPA or LipofectAMINE®), 1,2-bis(oleoyloxy)-3-(trim ethylammonio)propane (DOTAP), 1,2-dimystyloxypropyl-3-dimethylhydroxyethyl ammonium bromide (DMRE), 3β[N-(N’,N’-dimethylaminoethane)-carbomoyl] cholesterol (DC-CHOL), and dioctadecylamino-glycyl-spermine (DOGS or Transfectam®) (Mahato 2005).
Figure 6.
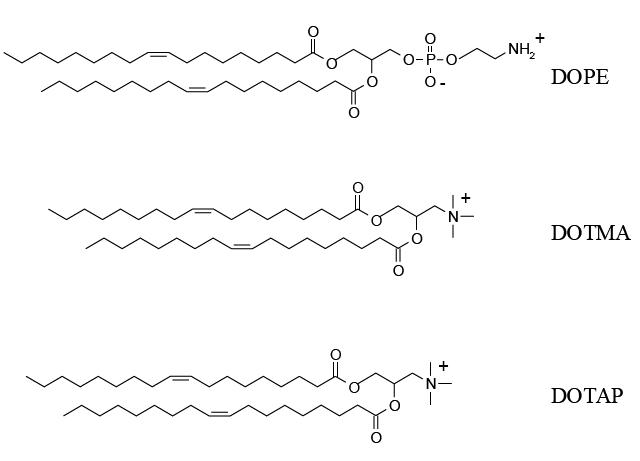
Chemical structures of the cationic lipids: dioleoylphophatidylethanolamine (DOPE), N-[1- dioleyloxy)propyl]-N,N,N-trimethylammonium (DOTMA) and 1,2-bis(oleoyloxy)-3-(trimethylammonio)p ropane (DOTAP).
Nevertheless, the clinical use of cationic liposomes is limited by their instability, rapid clearance, large particle size, toxicity at repeated administration, and induction of immunostimulation and complement activation. Water-soluble lipopolymers, obtained by conjugating different fatty acid chains to branched polethylenimine (PEI) of 25 kDa or above, have been shown to be effective for gene delivery; they can be delivered into the cytoplasm after endosomal disruption. Similarly, phosphatidyl ethylene glycol (PhosEG) has been linked to the amino group of branched PEI (Mahato 2005).
On the other hand, PEGylation of cationic liposomal vesicles is a promising alternative way to overcome these problems, prolonging circulation time in vivo and increasing accumulation at the disease site, even if the transfecting efficiency might be significantly reduced.
In liposomes composed of a cationic lipid (DOTAP, DOGS, dimethyldioctadecylammonium bromide-DDAB), a neutral lipid (DOPE) and a phospolipid derivative of PEG (PEG-PE), complexing 18-mer phosphothioate as a model for active oligodexyribonucleotide (ODN), surface modification with a relatively large amount of PEG (5.7 mol%) has been showed to improve ODN loading without losing structural activity or stability of the resulting complexes, retaining size without vesicle aggregation. Moreover, the hydrophilic shell of PEG enhances the in vitro stability by evading mononuclear phagocyte clearance, and retains a high level of the originally loaded ODN in the complex after plasma incubation. Only after modification of PEG cationic liposomes with targeting agents can cytoplasmatic delivery of DNA material be observed. The PEG-modified complex conjugated anti-HER2 F(ab’) dramatically enhanced cell uptake, increasing diffuse cytoplasmatic and nuclear localization of ODN in SK-BR-3 cells (Meyer et al 1998).
In liposomes composed of DODAC/DOPE, the inclusion of 5 mol% of PEG lipid conjugate did not inhibit uptake by the cell membrane of lipid/DNA complex, but substantially modified the ability of the cationic liposomal carrier to disrupt the endosomal membrane. Endosomal escape into the cytoplasm depended on the acyl chain of the lipid complex and on the molecular weight of the PEG. Optimizing the desorbtion rate of PEG-lipids may be one approach to overcoming the inhibitory effect on intracellular delivery of plasmid (Song et al 2002).
To contrast the low tranfection efficiency of PEG-modified cationic liposomes due to the absence of a net positive charge on the vesicle surface, a series of cationic PEG-lipids with one or more positive charges have been synthesized and designed for post-insertion in preformed stabilized plasmid-lipid particles (Figure 7). Incorporation of cationic-poly(ethylene glycol)-lipid conjugates (CPL4) in DOPE/DODAC/PEG-CerC20 liposomes resulted in both improved uptake into BHK cells and dramatically enhanced transfection potency in the presence of Ca2+, which assists in destabilizing the endosomal membrane following uptake. However, in this type of liposomal preparation, aggregation of vesicles was observed, probably due to formation of H-bonding between the amino and carbonyl groups present in the distal head-group at the end of the PEG chain (Palmer et al 2003). In order to optimize CLP-liposomes for systemic delivery, the length of PEG linker in the CPL can be modulated. When the PEG3400 linker extended beyond the PEG-CerC20 “cloud” was employed for liposomal insertion, charged liposomal systems were produced that rapidly cleared from circulation; it was suggested that a shorter PEG linker might be used, such as PEG1000, allowing the PEG-CerC20 to shield the positive charge of CPL. Moreover, PEG-CerC20 can be designed to slowly dissociate at the disease site, achieving exposure of the CPL at the target area with retention of long-circulation properties and interaction between liposomes and targeting cell (Chen et al. 2004).
Figure 7.
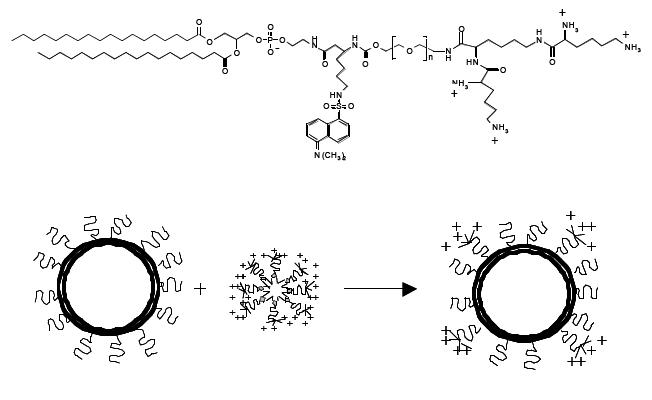
Structure of dansylated cationic-poly(ethylene glycol)-lipid (CPL4) and schematic representation of the post-insertion method for the production of CPL4 liposomes (redrawn from Palmer et al 2003).
Overall, the most suitable use of PEG is as a tether for a specific ligand on the surface of these systems, in order to obtain a target-specific gene delivery facilitating internalization in cells and endosomal escape. Cell-penetrating peptides (CPP), such as Trans-activating transcriptional activator (TAT), homeodomain of antennapedia (Antp), herpex simplex virus type I protein VP22 and transportan, have been reported to guarantee direct cytosolic delivery when coupled with several carriers, including liposomes. Multiple TATp molecules can be attached on the surface of liposomes via the spacer group of p-nitrophenylcarbonyl-PEG-phosphatidylethanolamine. TATp-liposomes-DNA complexes were found to be capable of transfection of both normal and cancer cells in vitro and in vivo with lower cytotoxicity that the commonly used lipid-based gene delivery systems (Gupta et al 2005).
Liposomes for diagnostic imaging
Actively or passively targeted liposomes can be used as carriers for contrast agents to increase the signal difference between areas of interest and background, and to specifically localize the contrast moieties in the target tissues or organs. The versatility of liposomal vesicles to carry different types of compound in the bilayer or in the aqueous compartment makes them suitable for all contrast procedures, including gamma-scintigraphy, magnetic resonance imaging (MRI), computed tomography imaging (CTI), and sonography. Using liposomes in diagnostic imaging leads to several advantages, owing to their capability to incorporate multiple contrast moieties, to specifically deliver the agent to the target area, and to enhance the contrasting signal.
In order to incorporate diagnostic agents (111In, 99Tc, Mn, Gd, etc) in liposomes, metals can be complexed with a soluble chelating agent (such as DTPA) that will be encapsulated in the aqueous core of the vesicles. Alternatively, the chelating compound complexing with the metal can be derivatized with a hydrophobic group for insertion in the lipid bilayer. Gd-DTPA complexes were the first to be incorporated in the aqueous core. Among the various lipophilic DTPA–conjugates that have been synthesized, DTPA-sterylamine (DTPA-SE) and DTPA-phosphatidyl ethanolamine (DTPA-PE) show reduced leakage and toxicity of potentially toxic metals. (DTPA-polylysil)glutaryl phosphatidyl ethanolamine (DTPA-PLL-NGPE) is a poly-chelating amphiphilic polymer suitable for liposome incorporation that drastically increases the number of metal ions attached to a single lipid conjugate. In these cases, metals are situated on the liposomal surface, directly exposed to the aqueous environment, thus enhancing the contrast proprieties.
To increase the stability and half-life of vesicles in the body after administration, liposomes for use as contrast agent can be modified with PEG. DTPA-PLL-NGPE liposomes with PEG5000 containing Gd improved visualization in the lymph nodes: PEG moieties increase the amount of water directly in contact with the Gd on the liposomal surface, and contrast phagocytic cell uptake at the injection site.
Long-circulating Gd liposomes have been successfully used for blood pool imaging, prolonging the presence of the contrast agent in the body. After systemic administration of Gd-DTPA-PLL-NGPE/PEG-liposomes, the signal was immediately clear and lasted for up to 4 hours (Torchilin 2000).
Incorporation of large amount of Gd-containing lipids in sterically stabilized PEGylated DSPC- or DOPC-based liposomes showed increased relaxivity compared with traditional Gd-DTPA; because of the higher accessibility of water, liposomes containing unsaturated phospholipids also showed increased relaxivity in comparison with liposomes composed of saturated phospholipids. These liposomes are therefore highly potent contrast agents for application in MR imaging (Strijkers et al 2005).
Liquid-filled liposomes have been demonstrated to be echogenic. The liquid-like composition of the vesicles makes them more resistant to pressure and mechanical stress than encapsulated gas microbubbles. Moreover, their long circulation characteristics and their small size are favorable in echography. Definity® (Bristol-Myers Squibb Medical Imaging, Inc. New York, NY, USA) is a contrast agent containing perfluoropropane with a phospholipid shell approved in the US for use in cardiology.
After lyophilization, liposomes can encapsulated small amounts of air, being echogenic upon rehydration. Is it possible to modulate the liposomal composition by changing the ratio between PC, PE, PG, and CHOL to produce agents that are echogenic in vitro and in vivo (Dayton and Ferrara 2002). Echogenic liposomes have also been utilized for intravascular ultrasound imaging, targeting the vesicles to the vascular signature associated with arteroma development (Morawski et al 2005).
A pH liposomal MRI contrast agent has recently been introduced as a potential marker of low pH in tumor interstitium. DPPE/DSPG/GdDTPA-BMA liposomes displayed increased relaxivity in the blood when the pH was below the physiological level, due to aggregation and leakage of GdDTPA-BMA. To optimize these liposomal formulations it is necessary that they retain pH sensitivity in the blood and accumulate in the tumor. Blood circulation time was prolonged by incorporating 1.5mol% in DPPE/ DSPG liposomal GdDTPA-BMA, but the pH-response was reduced. A compromise would be necessary between long blood retention and pH-sensitivity (Lokling et al 2004).
Liposomes for vaccines
Genetic vaccination-encoding antigens from bacteria, virus, and cancer has shown promise in protecting humoral and cellular immunity. The success of liposomes-based vaccines has been demonstrated in clinical trials and further human trials are also in progress. Liposomes are of interest as carriers of antigens, especially because they act as effective adjuvants for the immune system response, without causing granulomas at the injection site and producing no hypersensitivity reactions (Chen and Huang 2005). Liposome formulations would also protect their DNA content from deoxyribonuclease attack. Moreover, their transfection efficiency could be improved by modulating surface charge, size, and lipid composition of the vesicle and entrapping additional adjuvant or immunostimulator compounds in the antigen formulation. Several strategies have been followed to target liposomes to cell receptors, such as antibodies (or Fc-γ) or branched chain mannose moieties. Cationic or pH-sensitive liposomes that are able to release their contents into the cytoplasm following endocytosis have also been developed.
Two commercial vaccines based on virosome technology are currently on the market. Epaxal® (Berna Biotech Ltd, Bern, Switzerland), a hepatitis A vaccine, has inactivated hepatitis A virus particles adsorbed on the surface of the immunopotentiating reconstituted influenza virosomes (IRIV). In Inflexal® V (Berna Biotech Ltd) the virosome components themselves are the vaccine protective antigens (Copland et al 2005). Virosomes are liposomal formulations that have viral envelope proteins anchored to their lipid membrane. The lipid bilayer is composed of PC intercalated with the virus-derived proteins hemagglutinin and neuraminidase. These virus-like particles have proven to be effective immunogens with unique adjuvant properties (Felnerova et al 2004).
Liposome-encapsulated malaria vaccine contains monophosphoryl lipid A as adjuvant in the bilayer and the formulation is adsorbed on aluminum hydroxide. In a Phase I dose-escalating study, the formulation showed induction of higher level of anti-malaria antibody in human volunteers (Chen and Huang 2005). Some liposomal formulations are under investigation in preclinical studies against Yersina pestis, ricin toxin and Ebola Zaire virus. Liposomes against ricin or pestis, composed of PC/CHOL/DDA containing KWC vaccine, were administered intranasally to C57BL/6 mice; liposome formulations gave higher protection from infection than KWC in buffer. Liposomes composed of PC and CHOL containing ricin toxoid and ricin A-chain (rA) increased antibody responses to the rA chain. Liposomes composed of DMPC/DMPG/CHOL with or without lipid A containing Ebola Zaire virus have been tested in mice and cynomolgus monkeys (Bramwell et al 2005).
Liposome vaccination also has the potential to be a powerful weapon in cancer treatment. Chen et al (Chen and Huang 2005) developed a novel liposome-based system for the delivery of plasmid DNA. Lipid-polycation-DNA particles are formed by combining cationic liposomes and polycation-condensed DNA organized in a virus-like structure able to release its content in the cytoplasm. Cationic liposomes promote a much higher humoral and cytotoxic T lymphocyte immune response against the antigen encoded by the entrapped DNA vaccine. Liposome-stabilized prostate cancer vaccine is under investigation in a series of Phase I trials in patient with advanced prostate cancer. The new liposome-lipid A-prostate-specific antigen formulation showed greater safety and higher immunological potency than other formulations and has been transitioned to Phase II trials (Chen and Huang 2005).
Other pharmaceutical uses of stealth liposomes
Active research is in progress in the area of liposomes for use as vesicular containers, in particular for hemoglobin as blood substitute. Liposome-encapsulated hemoglobin (LEH) is being developed as an oxygen therapeutic. The spatial isolation of hemoglobin by a lipid bilayer potentially minimizes the cardiovascular/hemodynamic effects associated with other modified forms of hemoglobin; moreover, it is possible to co-encapsulate reductants, antioxidative enzyme systems, and oxygen-affinity modifiers with hemoglobin so as to artificially recreate the red blood cell environment. The circulation half-life is 65 hours for this PEG-LEH formulation; the results demonstrate that LEH circulates for a prolonged time after administration and that the animals tolerate at least 25% of blood exchange without any distress (Phillips et al 1999; Arifin and Palmer 2005).
Conclusions
The development of liposomes as carriers for therapeutic molecules is an ever-growing research area. The possibility of modulating the technological characteristics of the vesicles makes them highly versatile both as carriers of several types of drug (from conventional chemotherapeutics to proteins and peptides) and in therapeutic applications (from cancer therapy to vaccination). In recent years, several important formulations for the treatment of different diseases have been developed. Among these, PEG-coated liposomes are becoming increasingly important, giving technological and biological stability to liposomal systems. At present, few PEGylated liposomal formulations have been approved or are in advanced trials (DOXIL, SPI077, Lipoplatin, S-CKD-602), but stealth technology for different applications is destined to continue developing. PEG-derivatized liposomes with increased stability can easily be modified using a wide array of targeting moieties (MAb, ligands) to deliver the drug specifically to the target tissues with increasing accuracy. Moreover, PEG grafted onto the liposome surface can guide the liposome to a specific intracellular target, using for example cell-penetrating proteins and peptides as targeting agents. The development of liposome delivery to particular subcellular compartments is a field of great interest in different fields, such as gene therapy and vaccination. The interaction of stealth liposomes with cell membranes, and release of the drug in the neighborhood of target tissues are still under investigation, but some recent studies indicate that the use of detachable PEG may facilitate cell penetration and/or intracellular delivery of vesicles.
Taking into account these considerations and the great advantages of PEGylated liposomes in decreasing aspecific drug toxicity and in passively targeting the incorporated molecules to the site of action, new and “improved” stealth liposomal formulations designed for different therapeutic and diagnostic areas may soon be expected to arrive on the market.
References
- Adams GP, Weiner LM. Monoclonal antibody therapy of cancer. Nat Biotechnol. 2005;23:1147–57. doi: 10.1038/nbt1137. [DOI] [PubMed] [Google Scholar]
- Adlakha-Hutcheon G, Bally MB, Shew CR, et al. Controlled destabilization of a liposomal drug delivery system enhances mitoxantrone antitumor activity. Nat Biotechnol. 1999;17:775–9. doi: 10.1038/11710. [DOI] [PubMed] [Google Scholar]
- Agrawal AK, Gupta CM. Tuftsin-bearing liposomes in treatment of macrophage-based infections. Adv Drug Deliv Rev. 2000;41:135–46. doi: 10.1016/s0169-409x(99)00061-7. [DOI] [PubMed] [Google Scholar]
- Alberts DS, Muggia FM, Carmichael J, et al. Efficacy and safety of liposomal anthracyclines in phase I/II clinical trials. Semin Oncol. 2004;31(Suppl 13):53–90. doi: 10.1053/j.seminoncol.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Allen C, Dos SN, Gallagher R, et al. Controlling the physical behavior and biological performance of liposome formulations through use of surface grafted poly(ethylene glycol) Biosc Rep. 2002;22:225–50. doi: 10.1023/a:1020186505848. [DOI] [PubMed] [Google Scholar]
- Allen TM, Hansen C, Martin F, et al. Liposomes containing synthetic lipid derivatives of poly(ethylene glycol) show prolonged circulation half-lives in vivo. Biochim Biophys Acta. 1991;1066:29–36. doi: 10.1016/0005-2736(91)90246-5. [DOI] [PubMed] [Google Scholar]
- Allen TM, Hansen C, Rutledge J. Liposomes with prolonged circulation times: factors affecting uptake by reticuloendothelial and other tissues. Biochim Biophys Acta. 1989;981:27–35. doi: 10.1016/0005-2736(89)90078-3. [DOI] [PubMed] [Google Scholar]
- Allen TM, Hansen CB, Guo LSS. Subcutaneous administration of liposomes: a comparison with the intravenous and intraperitoneal routes of injection. Biochim Biophys Acta. 1993;1150:9–16. doi: 10.1016/0005-2736(93)90115-g. [DOI] [PubMed] [Google Scholar]
- Allen TM, Martin FJ. Advantages of liposomal delivery systems for anthracyclines. Semin Oncol. 2004;31(Suppl 13):5–15. doi: 10.1053/j.seminoncol.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Alving CR. Therapy of leishmaniasis: superior efficacies of liposome-encapsulated drugs. Proc Natl Acad Sci U S A. 1978;75:2959–63. doi: 10.1073/pnas.75.6.2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreff M, Estey EH. Method of cancer treatment. 2004057989. US patent. 2004
- Anonymous. Nystatin-liposomal. AR 121, Nyotran. Drugs R D. 1999;1:181–3. doi: 10.2165/00126839-199901020-00018. [DOI] [PubMed] [Google Scholar]
- Anonymous. Vincristine liposomal--INEX: lipid-encapsulated vincristine, onco TCS, transmembrane carrier system--vincristine, vincacine, vincristine sulfate liposomes for injection, VSLI. Drugs R D. 2004;5:119–23. doi: 10.2165/00126839-200405020-00012. [DOI] [PubMed] [Google Scholar]
- Arifin DR, Palmer AF. Physical properties and stability mechanisms of poly(ethylene glycol) conjugated liposome encapsulated hemoglobin dispersions. Artif Cells Blood Substit Immobil Biotechnol. 2005;33:137–62. doi: 10.1081/bio-200055880. [DOI] [PubMed] [Google Scholar]
- Arikan S, Rex JH. Nystatin LF (Aronex/Abbott) Curr Opin Investig Drugs. 2001;2:488–495. [PubMed] [Google Scholar]
- Asahi M, Rammohan R, Sumii T, et al. Antiactin-targeted immunoliposomes ameliorate tissue plasminogen activator-induced hemorrhage after focal embolic stroke. J Cereb Blood Flow Met. 2003;23:895–9. doi: 10.1097/01.WCB.0000072570.46552.DF. [DOI] [PubMed] [Google Scholar]
- Asai M, Oku N. Liposomalized oligopeptides in cancer therapy. Methods Enzymol. 2005;391:163–76. doi: 10.1016/S0076-6879(05)91009-4. [DOI] [PubMed] [Google Scholar]
- Awada A, Gil T, Sales F, et al. Prolonged schedule of temozolomide (Temodal) plus liposomal doxorubicin (Caelyx) in advanced solid cancers. Anticancer Drugs. 2004;15:499–502. doi: 10.1097/01.cad.0000127331.29310.8a. [DOI] [PubMed] [Google Scholar]
- Babai I, Samira S, Barenholz Y, et al. A novel influenza subunit vaccine composed of liposome-encapsulated haemagglutinin/neuraminidase and IL-2 or GM-CSF. I. Vaccine characterization and efficacy studies in mice. Vaccine. 1999;17:1223–38. doi: 10.1016/s0264-410x(98)00346-6. [DOI] [PubMed] [Google Scholar]
- Banerjee R, Tyagi P, Li S, et al. Anisamide-targeted stealth liposomes: a potent carrier for targeting doxorubicin to human prostate cancer cells. Int J Cancer. 2004;112:693–700. doi: 10.1002/ijc.20452. [DOI] [PubMed] [Google Scholar]
- Baselga J, Metselaar JM. Monoclonal antibodies: Clinical applications: Monoclonal antibodies directed against growth factor receptors. In: Rosenburg SA, editor. Principles and practice of biological therapy of cancer. Philadelphia: Lippincott Williams, Wilkins; 2000. pp. 475–89. [Google Scholar]
- Basu MK, Lala S. Macrophage specific drug delivery in experimental leishmaniasis. Curr Mol Med. 2004;4:681–9. doi: 10.2174/1566524043360186. [DOI] [PubMed] [Google Scholar]
- Blume G, C G. Molecular mechanism of the lipid vesicle longevity in vivo. Biochim Biophys Acta. 1993;1146:157–68. doi: 10.1016/0005-2736(93)90351-y. [DOI] [PubMed] [Google Scholar]
- Bomgaars L, Geyer JR, Franklin J, et al. Phase I trial of intrathecal liposomal cytarabine in children with neoplastic meningitis. J Clin Oncol. 2004;22:3916–21. doi: 10.1200/JCO.2004.01.046. [DOI] [PubMed] [Google Scholar]
- Boulikas T, Stathopoulos GP, Volakakis N, et al. Systemic Lipoplatin infusion results in preferential tumor uptake in human studies. Anticancer Res. 2005;25:3031–9. [PubMed] [Google Scholar]
- Bradley AJ, Devine DV, Ansell SM, et al. Inhibition of liposome-induced complement activation by incorporated poly(ethylene glycol)-lipids. Arch Biochem Biophys. 1998;357:185–94. doi: 10.1006/abbi.1998.0798. [DOI] [PubMed] [Google Scholar]
- Bramwell VW, Eyles JE, Oya AH. Particulate delivery systems for biodefense subunit vaccines. Adv Drug Del Rev. 2005;57:1247–65. doi: 10.1016/j.addr.2005.01.010. [DOI] [PubMed] [Google Scholar]
- Briasoulis E, Karavasilis V, Tzamakou E, et al. Interaction pharmacokinetics of pegylated liposomal doxorubicin (Caelyx) on coadministration with paclitaxel or docetaxel. Cancer Chemother Pharmacol. 2004;53:452–7. doi: 10.1007/s00280-003-0750-5. [DOI] [PubMed] [Google Scholar]
- Caliceti P, Veronese FM. Pharmacokinetic and biodistribution properties of poly(ethylene glycol)-protein conjugates. Adv Drug Del Rev. 2003;55:1261–77. doi: 10.1016/s0169-409x(03)00108-x. [DOI] [PubMed] [Google Scholar]
- Chen T, Palmer LR, Fenske DB, et al. Distal cationic poly(ethylene glycol) lipid conjugates in large unilamellar vesicles prepared by extrusion enhance liposomal cellular uptake. J Liposome Res. 2004;14:155–73. doi: 10.1081/lpr-200033437. [DOI] [PubMed] [Google Scholar]
- Chen WC, Huang L. Non-viral vector as vaccine carrier. Adv Genet. 2005;54:315–37. doi: 10.1016/S0065-2660(05)54013-6. [DOI] [PubMed] [Google Scholar]
- Chonn A, Cullis PR, Devine DV. The role of surface charge in the activation of the classical and alternative pathways of complement by liposomes. J Immunol. 1991;146:4234–41. [PubMed] [Google Scholar]
- Chonn A, Semple SC, Cullis PR. Association of blood proteins with large unilamellar liposomes in vivo. Relation to circulation lifetimes. J Biol Chem. 1992;267:18759–765. [PubMed] [Google Scholar]
- Chonn A, Semple SC, Cullis PR. Beta 2-glycoprotein I is a major protein associated with very rapidly cleared liposomes in vivo, suggesting a significant role in the immune clearance of “non-self” particles. J Biol Chem. 1995;270:25845–9. doi: 10.1074/jbc.270.43.25845. [DOI] [PubMed] [Google Scholar]
- Copland MJ, Rades T, Davies NM, et al. Lipid based particulate formulations for the delivery of antigen. Immunol Cell Biol. 2005;83:97–105. doi: 10.1111/j.1440-1711.2005.01315.x. [DOI] [PubMed] [Google Scholar]
- Corvo ML, Boerman OC, Oyen WJ, et al. Intravenous administration of superoxide dismutase entrapped in long circulating liposomes. II. In vivo fate in a rat model of adjuvant arthritis. Biochim Biophys Acta. 1999;1419:325–34. doi: 10.1016/s0005-2736(99)00081-4. [DOI] [PubMed] [Google Scholar]
- Crosasso P, Ceruti M, Brusa P, et al. Preparation, characterization and properties of sterically stabilized paclitaxel-containing liposomes. J Control Release. 2000;63:19–30. doi: 10.1016/s0168-3659(99)00166-2. [DOI] [PubMed] [Google Scholar]
- Dagar S, Krishnadas A, Rubinstein I, et al. VIP grafted sterically stabilized liposomes for targeted imaging of breast cancer: in vivo studies. J Control Release. 2003;91:123–33. doi: 10.1016/s0168-3659(03)00242-6. [DOI] [PubMed] [Google Scholar]
- Damen J. Transfer and exchange of phospholipid between small unilamellar liposomes and rat plasma high-density lipoproteins: dependence on cholesterol and phospholipid composition. Biochim Biophys Acta. 2005;665:538–45. doi: 10.1016/0005-2760(81)90268-x. [DOI] [PubMed] [Google Scholar]
- Dark GG, Calvert AH, Grimshaw R, et al. Randomized trial of two intravenous schedules of the topoisomerase I inhibitor liposomal lurtotecan in women with relapsed epithelial ovarian cancer: a trial of the national cancer institute of Canada clinical trials group. J Clin Oncol. 2005;23:1859–66. doi: 10.1200/JCO.2005.02.028. see comment. [DOI] [PubMed] [Google Scholar]
- Dayton PA, Ferrara KW. Targeted imaging using ultrasound. J Magn Reson Imaging. 2002;16:362–77. doi: 10.1002/jmri.10173. [DOI] [PubMed] [Google Scholar]
- de Gennes PG. Conformations of polymers attached to an interface. Macromolecules. 1980;13:1069–75. [Google Scholar]
- Devine DV, Wong K, Serrano K, et al. Liposome-complement interactions in rat serum: Implications for liposome survival studies. Biochim Biophys Acta. 1994;1191:43–51. doi: 10.1016/0005-2736(94)90231-3. [DOI] [PubMed] [Google Scholar]
- Dreborg S, Akerblom EB. Immunotherapy with monomethoxy-polyethylene glycol modified allergenes. Crit Rev Ther Drug Carrier Syst. 1990:315–65. [PubMed] [Google Scholar]
- Drummond DC, Meyer O, Hong K, et al. Optimizing liposomes for delivery of chemotherapeutic agents to solid tumors. Pharmacol Rev. 1999;51:691–743. [PubMed] [Google Scholar]
- Eichhorn ME, Becker S, Strieth S, et al. Paclitaxel encapsulated in cationic lipid complexes (MBT-0206) impairs functional tumor vascular properties as detected by dynamic contrast enhanced magnetic resonance imaging. Cancer Biol Ther. 2006;5 doi: 10.4161/cbt.5.1.2346. In press. [DOI] [PubMed] [Google Scholar]
- Falcone DJ. Fluorescent opsonization assay: binding of plasma fibronectin to fibrinderivatized fluorescent particles does not change their uptake by macrophages. J Leukoc Biol. 1986;39:1–12. doi: 10.1002/jlb.39.1.1. [DOI] [PubMed] [Google Scholar]
- Felnerova D, Viret JF, Gluck R, et al. Liposomes and virosomes as delivery systems for antigens, nucleic acids and drugs. Curr Opin Biotechnol. 2004;15:518–29. doi: 10.1016/j.copbio.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Funato KYRKH. Contribution of complement system on destabilization of liposomes composed of hydrogenated egg phosphatidylcholine in rat fresh plasma. Biochim Biophys Acta. 1992;1103:204. doi: 10.1016/0005-2736(92)90087-3. [DOI] [PubMed] [Google Scholar]
- Gabizon A, Papahadjopoulos D. Liposome formulations with prolonged circulation time in blood and enhanced uptake by tumors. Proc Natl Acad Sci U S A. 1988;85:6949–53. doi: 10.1073/pnas.85.18.6949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabizon AA, Muggia FM. Long circulating liposomes: old drugs, new therapeutics. Springer-Verlag and Landes Bioscience; 1998. [Google Scholar]
- Gulati M, Grover M, Singh S, et al. Lipophilic drug derivatives in liposomes. Int J Pharm. 1998;165:129–68. [Google Scholar]
- Gupta B, Levchenko TS, Torchilin VP. Intracellular delivery of large molecules and small particles by cell-penetrating proteins and peptides. Adv Drug Deliv Rev. 2005;57:637–51. doi: 10.1016/j.addr.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Hamaguchi T, Matsumura Y, Nakanishi Y, et al. Antitumor effect of MCC-465, pegylated liposomal doxorubicin tagged with newly developed monoclonal antibody GAH, in colorectal cancer xenografts. Cancer Sci. 2004;95:608–13. doi: 10.1111/j.1349-7006.2004.tb02495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han ILing YH, al-Baker S, et al. Cellular pharmacology of liposomal cis-bis-neodecanoato-trans-R,R-1,2-diaminocyclohexaneplatinum(II) in A2780/S and A2780/PDD cells. Cancer Res. 1993;53:4913–19. [PubMed] [Google Scholar]
- Harashima H, Sakata K, Funato K, et al. Enhanced hepatic uptake of liposomes through complement activation depending on the size of liposomes. Pharm Res. 1994;11:402–6. doi: 10.1023/a:1018965121222. [DOI] [PubMed] [Google Scholar]
- Harata M, Soda Y, Tani K, et al. CD19-targeting liposomes containing imatinib efficiently kill Philadelphia chromosome-positive acute lymphoblastic leukemia cells. Blood. 2004;104:1442–9. doi: 10.1182/blood-2004-02-0588. [DOI] [PubMed] [Google Scholar]
- Harrington KJ, Lewanski CR, Northcote AD, et al. Phase I-II study of pegylated liposomal cisplatin (SPI-077) in patients with inoperable head and neck cancer. Ann Oncol. 2001;12:493–6. doi: 10.1023/a:1011199028318. [DOI] [PubMed] [Google Scholar]
- Harrington KJ, Rowlinson-Busza G, Syrigos KN, et al. Influence of tumour size on uptake of(111)ln-DTPA-labelled pegylated liposomes in a human tumour xenograft model. Br J Cancer. 2000a;83:684–8. doi: 10.1054/bjoc.2000.1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington KJ, Rowlinson-Busza G, Syrigos KN, et al. Biodistribution and pharmacokinetics of 111In-DTPA-labelled pegylated liposomes in a human tumour xenograft model: implications for novel targeting strategies. Br J Cancer. 2000b;83:232–8. doi: 10.1054/bjoc.1999.1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JM. Poly(ethylene glycol) chemistry: biotechnical and biomedical applications. Plenum Press; 1992. [Google Scholar]
- Hau P, Fabel K, Baumgart U, et al. Pegylated liposomal doxorubicin-efficacy in patients with recurrent high-grade glioma. Cancer. 2004;100:1199–207. doi: 10.1002/cncr.20073. [DOI] [PubMed] [Google Scholar]
- Hussein MA, Anderson KC. Role of liposomal anthracyclines in the treatment of multiple myeloma. Semin Oncol. 2004;31(Suppl 13):147–60. doi: 10.1053/j.seminoncol.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Immordino ML, Brusa P, Arpicco S, et al. Preparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing docetaxel. J Control Release. 2003;91:417–29. doi: 10.1016/s0168-3659(03)00271-2. [DOI] [PubMed] [Google Scholar]
- Ishida O, Maruyama K, Tanahashi H, et al. Liposomes bearing polyethyleneglycol-coupled transferrin with intracellular targeting property to the solid tumors in vivo. Pharm Res. 2001;18:1042–8. doi: 10.1023/a:1010960900254. [DOI] [PubMed] [Google Scholar]
- Ishida T, Harada M, Wang XY, et al. Accelerated blood clearance of PEGylated liposomes following preceding liposome injection: effects of lipid dose and PEG surface-density and chain length of the first-dose liposomes. J Control Release. 2005;105:305–17. doi: 10.1016/j.jconrel.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Ishida T, Harashima H, Kiwada H. Liposome clearance. Biosci Rep. 2002;22:197–224. doi: 10.1023/a:1020134521778. [DOI] [PubMed] [Google Scholar]
- Jaeckle KA, Phuphanich S, Bent MJ, et al. Intrathecal treatment of neoplastic meningitis due to breast cancer with a slow-release formulation of cytarabine. Br J Cancer. 2001;84:157–63. doi: 10.1054/bjoc.2000.1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone SA, Masin D, Mayer L, et al. Surface associated serum proteins inhibit the uptake of phosphatidylserine and poly(ethylene glycol) liposomes by mouse macrophages. Biochim Biophys Acta. 2001;1513:25–37. doi: 10.1016/s0005-2736(01)00292-9. [DOI] [PubMed] [Google Scholar]
- Katsaros D, Oletti MV, Rigault de la Longrais A, et al. Clinical and pharmacokinetic phase II study of pegylated liposomal doxorubicin and vinorelbine in heavily pretreated recurrent ovarian carcinoma. Ann Oncol. 2005;16:300–6. doi: 10.1093/annonc/mdi055. [DOI] [PubMed] [Google Scholar]
- Kim ES, Lu C, Khuri FR, et al. A phase II study of STEALTH cisplatin (SPI-77) in patients with advanced non-small cell lung cancer. Lung Cancer. 2001;34:427–32. doi: 10.1016/s0169-5002(01)00278-1. [DOI] [PubMed] [Google Scholar]
- Kondo M, Asai T, Katanasaka Y, et al. Anti-neovascular therapy by liposomal drug targeted to membrane type-1 matrix metalloproteinase. Int J Cancer. 2004;108:301–6. doi: 10.1002/ijc.11526. [DOI] [PubMed] [Google Scholar]
- Kraut EH, Fishman MN, Lorusso PM, et al. Pharmacogenomic and pharmacokinetic assessment of liposome encapsulated SN-38 (LE-SN38) in advanced cancer patients. J Clin Oncol (Meeting Abstracts) 2004;22:2501. [Google Scholar]
- Krown SE, Northfelt DW, Osoba D, et al. Use of liposomal anthracyclines in Kaposi’s sarcoma. Semin Oncol. 2004;31(Suppl 13):36–52. doi: 10.1053/j.seminoncol.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Laverman P, Carstens MG, Boerman OC, et al. Factors affecting the accelerated blood clearance of polyethylene glycol liposomes upon repeated injection. J Pharmacol Exp Ther. 2001;298:607–12. [PubMed] [Google Scholar]
- Lee TY, Wu HC, Tseng YL, et al. A novel peptide specifically binding to nasopharyngeal carcinoma for targeted drug delivery. Cancer Res. 2004;64:8002–8. doi: 10.1158/0008-5472.CAN-04-1948. [DOI] [PubMed] [Google Scholar]
- Liu D, Mori A, Huang L. Role of liposome size and RES blockade in controlling biodistribution and tumor uptake of GM1-containing liposomes. Biochim Biophys Acta. 1992;1104:95–101. doi: 10.1016/0005-2736(92)90136-a. [DOI] [PubMed] [Google Scholar]
- Lokling KE, Fossheim SL, Klaveness J, et al. Biodistribution of pH-responsive liposomes for MRI and a novel approach to improve the pH-responsiveness. J Control Release. 2004;98:87–95. doi: 10.1016/j.jconrel.2004.04.015. [DOI] [PubMed] [Google Scholar]
- Lopes De, Menezes DE, Kirchmeier MJ, Gagne J-F, et al. Cellular trafficking and cytotoxicity of anti-CD19-targeted liposomal doxorubicin in B lymphoma cells. J Liposome Res. 1999;9:199–228. [Google Scholar]
- Lu C, Perez-Soler R, Piperdi B, et al. Phase II study of a liposome-entrapped cisplatin analog (L-NDDP) administered intrapleurally and pathologic response rates in patients with malignant pleural mesothelioma. J Clin Oncol. 2005;23:3495–501. doi: 10.1200/JCO.2005.00.802. [DOI] [PubMed] [Google Scholar]
- Lukyanov AN, Elbayoumi TA, Chakilam AR, et al. Tumor-targeted liposomes: doxorubicin-loaded long-circulating liposomes modified with anti-cancer antibody. J Control Release. 2004;100:135–44. doi: 10.1016/j.jconrel.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Lundberg BB, Griffiths G, Hansen HJ. Specific binding of sterically stabilized anti-B-cell immunoliposomes and cytotoxicity of entrapped doxorubicin. Int J Pharm. 2000;205:101–8. doi: 10.1016/s0378-5173(00)00492-0. [DOI] [PubMed] [Google Scholar]
- Lyass O, Uziely B, Ben-Yosef R, et al. Correlation of toxicity with pharmacokinetics of pegylated liposomal doxorubicin (DOXIL) in metastatic breast carcinoma. Cancer. 2000;89:1037–47. doi: 10.1002/1097-0142(20000901)89:5<1037::aid-cncr13>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Maeda H, Sawa T, Konno T. Mechanism of tumor targeted delivery of macromolecular drugs, including the EPR effect in solid tumor and clinical overview of the prototype polymeric drug SMANCS. J Control Release. 2001;74:47–61. doi: 10.1016/s0168-3659(01)00309-1. [DOI] [PubMed] [Google Scholar]
- Mahato RI. Water insoluble and soluble lipids for gene delivery. Adv Drug Deliv Rev. 2005;57:699–712. doi: 10.1016/j.addr.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Mahmood I, Green MD. Pharmacokinetic and pharmacodynamic considerations in the development of therapeutic proteins. Clin Pharmacokinet. 2005;44:331–47. doi: 10.2165/00003088-200544040-00001. [DOI] [PubMed] [Google Scholar]
- Mamot C, Drummond DC, Greiser U, et al. Epidermal growth factor receptor (EGFR)-targeted immunoliposomes mediate specific and efficient drug delivery to EGFR- and EGFRvIII-overexpressing tumor cells. Cancer Res. 2003;63:3154–61. [PubMed] [Google Scholar]
- Mantripragada S. A lipid based depot (DepoFoam technology) for sustained release drug delivery. Prog Lipid Res. 2002;41:392–406. doi: 10.1016/s0163-7827(02)00004-8. [DOI] [PubMed] [Google Scholar]
- Marjan J, Xie Z, Devine D. Liposome-induced activation of the classical complement pathway does not require immunoglobulin. Biochim Biophys Acta. 1994;1192:35–44. doi: 10.1016/0005-2736(94)90140-6. [DOI] [PubMed] [Google Scholar]
- Maruyama K, Ishida O, Kasaoka S, et al. Intracellular targeting of sodium mercaptoundecahydrododecaborate (BSH) to solid tumors by transferrin-PEG liposomes, for boron neutron-capture therapy (BNCT) J Control Release. 2004;98:195–207. doi: 10.1016/j.jconrel.2004.04.018. [DOI] [PubMed] [Google Scholar]
- Maruyama K, Okuizumi S, Ishida O, et al. Phosphatidyl polyglycerols prolong liposome circulation in vivo. Int J Pharm. 1994;111:103–7. [Google Scholar]
- Matsumura Y, Gotoh M, Muro K, et al. Phase I and pharmacokinetic study of MCC-465, a doxorubicin (DXR) encapsulated in PEG immunoliposome, in patients with metastatic stomach cancer. Ann Oncol. 2004;15:517–25. doi: 10.1093/annonc/mdh092. [DOI] [PubMed] [Google Scholar]
- Medina OP, Zhu Y, Kairemo K. Targeted liposomal drug delivery in cancer. Curr Pharm Des. 2004;10:2981–9. doi: 10.2174/1381612043383467. [DOI] [PubMed] [Google Scholar]
- Metselaar JM, Bruin P, de Boer LW, et al. A novel family of L-amino acid-based biodegradable polymer-lipid conjugates for the development of long-circulating liposomes with effective drug-targeting capacity. Bioconjug Chem. 2003;14:1156–64. doi: 10.1021/bc0340363. [DOI] [PubMed] [Google Scholar]
- Meyer O, Kirpotin D, Hong K, et al. Cationic liposomes coated with polyethylene glycol as carriers for oligonucleotides. J Biol Chem. 1998;273:15621–7. doi: 10.1074/jbc.273.25.15621. [DOI] [PubMed] [Google Scholar]
- Moghimi SM, Muir IS, Illum L, et al. Coating particles with a block copolymer (Poloxamine 908) suppresses opsonization but permits the activity of dysopsonins in the serum. Biochim Biophys Acta. 1993;1179:157–65. doi: 10.1016/0167-4889(93)90137-e. [DOI] [PubMed] [Google Scholar]
- Moghimi SM, Szebeni J. Stealth liposomes and long circulating nanoparticles: critical issues in pharmacokinetics, opsonization and protein-binding properties. Prog Lipid Res. 2003;42:463–78. doi: 10.1016/s0163-7827(03)00033-x. [DOI] [PubMed] [Google Scholar]
- Monfardini C, Veronese FM. Stabilization of substances in circulation. Bioconj Chem. 1998;9:418–50. doi: 10.1021/bc970184f. [DOI] [PubMed] [Google Scholar]
- Mora M, Sagrista ML, Trombetta D, et al. Design and characterization of liposomes containing long-chain N-acylPEs for brain delivery: penetration of liposomes incorporating GM1 into the rat brain. Pharm Res. 2002;19:1430–8. doi: 10.1023/a:1020440229102. [DOI] [PubMed] [Google Scholar]
- Morawski AM, Lanza GA, Wickline SA. Targeted contrast agents for magnetic resonance imaging and ultrasound. Curr Opin Biotechnol. 2005;16:89–92. doi: 10.1016/j.copbio.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Mukherjee A, Prasad TK, Rao NM, et al. Haloperidol-associated stealth liposomes: a potent carrier for delivering genes to human breast cancer cells. J Biol Chem. 2005;280:15619–27. doi: 10.1074/jbc.M409723200. [DOI] [PubMed] [Google Scholar]
- Murai M, Aramki Y, Tsuchiya S. Identification of the serum factor required for liposome primed activation of mouse peritoneal macrophages: modified 2-macroglobulin enhances Fc gamma receptor mediated phagocytosis of opsonized sheep red blood cells. Immunology. 1995;86:64–70. [PMC free article] [PubMed] [Google Scholar]
- Needham D, McIntosh TJ, Lasic DD. Repulsive interactions and mechanical stability of polymer-grafted lipid membranes. Biochim Biophys Acta. 1992;1108:40–8. doi: 10.1016/0005-2736(92)90112-y. [DOI] [PubMed] [Google Scholar]
- Nellis DF, Ekstrom DL, Kirpotin DB, et al. Preclinical manufacture of an anti-HER2 scFv-PEG-DSPE, liposome-inserting conjugate. 1. Gram-scale production and purification. Biotechnol Prog. 2005a;21:205–20. doi: 10.1021/bp049840y. [DOI] [PubMed] [Google Scholar]
- Nellis DF, Giardina SL, Janini GM, et al. Preclinical manufacture of anti-HER2 liposome-inserting, scFv-PEG-lipid conjugate. 2. Conjugate micelle identity, purity, stability, and potency analysis. Biotechnol Prog. 2005b;21:221–32. doi: 10.1021/bp049839z. [DOI] [PubMed] [Google Scholar]
- Nishikawa KAHIK. Scavanger receptor-mediated uptake and metabolism of lipid vesicles containing acidic phospholipids by mouse peritoneal macrophages. J Biol Chem. 1990;265:5226–31. [PubMed] [Google Scholar]
- Oja CDM, Semple SC, Chonn A, et al. Influence of dose on liposome clearance: critical role of blood proteins. Biochim Biophys Acta. 1996;1281:31–7. doi: 10.1016/0005-2736(96)00003-x. [DOI] [PubMed] [Google Scholar]
- Oku N, Namba Y. Glucuronate-modified, long-circulating liposomes for the delivery of anticancer agents. Methods Enzymol. 2005;391:145–62. doi: 10.1016/S0076-6879(05)91008-2. [DOI] [PubMed] [Google Scholar]
- Oussoren C, Storm G. Lymphatic uptake and biodistribution of liposomes after subcutaneous injection: III. Influence of surface modification with poly(ethyleneglycol) Pharm Res. 1997;14:1479–84. doi: 10.1023/a:1012145410859. [DOI] [PubMed] [Google Scholar]
- Ozpolat B, Lopez-Berestein G, Adamson P, et al. Pharmacokinetics of intravenously administered liposomal all-trans-retinoic acid (ATRA) and orally administered ATRA in healthy volunteers. J Pharm Pharm Sc. 2003;6:292–301. [PubMed] [Google Scholar]
- Palmer LR, Chen T, Lam AM, et al. Transfection properties of stabilized plasmid-lipid particles containing cationic PEG lipids. Biochim Biophys Acta. 2003;1611:204–16. doi: 10.1016/s0005-2736(03)00058-0. [DOI] [PubMed] [Google Scholar]
- Park JW, Hong K, Kirpotin DB, et al. Anti-HER2 immunoliposomes: enhanced efficacy attributable to targeted delivery. Clin Cancer Res. 2002;8:1172–81. [PubMed] [Google Scholar]
- Park YS, Maruyama K, Huang L. Some negatively charged phospholipid derivatives prolong the liposome circulation in vivo. Biochim Biophys Acta. 1992;1108:257–60. doi: 10.1016/0005-2736(92)90034-j. [DOI] [PubMed] [Google Scholar]
- Pastorino F, Brignole C, Marimpietri D, et al. Doxorubicin-loaded Fab’ fragments of anti-disialoganglioside immunoliposomes selectively inhibit the growth and dissemination of human neuroblastoma in nude mice. Cancer Res. 2003;63:86–92. [PubMed] [Google Scholar]
- Patel HM. Serum opsonins and liposomes: their interaction and opsonophagocytosis. Crit Rev Ther Drug Carrier Syst. 1992;9:39–90. [PubMed] [Google Scholar]
- Phillips WT, Klipper RW, Awasthi VD, et al. Polyethylene glycol-modified liposome-encapsulated hemoglobin: a long circulating red cell substitute. J Pharmacol Exp Ther. 1999;288:665–70. [PubMed] [Google Scholar]
- Powell GM. Polyethylene glycol. In: Davidson RL, editor. Handbook of water soluble gums and resins. McGraw-Hill: 1980. pp. 18–31. [Google Scholar]
- Price ME, Cornelius RM, Brash JL. Protein adsorption to polyethylene glycol modified liposomes from fibrinogen solution and from plasma. Biochim Biophys Acta. 2001;1512:191–205. doi: 10.1016/s0005-2736(01)00330-3. [DOI] [PubMed] [Google Scholar]
- Priebe W, Perez-Soler R. Design and tumor targeting of anthracyclines able to overcome multidrug resistance: a double-advantage approach. Pharmacol Ther. 1993;60:215–34. doi: 10.1016/0163-7258(93)90007-z. [DOI] [PubMed] [Google Scholar]
- Robert NJ, Vogel CL, Henderson IC, et al. The role of the liposomal anthracyclines and other systemic therapies in the management of advanced breast cancer. Semin Oncol. 2004;31(Suppl 13):106–46. doi: 10.1053/j.seminoncol.2004.09.018. [DOI] [PubMed] [Google Scholar]
- Rose PG. Pegylated liposomal doxorubicin: optimizing the dosing schedule in ovarian cancer. Oncologist. 2005;10:205–14. doi: 10.1634/theoncologist.10-3-205. [DOI] [PubMed] [Google Scholar]
- Sapra P, Allen TM. Ligand-targeted liposomal anticancer drugs. Prog Lipid Res. 2003;42:439–62. doi: 10.1016/s0163-7827(03)00032-8. [DOI] [PubMed] [Google Scholar]
- Scherphof GL. Uptake and intracellular processing of targeted and nontargeted liposomes by rat Kupffer cells in vivo and in vitro. Ann N Y Acad Sci. 1985;446:368–84. doi: 10.1111/j.1749-6632.1985.tb18414.x. [DOI] [PubMed] [Google Scholar]
- Seiden MV, Muggia F, Astrow A, et al. A phase II study of liposomal lurtotecan (OSI-211) in patients with topotecan resistant ovarian cancer. Gynecol Oncol. 2004;93:229–32. doi: 10.1016/j.ygyno.2003.12.037. [DOI] [PubMed] [Google Scholar]
- Semple SC, Leone R, Wang J, et al. Optimization and characterization of a sphingomyelin/cholesterol liposome formulation of vinorelbine with promising antitumor activity. J Pharm Sci. 2005;94:1024–38. doi: 10.1002/jps.20332. [DOI] [PubMed] [Google Scholar]
- Senior J. Is half-life of circulating liposomes determined by changes in their permeability? FEBS Lett. 1982;145:109–14. doi: 10.1016/0014-5793(82)81216-7. [DOI] [PubMed] [Google Scholar]
- Senior JH. Fate and behavior of liposomes in vivo: a review of controlling factors. Crit Rev Ther Drug Carrier Syst. 1987;3:123–93. [PubMed] [Google Scholar]
- Song LY, Ahkong QF, Rong Q, et al. Characterization of the inhibitory effect of PEG-lipid conjugates on the intracellular delivery of plasmid and antisense DNA mediated by cationic lipid liposomes. Biochim Biophys Acta. 2002;1558:1–13. doi: 10.1016/s0005-2736(01)00399-6. [DOI] [PubMed] [Google Scholar]
- Stathopoulos GP, Boulikas T, Vougiouka M, et al. Pharmacokinetics and adverse reactions of a new liposomal cisplatin (Lipoplatin): phase I study. Oncol Rep. 2005;13:589–95. [PubMed] [Google Scholar]
- Stephenson SM, Low PS, Lee RJ. Folate receptor-mediated targeting of liposomal drugs to cancer cells. Methods Enzymol. 2004;387:33–50. doi: 10.1016/S0076-6879(04)87003-4. [DOI] [PubMed] [Google Scholar]
- Stephenson SM, Yang W, Stevens PJ, et al. Folate receptor-targeted liposomes as possible delivery vehicles for boron neutron capture therapy. Anticancer Res. 2003;23:3341–45. [PubMed] [Google Scholar]
- Strijkers GJ, Mulder WJM, van Heeswijk RB, et al. Relaxivity of liposomal paramagnetic MRI contrast agents. MAGMA. 2005;18:186–92. doi: 10.1007/s10334-005-0111-y. [DOI] [PubMed] [Google Scholar]
- Sugano M, Egilmez NK, Yokota SJ, et al. Antibody targeting of doxorubicin-loaded liposomes suppresses the growth and metastatic spread of established human lung tumor xenografts in severe combined immunodeficient mice. Cancer Res. 2000;60:6942–9. [PubMed] [Google Scholar]
- Taira MC, Chiaramoni NS, Pecuch KM, et al. Stability of liposomal formulations in physiological conditions for oral drug delivery. Drug Deliv. 2004;11:123–8. doi: 10.1080/10717540490280769. [DOI] [PubMed] [Google Scholar]
- Takeuchi H, Kojima H, Yamamoto H, et al. Evaluation of circulation profiles of liposomes coated with hydrophilic polymers having different molecular weights in rats. J Control Release. 2001;75:83–91. doi: 10.1016/s0168-3659(01)00368-6. [DOI] [PubMed] [Google Scholar]
- Tardi P, Choice E, Masin D, et al. Liposomal encapsulation of topotecan enhances anticancer efficacy in murine and human xenograft models. Cancer Res. 2000;60:3389–93. [PubMed] [Google Scholar]
- Torchilin VP. Polymeric contrast agents for medical imaging. Curr Pharm Biotechnol. 2000;1:183–215. doi: 10.2174/1389201003378960. [DOI] [PubMed] [Google Scholar]
- Torchilin VP, Levchenko TS, Whiteman KR, et al. Amphiphilic poly-N-vinylpyrrolidones: synthesis, properties and liposome surface modification. Biomaterials. 2001;22:3035–44. doi: 10.1016/s0142-9612(01)00050-3. [DOI] [PubMed] [Google Scholar]
- Torchilin VP, Omelyanenko VG, Papisov MI, et al. Poly(ethylene glycol) on the liposome surface: on the mechanism of polymer-coated liposomes longevity. Biochim Biophys Acta. 1994a;1195:11–20. doi: 10.1016/0005-2736(94)90003-5. [DOI] [PubMed] [Google Scholar]
- Torchilin VP, Papisov MI. Why do polyethylene glycol-coated liposomes circulate so long ? J Liposome Res. 1994;4:725–39. [Google Scholar]
- Torchilin V, Shtilman M, Trubetskoy V, et al. Amphiphilic vinyl polymers selectively prolong liposome circulation time in vivo. Biochim Biophys Acta. 1994c;1195:181–4. doi: 10.1016/0005-2736(94)90025-6. [DOI] [PubMed] [Google Scholar]
- Torchilin V, Trubetskoy V, Whiteman K, et al. New synthetic amphiphilic polymers for steric protection of liposomes in vivo. J Pharm Sci. 1995;84:1049–53. doi: 10.1002/jps.2600840904. [DOI] [PubMed] [Google Scholar]
- Tseng YL, Hong RL, Tao MH, et al. Sterically stabilized anti-idiotype immunoliposomes improve the therapeutic efficacy of doxorubicin in a murine B-cell lymphoma model. Int J Cancer. 1999;80:723–30. doi: 10.1002/(sici)1097-0215(19990301)80:5<723::aid-ijc16>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Ugwu S, Zhang A, Parmar M, et al. Preparation, characterization, and stability of liposome-based formulations of mitoxantrone. Drug Dev Ind Pharm. 2005;31:223–9. doi: 10.1081/ddc-200047850. [DOI] [PubMed] [Google Scholar]
- Veerareddy PR, Vobalaboina V. Lipid-based formulations of amphotericin B. Drugs Today. 2004;40:133–45. doi: 10.1358/dot.2004.40.2.799425. [DOI] [PubMed] [Google Scholar]
- Verschraegen CF, Kumagai S, Davidson R, et al. Phase I clinical and pharmacological study of intraperitoneal cis-bis-neodecanoato (trans-R, R-1, 2-diaminocyclohexane)-platinum II entrapped in multilamellar liposome vesicles. J Cancer Res Clin Oncol. 2003;129:549–55. doi: 10.1007/s00432-003-0481-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vert M, Domurado D. Poly(ethylene glycol): protein-repulsive or albumin-compatible? J Biomater Sci Polym Ed. 2000;11:1307–17. doi: 10.1163/156856200744345. [DOI] [PubMed] [Google Scholar]
- Volanakis JE, Narkates AJ. Interaction of C-reactive protein with artificial phosphatidylcholine bilayers and complement. J Immunol. 1981;126:1820–5. [PubMed] [Google Scholar]
- Vorobiof DA, Rapoport BL, Chasen MR, et al. First line therapy with paclitaxel (Taxol) and pegylated liposomal doxorubicin (Caelyx) in patients with metastatic breast cancer: a multicentre phase II study. Breast. 2004;13:219–26. doi: 10.1016/j.breast.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Whiteman KR, Subr V, Ulbrich K, et al. Poly(HPMA)-coated liposomes demonstrate prolonged circulation in mice. J Liposome Res. 2001;11:153–64. doi: 10.1081/LPR-100108459. [DOI] [PubMed] [Google Scholar]
- Woodle M, Engbers C, Zalipsky S. New amphipatic polymer-lipid conjugates forming long-circulating reticuloendothelial system-evading liposomes. Bioconjug Chem. 1994;5:493–6. doi: 10.1021/bc00030a001. [DOI] [PubMed] [Google Scholar]
- Yamaoka T, Tabata Y, Ikada Y. Distribution and tissue uptake of poly(ethylene glycol) with different molecular weights after intravenous administration in mice. J Pharm Sci. 1994;83:601–6. doi: 10.1002/jps.2600830432. [DOI] [PubMed] [Google Scholar]
- Yamauchi H, Yano T, Kato T, et al. Effects of sialic acid derivative on long circulating time and tumor concentration of liposomes. Int J Pharmacol. 1994;113:141–8. [Google Scholar]
- Zalipsky S, Qazen M, Walker JA, et al. New detachable poly(ethylene glycol) conjugates: cysteine-cleavable lipopolymers regenerating natural phospholipid, diacyl phosphatidylethanolamine. Bioconjug Chem. 1999;10:703–7. doi: 10.1021/bc990031n. [DOI] [PubMed] [Google Scholar]
- Zamboni WC, Gervais AC, Egorin MJ, et al. Systemic and tumor disposition of platinum after administration of cisplatin or STEALTH liposomal-cisplatin formulations (SPI-077 and SPI-077 B103) in a preclinical tumor model of melanoma. Cancer Chemother Pharmacol. 2004;53:329–36. doi: 10.1007/s00280-003-0719-4. [DOI] [PubMed] [Google Scholar]
- Zamboni WC, Ramalingam S, Friedland DM, et al. Phase I and pharmacokinetic (PK) study of STEALTH liposomal CKD-602 (S-CKD602) in patients with advanced solid tumors. J Clin Oncol (Meeting Abstracts) 2005;23:2069. [Google Scholar]
- Zhang JA, Anyarambhatla G, Ma L, et al. Development and characterization of a novel Cremophor EL free liposome-based paclitaxel (LEP-ETU) formulation. Eur J Pharm Biopharm. 2005;59:177–87. doi: 10.1016/j.ejpb.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Zhang JA, Xuan T, Parmar M, et al. Development and characterization of a novel liposome-based formulation of SN-38. Int JPharm. 2004;270:93–107. doi: 10.1016/j.ijpharm.2003.10.015. [DOI] [PubMed] [Google Scholar]