

Abstract
Cutaneous squamous cell carcinomas (SCC) are the second most commonly diagnosed cancers in fair-skinned people; yet the genetic mechanisms involved in SCC tumorigenesis remain poorly understood. We have used single nucleotide polymorphism (SNP) microarray analysis to examine genome-wide allelic imbalance in 16 primary and 2 lymph node metastatic SCC using paired non-tumour samples to counteract normal copy number variation. The most common genetic change was loss of heterozygosity (LOH) on 9p, observed in 13 of 16 primary SCC. Other recurrent events included LOH on 3p (9 tumors), 2q, 8p, and 13 (each in 8 SCC) and allelic gain on 3q and 8q (each in 6 tumors). Copy number-neutral LOH was observed in a proportion of samples, implying that somatic recombination had led to acquired uniparental disomy, an event not previously demonstrated in SCC. As well as recurrent patterns of gross chromosomal changes, SNP microarray analysis revealed, in 2 primary SCC, a homozygous microdeletion on 9p23 within the protein tyrosine phosphatase receptor type D (PTPRD) locus, an emerging frequent target of homozygous deletion in lung cancer and neuroblastoma. A third sample was heterozygously deleted within this locus and PTPRD expression was aberrant. Two of the 3 primary SCC with PTPRD deletion had demonstrated metastatic potential. Our data identify PTPRD as a candidate tumor suppressor gene in cutaneous SCC with a possible association with metastasis.
INTRODUCTION
Nonmelanoma skin cancers (NMSC), comprising basal cell carcinoma (BCC) and squamous cell carcinoma (SCC), have become a significant health problem in fair-skinned people worldwide and their incidence is rising rapidly (de Vries et al., 2005). UV radiation (UVR) is the principal carcinogen implicated in the development of SCC (Armstrong and Kricker, 2001). Although UVR-induced genetic changes in potential target genes including TP53 (Brash et al., 1991), CDKN2A (Soufir et al., 1999) and RAS (Pierceall et al., 1991) have been demonstrated in cutaneous SCC, other key molecular events in tumor initiation and progression are yet to be defined. Microarray-based single nucleotide polymorphism (SNP) analysis facilitates the accurate and rapid identification of genome-wide allelic changes in tumor DNA samples. It has been used to investigate the molecular pathogenesis of various human cancers, including BCC (Teh et al., 2005) and acute myeloid leukemia (Raghavan et al., 2005). Here, this technique has been applied to cutaneous SCC for the first time.
MATERIALS AND METHODS
Sixteen primary cutaneous SCC and 2 lymph node metastatic SCC were obtained from 16 patients, 8 of whom were immunosuppressed (1 cardiac transplant and 7 renal transplant recipients). Ethical approval for this investigation was obtained from the East London and City Health Authority local ethics committee and all patients provided informed consent. Samples were enriched for SCC keratinocytes by short-term culture as previously described (Proby et al., 2000) or by microdissection of frozen lesional tissue sections using a H&E-stained section as a reference. Non-tumour control DNA was obtained from blood or from dermal fibroblasts isolated from perilesional skin by incubation in 0.05 mg/ml collagenase D (Roche, Applied Science, Indianapolis, IN) for 16 hr at 37°C.
DNA was extracted from cultured cells and blood samples using the Nucleon BACC3 Genomic DNA Extraction kit and from lesional tissue using the Nucleon HT Genomic DNA Extraction kit (both from Amersham Biosciences, Buckinghamshire, UK). Samples were subjected to the GeneChip Human Mapping 10K (V2.0) Xba or 250K Nsp assay (Affymetrix, Santa Clara, CA) according to the manufacturer's protocol with the modification that PCR products were purified using the Ultrafree-MC filtration column (Millipore, Watford, UK). Processing was performed as previously described (Teh et al., 2005) using the Genome Oriented Laboratory File (GOLF) system for the analysis and display of SNP call data.
The Qiagen RNeasy Mini kit (Qiagen, Crawley, UK) was used to extract RNA from primary and metastatic SCC tissue and from normal skin samples removed during cosmetic surgery from 7 different individuals with no known skin cancer history. cDNA was synthesized from RNA using Superscript II Reverse Transcriptase and oligo (dT)12–18 primers (both from Invitrogen, Paisley, UK). Samples were subjected to RT-PCR using the PTPRD primers described previously (Sato et al., 2005). Poly (A+) RNA (Ambion, Austin, TX) from brain tissue, which expresses both S and L PTPRD isoforms (Sato et al., 2005), was used as a positive control.
RESULTS AND DISCUSSION
SNP microarray analysis was performed on 16 primary and 2 lymph node metastatic SCC with corresponding non-tumour samples analyzed concomitantly to overcome the recently highlighted problem of normal copy number variation (Redon et al., 2006) thereby allowing accurate identification of acquired genome-wide genetic events that may be important in SCC tumorigenesis. Allelic loss: Of 16 primary SCC, 13 exhibited extensive loss of heterozygosity (LOH) at 9p (Fig. 1a, Table 1). Analysis of SNP call signal intensity data (Fig. 1b) revealed that in 23% (3/13) of samples, LOH regions were not accompanied by loss of copy number, suggesting that LOH was due to acquired uniparental disomy (UPD). UPD occurs as a result of a somatic recombination event, and has recently been recognized as a key mechanism of LOH in several types of solid tumors including BCC (Teh et al., 2005) but our data provide the first evidence of UPD in SCC tumorigenesis. Previous studies have linked the acquisition of UPD in leukemia with homozygosity for certain gene mutations (Fitzgibbon et al., 2005) suggesting that it may play a similar role in the evolution of SCC. Other frequent genetic events including 3p LOH (observed in 9 primary SCC) and LOH on 2q, 8p, and 13 (each in 8 primary tumors) are listed in Table 2. Allelic gain: Analysis of SNP call signal intensity data also revealed recurrent regions of increased allelic copy number in the absence of LOH (Table 1). The most frequent of these occurred on 3q and 8q, each in 6 of 16 primary tumors. Other recurrent regions of allelic gain are listed in Table 2.
Figure 1.
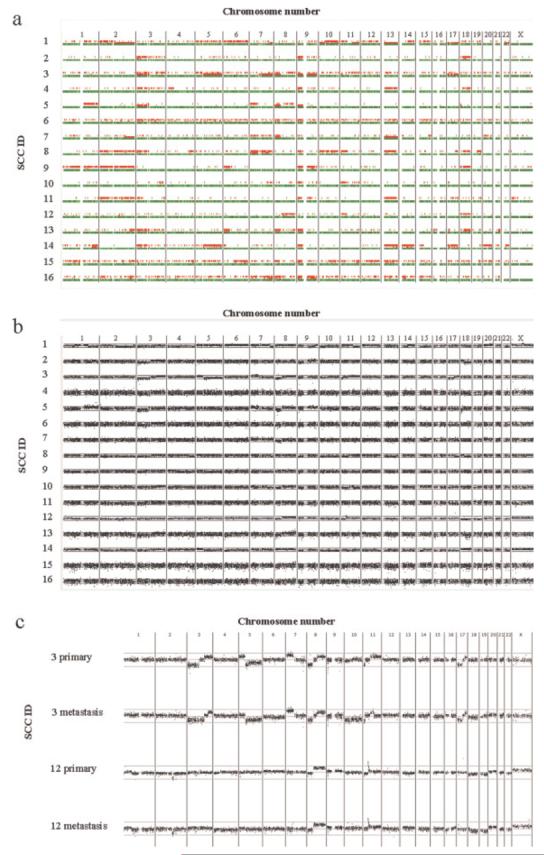
Allelic imbalances demonstrated by 10K SNP microarray analysis. (a) Comparison of SNP genotypes in 16 primary SCC and paired non-tumour samples. SNP loci shown in green indicate calls unchanged between tumor and non-tumour samples, loci shown in red immediately above indicate LOH in the tumor sample and loci shown in red on the top row indicate SNPs not called in the tumor samples. Localized areas of low call rate in the tumor sample may imply LOH in a tumor subpopulation or allelic gain. (b,c) DNA copy number alterations in 16 primary SCC (b) and in 2 primary-metastatic SCC pairs (c). A running average of 2 consecutive ratios of signal values is plotted on a log2 scale according to chromosomal position. Upper line represents log2(2) and lower line represents log2(0.5).
TABLE 1.
Clinicohistopathogical Data and Chromosomal Aberrations of the 18 Squamous Cell Carcinomas
| Aberrations |
||||||||
|---|---|---|---|---|---|---|---|---|
| ID | Immune status |
Sex | Age | Location | Histological diagnosis |
Loss | Uniparental disomy | Gain |
| 1a | RT | M | 67 | ear | poorly differentiatedb | 1q25.1-qter | 13 | 1p31.1-1q25.1 |
| 2q11.2-2qter | 17 | 6 | ||||||
| 7q33-7qter | 9q33.2-9qter | |||||||
| 9qcen-9q33.2 | 14q | |||||||
| 10pter-10pcen | 20q | |||||||
| 11q14.1-11qter | Xpter-Xp21.1 | |||||||
| 14p | Xq13.1-Xq21.31 | |||||||
| 22 | ||||||||
| 2a | CT | M | 66 | hand | well differentiatedc | 3pter-3pcen | – | 3qcen-3qter |
| 9pter-9p13.2 | 9qcen-3qter | |||||||
| 18qcen-18qter | ||||||||
| 3a | RT | M | 55 | 1. hand | moderately differentiated | 3pter-3p12.1 | 7q11.21-7qter | 3q22.1-3qter |
| 5q11.2-5qter | 9 | 5pter-5p12 | ||||||
| 8pter-8p21.3 | 17q21.31-17q24.1 | 7pter-7pcen | ||||||
| 8p21.2-8p11.21 | 8q21.11-8qter | |||||||
| 9p23 | 11p11.12-11qter | |||||||
| 11pter-11p15.3 | 17q24.1-17qter | |||||||
| 17pter-17q21.31 | ||||||||
| 2. axillary lymph node | metastaticd | 3pter-3q21.3 | 7q11.21-7qter | 3q25.33-3qter | ||||
| 5q11.2-5qter | 9 | 7pter-7pcen | ||||||
| 8pter-8p21.3 | 17q21.31-17q24.1 | 8q21.11-8qter | ||||||
| 8p21.2-8p11.21 | 11p11.12-11q14.1 | |||||||
| 9p23 | 17q24.1-17qter | |||||||
| 10 | 11pter-11p15.3 | |||||||
| 17pter-17q21.31 | ||||||||
| 4 | RT | M | 66 | ear | well differentiated | 3pter-3pcen | – | 3qcen-3qter |
| 9pter-9pcen | 9qcen-9qter | |||||||
| 13 | ||||||||
| 5a | RT | M | 61 | arm | well differentiated | 3pter-3p12.1 | 8pter-8p23.1 | 1qcen-1qter |
| 8p23.1-8p11.21 | 9pter-9pcen | 3p12.1-3p11.1 | ||||||
| 18pter-18p11.21 | 7pter-7p14.3 | |||||||
| 7p14.1-7p11.1 | ||||||||
| 8q11.21-8qter | ||||||||
| 9qcen-9qter | ||||||||
| 6 | RT | F | 71 | scalp | well differentiated | 3pter-3pcen | – | 3qcen-3qter |
| 8pter-8p12 | ||||||||
| 9pter-9pcen | ||||||||
| 7 | RT | M | 42 | scalp | well differentiated | 8pter-8p11.21 | 2q31.1-2qter | 7 |
| 13 | 20 | |||||||
| 8 | RT | F | 66 | wrist | moderately differentiated | 2 | 7pter-7q34 | – |
| 3pter-3p12.1 | ||||||||
| 4q31.3-4qter | ||||||||
| 7q34-7qter | ||||||||
| 9pter-9p13.2 | ||||||||
| 10 | ||||||||
| 11pter-11pcen | ||||||||
| 13pter-13q33.1 | ||||||||
| 17q24.1-17qter | ||||||||
| 18q12.1-18qter | ||||||||
| 9 | IC | M | 83 | forehead | poorly differentiated | 1 | 2 | |
| 9p23 | 6pter-6p11.2 | |||||||
| 9 | ||||||||
| 18 | ||||||||
| 10 | IC | M | 75 | forehead | moderately differentiated | 7q31.1-7qter | – | 1qcen-1qter |
| 8pter-8p11.21 | 3q23-3qter | |||||||
| 9pter-9pcen | 7pter-7p21.1 | |||||||
| 11pter-11p11.12 | 7q21.11-7q31.1 | |||||||
| 11q21-11qter | 8q11.1-8qter | |||||||
| 19pter-19p12 | 11q12.1-11q14.2 | |||||||
| 15 | ||||||||
| 11 | IC | F | 79 | forehead | well differentiated | 2 | 3q28-3q29 | 19p13.3-19p12 |
| 3pter-3p21.31 | 19q13.32-19qter | |||||||
| 9pter-9q22.33 | ||||||||
| 9q33.2-9qter | ||||||||
| 13 | ||||||||
| 17q11.1-17qter | ||||||||
| Xpter-Xp11.22 | ||||||||
| 12a | IC | M | 77 | 1. temple | moderately differentiated | 2q14.3-2q22.1 | – | 8q11.1-8qter |
| 2q24.1 | 11p13-11p12 | |||||||
| 9p24.1-9p23 | X | |||||||
| 18 | ||||||||
| 19 | ||||||||
| 2. pre-auricular lymph node | metastatic | 2q14.3-2q22.1 | – | 8q11.1-8qter | ||||
| 2q24.1 | 11p13-11p12 | |||||||
| 4 | X | |||||||
| 9p24.1-9p23 | ||||||||
| 18q11.2-18qter | ||||||||
| 19 | ||||||||
| 13 | IC | M | 64 | scalp | moderately differentiated | 2q33.3-2qter | 17p13.1-17p11.2 | 3q12.1-3qter |
| 3pter-3p12.1 | 8q11.21-8qter | |||||||
| 6pter-6p21.1 | 12pter-12p11.1 | |||||||
| 8pter-8p11.21 | 19pter-19p31.12 | |||||||
| 9pter-9p13.2 | 19q13.41-19q13.43 | |||||||
| 16pter-16p11.2 | 21q22.11-21qter | |||||||
| 18q12.1-18qter | ||||||||
| 14 | IC | M | 92 | ear | moderately differentiated | 3pter-3p11.1 | 1q32.2-1q43 | 5pter-5pcen |
| 6pter-6p22.3 | 5qcen-5qter | 18 | ||||||
| 9pter-9p13.2 | 13 | X | ||||||
| 10 | 14 | |||||||
| 15pter-15q12 | ||||||||
| 16p | ||||||||
| 17 | ||||||||
| 20 | ||||||||
| 22q11.22-22qter | ||||||||
| 15 | IC | M | 84 | scalp | moderately differentiated | 1p31.1-1p22.2 | 2q33.1-2qter | 1p12-1q32.1 |
| 4q31.3-4qter | 10q21.1-10qter | 2q24.1-2q33.1 | ||||||
| 6q12-6qter | 11q24.2-11qter | 8q24.12-8qter | ||||||
| 8pter-8p21.1 | 19q13.2-19q13.32 | 15 | ||||||
| 9pter-9p13.2 | 19p13.2-19q13.12 | |||||||
| 11pter-11p13 | 20q11.21-20qter | |||||||
| 13 | 21q22.3 | |||||||
| 16 | IC | M | 70 | scalp | moderately differentiated | 2q33.2-2qter | 7q11.22-7qter | 1 |
| 8pter-8q12.3 | 9q | 2pter-2p21 | ||||||
| 9pter-9p21.1 | 7pter-7q11.22 | |||||||
| 13pter-9p21.1 | 12q21.32-12q23.3 | |||||||
| 14q11.2-14q21.1 | 19p13.2-19p13.12 | |||||||
| X | ||||||||
Indicates that short-term cultured cells were used for the tumour DNA sample.
All primary tumours were invasive SCC.
Well differentiated SCC did not include keratoacanthomas.
Both metastatic SCC were lymph node metastases.
RT, renal transplant recipient; CT, cardiac transplant recipient; IC immunocompetent individual.
TABLE 2.
Recurrent Aberrations in 16 Primary Squamous Cell Carcinomas
| Chromosomal location | Frequency | Copy number |
|---|---|---|
| 1q | 5 | gain |
| 2q | 8 | deletion, UPD |
| 3p | 9 | deletion |
| 3q | 6 | gain |
| 7p | 5 | gain |
| 7q | 5 | deletion, UPD |
| 8p | 8 | deletion, UPD |
| 8q | 6 | gain |
| 9p | 13 | deletion, UPD |
| 9q | 4 | UPD, deletion |
| 9q | 4 | gain |
| 11p | 4 | deletion |
| 13 | 8 | deletion, UPD |
| 17q | 4 | deletion, UPD |
| 18q | 5 | deletion, UPD |
| X | 4 | gain |
Paired primary and lymph node metastatic SCC were available for two patients in this study, thereby presenting an opportunity to investigate genetic changes associated with acquisition of metastatic potential. The primary and metastatic tumors from patient 3 displayed identical patterns of LOH accompanied by allelic loss on 5q, 8p, 11p, and 17 together with LOH due to UPD on 7q, 9, and 17q and allelic gain on 7p, 8q, and 17q (Table 1, Fig. 1c). However, only the metastatic lesion exhibited allelic loss on chromosome 10 while the 3p deletion observed in the primary lesion extended to 3q21.3 in the metastatic sample. Conversely, allelic gain at 5p was observed in the primary SCC alone together with increased regions of allelic gain on 3q and 11q in comparison to the metastatic tumor. The primary and metastatic tumors from patient 12 demonstrated identical patterns of allelic loss on 2q, 9p, and 18q with allelic gain on 8q, 11p, and X but the metastatic sample alone showed allelic loss on chromosome 4 while 18p loss was observed only in the primary SCC. These data indicate that, in both patients, primary and metastatic SCC were of common origin, with further genetic events occurring in each tumor after metastasis.
Although gross chromosomal changes constitute the majority of the aberrations revealed by this technique, we have identified a microdeletion on 9p23 in both primary-metastatic SCC pairs as well as the primary SCC from patient 9* (Table 1). The microdeletion was detected by comparison of SNP genotypes in tumors and paired non-tumor samples and therefore was clearly somatically acquired. This region was homozygously deleted in the samples from patients 3 and 9 whereas both primary and metastatic SCC from patient 12 contained a heterozygous deletion. Using 10K SNP microarray analysis all microdeletions were mapped to a region spanning 8.45–11.0 Mb, within the locus of the recently identified PTPRD (protein tyrosine phosphatase receptor type D) L isoform (Sato et al., 2005). The physiological function of PTPRD has not been fully characterized, although it is known to interact with the putative metastasis suppressor protein MIM to mediate actin cytoskeletal reorganization (Gonzalez-Quevedo et al., 2005). To date, a tumor suppressor role for PTPRD remains speculative, although several other protein tyrosine phosphatase receptors have been postulated as tumor suppressor genes including PTPRO in lung cancer (Motiwala et al., 2004) and HPTPη, implicated in several carcinomas, most recently pancreatic cancer (Trapasso et al., 2004). Interestingly, PTPRD has been revealed as a recurrent target of homozygous deletion in lung carcinoma (Sato et al., 2005; Zhao et al., 2005) as well as in neuroblastoma (Stallings et al., 2006), where a possible association with metastasis was suggested. Furthermore, cancer genome mutation analysis has recently identified PTPRD as a candidate cancer gene in colorectal carcinoma (Sjöblom et al., 2006). To compare microdeletions in the two primary SCC with demonstrated metastatic potential, samples were subjected to higher resolution 250K Nsp SNP microarray analysis (Fig. 2a). The sample from patient 3 demonstrated two regions of homozygous deletion at 9.08–9.22 Mb and 10.42–10.51 Mb, while an overlapping region of deletion at 8.55–9.38 Mb was observed on one allele in the patient 12 sample. DNA sequencing (primers listed in supplementary information (Supplementary material for this article can be found at http://www.interscience.wiley.com/jpages/1045-2257/suppmat)) revealed the second copy of PTPRD to be wild type. RT-PCR was performed to compare PTPRD expression in the primary and metastatic SCC from patient 12 with the pattern observed in normal skin. Two primer sets were used to amplify sequences common to S and L isoforms, with the forward primer located either within or outside the region of heterozygous deletion (Fig. 2b). Products of the expected size were observed in all cases (Fig. 2c), suggesting that wild type PTPRD transcripts were present in the SCC samples. Additional RT-PCR reactions with an L-isoform-specific forward primer located within the region of heterozygous deletion yielded no products in any samples, whereas the use of an L-isoform-specific forward primer located outside the region of heterozygous deletion yielded products solely in the SCC samples (Fig. 2d), implying that normal skin expresses the S isoform alone. In both SCC samples, only a single band of smaller size than the predicted full-length product was detected, pointing to the absence of wild type PTPRD L isoform transcripts. Sequencing revealed that exon B8 was fused to exon 5 within this amplimer (Fig. 2d), thereby corresponding to the region of heterozygous deletion observed by SNP analysis. A similar PTPRD deletion involving part of the 5′-UTR together with coding sequence has been reported previously in lung cancer (Sato et al., 2005), but our data provide the first reported example of transcription of this deleted gene. Although the wild type PTPRD start codon in exon 2 is located within the deleted region, the shortened transcripts contain another start codon within exon 5. Translation from this codon would yield a truncated product in frame with full-length PTPRD, suggesting a possible dominant negative mechanism. Further study is clearly required to investigate this hypothesis.
Figure 2.
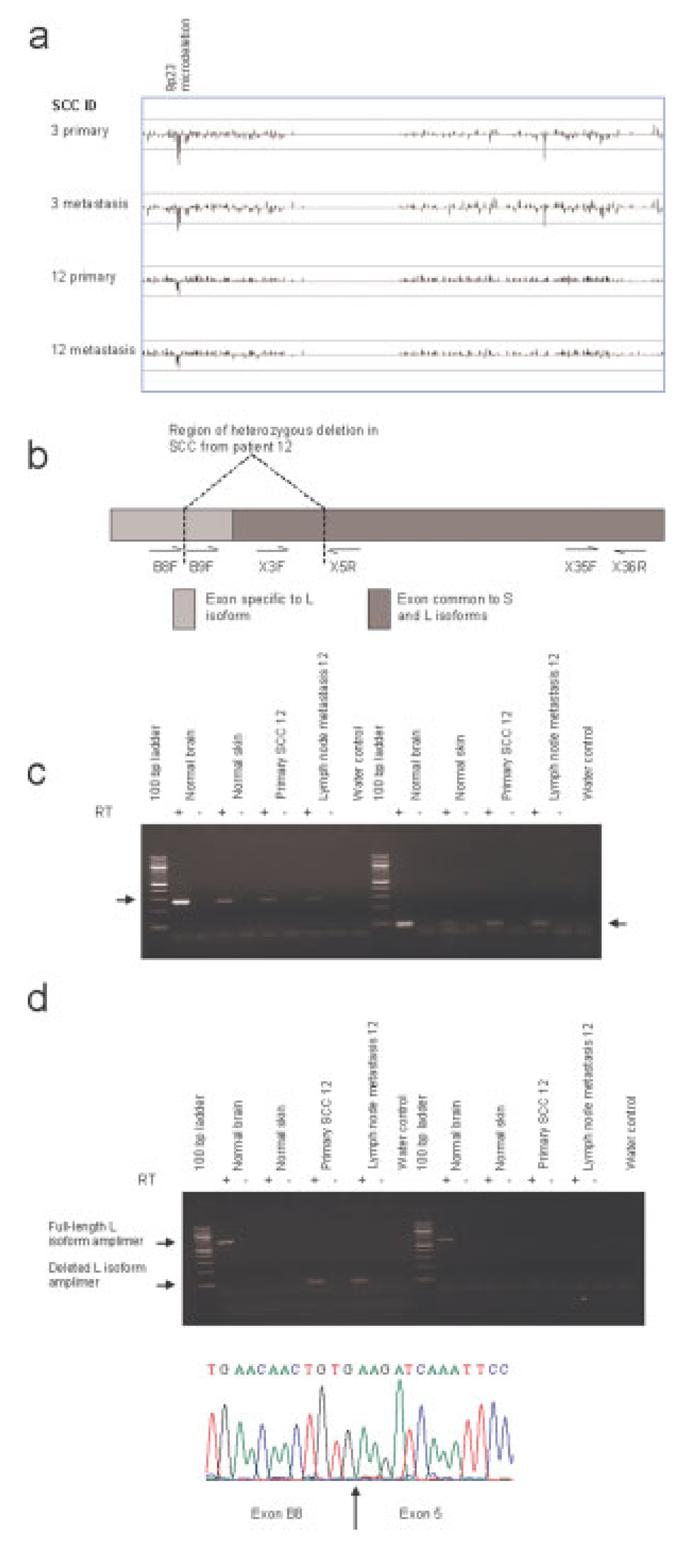
Chromosome band 9p23 microdeletion and aberrant PTPRD expression observed in SCC with metastatic potential. (a) Display of copy number values on chromosome 9 reveals 9p23 microdeletion within the PTPRD locus. A running average of 2 consecutive tumor:non-tumour signal value ratios is plotted on a log2 scale according to chromosomal position. Upper line represents log2(2) and lower line represents log2(0.5). For clarity each value is represented by a vertical line from log2(1). (b–d) Aberrant PTPRD expression in patient 12 SCC with heterozygous 9p23 microdeletion. One representative set of data summarizing the pattern of PTPRD expression observed in 7 normal skin samples is shown. RT indicates RNA was subjected to reverse transcription. Arrows indicate PTPRD-specific amplimers. (b) Schematic representation of location of RT-PCR primers as described previously (Sato et al., 2005). (c) RT-PCR amplification of PTPRD using forward primer in exon 3 within the region of heterozygous deletion and reverse primer in exon 5 (left panel) or primers in exons 35 and 36 outside the deleted region (right panel). (d) L isoform-specific RT-PCR with forward primer located in exon B8 outside (left panel) or in exon B9 within (right panel) the region of heterozygous deletion. The reverse primer was located in exon 5. Sequence trace shows splicing within the deleted L isoform amplimer. Arrow marks junction between B8 and exon 5. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com]
Our data show similarities with the results of early research where polymorphic microsatellite markers have revealed widespread allelic loss in SCC. In one study (Quinn et al., 1994) of 47 tumors frequent LOH of markers was observed on 9p (41%), 13q (46%), 17p (33%), 17q (33%), and 3p (23%). More recently, comparative genomic hybridization (CGH) has been used to identify genome-wide chromosomal imbalances within SCC (Popp et al., 2002; Ashton et al., 2003; Clausen et al., 2006). Despite considerable variation, the combined data from these three studies suggest a characteristic pattern of genetic aberrations within SCC. Recurrent allelic loss was reported on 3p, 4q, 5q, 8p, 9p, 11p, 13q, 17p, and 18 while recurrent allelic gain occurred on 1q, 3q, 5p, 7, 8q, 9q, 14q, 17q, 20q and Xq. In general, the allelic imbalance frequencies detected in the current study fall within the ranges of these earlier data. However, LOH was observed on 2q, 7q and 13q at higher frequencies than previously reported. This suggests that SNP microarray mapping may provide greater sensitivity than the other techniques, possibly due to its ability to detect copy-number-neutral LOH. Despite the small sample number, all of the allelic imbalances reported previously were detected in at least 1 of the 16 primary SCC examined here.
Organ transplant recipients (OTR) receiving immunosuppression are at a 100-fold increased risk of SCC compared with immunocompetent (IC) individuals, and tumors are frequently more aggressive (Glover et al., 1997). One early study (Rehman et al., 1997) suggested a different molecular pathogenesis for OTR SCC based on a rate of LOH less than half of that observed in IC tumors. In the current study, we observed allelic imbalance at 3–13 chromosomes (median value 5.5) in the 8 primary OTR SCC compared with 5–15 chromosomes (median value 9) in the 8 IC samples. Although numbers were small, our results suggested that the frequency of aberrations may be correlated with the degree of differentiation rather than immune status of the tumor: overall, allelic imbalance was observed at 3–7 chromosomes (median of 4) in well-differentiated tumors compared with 5–15 chromosomes (median of 10) in moderately or poorly differentiated tumors. Furthermore, despite a lower rate of allelic imbalance, some specific aberrations were more frequent in well differentiated than in moderately or poorly differentiated tumors. For example, 3p LOH was observed in 83% (5/6) of well-differentiated SCC compared with only 40% (4/10) of other SCC and allelic gain at 9q occurred in 50% (3/6) of well-differentiated but only 10% (1/10) of moderately and poorly differentiated tumors. These data suggest that well differentiated tumors may constitute a genetically distinct subpopulation of cutaneous SCC.
Sample purity is of particular importance when performing SNP microarray analysis as this technique is reliant on high-level PCR amplification. Indeed, it has been shown that normal DNA contamination of only 10% may decrease the accuracy of LOH detection in tumor samples, and that accuracy drops markedly once contamination approaches 30% (Lindblad-Toh et al., 2000). Normal DNA contamination is a major issue for solid epithelial tumors such as SCC where cancer cells are generally intermixed with stroma and inflammatory cells. In this study we have examined the efficacy of using short-term cell culture or lesional tissue microdissection to increase the purity of the tumor DNA sample. For one pair of primary and lymph node metastatic SCC samples from patient 12, analysis was performed in triplicate using DNA extracted from whole lesional biopsy, microdissected lesional tissue and short-term (passage 3) cultured cells. A comparison of the tumor to blood SNP signal ratios revealed that, while the method of tumor DNA preparation did not affect the pattern of allelic copy number changes observed for either primary or metastatic lesion, the cultured cell DNA sample yielded the clearest results and the use of microdissection also reduced the level of “noise” in comparison with the whole biopsy sample. Hence, the use of short-term culture did not change the composition of the original tumor sample by selecting for a subpopulation of cells. Furthermore, we found no evidence for the acquisition of culture-specific genetic aberrations in our samples. This is in line with the results of an earlier study (Popp et al., 2000), where few cell-line specific aberrations were observed after much longer periods of culture. Overall, a higher call rate was obtained with the DNA sample extracted from cultured cells rather than microdissected lesional tissue, suggesting that, where possible, this may be the better method of obtaining a pure tumor sample.
In summary, we have used SNP microarray analysis to reveal UPD in SCC for the first time and to show frequent 3p and 9p loss, consistent with previous microsatellite and CGH studies. While 9p loss may involve the CDKN2A locus, epigenetic events seem more common than genetic mechanisms for CDKN2A inactivation (Brown et al., 2004). Consequently, the identification in this study of a microdeletion at 9p23, within the PTPRD locus, possibly associated with acquisition of metastatic potential may be relevant. Our findings highlight the power of SNP array genotyping for investigating the global genetic changes underlying tumorigenesis.
Acknowledgments
Supported by: Cancer Research UK; British Skin Foundation Small Grant Award; Grant number: S/BS3; Research Advisory Board of St. Bartholomews; The Royal London Charitable Foundation; Grant number: RAC 404.
Footnotes
This article contains Supplementary Material available at http://www.interscience.wiley.com/jpages/1045-2257/suppmat.
The primary SCC from patient 9 was particularly aggressive, despite apparent complete excision two recurrences occurred within 12 months. Furthermore, the presence of metastatic SCC cannot be excluded; a suspicious pulmonary nodule was observed by CT scan 21 months after excision of the primary tumor but not confirmed as the patient died shortly afterwards of chest infection.
REFERENCES
- Armstrong BK, Kricker A. The epidemiology of UV induced skin cancer. J Photochem Photobiol B. 2001;63:8–18. doi: 10.1016/s1011-1344(01)00198-1. [DOI] [PubMed] [Google Scholar]
- Ashton KJ, Weinstein SR, Maguire DJ, Griffiths LR. Chromosomal aberrations in squamous cell carcinoma and solar keratoses revealed by comparative genomic hybridization. Arch Dermatol. 2003;139:876–882. doi: 10.1001/archderm.139.7.876. [DOI] [PubMed] [Google Scholar]
- Brash DE, Rudolph JA, Simon JA, Lin A, McKenna GJ, Baden HP, Halperin AJ, Ponten J. A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinoma. Proc Natl Acad Sci USA. 1991;88:10124–10128. doi: 10.1073/pnas.88.22.10124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown VL, Harwood CA, Crook T, Cronin JG, Kelsell DP, Proby CM. p16INK4a and p14ARF tumor suppressor genes are commonly inactivated in cutaneous squamous cell carcinoma. J Invest Dermatol. 2004;122:1284–1292. doi: 10.1111/j.0022-202X.2004.22501.x. [DOI] [PubMed] [Google Scholar]
- Clausen OP, Aass HC, Beigi M, Purdie KJ, Proby CM, Brown VL, Mattingsdal M, Micci F, Kolvraa S, Bolund L, DeAngelis PM. Are keratoacanthomas variants of squamous cell carcinomas? A comparison of chromosomal aberrations by comparative genomic hybridization. J Invest Dermatol. 2006;126:2308–2315. doi: 10.1038/sj.jid.5700375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries E, van de Poll-Franse LV, Louwman WJ, de Gruijl FR, Coebergh JW. Predictions of skin cancer incidence in the Netherlands up to 2015. Br J Dermatol. 2005;152:481–488. doi: 10.1111/j.1365-2133.2005.06386.x. [DOI] [PubMed] [Google Scholar]
- Fitzgibbon J, Smith LL, Raghavan M, Smith ML, Debernardi S, Skoulakis S, Lillington D, Lister TA, Young BD. Association between acquired uniparental disomy and homozygous gene mutation in acute myeloid leukemias. Cancer Res. 2005;65:9152–9154. doi: 10.1158/0008-5472.CAN-05-2017. [DOI] [PubMed] [Google Scholar]
- Glover MT, Deeks JJ, Raftery MJ, Cunningham J, Leigh IM. Immunosuppression and risk of non-melanoma skin cancer in renal transplant recipients. Lancet. 1997;349:398. doi: 10.1016/S0140-6736(97)80015-3. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Quevedo R, Shoffer M, Horng L, Oro AE. Receptor tyrosine phosphatase-dependent cytoskeletal remodeling by the hedgehog-responsive gene MIM/BEG4. J Cell Biol. 2005;168:453–463. doi: 10.1083/jcb.200409078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindblad-Toh K, Tanenbaum DM, Daly MJ, Winchester E, Lui WO, Villapakkam A, Stanton SE, Larsson C, Hudson TJ, Johnson BE, Lander ES, Meyerson M. Loss-of-heterozygosity analysis of small-cell lung carcinomas using single-nucleotide polymorphism arrays. Nat Biotechnol. 2000;18:1001–1005. doi: 10.1038/79269. [DOI] [PubMed] [Google Scholar]
- Motiwala T, Kutay H, Ghoshal K, Bai S, Seimiya H, Tsuruo T, Suster S, Morrison C, Jacob ST. Protein tyrosine phosphatase receptor-type O (PTPRO) exhibits characteristics of a candidate tumor suppressor in human lung cancer. Proc Natl Acad Sci USA. 2004;101:13844–13849. doi: 10.1073/pnas.0405451101. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Pierceall WE, Goldberg LH, Tainsky MA, Mukhopadhyay T, Ananthaswamy HN. Ras gene mutation and amplification in human nonmelanoma skin cancers. Mol Carcinogen. 1991;4:196–202. doi: 10.1002/mc.2940040306. [DOI] [PubMed] [Google Scholar]
- Popp S, Waltering S, Holtgreve-Grez H, Jauch A, Proby C, Leigh IM, Boukamp P. Genetic characterization of a human skin carcinoma progression model: From primary tumor to metastasis. J Invest Dermatol. 2000;115:1095–1103. doi: 10.1046/j.1523-1747.2000.00173.x. [DOI] [PubMed] [Google Scholar]
- Popp S, Waltering S, Herbst C, Moll I, Boukamp P. UV-B-type mutations and chromosomal imbalances indicate common pathways for the development of Merkel and skin squamous cell carcinomas. Int J Cancer. 2002;99:352–360. doi: 10.1002/ijc.10321. [DOI] [PubMed] [Google Scholar]
- Proby CM, Purdie KJ, Sexton CJ, Purkis P, Navsaria HA, Stables JN, Leigh IM. Spontaneous keratinocyte cell lines representing early and advanced stages of malignant transformation of the epidermis. Exp Dermatol. 2000;9:104–117. doi: 10.1034/j.1600-0625.2000.009002104.x. [DOI] [PubMed] [Google Scholar]
- Quinn AG, Sikkink S, Rees JL. Basal cell carcinomas and squamous cell carcinomas of human skin show distinct patterns of chromosome loss. Cancer Res. 1994;54:4756–4759. [PubMed] [Google Scholar]
- Raghavan M, Lillington DM, Skoulakis S, Debernardi S, Chaplin T, Foot NJ, Lister A, Young BD. Genome-wide single nucleotide polymorphism analysis reveals frequent partial uniparental disomy due to somatic recombination in acute myeloid leukemias. Cancer Res. 2005;65:375–378. [PubMed] [Google Scholar]
- Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AW, Chen W, Cho EK, Dallaire S, Freeman JL, Gonzalez JR, Gratacos M, Huang J, Kalaitzopoulos D, Komura D, MacDonald JR, Marshall CR, Mei R, Montgomery L, Nishimura K, Okamura K, Shen F, Somerville MJ, Tchinda J, Valsesia A, Woodwark C, Yang F, Zhang J, Zerjal T, Zhang J, Armengol L, Conrad DF, Estivill X, Tyler-Smith C, Carter NP, Aburatani H, Lee C, Jones KW, Scherer SW, Hurles ME. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehman I, Quinn AG, Takata M, Taylor AE, Rees JL. Low frequency of allelic loss in skin tumours from immunosuppressed individuals. Br J Cancer. 1997;76:757–759. doi: 10.1038/bjc.1997.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Takahashi K, Nagayama K, Arai Y, Ito N, Okada M, Minna JD, Yokota J, Kohno T. Identification of chromosome arm 9p as the most frequent target of homozygous deletions in lung cancer. Genes Chromosomes Cancer. 2005;44:405–414. doi: 10.1002/gcc.20253. [DOI] [PubMed] [Google Scholar]
- Sjöblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buckhaults P, Farrell C, Meeh P, Markowitz SD, Willis J, Dawson D, Willson JKV, Gazdar AF, Hartigan J, Wu L, Liu C, Parmigiani G, Park BH, Bachman KE, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- Soufir N, Moles JP, Vilmer C, Moch C, Verola O, Rivet J, Tesniere A, Dubertret L, Basset-Seguin N. p16 UV mutations in human skin epithelial tumours. Oncogene. 1999;18:5477–5481. doi: 10.1038/sj.onc.1202915. [DOI] [PubMed] [Google Scholar]
- Stallings RL, Nair P, Maris JM, Catchpoole D, McDermott M, O'Meara A, Breatnach F. High-resolution analysis of chromosomal breakpoints and genomic instability identifies PTPRD as a candidate tumor suppressor gene in neuroblastoma. Cancer Res. 2006;66:3673–3680. doi: 10.1158/0008-5472.CAN-05-4154. [DOI] [PubMed] [Google Scholar]
- Teh MT, Blaydon D, Chaplin T, Foot NJ, Skoulakis S, Raghavan M, Harwood CA, Proby CM, Philpott MP, Young BD, Kelsell DP. Genomewide single nucleotide polymorphism microarray mapping in basal cell carcinomas unveils uniparental disomy as a key somatic event. Cancer Res. 2005;65:8597–8603. doi: 10.1158/0008-5472.CAN-05-0842. [DOI] [PubMed] [Google Scholar]
- Trapasso F, Yendamuri S, Dumon KR, Iuliano R, Cesari R, Feig B, Seto R, Infante L, Ishii H, Vecchione A, During M, Croce C, Fusco A. Restoration of receptor-type protein tyrosine phosphatase eta function inhibits human pancreatic carcinoma cell growth in vitro and in vivo. Carcinogenesis. 2004;25:2107–2114. doi: 10.1093/carcin/bgh224. [DOI] [PubMed] [Google Scholar]
- Zhao X, Weir BA, LaFramboise T, Lin M, Beroukhim R, Garraway L, Beheshti J, Lee JC, Naoki K, Richards WG, Sugarbaker D, Chen F, Rubin MA, Janne PA, Girard L, Minna J, Christiani D, Li C, Sellers WR, Meyerson M. Homozygous deletions and chromosome amplifications in human lung carcinomas revealed by single nucleotide polymorphism array analysis. Cancer Res. 2005;65:5561–5570. doi: 10.1158/0008-5472.CAN-04-4603. [DOI] [PubMed] [Google Scholar]