

Abstract
Runx2, an essential transactivator for osteoblast differentiation, is tightly regulated at both the transcriptional and posttranslational levels. In this paper, we report that CHIP (C terminus of Hsc70-interacting protein)/STUB1 regulates Runx2 protein stability via a ubiquitination-degradation mechanism. CHIP interacts with Runx2 in vitro and in vivo. In the presence of increased Runx2 protein levels, CHIP expression decreases, whereas the expression of other E3 ligases involved in Runx2 degradation, such as Smurf1 or WWP1, remains constant or increases during osteoblast differentiation. Depletion of CHIP results in the stabilization of Runx2, enhances Runx2-mediated transcriptional activation, and promotes osteoblast differentiation in primary calvarial cells. In contrast, CHIP overexpression in preosteoblasts causes Runx2 degradation, inhibits osteoblast differentiation, and instead enhances adipogenesis. Our data suggest that negative regulation of the Runx2 protein by CHIP is critical in the commitment of precursor cells to differentiate into the osteoblast lineage.
Introduction
Runx2, a runt domain family protein, is an essential transactivator for osteoblast differentiation and bone formation. In Runx2-null mutant mice, both intramembraneous and endochondral ossification are absent (Komori et al., 1997; Otto et al., 1997). The expression of Runx2 is a milestone for mesenchymal cells' commitment to osteoblasts (Ducy et al., 1997; Komori et al., 1997; Otto et al., 1997). During the differentiation of precursor cells into osteoblasts, Runx2 is activated and induces osteoblast marker gene expression by binding to a cis-acting element, OSE2 (Ducy et al., 1997). Several studies indicate that Runx2 is involved in the commitment and differentiation of cells in the osteoblast and adipocyte lineages (Chen et al., 1998; Gori et al., 1999; Lecka-Czernik et al., 1999; Enomoto et al., 2004; Hong et al., 2005). Multiple biological functions of Runx2 have been demonstrated recently (Nam et al., 2002; Taniuchi et al., 2002; Ito and Miyazono, 2003; Woolf et al., 2003; Aberg et al., 2004; Yoshida et al., 2004; Yoshida and Komori, 2005; Hinoi et al., 2006; Pratap et al., 2006; Whiteman and Farrell, 2006; Young et al., 2007).
Runx2 is tightly regulated at both the transcriptional and posttranslational levels. In particular, Runx2 activity can be regulated through a ubiquitin–proteasome-mediated protein degradation mechanism. Smurf1 was the first factor identified as an E3 ligase for Runx2 ubiquitination and degradation (Zhao et al., 2003; Zhao et al., 2004). Smurf1-induced Runx2 degradation can be enhanced by Smad6 (Shen et al., 2006) and TNF (Kaneki et al., 2006), as Smad6 interacts with Runx2 and TNF promotes the expression of Smurf1 expression in osteoblasts. Recently, Schnurr-3 (Shn3) was reported to recruit the E3 ligase WWP1 to mediate Runx2 degradation (Jones et al., 2006). The osteoblast activity is augmented in the Shn3 deficiency mice that generate adult onset osteosclerosis with increased bone mass.
CHIP (C terminus of Hsc70-interacting protein) is a cochaperone protein identified through its interaction with Hsc/Hsp70 (Ballinger et al., 1999). CHIP promotes the ubiquitination and degradation of chaperone-bound proteins, such as receptor tyrosine kinase ErbB2, glucocorticoid receptor, and the misfolded cystic fibrosis transmembrane conductance regulator protein (Wiederkehr et al., 2002). In a recent study using a CHIP−/− cell model, CHIP has been reported to elicit the transcriptional activation of HSF1 during stress recovery (Qian et al., 2006). We have recently reported that CHIP regulates bone morphogenetic protein (BMP) and TGF-β signals by enhancing Smad protein ubiquitination and degradation (Li et al., 2004; Xin et al., 2005; Li et al., 2007). Here, we show that CHIP promotes Runx2 ubiquitination and degradation and thereby negatively regulates osteoblast differentiation.
Results
Runx2 interacts with CHIP as identified by yeast two-hybrid and coimmunoprecipitation analyses in culture mammalian cells
To search for additional potential regulators of Runx2, we performed a yeast two-hybrid screening experiment using the full-length mouse type II isoform of Runx2. CHIP was identified as a Runx2-interacting protein. The interaction was confirmed by a GST pull-down experiment (Fig. 1 A). To show the interaction in mammalian cells, constructs expressing Myc-Runx2 and HA-CHIP were transfected into 293T cells. Because CHIP is reported to mostly localize to the cytoplasm (Ballinger et al., 1999; Hatakeyama et al., 2001) and Runx2 is in the nucleus (Zaidi et al., 2001), we determined whether the two proteins have any opportunity to interact in intact cells. Separation of nuclear and cytoplasm proteins indicates that although the majority of Myc-Runx2 is present in the nucleus, HA-CHIP is present in both the cytoplasm and the nucleus (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200711044/DC1), which is consistent with a previous study (Huang et al., 2004). Coimmunoprecipitation experiments using cytoplasmic and nuclear fractions show that Myc-Runx2 is precipitated by HA-CHIP protein in the nuclear extracts (Fig. 1 B, left) but not from the cytoplasmic fraction (Fig. 1 B, right). These results suggest that CHIP is capable of interacting with Runx2 in the nucleus of mammalian cells. Endogenous Runx2 and CHIP protein interaction was observed in MC3T3-E1 cells, a preosteoblast cell line (Fig. 1 C). This result firmly confirmed that Runx2 specifically interacts with CHIP under physiological conditions.
Figure 1.

Runx2 interacts with CHIP. (A) Runx2 physically interacts with CHIP in vitro. A GST pull-down assay was performed with the purified GST or GST-CHIP protein and Myc-Runx2 protein expressed by 293T cells. (B) Interaction of Runx2 with CHIP occurs in the nucleus of mammalian cells. Cytoplasmic (Cy) and nuclear (Nu) extracts were used in coimmunoprecipitation assays with anti-HA antibody, and the precipitated complexes were analyzed with an anti-Myc antibody against Myc-Runx2. (C) Endogenous Runx2 and CHIP proteins interact. MC3T3-E1 cells were treated with 10 μM MG132 for 4 h before lysis. (top) Whole cell lysates were immunoprecipitated with an anti-CHIP antiserum or a preimmune serum (Pre). The immunoprecipitates were visualized with an anti-Runx2 antibody (65 kD*; Santa Cruz Biotechnology, Inc.). (bottom) Reversed coimmunoprecipitation assays were performed using anti-Runx2 antibodies (MBL International) and IgG. (D) The C terminus of Runx2 interacts with CHIP. Schematic illustration of Runx2 and its truncated constructs is shown on top. QA, QA domain; RHD, runt homology domain; NMTS, nuclear matrix targeting signal. A GST pull-down assay was performed using the purified GST-CHIP protein and different deletions of the hRunx2 protein expressed in mammalian cells. (E) TPR domain of CHIP is critical for the interaction with Runx2. Three main functional domains of CHIP (TPR, tetratricopeptide repeats; U-box, U-box domain; Charged, charged domain) are schematically illustrated on top. A GST pull-down assay was performed using GST-fused CHIP, and the deletion proteins to pull down the mammalian cell expressed Myc-Runx2 protein. (F) Mutation CHIP(K31A) impairs the ability of CHIP to interact with Runx2. Lysates were prepared from 293T cells expressing HA-tagged CHIP, CHIP(K31A), or CHIP(H261Q) together with the indicated Myc-Runx2. Coimmunoprecipitation and immunoblotting were performed as described in A. The result from a longer exposure (5×) for the immunoblot is also presented (top).
The C terminus of Runx2 and the TPR domain of CHIP are critical for interaction
Runx2 has two protein isoforms with the glutamine–arginine-rich domain (QA), DNA-binding runt homology domain, NLS, nuclear matrix targeting signal, and the transducin-like enhancer of split/groucho-interacting C-terminal pentapeptide VWRPY (for review see Stein et al., 2004). To map the interaction domains between Runx2 and CHIP proteins, we expressed the human type I isoform of Runx2 and its deletion mutants (Zhang et al., 2000) in 293T cells. The GST pull-down experiment results show that among the different deletions of Runx2 (Fig. 1 D, top), the full-length (hRunx2 1–507) and the deletion mutant hRunx2(88–507) show strong interactions with GST-CHIP (Fig. 1 D, bottom; third and fourth lanes), whereas the deletion mutant hRunx2(1–340) has completely lost, and hRunx2(1–424) only has a weak interaction with GST-CHIP. These experiments suggest that the C-terminal end of Runx2, which is around the nuclear matrix–targeting signal motif, contributes to the interaction with CHIP. As CHIP contains a tetratricopeptide repeat (TPR) domain responsible for chaperone binding, a U-box domain important for E3 ubiquitin ligase activity, and a central domain rich in charged residues (Ballinger et al., 1999; Murata et al., 2001), interaction with Runx2 was further mapped using a series of GST-CHIP deletion mutants (Fig. 1 E, top). The data show that both GST-TPR and GST–ΔU-box fusion proteins interact with Myc-Runx2, but GST–U-box and GST-ΔTPR do not (Fig. 1 E, bottom). This suggests that the TPR domain is necessary and sufficient for the interaction with Runx2.
The chaperone activity of CHIP is required for its interaction with Runx2
Next, we investigated whether the Runx2–CHIP interaction is associated with the activity of CHIP. For this, we compared the interaction of Runx2 with the mouse CHIP(K31A) and CHIP(H261Q) mutants, as the K31A mutant (in the TPR domain) binds neither Hsp90 nor Hsp/Hsc70 chaperone (Xu et al., 2002), whereas H261Q mutant (in the U box) fails to bind the cognate E2 and abolishes its E3 ligase activity (Hatakeyama et al., 2001). The coimmunoprecipitation assay results show that the interaction of HA-CHIP(K31A) with Myc-Runx2 is barely detectable on a short exposure and is still only weak with a long exposure, and HA-CHIP(H261Q) demonstrates a strong interaction with Myc-Runx2 (Fig. 1 F). These results suggest that the interaction of CHIP with Runx2 is dependent on the K residue in the TPR domain, which is critical for the chaperone activity, as the CHIP mutation that abolishes interactions with chaperones fails to interact with Runx2.
CHIP enhances Runx2 degradation and ubiquitination
CHIP has been reported to be an E3 ubiquitin ligase mediating the ubiquitination and proteasome-dependent degradation of several substrates (Wiederkehr et al., 2002). In our experiments, we have observed that expression of CHIP significantly reduced Runx2 protein levels (Fig. S1, top; and Fig. 1, B [third blot] and F [fourth blot, compare the third lane with the first, fourth, and fifth lanes]). We speculated that CHIP might enhance Runx2 turnover. To examine this hypothesis, Myc-Runx2 (mouse type II) was coexpressed with increasing amounts of HA-CHIP protein in 293T cells in the presence or absence of MG132, a proteasome inhibitor. Immunoblotting results demonstrate that with increasing amounts of HA-CHIP, the Myc-Runx2 protein levels decrease in the absence of MG132 but remain constant in the presence of MG132 (Fig. 2 A). In a parallel experiment, we observed that the protein levels of the two C terminus–truncated forms of Runx2, which show little or no interaction with CHIP, remained unchanged (Fig. 2 B). These results suggest that CHIP participates in Runx2 degradation.
Figure 2.

CHIP prompts Runx2 degradation and ubiquitination. (A) CHIP mediates the degradation of Runx2 in a dose-dependent manner. Myc-Runx2 was coexpressed with increasing amounts of HA-CHIP in 293T cells treated with (right) or without (left) 10 μM MG132 for 4 h. Protein levels of Myc-Runx2 were measured by immunoblotting with an anti-Myc antibody. EGFP was used as a control. (B) CHIP does not induce degradation of the C-terminal truncated Runx2. Myc-tagged Runx2 (full length), Runx2(1–424), and Runx2(1–340) were expressed with increasing amounts of HA-CHIP in 293T cells. Levels of the Myc-tagged proteins were measured by immunoblotting with an anti-Myc antibody. (C and D) CHIP accelerates turnover of the Runx2 protein. A pulse-chase assay (C) was performed in COS7 cells transfected with Runx2 together with empty vector, CHIP, or Smurf1. Quantitative presentation of the results with SEM (error bars; three repeats) is shown in D. (E and F) Knocking down CHIP by siRNA increases Runx2 protein levels. pBS/U6 vector, vector-based siRNA targeting CHIP (CHIP RNAi), or EGFP (EGFP RNAi) was transfected with or without Myc-Runx2 vector into UMR106 cells. Myc-Runx2 (E) or endogenous Runx2 protein (F) levels were measured by immunoblotting with an anti-Myc antibody or an anti-Runx2 antibody (65 kD*; Santa Cruz Biotechnology, Inc.). (G) CHIP prompts Runx2 ubiquitination. Myc-Runx2 and His-ubiquitin (His-Ubi) were coexpressed with or without HA-CHIP in 293T cells treated with 25 μM MG132 for 4 h before lysis. Total ubiquitinated proteins were precipitated from whole cell lysates by Ni–nitrilotriacetic acid resin and analyzed by immunoblotting with an anti-Runx2 antibody. (H) Runx2 ubiquitination by CHIP occurs in the nucleus. Myc-Runx2 and His-ubiquitin were coexpressed with HA-tagged CHIP, CHIP(K31A), or CHIP(H261Q) in 293T cells. The nuclear exacts were separated and used for the ubiquitination experiment.
Because Smurf1 has been reported to enhance Runx2 degradation (Zhao et al., 2003, 2004), we compared whether CHIP and Smurf1 have a similar effect on turnover rates of the Runx2 protein. A pulse-chase analysis using COS7 cells expressing Myc-Runx2 alone or together with CHIP or Smurf1 show that overexpression of CHIP increases the Runx2 degradation rate (Fig. 2, C and D [line with open squares]) compared with Smurf1 (Fig. 2, C and D [line with closed triangles]). When the two E3 ligases were coexpressed, we observed an additive effect on Runx2 degradation (unpublished data). These results indicate that CHIP is a potent factor enhancing Runx2 degradation.
To determine the role of CHIP on Runx2 degradation under physiological conditions, we knocked down CHIP expression using siRNA in a rat osteosarcoma cell line, UMR106 (Fig. 2, E [third blot, last lane] and F [bottom, right lane]), in which endogenous CHIP is moderately expressed (see Fig. 4 G). This results in a significant increase in the amount of either overexpressed Myc-Runx2 protein (Fig. 2 E) or endogenous Runx2 protein (Fig. 2 F). These results suggest that the endogenous CHIP protein degrades Runx2 in osteoblast-like cells.
Figure 4.

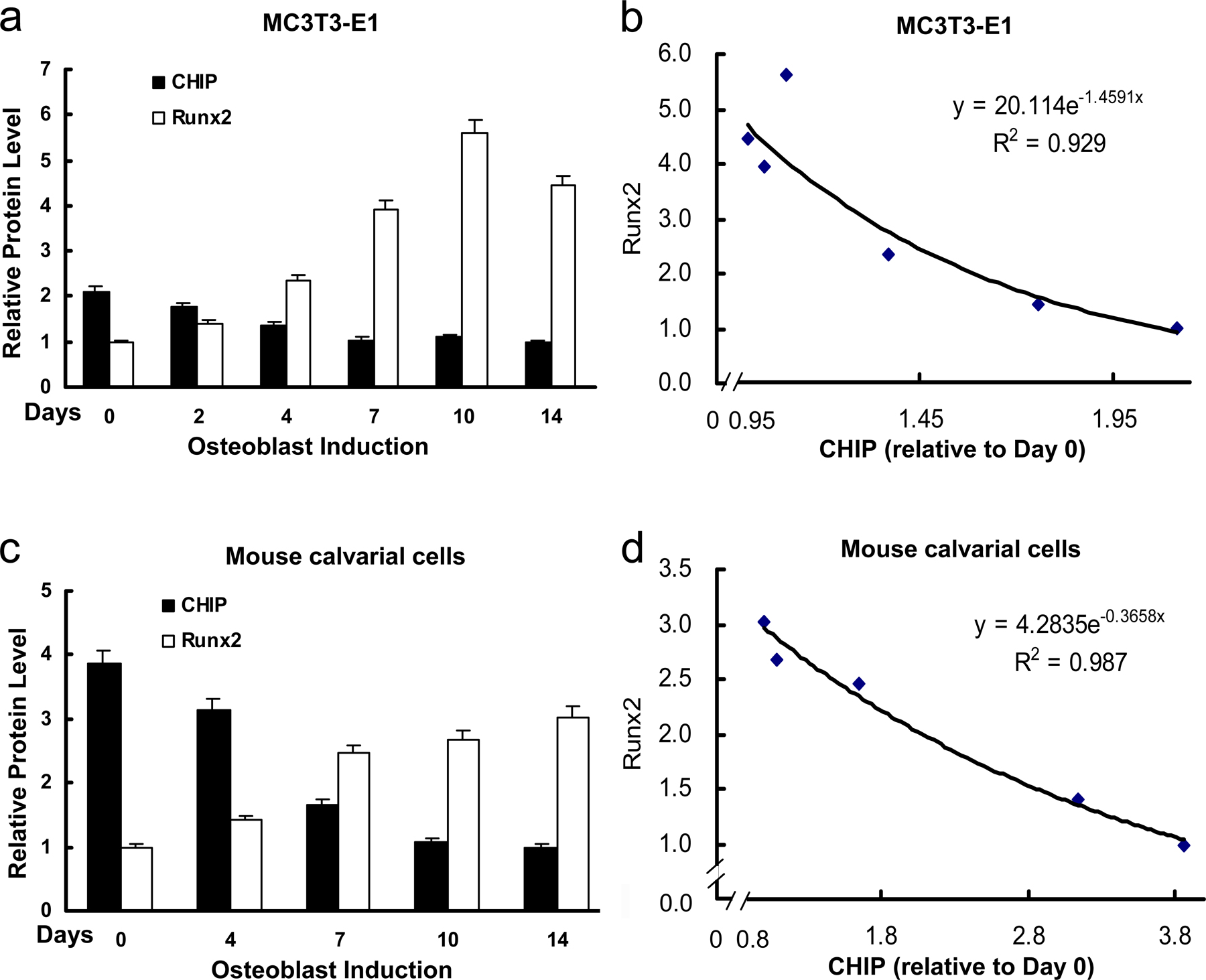
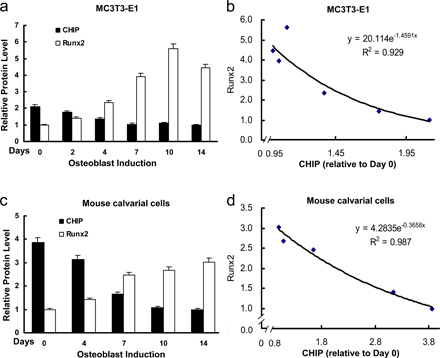
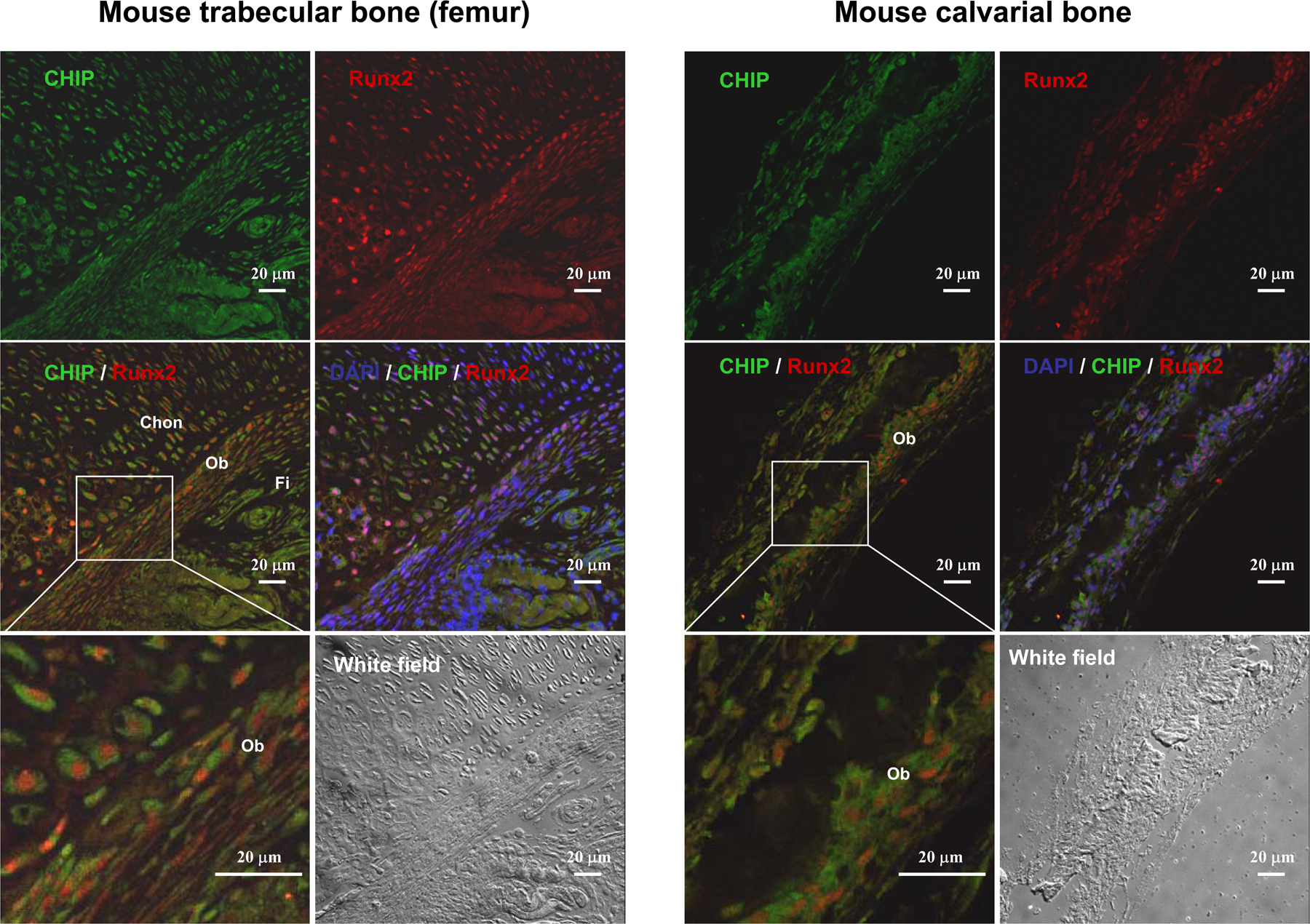
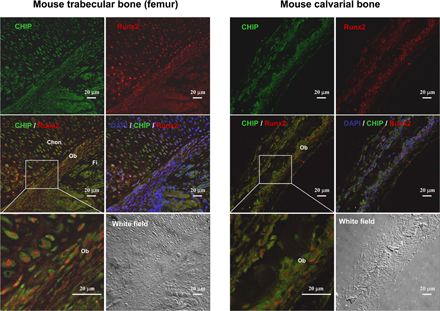
Up-regulation of Runx2 protein level parallels reduced expression of CHIP during osteoblast differentiation. (A) von Kossa staining for MC3T3-E1 cells cultured for 3 wk in the presence of AA/β-GP was performed to demonstrate the cellular phenotype of osteoblasts. (B) mRNA and protein levels of Runx2 and CHIP during the osteoblast differentiation were compared. RT-PCR and Western blot (see quantitative presentations in Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200711044/DC1) analyses were performed on MC3T3-E1 cells induced by AA/β-GP for the indicated number of days. (C) Smurf1 expression is unchanged, and WWP1/Shn3 is increased during osteoblast differentiation. Real-time RT-PCR analysis was performed for the expression of the indicated genes during osteoblast differentiation of MC3T3-E1 cells. Values are shown relative to cells on day 0. (D) Alizarin red staining for mouse calvarial cells cultured for 2 wk in the presence of AA/β-GP. (E and F) The expression of Runx2, CHIP, WWP1, Shn3, and Smurf1 in mouse calvarial cells was analyzed as in B and C. (G) Real-time RT-PCR was performed to show expression of the indicated genes in different lineages of mesenchyma-originated cells. Fold induction was calculated based on the mRNA level of each gene in NIH3T3. (H) Expression of the CHIP (green) and Runx2 (red) proteins in osteoblasts in vivo. Double immunofluorescence staining was performed using sections of trabecular (femur; a and b) and calvarial (c and d) bone from newborn mice. Enlarged osteoblasts (boxed) are shown on the bottom (b and d). Ob, osteoblast; Chon, chondrocyte; Fi, fibroblast-like or other types of cells. Error bars represent SEM.
As CHIP affects protein degradation through the ubiquitination process, we examined the effect of CHIP on Runx2 ubiquitination. We coexpressed Runx2 and polyhistidine-tagged ubiquitin in 293T cells with HA-CHIP. Ubiquitinated proteins were precipitated from cell lysates under denatured conditions and analyzed by immunoblotting with the anti-Runx2 antibody. Our results show that coexpression of CHIP is associated with a marked increase in the ubiquitination of Runx2 (Fig. 2 G), suggesting that CHIP is involved in Runx2 ubiquitination. In an experiment with the CHIP mutants, we observed that CHIP(K31A) did not alter the level of Runx2 ubiquitination (Fig. 2 H, fourth lane), presumably because it fails to interact with Runx2. However, the expression of CHIP(H261Q) caused a decrease in Runx2 ubiquitination compared with wild-type CHIP (Fig. 2 H, compare the fifth and third lanes) but a slight increase in Runx2 ubiquitination compared with the control vector (Fig. 2 H, compare the fifth and second lanes). Because the later experiments were performed using nuclear proteins, the results suggest that CHIP enhances Runx2 ubiquitination in the nucleus.
CHIP decreases Runx2-induced transcriptional activation
We analyzed how CHIP influences Runx2 biological activity using a luciferase reporter assay in which the firefly luciferase gene is under control of a Runx2-response element (Zhang et al., 2000). Our data demonstrate that in both NIH3T3 and COS7 cells, the overexpression of Runx2 alone activates reporter expression four- to sixfold and that overexpression of CHIP significantly blunts the enhancing effect of Runx2 (Fig. 3 A). When the inhibitory effects of wild-type CHIP, CHIP mutants, and Smurf1 were compared, we observed that both of the CHIP mutants could not inhibit Runx2-induced transcriptional activation (Fig. 3 B, third and fourth bar groups) but that Smurf1 had a similar scale of inhibitory effect as wild-type CHIP (Fig. 3 B, last bar group). CHIP(H261Q) may have a slightly enhanced effect on the luciferase activity (Fig. 3 B, fourth bar group). CHIP-induced attenuation of Runx2 transcriptional activity was also observed in the UMR106 cells (Fig. 3 C, second bar group). Furthermore, when the endogenous CHIP protein was depleted in the UMR106 cells by CHIP siRNA (Li et al., 2004; Xin et al., 2005), we observed an increase in the luciferase activity either at basal (without overexpression of Runx2) or under Runx2 overexpression (Fig. 3 C, last bar group). As CHIP did not affect the activity of the nonrelated reporter pGL3–acute phase response element (this reporter responds to interleukin 6 through the activation of STAT3; Fig. 3 D), these data suggest that CHIP decreases Runx2-induced transcriptional activation.
Figure 3.

CHIP inhibits Runx2-induced transcriptional activity. (A) Overexpression of CHIP inhibits Runx2-induced transcriptional activity. Luciferase assays were performed using NIH3T3 or COS7 cells transfected with the indicated expression vectors along with a Runx2-responsive reporter construct, 6xOSE2-OC/pGL3 and pRL-TK (internal control). Values were normalized using the internal control and presented relative to basal activity (NIH3T3, first bar; COS7, fifth bar) with the mean from three independent repeats. (B) Mutants of CHIP fail to inhibit Runx2-induced transcriptional activity. CHIP, CHIP mutants, and Flag-Smurf1 were compared for the inhibitory effect on Runx2 activity in NIH3T3 cells. (C) Knocking down CHIP by siRNA facilitates Runx2-mediated transcription in UMR106 cells. Luciferase assays were performed in UMR106 cells transfected with Runx2 in the presence of CHIP, EGFP RNAi, or CHIP RNAi. (D) CHIP did not affect the transcriptional activity of STAT3. NIH3T3 cells transfected with STAT3 in the presence or absence of CHIP along with pGL3–acute phase response element and treated with 10 ng/ml interleukin-6. All of the data are presented as mean with SEM (error bars) from three independent experiments. The same letter represents a significant difference within this group in a meaningful comparison (P < 0.05).
CHIP is down-regulated during osteoblast differentiation
To investigate the physiological role of CHIP on Runx2 protein turnover, we examined CHIP expression during osteoblast differentiation. Treatment with ascorbic acid (AA) and β-glycerophosphate (β-GP) induced the differentiation of MC3T3-E1 cells into osteoblast-like cells, as indicated by von Kossa–positive staining (Fig. 4 A) and increased AP activity examination (not depicted). We next analyzed CHIP and Runx2 mRNA and protein levels at different time points during early osteoblast differentiation. The results show that Runx2 mRNA levels remained constant during 2 wk of induction (Fig. 4 B, second blot), whereas both CHIP mRNA and protein levels significantly decreased over time (Fig. 4 B). In contrast, the levels of Runx2 protein increased during this culture period (Fig. 4 B, fifth blot; and Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200711044/DC1) concurrent with decreased CHIP protein expression. These results indicate that decreased CHIP protein levels correlate with increased Runx2 protein levels despite the maintenance of constant Runx2 mRNA levels (obviously increased in the later stage) during early osteoblast differentiation. To determine the potential involvement of other E3 ligases, we also examined the expression of Smurf1 and WWP1 (and its adaptor Shn3), two known E3 ligases that regulate Runx2 protein turnover, and found that the Smurf1 mRNA levels were unchanged and that WWP1 (also Shn3) mRNA levels became elevated (Fig. 4 C).
To study the expression of the aforementioned genes in the later osteoblast differentiation, we isolated mouse calvarial cells. When these cells were cultured for further differentiation into mature osteoblasts (indicated by Alizarin red staining in Fig. 4 D), we observed that CHIP expression decreased, whereas Runx2 protein levels increased together with its mRNA levels (Fig. 4, E and F). Changes in WWP1, Shn3, and Smurf1 mRNA levels remained the same as patterns observed in MC3T3-E1 cells, the early osteoblasts.
Further analysis of the expression levels of Runx2, CHIP, WWP1, Shn3, and Smurf1 in different cell lines derived from mesenchymal cells was performed. We observed that Runx2 is not expressed in NIH3T3 (a fibroblast cell line), C3H10T1/2 (a pluripotent mesenchymal cell line), and C2C12 (a progenitor cell line for myoblasts), is moderately expressed in MC3T3-E1 (a progenitor cell line for osteoblasts), and is abundantly expressed in UMR106 (an osteosarcoma cell line; Fig. 4 G). In contrast, CHIP is expressed at high levels in C2C12, NIH3T3, and C3H10T1/2 cells but moderately expressed in MC3T3-E1 and UMR106 cells (Fig. 4 G). Furthermore, Smurf1 is expressed ubiquitously in these cell lines, except it is higher in C2C12 cells. WWP1, similar to Shn3, is barely expressed in C2C12 cells, moderately expressed in NIH3T3 and C3H10T1/2 cells, and most highly expressed in MC3T3-E1 and UMR106 cells (Fig. 4 G). As these cell lines represent different differentiation stages of mesenchymal cells, the expression patterns of these genes are potentially related to cell differentiation. We were particularly intrigued by the reversed pattern of Runx2 and CHIP expression in nonosteoblast (NIH3T3, C3H10T1/2, and C2C12) and osteoblast (MC3T3-E1 and UMR106) lineages. Collectively, these data suggest that CHIP is down-regulated during osteoblast differentiation, and this down-regulation maintains Runx2 protein stability. The coexpression of CHIP and Runx2 in osteoblasts in vivo was finally confirmed by double immunofluorescence staining using sections of calverial and trabecular bone (femur) from newborn mice (Fig. 4 H and Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200711044/DC1).
CHIP affects osteoblast differentiation and mineralization of MC3T3-E1 cells
To further investigate the potential functions of CHIP on osteoblasts, we established stable MC3T3-E1 cell lines with the overexpression of HA-CHIP (Fig. 5 A) or depletion of endogenous CHIP (Fig. 5 C, left). The stable cell lines were treated with AA/β-GP for different periods of time and analyzed for Runx2 expression. AA/β-GP induced an elevation of Runx2 protein levels but not mRNA levels in the mock cells (Fig. 5 B, left lanes), whereas Runx2 protein levels decreased in HA-CHIP–expressing clones (Fig. 5 B). These results indicate that overexpression of CHIP decreases Runx2 protein levels but has no effect on its mRNA levels in the MC3T3-E1 cells. On the other hand, we observed that CHIP depletion by siRNA increased Runx2 protein levels but did not affect its mRNA (Fig. 5 C). These data indicated the role of endogenous CHIP in the regulation of Runx2 protein.
Figure 5.

Stable expression of CHIP inhibits osteoblast differentiation and mineralization. (A) Establishment of cell lines stably expressing HA-CHIP in MC3T3-E1 cells (#2 and #8). Endogenous CHIP and HA-CHIP expression was measured by immunoblotting with an anti-CHIP antiserum. (B) Stable expression of HA-CHIP in MC3T3-E1 cells reduces the endogenous Runx2 protein levels. The cell lines were treated with AA/β-GP for the indicated days. The mRNA (top) and protein (bottom) levels of Runx2 were measured by RT-PCR and Western blotting, respectively. (C) Stable depletion of CHIP using siRNA in MC3T3-E1 cells increased Runx2 protein levels. The cell lines with depletion of CHIP (#9 and #20) were treated with AA/β-GP for the indicated days. mRNA (left) and protein (right) levels of Runx2 were measured by RT-PCR and Western blotting, respectively. (D–F) CHIP inhibits the differentiation of MC3T3-E1 cells into osteoblast-like cells. CHIP- or CHIP siRNA–expressing cell lines were induced to differentiate into osteoblast-like cells in the presence of AA/β-GP for 4–21 d followed by the determination of AP activity (D), calcium accumulation by Alizarin red staining (E), and expression of the osteoblast marker genes (BSP and OCN) by real-time RT-PCR (F). Error bars represent SEM.
To investigate whether the overexpression of CHIP affects the differentiation of cells into osteoblasts, we characterized the cells by induction with AA/β-GP for different periods of time. First, we observed that AP activity was dramatically decreased in the two HA-CHIP–expressing clones but significantly increased in the two CHIP knockdown clones compared with the two mock cells (Fig. 5 D). Alizarin red staining demonstrated similar results (Fig. 5 E). Furthermore, real-time RT-PCR was performed to examine expression of the osteoblast differentiation marker genes (bone sialoprotein [BSP] and osteocalcin [OCN]). Expression of these genes was delayed in the overexpression clones or enhanced in the depletion clones (Fig. 5 F), whereas the other E3 ligase genes known to participate in Runx2 degradation were not affected (not depicted). These results indicate that increased CHIP overexpression prevents or delays cell differentiation into mature osteoblasts, and the depletion of CHIP promotes or enhances this process.
Overexpression of CHIP drives MC3T3-E1 cells to differentiate into adipocytes but has no effect on the adipocytic differentiation of C3H10T1/2 cells
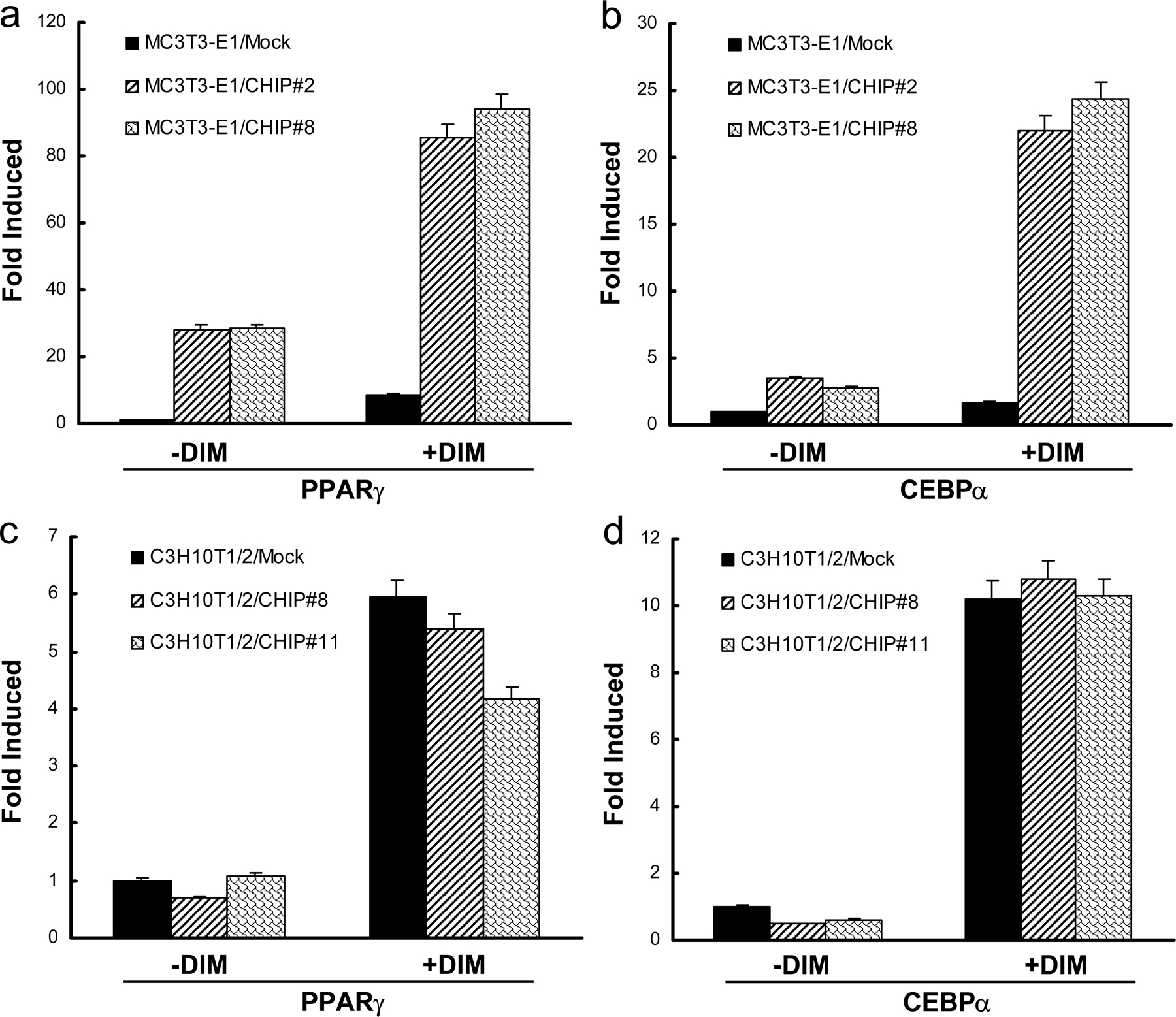
MC3T3-E1 cells are committed to differentiate into osteoblasts by the expression of Runx2 (Prince et al., 2001). Based on the observation that the overexpression of CHIP decreased Runx2 protein levels, we hypothesized that CHIP may prevent precursor cells from differentiating into osteoblasts but may promote cells to differentiate into adipocytes. When the two MC3T3-E1–CHIP clones were cultured under confluence for long times, cells appeared with features of adipocytes, including the accumulation of triglyceride lipid droplets (Oil Red O staining positive; Fig. 6 A, left; #2 and #8). When these cells were induced using the adipogenesis inducer DIM (dexamethasone, 3-isobutyl-1-methylxanthine, and insulin) mixture, the percentages of the triglyceride-containing cells increased to 37.8 ± 6.7% (#2) and 28.6 ± 4.3% (#8) compared with the noninduced 6.0 ± 1.9% (#2) and 3.4 ± 1.4% (#8; Fig. 6, A and B). No triglyceride-containing cells could be detected in mock cells cultured in the absence or presence of DIM mixture.
Figure 6.

Overexpression of CHIP drives MC3T3-E1 cells to differentiate into adipocytes but has no effect on adipocyte differentiation in C3H10T1/2 cells. (A–C) Overexpression of CHIP results in the facilitated differentiation of MC3T3-E1 cells into adipocytes. MC3T3-E1-CHIP cell lines (#2 and #8) and the mock cells were cultured in the presence or absence of DIM mixture for 7 or 14 d. Accumulation of cytoplasmic triglyceride was detected by Oil Red O staining (A). A quantitative representation of positive Oil Red O staining adipocytes. Cell numbers were counted on three randomized fields with a phase-contrast microscope. The percentages of triglyceride-containing cells are shown with mean and SD (error bars; B). The overexpression of CHIP induces the expression of adipocyte marker genes PPARγ and CEBPα. RT-PCR was performed on day 7 (C). (D) Establishment of cell lines stably expressing HA-CHIP in C3H10T1/2 cells (#8 and #11). Protein levels of the endogenous CHIP and HA-CHIP are shown. (E–G) CHIP does not affect the ability of C3H10T1/2 cells to differentiate into adipocytes. C3H10T1/2-CHIP cell lines (#8 and #11) were used in adipocyte differentiation experiments. Oil Red O staining shows accumulation of cytoplasmic triglyceride on day 14 (E) with a quantitative representation of positive Oil Red O staining adipocytes (F) and expression of PPARγ and CEBPα on day 7 (G).
In addition to cell morphology changes, we also examined the expression of the adipocyte marker genes PPARγ and CEBPα. Our data show that expression of these genes is dramatically increased after 7 d of DIM treatment in the MC3T3-E1–CHIP clones (Fig. 6 C and Fig. S4, a and b; available at http://www.jcb.org/cgi/content/full/jcb.200711044/DC1). Significantly elevated PPARγ and CEBPα mRNA levels were even observed in the MC3T3-E1–CHIP cell clones without treatment (Fig. 6 C and Fig. S4, a and b). These data suggest that exogenous expression of CHIP directs MC3T3-E1 cells to differentiate into adipocytes.
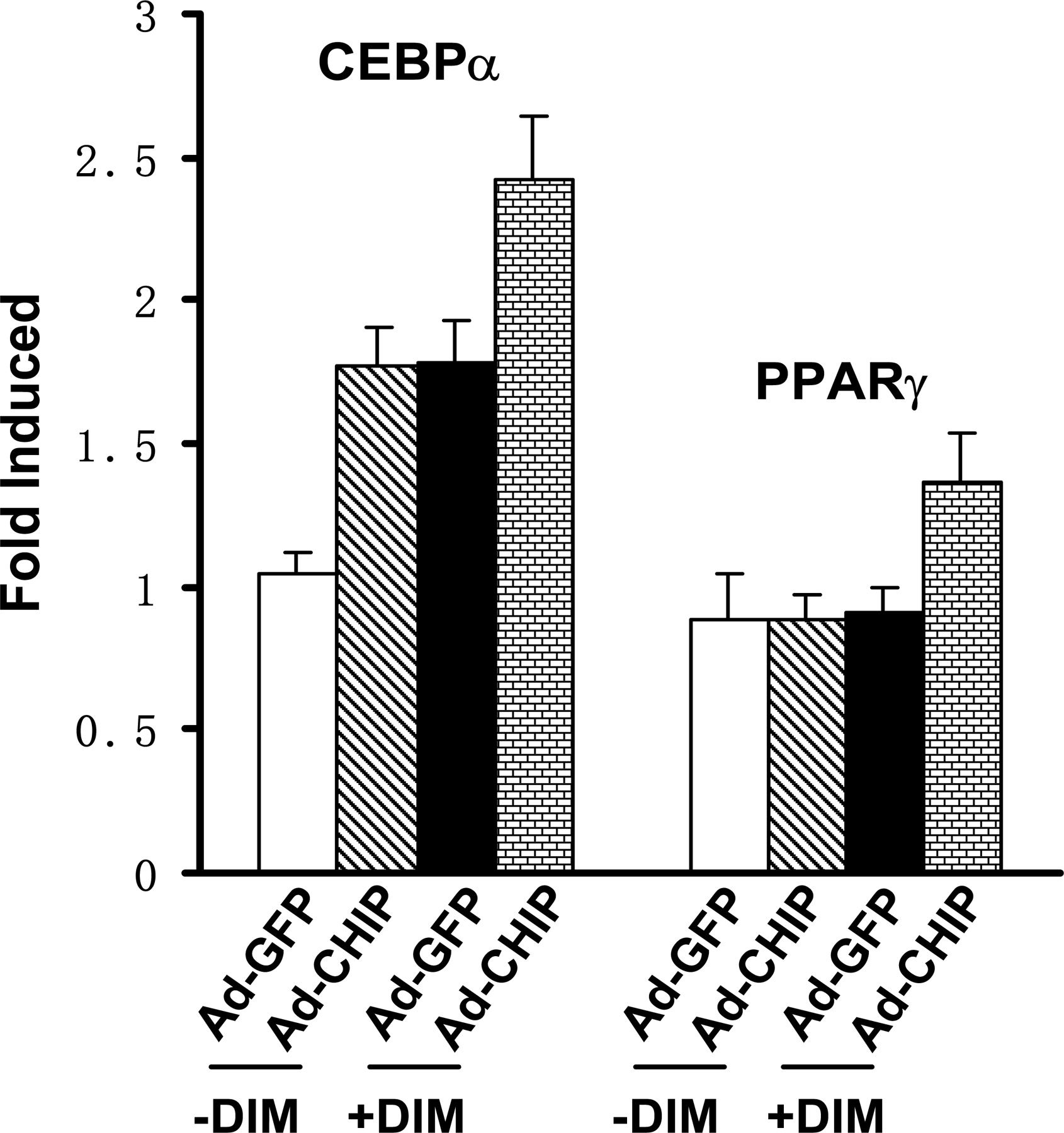
To determine whether the CHIP-induced reversed differentiation of preosteoblast to adipocyte is dependent on CHIP-mediated Runx2 protein turnover, we used C3H10T1/2 cells, a pluripotent cell line with features of mesenchymal stem cells. Consistent with previous studies (for review see Rosen and Spiegelman, 2000; Saito et al., 2002), we found that this cell line does not express Runx2 (Fig. 4 G) and can be induced by DIM into adipocytes (Fig. 6 E, mock). We then established cell lines that stably overexpress HA-CHIP in C3H10T1/2 cells (Fig. 6 D). Our data show that DIM treatment results in similar percentages of differentiated adipocytes in both the mock and stable cell lines (Fig. 6, E and F), suggesting that the overexpression of CHIP has no effect on the commitment of C3H10T1/2 cells to the adipocyte lineage. The mRNA levels of PPARγ and CEBPα also showed no significant change between the mock and C3H10T1/2-CHIP cell lines after DIM treatment for 7 d (Fig. 6 G and Fig. S4, c and d). These results suggest that CHIP does not affect adipogenesis in uncommitted mesenchymal cells (without Runx2 expression) and only functions in the osteoblastic cell lineage (with Runx2 expression). Indeed, we also observed that the overexpression of CHIP by adenovirus starts to enhance PPARγ and CEBPα gene expression in primary calvarial cells (Fig. S5, available at http://www.jcb.org/cgi/content/full/jcb.200711044/DC1). Collectively, we propose that by degrading Runx2 protein, CHIP is an inhibitor for osteoblast differentiation. The down-regulation of CHIP expression is an important event to initiate osteoblast differentiation, which releases Runx2 protein to allow further progress in osteoblast differentiation.
CHIP functions on the primary osteoblasts
Finally, we address whether CHIP functions in primary osteoblasts. For this purpose, we generated adenovirus to overexpress (Ad-CHIP) or deplete (Ad-CHIPi) CHIP in isolated calvarial cells. After we tested the efficacy of the virus infection (not depicted), we infected calvarial cells with the adenovirus (Fig. 7 A). Western blots demonstrated that the Runx2 protein levels were largely depleted in the presence of the Ad-CHIP virus but increased in the presence of the Ad-CHIPi virus (Fig. 7 B). The Alizarin red staining experiments indicated that the overexpression of CHIP blocked differentiation of the cells into mature osteoblasts but that depletion of CHIP significantly enhanced osteoblast maturation compared with the control virus (Ad-EGFP; Fig. 7 C). The inhibited osteoblast differentiation by overexpression of CHIP with the virus was further confirmed by examining the expression of osteoblast-specific marker genes BSP and OCN (Fig. 7 D, middle bar groups). Reversely, depletion of CHIP with the Ad-CHIPi virus resulted in a higher level of the marker gene expression (Fig. 7 D, right bar groups). These data strongly indicate that CHIP has an inhibitory role during osteoblast differentiation.
Figure 7.

CHIP inhibits osteoblast differentiation. (A) Microscopic analysis of GFP expression 4 d after adenoviral infection shows equally efficient transduction (up to 70%) of primary mouse calvarial cells. GFP is expressed from the same viral vector as CHIP or CHIP siRNA. (B) Examination of the adenovirus expressing CHIP (Ad-CHIP) or CHIP siRNA (Ad-CHIPi) on endogenous Runx2 protein levels in calvarial cells. GFP levels indicate amounts of virus infection. (C) Alizarin red staining of mouse calvarial osteoblasts transfected with the adenovirus expressing GFP, CHIP, or CHIP siRNA and cultured in the presence of AA/β-GP for 2 wk. (D) Real-time RT-PCR demonstrates that the mRNA of bone marker genes (BSP and OCN) in the mouse primary calvarial cells infected the virus in the condition of osteoblast differentiation. Error bars represent SEM. (E) A schematic presentation of the functions of CHIP on osteoblast differentiation. Although Runx2 mRNA is expressed in preosteoblast cells, the Runx2 protein level remains at a low level because high CHIP expression leads to its degradation. When cells commit to differentiate into osteoblasts, CHIP expression decreases, leading to increased Runx2 protein levels, which drives the cells to become mature osteoblasts. WWP1 seems to enhance Runx2 degradation in the late stage of osteoblast differentiation (Jones et al., 2006). The question mark indicates an undefined stage of the function. This diagram has been modified from Komori, 2005.
Discussion
Runx2 is a critical transactivator in osteoblast differentiation. The activity of Runx2 is tightly regulated at different levels, including transcription (Sudhakar et al., 2001a), translation (Sudhakar et al., 2001b), phosphorylation (Kim et al., 2006; Rajgopal et al., 2006), acetylation (Jeon et al., 2006), protein–protein interactions (Hong et al., 2005; Dobreva et al., 2006), and ubiquitin-mediated degradation (Zhao et al., 2003; Jones et al., 2006). The important role of the posttranslational regulation of Runx2 has been demonstrated by experiments showing that dexamethasone treatment yields a significant increase in Runx2 protein levels without affecting its mRNA expression (Prince et al., 2001). In this study, we show that CHIP is a novel posttranslational regulator of Runx2 through several lines of evidence. Our findings suggest that CHIP has an important role in the regulation of Runx2 protein levels and cell commitment in osteoblast precursor cells.
Previously, several other E3 ligases have been reported to regulate Runx2 protein turnover, including Smurf1, a HECT domain E3 ubiquitin ligase. Smurf1 has been reported to directly interact with Runx2 and to induce ubiquitin-dependent degradation of Runx2 protein (Zhao et al., 2003). In this study, we compared the influences of Smurf1 and CHIP in mediating Runx2 degradation. We found that both overexpressed CHIP and Smurf1 mediate Runx2 degradation (Fig. 2, C and D) and thus inhibit Runx2-mediated transcription (Fig. 3 B). However, in wild-type MC3T3-E1 cells induced by AA/β-GP, we observed that CHIP expression decreases significantly with cell differentiation but that Smurf1 expression remains unchanged (Fig. 4, C and F). Moreover, in comparison with Smurf1, which is ubiquitously expressed in different cell lines, CHIP expression is more restricted to preosteoblast cells (Fig. 4). These observations indicate that CHIP-facilitated Runx2 degradation is closely related to its role in osteoblast differentiation.
Recently, WWP1, another Nedd4 family E3 ligase, has been reported to be responsible for Runx2 ubiquitination and degradation. WWP1 is recruited to Runx2 by an adaptor protein, Shn3, and mice lacking Shn3 indeed display adult onset osteosclerosis with increased bone mass as a result of augmented osteoblast activity (Jones et al., 2006). In this study, we show that the expression level of WWP1 is up-regulated during osteoblast (from both cell lines and primary cultured calvarial cells) differentiation in vitro, which is consistent with a previous study (Jones et al., 2006). Also, WWP1 is expressed at higher levels in the osteoblast lineages (Fig. 4 G, MC3T3-E1 and UMR106). The increased levels of WWP1/Shn3 together with the increased Runx2 proteins during the osteoblast differentiation are not concordant with the role of WWP1/Shn3 on Runx2 protein degradation. One possible explanation is that WWP1 may not play a role in Runx2 protein degradation at this stage of osteoblast differentiation. Indeed, knocking out Shn3, a WWP1 adaptor, leads to enhanced mineralization in calvarial osteoblasts but has no effect on AP induction (Jones et al., 2006). Because elevation of AP activity occurs before the formation of mineralized matrix nodules, it could be concluded that Shn3 and WWP1 function at the mineralization stage, as Runx2 has to be turned off in fully matured osteoblasts. In our study, we observed impaired AP induction in CHIP-overexpressing MC3T3-E1 cells from an early stage of differentiation. Therefore, we suggest that both CHIP and WWP1 are responsible for the degradation of Runx2 but function at different stages of osteoblast differentiation. We propose that CHIP maintains the low levels of Runx2 protein in preosteoblasts and is turned down when differentiation starts to ensure that Runx2 protein can be quickly elevated to the level high required to initiate further osteoblast differentiation. When cells have fully differentiated to mature osteoblasts, WWP1 (or Shn3) expression is elevated to maintain the common features of osteoblasts. Our data depict a complex relationship among the different E3 ligases in regulating Runx2 protein levels during preosteoblast differentiation.
It still remains to be determined whether there is an adaptor involved in the regulation of CHIP–Runx2 interaction, as in the case of WWP1 and Shn3. In our study, we observed that the H261Q mutant of CHIP causes an increase in Runx2 protein level (Fig. 1 F). It is conceivable that CHIP(H261Q) loses its E3 activity because it lacks the capability to bind its cognate E2 enzyme. In addition, the H261Q mutant was found to retain, rather than lose, the ability to ubiquitinate Runx2 (Fig. 2 H, last lane). Similar results were also observed on CHIP-mediated E47 degradation (Huang et al., 2004). On the other hand, CHN-1, the Caenorhabditis elegans orthologue of CHIP, was reported to form a complex with UFD-2, an enzyme known to have ubiquitin-conjugating E4 activity in yeast, and this complex is necessary and sufficient to multiubiquitylate UNC-45 in vitro (Hoppe et al., 2004). We presume that other factors such as the Hsc70–Skp2 complex might also be involved in the Runx2 ubiquitination process mediated by CHIP.
It is not clear how CHIP expression is regulated at the transcriptional level during osteoblast differentiation. Our data indicate that CHIP is down-regulated when cells differentiate into osteoblasts and is maintained at high levels in nonosteoblast lineages. Conversely, WWP1 (also Shn3) is up-regulated when cells differentiate into osteoblasts but is maintained at a basal level at the early stage of differentiation (in uninduced MC3T3-E1, NIH3T3, or C3H10T1/2 cells) or is completely turned off in C2C12 cells. These observations implicate that the regulation of CHIP gene expression together with that of WWP1 and Smurf1 yields a specific pattern for different fates of the cells and may be controlled by common factors during cell differentiation.
We have reported that CHIP facilitates the degradation of Smad proteins in several cell lines (Li et al., 2004; Xin et al., 2005). Because Smad proteins are critical mediators of BMP/TGF-β signals, which play important roles in osteoblast differentiation, and activation of BMP signaling has been reported to activate Runx2 mRNA expression (Ito and Miyazono, 2003), we questioned whether CHIP overexpression would also block these signals in osteoblasts. In our experiments, we adopted AA/β-GP (rather than BMP2) to induce MC3T3-E1 and calvarial cell differentiation. Our observations on the cell differentiation affected by CHIP suggest that AA/β-GP induces cell differentiation through a pathway not directly related to BMP signaling. Whether CHIP plays a role in BMP signaling to regulate osteoblast differentiation in vivo remains to be studied.
The biological significance of CHIP-mediated Runx2 regulation on preosteoblast differentiation is underscored by its dependence on preosteoblast-specific cell types. In contrast to the MC3T3-E1 cells, which express Runx2 and are committed to the osteoblast lineage, overexpression of CHIP in C3H10T1/2 cells that are deficient in Runx2 expression did not change the differentiation potential (to become adipocytes) of these cells. Based on these and previous work (Komori, 2005) on Runx2, we propose a model by which CHIP interacts with Runx2 and regulates preosteoblast differentiation (Fig. 7 E). In this model, CHIP regulates Runx2 protein levels and thereafter regulates the commitment of preosteoblast differentiation to osteoblasts and adipocytes. CHIP levels decrease steadily during preosteoblast differentiation accompanied by a steady increase in Runx2 protein levels. This model can explain our observations that the forced expression of CHIP prevented the committed differentiation of preosteoblasts to osteoblasts, resulting in adipocyte differentiation. This is consistent with previous studies on the effect of Runx2 on adipocyte differentiation, as Runx2 has been shown to be an inhibitor that prevents mesenchymal cells from entering into the adipocyte lineage (Lecka-Czernik et al., 1999; Kobayashi et al., 2000; Jeon et al., 2003), and Runx2 deficiency in chondrocytes stimulates their differentiation to adipocytes (Enomoto et al., 2004). Collectively, our data identifies for the first time an important role of CHIP in regulating preosteoblast differentiation by modulating the protein levels of Runx2 through ubiquitination and protein degradation. This enriches our knowledge of the regulation of Runx2 protein during the osteoblast differentiation in addition to the known negative regulators WWP1/Shn3 and Smurf1/2.
Materials and methods
Antibodies
The antiserum against CHIP was generated by immunizing rabbits with purified GST-CHIP(1–161) in this laboratory. Anti–β-actin and anti-Runx2 (8G5) were purchased from Sigma-Aldrich and MBL International, respectively. Other antibodies (including anti-PEBPαA and M70) were purchased from Santa Cruz Biotechnology, Inc.
Plasmids, cell lines, and cultures
Type II mouse Runx2 cDNA was cloned from pBSA3 (provided by G. Karsenty, Columbia University, New York, NY; Ducy et al., 1997) and inserted into the pGBKT7 and pCMV/Myc vectors (Clontech Laboratories, Inc.). Constructs expressing deletions of type I human Runx2 (PEBP2αA) were provided by Y. Ito (Institute of Molecular and Cell Biology, Singapore; Zhang et al., 2000). pGL3/6xOSE2-OC-luc was described previously (Zhao et al., 2003). pGEX-5X-3/CHIP and its deletions, pcDNA6/HA-CHIP, pcDNA6/HA-CHIP(K30A), pcDNA6/HA-CHIP(H260Q), and pACT-2/CHIP, together with CHIPi constructs were preserved in our laboratory (Li et al., 2004; Xin et al., 2005). The siRNA target sequences for mouse CHIP is 5′-AACAGGCACTTGCTGACTG-3′ and for human CHIP is 5′-AGCAGGCCCTGGCCGACTG-3′. His-ubiquitin and Flag-Smurf1 were provided by Y. Chen (Tsinghua University, Beijing, China). pEFNue/HA-CHIP was generated from pcDNA6/HA-CHIP. The recombinant adenoviruses expressing GFP, CHIP-GFP, and CHIPi-GFP were constructed using the pAd Easy system.
HEK293T, COS7, and C2C12 cells were cultured in DME supplemented with 10% FBS. NIH3T3 cells were maintained in DME supplemented with 10% newborn calf serum, which was replaced with 10% FBS the day before transfection. MC3T3-E1 and UMR106 cells were cultured in αMEM supplemented with 10% FBS. C3H10T1/2 cells were cultured in basal medium Eagle supplemented with 10% FBS. All of the cells were kept at 37°C in a 5% CO2-containing atmosphere. The stable cell lines were selected by 1 mg/ml G418 (Sigma-Aldrich) after transfection of the related plasmids, and the obtained clones were maintained in media containing 400 μg/ml G418.
In the induction of osteoblast differentiation, cells were treated with AA/β-GP (50 μg/ml AA and 10 mM β-GP for primary mouse calvarial osteoblastic cells using 5 mM β-GP) for the indicated days. In the adipogenesis experiments, cells were cultured in the presence of DIM (1 μM dexamethasone, 0.5 mM 3-isobutyl-1-methylxanthine, and 1 μg/ml insulin) for up to 14 d. Media were changed every 3 d.
Protein assays and luciferase assay
Protein extraction, immunoblotting, coimmunoprecipitation, ubiquitination, GST pull down, pulse-chase assays, and luciferase assay were performed according to previous studies (Li et al., 2004; Xin et al., 2005).
Reverse transcription/real-time PCR
DNase-treated RNA was reverse transcribed with SuperScript II reverse transcription (Invitrogen) using random hexamer primer (Promega). The resulting cDNA was subsequently used for PCR or quantitative real-time PCR analyses. Primer sequences are listed in Table S1 (available at http://www.jcb.org/cgi/content/full/jcb.200711044/DC1).
AP activity assay
Cells were plated at a density of 104 cells/cm2 for osteoblast differentiation. Cultures from indicated days were lysed with 250 μl of 0.1% Triton X-100/well. 500 μl of freshly prepared 20-mM p-nitrophenyl-phosphate in AMP buffer (1.5 M AMP and 0.1 M MgCl2, pH 10.3) was mixed with 100 μl of cell lysate and incubated for 30 min at 37°C until the reaction was terminated with 2 ml of 0.25-M NaOH solution. The product p-nitrophenol had a colorimetric determination at 405 nm (Coelho and Fernandes, 2000). Protein concentration of the lysate was determined using the BCA protein assay kit (Thermo Fisher Scientific). AP activity was standardized as OD405/OD562. The experiment was repeated three times, and SEM was calculated.
Histochemical assays
Cultures were fixed with 95% ethanol for 10 min and rinsed with distilled water. Phosphate deposits were assessed by von Kossa staining. The fixed cultures were covered with a 1.0% AgNO3 solution and kept for 1 h under UV light. After rinsing, a 5.0% NaS2O3 solution was added for 2 min, and cultures were washed again. For calcium staining, the fixed cultures were covered with a 1.0% Alizarin red solution (0.028% NH4OH, pH 6.4) for 2 min and rinsed with distilled water and acid ethanol (ethanol and 0.01% HCl). Lipid droplets were examined with Oil Red O staining, and the positive cells were counted with a microscope (BX-51; Olympus) and CCD camera (DP70; Olympus).
Immunofluorescence staining
The bones were fixed in 10% formalin and embedded with paraffin. Sections were incubated with anti-CHIP and anti-Runx2 antibodies overnight at 4°C followed by FITC- and TRITC-labeled second antibodies for 60 min at RT. Nuclei were labeled with DAPI nuclear dye (blue). Sections were mounted with glycerol and photographed with a microscope (Eclipse C1si; Nikon).
Preparation of primary mouse calvarial osteoblastic cells
Calvariae from newborn mice were dissected aseptically and treated with 0.2% collagenase (Sigma-Aldrich) for 20 min with shaking. Cell suspension in collagenase was collected and transferred into a 50-ml tube by adding FBS to 10% (to inactive collagenase). Then, fresh collagenase was added with shaking for second digestion. Digestion was repeated six times to obtain as many cells as possible (the cells from the first digestion were discarded). Cells were plated at a density of 1.5 × 104 cells/cm2 and cultured in αMEM supplemented with 10% FBS to confluent.
Adenoviral infection of calvarial osteoblasts
Calvarial cells were transduced with adenoviruses expressing GFP, CHIP, or CHIP siRNA or the empty virus (pAdTrack-cytomegalovirus). Virus particles were administered at 50 plaque-forming units/cell in α-MEM with 1% FBS and incubated for 1 h at 37°C. After 1 h, medium was aspirated, cultures were rinsed twice with serum-free medium, and fresh medium supplemented with 10% FBS was added to the dishes.
Online supplemental material
Fig. S1 shows the nuclear and cytoplasmic localization of Runx2 and CHIP proteins. Fig. S2 shows a quantitative analysis of Runx2 and CHIP protein levels in the induced MC3T3-E1 cells and mouse calvarial cells. Fig. S3 shows the coexpression of CHIP and Runx in mouse osteoblasts in femur and calvaria. Fig. S4 quantitatively presents adipocyte-specific marker gene expression in MC3T3-E1 and C3H10T1/2 cells. Fig. S5 shows the expression of adipocyte markers in the primary calvarial cells infected with Ad-CHIP or Ad-EGFP in the presence or absence of DIM. Table S1 shows the primer sequences used in real-time PCR analyses. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200711044/DC1.
Supplementary Material
Acknowledgments
We are grateful to Xin-Yuan Fu (Indiana University, Bloomington, IN) and Xiaofan Wang (Duke University, Durham, NC) for their support and suggestions. We thank Gerard Karsenty, Yeguang Chen, and Yoshiaki Ito for providing constructs.
This work was supported by the Tsinghua Yu-Yuan Medical Sciences Fund and grants from the National Nature Science Foundation of China (30530420, 30470888, and 30518002), Chinese National Support Project (2006CB910102), and 973 Project (2002CB513007).
X. Li, M. Huang, and H. Zheng contributed equally to this paper.
Abbreviations used in this paper: AA, ascorbic acid; β-GP, β-glycerophosphate; BMP, bone morphogenetic protein; BSP, bone sialoprotein; OCN, osteocalcin; TPR, tetratricopeptide repeat.
References
- Aberg, T., X.P. Wang, J.H. Kim, T. Yamashiro, M. Bei, R. Rice, H.M. Ryoo, and I. Thesleff. 2004. Runx2 mediates FGF signaling from epithelium to mesenchyme during tooth morphogenesis. Dev. Biol. 270:76–93. [DOI] [PubMed] [Google Scholar]
- Ballinger, C.A., P. Connell, Y. Wu, Z. Hu, L.J. Thompson, L.Y. Yin, and C. Patterson. 1999. Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol. Cell. Biol. 19:4535–4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, D., X. Ji, M.A. Harris, J.Q. Feng, G. Karsenty, A.J. Celeste, V. Rosen, G.R. Mundy, and S.E. Harris. 1998. Differential roles for bone morphogenetic protein (BMP) receptor type IB and IA in differentiation and specification of mesenchymal precursor cells to osteoblast and adipocyte lineages. J. Cell Biol. 142:295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho, M.J., and M.H. Fernandes. 2000. Human bone cell cultures in biocompatibility testing. Part II: effect of ascorbic acid, beta-glycerophosphate and dexamethasone on osteoblastic differentiation. Biomaterials. 21:1095–1102. [DOI] [PubMed] [Google Scholar]
- Dobreva, G., M. Chahrour, M. Dautzenberg, L. Chirivella, B. Kanzler, I. Farinas, G. Karsenty, and R. Grosschedl. 2006. SATB2 is a multifunctional determinant of craniofacial patterning and osteoblast differentiation. Cell. 125:971–986. [DOI] [PubMed] [Google Scholar]
- Ducy, P., R. Zhang, V. Geoffroy, A.L. Ridall, and G. Karsenty. 1997. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 89:747–754. [DOI] [PubMed] [Google Scholar]
- Enomoto, H., T. Furuichi, A. Zanma, K. Yamana, C. Yoshida, S. Sumitani, H. Yamamoto, M. Enomoto-Iwamoto, M. Iwamoto, and T. Komori. 2004. Runx2 deficiency in chondrocytes causes adipogenic changes in vitro. J. Cell Sci. 117:417–425. [DOI] [PubMed] [Google Scholar]
- Gori, F., T. Thomas, K.C. Hicok, T.C. Spelsberg, and B.L. Riggs. 1999. Differentiation of human marrow stromal precursor cells: bone morphogenetic protein-2 increases OSF2/CBFA1, enhances osteoblast commitment, and inhibits late adipocyte maturation. J. Bone Miner. Res. 14:1522–1535. [DOI] [PubMed] [Google Scholar]
- Hatakeyama, S., M. Yada, M. Matsumoto, N. Ishida, and K.I. Nakayama. 2001. U box proteins as a new family of ubiquitin-protein ligases. J. Biol. Chem. 276:33111–33120. [DOI] [PubMed] [Google Scholar]
- Hinoi, E., P. Bialek, Y.T. Chen, M.T. Rached, Y. Groner, R.R. Behringer, D.M. Ornitz, and G. Karsenty. 2006. Runx2 inhibits chondrocyte proliferation and hypertrophy through its expression in the perichondrium. Genes Dev. 20:2937–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong, J.H., E.S. Hwang, M.T. McManus, A. Amsterdam, Y. Tian, R. Kalmukova, E. Mueller, T. Benjamin, B.M. Spiegelman, P.A. Sharp, et al. 2005. TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science. 309:1074–1078. [DOI] [PubMed] [Google Scholar]
- Hoppe, T., G. Cassata, J.M. Barral, W. Springer, A.H. Hutagalung, H.F. Epstein, and R. Baumeister. 2004. Regulation of the myosin-directed chaperone UNC-45 by a novel E3/E4-multiubiquitylation complex in C. elegans. Cell. 118:337–349. [DOI] [PubMed] [Google Scholar]
- Huang, Z., L. Nie, M. Xu, and X.H. Sun. 2004. Notch-induced E2A degradation requires CHIP and Hsc70 as novel facilitators of ubiquitination. Mol. Cell. Biol. 24:8951–8962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito, Y., and K. Miyazono. 2003. RUNX transcription factors as key targets of TGF-beta superfamily signaling. Curr. Opin. Genet. Dev. 13:43–47. [DOI] [PubMed] [Google Scholar]
- Jeon, E.J., K.Y. Lee, N.S. Choi, M.H. Lee, H.N. Kim, Y.H. Jin, H.M. Ryoo, J.Y. Choi, M. Yoshida, N. Nishino, et al. 2006. Bone morphogenetic protein-2 stimulates Runx2 acetylation. J. Biol. Chem. 281:16502–16511. [DOI] [PubMed] [Google Scholar]
- Jeon, M.J., J.A. Kim, S.H. Kwon, S.W. Kim, K.S. Park, S.W. Park, S.Y. Kim, and C.S. Shin. 2003. Activation of peroxisome proliferator-activated receptor-gamma inhibits the Runx2-mediated transcription of osteocalcin in osteoblasts. J. Biol. Chem. 278:23270–23277. [DOI] [PubMed] [Google Scholar]
- Jones, D.C., M.N. Wein, M. Oukka, J.G. Hofstaetter, M.J. Glimcher, and L.H. Glimcher. 2006. Regulation of adult bone mass by the zinc finger adapter protein Schnurri-3. Science. 312:1223–1227. [DOI] [PubMed] [Google Scholar]
- Kaneki, H., R. Guo, D. Chen, Z. Yao, E.M. Schwarz, Y.E. Zhang, B.F. Boyce, and L. Xing. 2006. Tumor necrosis factor promotes Runx2 degradation through up-regulation of Smurf1 and Smurf2 in osteoblasts. J. Biol. Chem. 281:4326–4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, B.G., H.J. Kim, H.J. Park, Y.J. Kim, W.J. Yoon, S.J. Lee, H.M. Ryoo, and J.Y. Cho. 2006. Runx2 phosphorylation induced by fibroblast growth factor-2/protein kinase C pathways. Proteomics. 6:1166–1174. [DOI] [PubMed] [Google Scholar]
- Kobayashi, H., Y. Gao, C. Ueta, A. Yamaguchi, and T. Komori. 2000. Multilineage differentiation of Cbfa1-deficient calvarial cells in vitro. Biochem. Biophys. Res. Commun. 273:630–636. [DOI] [PubMed] [Google Scholar]
- Komori, T. 2005. Regulation of skeletal development by the Runx family of transcription factors. J. Cell. Biochem. 95:445–453. [DOI] [PubMed] [Google Scholar]
- Komori, T., H. Yagi, S. Nomura, A. Yamaguchi, K. Sasaki, K. Deguchi, Y. Shimizu, R.T. Bronson, Y.H. Gao, M. Inada, et al. 1997. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 89:755–764. [DOI] [PubMed] [Google Scholar]
- Lecka-Czernik, B., I. Gubrij, E.J. Moerman, O. Kajkenova, D.A. Lipschitz, S.C. Manolagas, and R.L. Jilka. 1999. Inhibition of Osf2/Cbfa1 expression and terminal osteoblast differentiation by PPARgamma2. J. Cell. Biochem. 74:357–371. [PubMed] [Google Scholar]
- Li, L., H. Xin, X. Xu, M. Huang, X. Zhang, Y. Chen, S. Zhang, X.Y. Fu, and Z. Chang. 2004. CHIP mediates degradation of Smad proteins and potentially regulates Smad-induced transcription. Mol. Cell. Biol. 24:856–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, R.F., Y. Shang, D. Liu, Z.S. Ren, Z. Chang, and S.F. Sui. 2007. Differential ubiquitination of Smad1 mediated by CHIP: implications in the regulation of the bone morphogenetic protein signaling pathway. J. Mol. Biol. 374:777–790. [DOI] [PubMed] [Google Scholar]
- Murata, S., Y. Minami, M. Minami, T. Chiba, and K. Tanaka. 2001. CHIP is a chaperone-dependent E3 ligase that ubiquitylates unfolded protein. EMBO Rep. 2:1133–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam, S., Y.H. Jin, Q.L. Li, K.Y. Lee, G.B. Jeong, Y. Ito, J. Lee, and S.C. Bae. 2002. Expression pattern, regulation, and biological role of runt domain transcription factor, run, in Caenorhabditis elegans. Mol. Cell. Biol. 22:547–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto, F., A.P. Thornell, T. Crompton, A. Denzel, K.C. Gilmour, I.R. Rosewell, G.W. Stamp, R.S. Beddington, S. Mundlos, B.R. Olsen, et al. 1997. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 89:765–771. [DOI] [PubMed] [Google Scholar]
- Pratap, J., J.B. Lian, A. Javed, G.L. Barnes, A.J. van Wijnen, J.L. Stein, and G.S. Stein. 2006. Regulatory roles of Runx2 in metastatic tumor and cancer cell interactions with bone. Cancer Metastasis Rev. 25:589–600. [DOI] [PubMed] [Google Scholar]
- Prince, M., C. Banerjee, A. Javed, J. Green, J.B. Lian, G.S. Stein, P.V. Bodine, and B.S. Komm. 2001. Expression and regulation of Runx2/Cbfa1 and osteoblast phenotypic markers during the growth and differentiation of human osteoblasts. J. Cell. Biochem. 80:424–440. [DOI] [PubMed] [Google Scholar]
- Qian, S.B., H. McDonough, F. Boellmann, D.M. Cyr, and C. Patterson. 2006. CHIP-mediated stress recovery by sequential ubiquitination of substrates and Hsp70. Nature. 440:551–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajgopal, A., D.W. Young, K.A. Mujeeb, J.L. Stein, J.B. Lian, A.J. van Wijnen, and G.S. Stein. 2006. Mitotic control of RUNX2 phosphorylation by both CDK1/cyclin B kinase and PP1/PP2A phosphatase in osteoblastic cells. J. Cell. Biochem. 100:1509–1517. [DOI] [PubMed] [Google Scholar]
- Rosen, E.D., and B.M. Spiegelman. 2000. Molecular regulation of adipogenesis. Annu. Rev. Cell Dev. Biol. 16:145–171. [DOI] [PubMed] [Google Scholar]
- Saito, Y., T. Yoshizawa, F. Takizawa, M. Ikegame, O. Ishibashi, K. Okuda, K. Hara, K. Ishibashi, M. Obinata, and H. Kawashima. 2002. A cell line with characteristics of the periodontal ligament fibroblasts is negatively regulated for mineralization and Runx2/Cbfa1/Osf2 activity, part of which can be overcome by bone morphogenetic protein-2. J. Cell Sci. 115:4191–4200. [DOI] [PubMed] [Google Scholar]
- Shen, R., M. Chen, Y.J. Wang, H. Kaneki, L. Xing, R.J. O'Keefe, and D. Chen. 2006. Smad6 interacts with Runx2 and mediates Smad ubiquitin regulatory factor 1-induced Runx2 degradation. J. Biol. Chem. 281:3569–3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein, G.S., J.B. Lian, A.J. van Wijnen, J.L. Stein, M. Montecino, A. Javed, S.K. Zaidi, D.W. Young, J.Y. Choi, and S.M. Pockwinse. 2004. Runx2 control of organization, assembly and activity of the regulatory machinery for skeletal gene expression. Oncogene. 23:4315–4329. [DOI] [PubMed] [Google Scholar]
- Sudhakar, S., M.S. Katz, and N. Elango. 2001. a. Analysis of type-I and type-II RUNX2 protein expression in osteoblasts. Biochem. Biophys. Res. Commun. 286:74–79. [DOI] [PubMed] [Google Scholar]
- Sudhakar, S., Y. Li, M.S. Katz, and N. Elango. 2001. b. Translational regulation is a control point in RUNX2/Cbfa1 gene expression. Biochem. Biophys. Res. Commun. 289:616–622. [DOI] [PubMed] [Google Scholar]
- Taniuchi, I., M. Osato, T. Egawa, M.J. Sunshine, S.C. Bae, T. Komori, Y. Ito, and D.R. Littman. 2002. Differential requirements for Runx proteins in CD4 repression and epigenetic silencing during T lymphocyte development. Cell. 111:621–633. [DOI] [PubMed] [Google Scholar]
- Whiteman, H.J., and P.J. Farrell. 2006. RUNX expression and function in human B cells. Crit. Rev. Eukaryot. Gene Expr. 16:31–44. [DOI] [PubMed] [Google Scholar]
- Wiederkehr, T., B. Bukau, and A. Buchberger. 2002. Protein turnover: a CHIP programmed for proteolysis. Curr. Biol. 12:R26–R28. [DOI] [PubMed] [Google Scholar]
- Woolf, E., C. Xiao, O. Fainaru, J. Lotem, D. Rosen, V. Negreanu, Y. Bernstein, D. Goldenberg, O. Brenner, G. Berke, et al. 2003. Runx3 and Runx1 are required for CD8 T cell development during thymopoiesis. Proc. Natl. Acad. Sci. USA. 100:7731–7736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin, H., X. Xu, L. Li, H. Ning, Y. Rong, Y. Shang, Y. Wang, X.Y. Fu, and Z. Chang. 2005. CHIP controls the sensitivity of transforming growth factor-beta signaling by modulating the basal level of Smad3 through ubiquitin-mediated degradation. J. Biol. Chem. 280:20842–20850. [DOI] [PubMed] [Google Scholar]
- Xu, W., M. Marcu, X. Yuan, E. Mimnaugh, C. Patterson, and L. Neckers. 2002. Chaperone-dependent E3 ubiquitin ligase CHIP mediates a degradative pathway for c-ErbB2/Neu. Proc. Natl. Acad. Sci. USA. 99:12847–12852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida, C.A., and T. Komori. 2005. Role of Runx proteins in chondrogenesis. Crit. Rev. Eukaryot. Gene Expr. 15:243–254. [DOI] [PubMed] [Google Scholar]
- Yoshida, C.A., H. Yamamoto, T. Fujita, T. Furuichi, K. Ito, K. Inoue, K. Yamana, A. Zanma, K. Takada, Y. Ito, and T. Komori. 2004. Runx2 and Runx3 are essential for chondrocyte maturation, and Runx2 regulates limb growth through induction of Indian hedgehog. Genes Dev. 18:952–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young, D.W., M.Q. Hassan, J. Pratap, M. Galindo, S.K. Zaidi, S.H. Lee, X. Yang, R. Xie, A. Javed, J.M. Underwood, et al. 2007. Mitotic occupancy and lineage-specific transcriptional control of rRNA genes by Runx2. Nature. 445:442–446. [DOI] [PubMed] [Google Scholar]
- Zaidi, S.K., A. Javed, J.Y. Choi, A.J. van Wijnen, J.L. Stein, J.B. Lian, and G.S. Stein. 2001. A specific targeting signal directs Runx2/Cbfa1 to subnuclear domains and contributes to transactivation of the osteocalcin gene. J. Cell Sci. 114:3093–3102. [DOI] [PubMed] [Google Scholar]
- Zhang, Y.W., N. Yasui, K. Ito, G. Huang, M. Fujii, J. Hanai, H. Nogami, T. Ochi, K. Miyazono, and Y. Ito. 2000. A RUNX2/PEBP2alpha A/CBFA1 mutation displaying impaired transactivation and Smad interaction in cleidocranial dysplasia. Proc. Natl. Acad. Sci. USA. 97:10549–10554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, M., M. Qiao, B.O. Oyajobi, G.R. Mundy, and D. Chen. 2003. E3 ubiquitin ligase Smurf1 mediates core-binding factor alpha1/Runx2 degradation and plays a specific role in osteoblast differentiation. J. Biol. Chem. 278:27939–27944. [DOI] [PubMed] [Google Scholar]
- Zhao, M., M. Qiao, S.E. Harris, B.O. Oyajobi, G.R. Mundy, and D. Chen. 2004. Smurf1 inhibits osteoblast differentiation and bone formation in vitro and in vivo. J. Biol. Chem. 279:12854–12859. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}