

Abstract
Plectin is a versatile intermediate filament (IF)–bound cytolinker protein with a variety of differentially spliced isoforms accounting for its multiple functions. One particular isoform, plectin 1b (P1b), remains associated with mitochondria after biochemical fractionation of fibroblasts and cells expressing exogenous P1b. Here, we determined that P1b is inserted into the outer mitochondrial membrane with the exon 1b–encoded N-terminal sequence serving as a mitochondrial targeting and anchoring signal. To study P1b-related mitochondrial functions, we generated mice that selectively lack this isoform but express all others. In primary fibroblasts and myoblasts derived from these mice, we observe a substantial elongation of mitochondrial networks, whereas other mitochondrial properties remain largely unaffected. Normal morphology of mitochondria could be restored by isoform-specific overexpression of P1b in P1b-deficient as well as plectin-null cells. We propose a model where P1b both forms a mitochondrial signaling platform and affects organelle shape and network formation by tethering mitochondria to IFs.
Introduction
Mitochondria perform a multitude of cellular activities that are essential for a cell's life and death. There is evidence that mitochondrial morphology and distribution depend on interactions with the cytoskeleton, although the molecular mechanisms involved are hardly understood (Toivola et al., 2005; Anesti and Scorrano, 2006). A connection of mitochondria with intermediate filaments (IFs) was suggested some 25 years ago (Toh et al., 1980), and several IF proteins have been associated with mitochondrial functions since then. Mutations in the neurofilament protein NF-L gene have been shown to affect mitochondrial distribution (Perez-Olle et al., 2005), and an aberrant mitochondrial distribution in keratinocytes was observed in some patients with epidermolysis bullosa simplex caused by mutations in keratins 5 and 14 genes (Uttam et al., 1996). Furthermore, the ablation of desmin in the mouse results in characteristic alterations in distribution, number, morphology, and respiratory activity of mitochondria (for review see Capetanaki et al., 2007). The question of whether the interaction between IFs and mitochondria occurs directly or is mediated by linker proteins remains to be solved.
The highly versatile IF-based cytolinker protein plectin (Wiche, 1998) would be an interesting candidate for mediating the interactions between IFs and mitochondria. The versatility of plectin is largely caused by complex splicing events in the N-terminal region of its gene that give rise to 11 alternatively spliced isoforms containing different first exons (1–1j; Elliott et al., 1997; Fuchs et al., 1999). The expression patterns of these isoforms are cell type–dependent, and some of the expressed variants have been shown to differ in their subcellular localization (Rezniczek et al., 2003). By forced expression in fibroblasts, isoform plectin 1b (P1b) was found to be specifically targeted to mitochondria (Rezniczek et al., 2003). Here, we analyzed the mode of P1b–mitochondrion interaction and show that this interaction affects the shape and network formation of mitochondria.
Results and Discussion
Mitochondrion-associated P1b is an outer membrane–anchored protein facing the cytosol
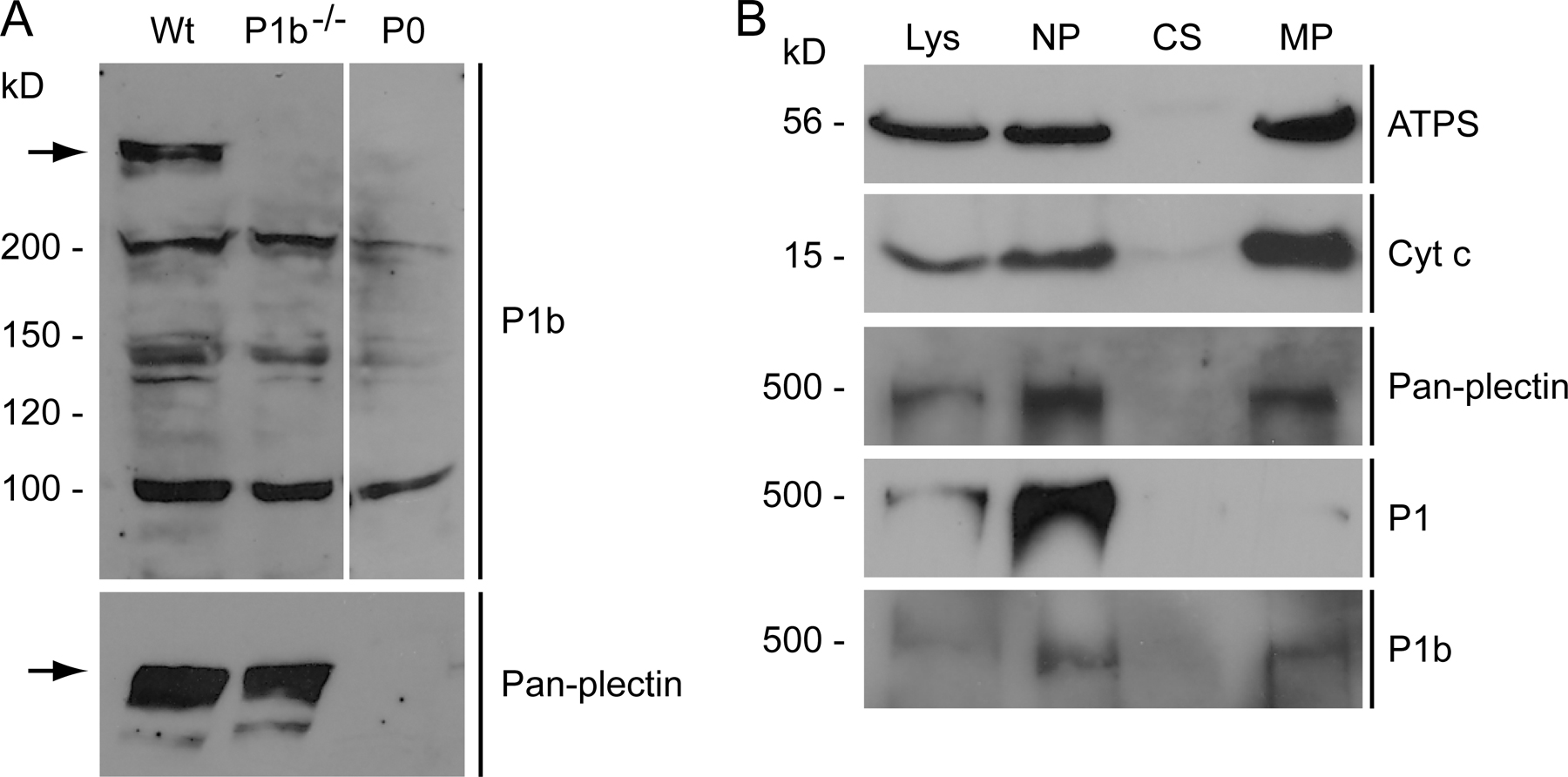
First, we analyzed the mode of P1b interaction with mitochondria and the topology of its molecular subdomains. After subcellular fractionation of mouse fibroblasts, the distribution of P1b was found to be very similar to that of genuine mitochondrial proteins but different from other plectin isoforms (Fig. S1, A and B, available at http://www.jcb.org/cgi/content/full/jcb.200710151/DC1). To determine the submitochondrial localization of P1b, we generated a stable cell line expressing a truncated version of P1b (encoded by exons 1b–8 and carrying a C-terminal EGFP tag; P1b–8 EGFP) in an otherwise plectin null (P0) background. Cells expressing a similar fusion protein of plectin isoform 1 (P1–8 EGFP) or EGFP alone were generated as controls. The distribution of P1b–8 EGFP in these cells was indistinguishable from that of mitochondria (Fig. 1 A). Upon subcellular fractionation, P1b–8 EGFP was found in the mitochondrial pellet, in contrast to P1–8 EGFP, which remained in the cytosolic fraction (Fig. 1 B). To distinguish nonspecific association from mitochondrial membrane insertion, isolated mitochondria were subjected to alkaline extraction (Ryan et al., 2001), whereupon peripheral proteins such as cytochrome c (cyt c) become soluble, whereas integral membrane proteins (porin) prove to be extraction resistant. Because P1b–8 EGFP, in contrast to EGFP, remained insoluble, it could be classified as a mitochondrial membrane protein (Fig. 1 B). Proteins residing entirely inside of mitochondria such as ATP synthase (ATPS) and cyt c are not accessible to proteinase K but become degraded after membrane permeabilization with detergent. We observed that P1b–8 EGFP was degraded by this proteinase, as monitored using P1b-specific antibodies (Fig. 1 C). When anti-GFP antibodies were used, a 27-kD fragment was detected, which corresponds to protease-resistant GFP (Cubitt et al., 1995). These results suggested that P1b was inserted into the outer mitochondrial membrane via its N-terminal portion containing the isoform-specific 1b sequence, whereas the bulk of the protein was exposed to the cytosol.
Figure 1.

Submitochondrial localization of P1b. (A) Colocalization of P1b–8 EGFP with mitochondria in stably transfected P0 fibroblasts visualized using anti-GFP antibodies and MitoTracker. Bar, 20 μm. (B) Cell lysates and cytosolic and mitochondrial fractions were obtained from fibroblasts stably expressing P1b–8 EGFP, P1–8 EGFP, or EGFP. Mitochondria were extracted with sodium carbonate, and insoluble pellet (Na2CO3-P) and soluble supernatant (Na2CO3-S) fractions were subjected to immunoblotting. White lines indicate that intervening lanes have been spliced out. (C) Mitochondria were left untreated or lysed with Triton X-100 before the addition of indicated amounts of proteinase K.
The isoform 1b-specific sequence serves as signal-anchor domain
To investigate whether the exon 1b–specific sequence was sufficient for mitochondrial targeting, corresponding cDNA expression constructs with N- or C-terminal EGFP tags (EGFP-E1b and E1b-EGFP) were transfected into fibroblasts. As shown in Fig. 2 A, the staining pattern of E1b-EGFP clearly coincided with that of MitoTracker, whereas EGFP-E1b was evenly distributed throughout the cytoplasm without showing mitochondrial accumulation. Deletion mutagenesis (Fig. 2 B) revealed that the isoform-specific (exon 1b–encoded) sequence of P1b was indeed sufficient for mitochondrial targeting, as long as it was located at the N terminus of the protein.
Figure 2.

The amino acid sequence encoded by plectin exon 1b serves as a mitochondrion-targeting signal. (A) Fibroblasts expressing the exon 1b–specific sequence, C- or N-terminally fused to EGFP (E1b-EGFP and EGFP-E1b, respectively), were visualized using MitoTracker and anti-GFP antibodies. Bar, 20 μm. (B) Schematic representation of plectin fragments tested and their mitochondrial targeting potential. Plectin's domain structure is shown on top, and actin- (ABD) and IF-binding (IF-BD) domains, the rod domain (ROD), and C-terminal plectin repeat domains 1–6 (black circles) are indicated. (C) Amino acid sequence (residues 1 to 38) encoded by exon 1b. Positively charged residues are depicted in bold letters and negatively charged residues in gray; the transmembrane domain is shaded gray.
One class of mitochondrial outer membrane proteins, referred to as signal-anchored proteins (Shore et al., 1995), is targeted and anchored to the organelle by a single transmembrane domain (residing in the N-terminal regions of the proteins) and its flanking sequences. Although such proteins do not share any sequence similarity in their signal-anchor domain (Rapaport, 2003), a moderate transmembrane domain hydrophobicity and a net positive charge at the C-terminal flanking region of the membrane-spanning segment were found to be crucial (Waizenegger et al., 2003). Based on the membrane protein topology prediction method TMHMM (Krogh et al., 2001), P1b's transmembrane domain spans from aa 5 to 27 (Fig. 2 C). The mean hydropathicity of this transmembrane domain, calculated according to the scale of Kyte and Doolittle (ExPASy, ProtParam tool; http://ca.expasy.org/tools/protparam.html), was 1.491, which corresponds to moderate hydrophobicity. Furthermore, the net charge of the C-terminal flanking region of this transmembrane domain is positive, making the isoform-specific sequence of P1b fit perfectly to this model.
P1b links IFs to mitochondria
To assess whether mitochondria-anchored P1b serves as a docking site for IFs, primary fibroblasts were transiently transfected with EGFP-tagged full-length P1b and analyzed using immunofluorescence microscopy. EGFP-tagged P1b was found to colocalize with mitochondria and to accumulate in discrete regions of vimentin (vim)-positive filaments (Fig. 3 A, a–e). In cells with high level expression of P1b, a collapse of mitochondrial as well as of vim networks was observed (Fig. 3 A, f–j). Forced expression of P1, an isoform not associating with mitochondria, led to a collapse of IFs but not of mitochondria, which supports the notion that the latter phenotype was isoform 1b–specific (Fig. 3 B).
Figure 3.

Association of P1b with vim IFs. (A) Fibroblasts expressing P1b EGFP were immunolabeled using antibodies to vim (a and f), GFP (b and g), and cyt c (c and h). Note the P1b colocalization with both mitochondria and IF networks in defined regions (e). In cells expressing high levels of the fusion protein (f–j), a collapse of mitochondrial as well as IF networks was observed. Boxed areas in panels d and i are shown enlarged in panels e and j, respectively. Bars: (a–d and f–i) 20 μm; (e and j) 5 μm. (B) Cells expressing P1 instead of P1b. Bar, 20 μm. (C) Immortalized wt and P0 fibroblasts were fractionated, and equal amounts of total lysates and cytosolic and mitochondrial fractions were analyzed. (D and E) Signal intensities of protein bands in C were densiometrically measured and normalized to the signals of actin (D) or ATPS (E). Values (mean ± SEM) represent protein levels of cytosolic (D) and mitochondrial fractions (E). (F) Fibroblasts derived from wt and P1b−/− mice were subjected to subcellular fractionation and analyzed by immunoblotting. White lines indicate that intervening lanes have been spliced out. (G) Statistical analyses of data shown in F (mean ± SEM). **, P < 0.01; ***, P < 0.001.
To confirm that the linkage of IFs to mitochondria was mediated by plectin, immortalized P0 fibroblasts (Osmanagic-Myers and Wiche, 2004) and their wild-type (wt) counterparts were subjected to subcellular fractionation (Fig. 3 C). Comparable amounts of ATPS, actin, and vim were detected in the lysates from both cell types. Actin, a cytoskeleton marker protein, was also detected in the cytosolic fraction without showing differences. In contrast, a >20-fold increase in soluble vim was observed in the cytosolic fraction of P0 compared with wt fibroblasts (Fig. 3 D). Even more importantly, the amount of vim found in mitochondrial extracts from P0 cells was significantly reduced to nearly 40% of wt levels (Fig. 3 E). Although these results clearly indicated a role of plectin in linking cytoskeletal networks to mitochondria, they fell short of enabling a distinction between effects of general plectin deficiency and specific loss of P1b.
Generation of P1b-deficient mice enables isoform-specific assessment of mitochondrion–IF linkage
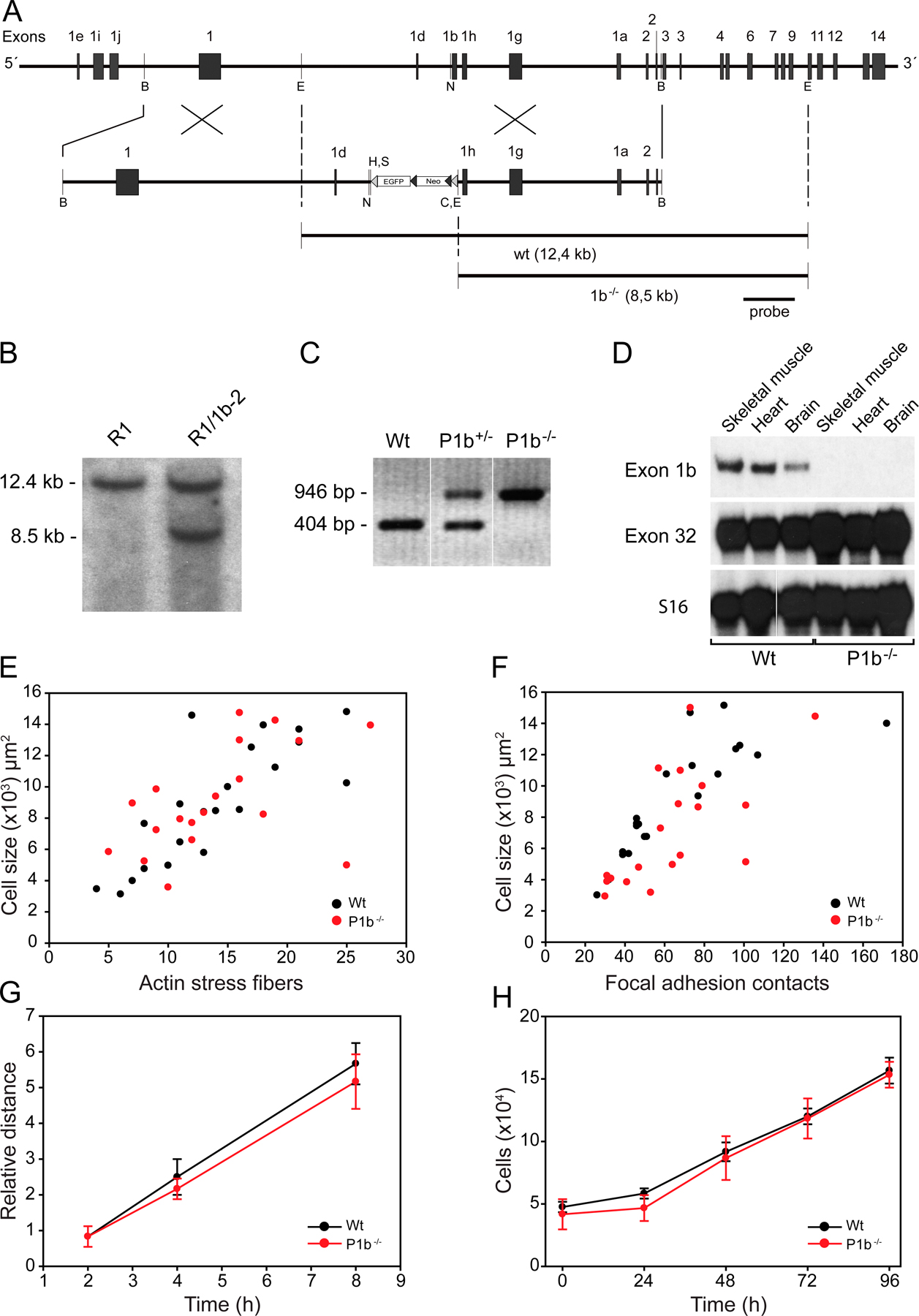
To prove that the interaction of mitochondria with IF networks was P1b-specific, we generated a mouse line that selectively lacks isoform 1b but expresses all others (Fig. S2, A–D, available at http://www.jcb.org/cgi/content/full/jcb.200710151/DC1). P1b-deficient (P1b−/−) mice were viable and fertile, and revealed no obvious phenotype when compared with their wt littermates (not depicted; for phenotypic analyses of P1b−/− fibroblasts, see Fig. S2, E–H). When fibroblasts isolated from these mutant mice were subjected to subcellular fractionation (Fig. 3 F), ATPS, actin, and vim were maintained in cell lysates at levels that were comparable to those of wt cells, which is reminiscent of the situation of P0 fibroblasts (Fig. 3 C). Contrary to P0 fibroblasts, the cytosolic fractions of P1b−/− cells contained no soluble vim. In the mitochondrial fraction of P1b−/− cells, the amount of vim was found decreased to 66% of the corresponding wt level (Fig. 3 G), validating that the interaction of mitochondria with the IF network was mediated by P1b. As the reduction was more prominent in P0 compared with P1b−/− mitochondrial fractions (39 vs. 66%), the additional lack of isoforms other than P1b in P0 cells apparently contributed to this reduction. To our knowledge, P1b is the first protein shown to act as a direct linker between IFs and the mitochondrial network.
P1b deficiency leads to elongation of mitochondria
Interactions between IFs and mitochondria have been suggested to contribute to organelle morphology and cytoplasmic distribution (Anesti and Scorrano, 2006). Thus, we monitored the shape of mitochondria in P1b−/−, P0, and wt fibroblasts using cyt c–specific immunostaining. As exemplified in Fig. 4 A, mitochondrial networks were classified as fragmented, intermediate, or tubular (for detailed classification criteria, see Materials and methods). Interestingly, although in wt fibroblasts, 63, 30, and only 7% of the mitochondria exhibited intermediate, fragmented, and tubular morphology, respectively, in P1b−/− fibroblasts, the fragmented type was reduced to 9% and the tubular type increased to 37% (Fig. 4 B). Thus, in P1b−/− cells, there was a dramatic shift from the small and fragmented mitochondrial network type to the elongated type. In P0 fibroblasts, we found very similar alterations in mitochondrial morphology, with 13 and 36% of cells exhibiting fragmented and tubular mitochondrial networks, respectively (Fig. 4 B). This suggested that the observed alterations in mitochondrial morphology were entirely due to the lack of P1b, with no other isoform of plectin being involved. A similar phenotype was revealed in primary P1b−/− myoblasts (Fig. 4 C).
Figure 4.

Morphology of mitochondria in wt, P1b−/−, and P0 fibroblasts. (A) Mitochondria of wt (a and d), P1b−/− (b and e), and P0 fibroblasts (c and f) were visualized using antibodies to cyt c. Morphologies were classified as fragmented, tubular, or intermediate, as exemplified in panels d, e, and f, which represent magnifications of the boxed areas shown in a, b, and c, respectively. Bars: (a–c) 20 μm; (d–f) 5 μm. (B) Morphologies of wt (n = 239), P1b−/− (n = 226), and P0 (n = 204) mitochondria (mean ± SEM). *, P < 0.05; **, P < 0.01; ***, P < 0.001. (C) Statistical evaluation of mitochondrial morphologies in primary myoblasts derived from wt (n = 238 cells) and P1b−/− (n = 253 cells) mice. Values represent mean ± SEM. (D) P1b−/− fibroblasts expressing P1b EGFP were immunolabeled using anti–cyt c and anti-GFP antibodies. Magnifications of boxed areas in panel c are shown in panels d and e. Note the reversion of the mitochondrial phenotype characteristic of P1b−/− cells in the transfected cell (e) but not in the adjacent untransfected cell (d). Bars: (a–c) 20 μm; (d and e) 10 μm. (E and F) Rescue efficiencies (mean ± SEM) determined by analyzing P1b−/− (E) or P0 cells (F) expressing P1b (n = 165 and n = 109, respectively) or P1 (n = 69 and n = 82, respectively).
Next, we assessed the potential of full-length P1b to revert the mitochondrial phenotype of P1b−/− fibroblasts (Fig. 4 D). In parallel, we performed similar experiments with the alternative isoform P1, which in fibroblasts is expressed at 2× higher levels than P1b (Abrahamsberg et al., 2005). Quantitative analyses revealed that 32% of transfected cells harbored fragmented mitochondria, whereas only 11% contained mitochondria of tubular morphology (Fig. 4 E). As the corresponding values measured in wt cells were 30 and 7%, respectively (Fig. 4 B), this analysis clearly showed that P1b expressed by force led to a reversion of the mitochondrial phenotype characteristic of P1b−/− fibroblasts. For P1, hardly any rescue potential was observed (Fig. 4 E). When, in a similar experiment, full-length P1b was introduced in P0 fibroblasts, the rescue potential of P1b on a P0 background was practically undiminished compared with the P1b−/− background (Fig. 4 F).
P1b, a mitochondrial platform for signaling complexes?
It was conceivable that mitochondria become anchored to IFs via P1b primarily in those subcellular regions where they are needed most for metabolic or homeostatic reasons. To investigate P1b-dependent mitochondrial parameters, the membrane potential and mass of mitochondria in P1b−/−, wt, and P0 primary fibroblasts were comparatively assessed using MitoTracker and 10-n-nonyl-acridine orange (NAO) staining, respectively. No significant differences between the cell types were revealed, however (Fig. 5, A and B). The same was true when the apoptotic predisposition of cells were analyzed after incubation with staurosporine (Fig. 5, C and D).
Figure 5.

Analysis of mitochondrial parameters in P1b-deficient cells. (A) FACS measurements of mitochondrial membrane potentials in wt, P1b−/−, and P0 cells exposed to MitoTracker. (B) Mitochondrial mass monitored using the fluorescent dye NAO. (C) Analysis of apoptotic predisposition of wt, P1b−/−, and P0 cells (only wt cells are shown). After incubation with 1 μM staurosporine (STS) for 6 h, nuclei were analyzed by confocal microscopy using Hoechst dye. Bar, 20 μm. (D) Statistical analyses of apoptotic cells containing condensed or fragmented DNA. (E) Statistical analyses of mitochondrial motility. Values represent mean ± SEM. (F) Analyses of mitochondrial movement according to the covered distance (classified as movement over distances of 1–5, 5–10, and >10 μm). (G) ROIs (white circles) were activated in fibroblasts expressing mito-PAGFP with 405-nm light followed by time-lapse confocal microscopy. To highlight the fluorescence decrease, images are depicted as false-colored projections. Bar, 20 μm. (H) Changes in fluorescence intensity in nonactivated (black) and activated (red) ROIs. (I) Evaluation of mitochondrial fluorescence decrease over time. Values represent mean ± SEM. n = 50 (wt), 47 (P1b−/−), and 53 (P0) ROIs analyzed. (J) Cells expressing P1b EGFP were immunolabeled using antibodies to Drp-1 (a) and GFP (b). Mitochondrial fission sites were characterized by Drp-1 staining (arrowheads) as depicted in panels d, e, and f, which represent magnifications of the boxed areas shown in a, b, and c, respectively. Bars: (a–c) 20 μm; (d–f) 5 μm.
Potentially, increased mitochondrial motility could indirectly lead to an increase in mitochondrial length. However, when wt, P1b−/−, and P0 fibroblasts were transfected with cDNA encoding an ATPS subunit 9 presequence fused to EGFP (mito-GFP) and the velocities of mitochondrial movement were analyzed by time-lapse video microscopy, no significant differences were found, except for a trend toward a higher velocity in P0 cells (Fig. 5, E and F). The velocity increase of P0 compared with P1b−/− mitochondria was probably caused by abnormalities of IF network cytoarchitecture typical for P0 but not P1b−/− cells (Osmanagic-Myers et al., 2006; unpublished data). Nevertheless, these differences in cytoarchitecture are unlikely to contribute to the observed mitochondrial phenotype, as P1b−/− and P0 cells displayed similar mitochondrial morphology (Fig. 4 B).
To assess whether changes in mitochondrial fusion rates were a cause for differences in mitochondrial length, wt, P1b−/−, and P0 fibroblasts were transfected with a mitochondrial matrix–targeted photoactivatable GFP (mito-PAGFP; Karbowski et al., 2004). Selected regions of interest (ROIs) were activated using a short laser beam, and redistribution of the photoactivated protein out of the activated ROIs was monitored (Fig. 5 G). When changes of fluorescence intensities within activated and nonactivated ROIs were measured over a time period of 60 min, P1b−/− and P0 fibroblasts showed unaltered mitochondrial fusion dynamics compared with wt cells (Fig. 5, H and I). Furthermore, when fibroblasts expressing GFP-tagged full-length P1b were immunolabeled using antibodies to the mitochondrial fission site marker Drp-1, P1b showed partial colocalization with Drp-1-marked sites but did not appear to accumulate there (Fig. 5 J).
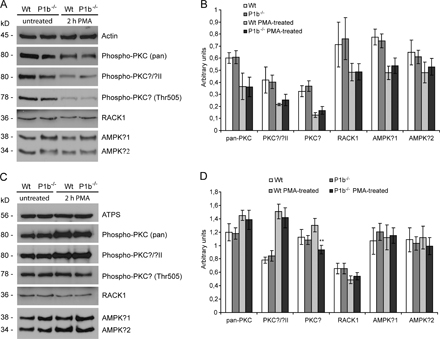
Increasing evidence indicates that plectin serves the cell not merely as a structural element but also as a scaffolding platform of proteins involved in signaling. Among others, plectin was shown to bind and sequester the receptor for activated C kinase 1 (RACK1) to the cytoskeleton, thereby influencing PKC signaling pathways (Osmanagic-Myers and Wiche, 2004). Given that the various plectin isoforms are targeted to different cellular locations, it is quite conceivable that P1b, as a mitochondrial outer membrane protein, could act as a scaffolding platform for signaling molecules proximal to mitochondria. To test this hypothesis, we prepared cellular subfractions from P1b−/− and wt fibroblasts that had been exposed (or not) to the PKC activator PMA. A quantitative analysis of autophosphorylated (activated) PKC (pan), PKCα/βII, and PKCδ, as well as of RACK1 and AMP-activated protein kinase (AMPK) β1/2, levels in total lysate fractions revealed no differences between the two cell types (Fig. S3, A and B, available at http://www.jcb.org/cgi/content/full/jcb.200710151/DC1). Interestingly, however, in the mitochondrial fraction from PMA-treated P1b−/− cells, activated PKCδ was decreased to nearly 70% of wt levels (Fig. S3, C and D), whereas all other proteins tested remained unaffected. Thus, P1b seemed to be at least partially involved in recruiting activated PKCδ to mitochondria.
Conclusions
With P1b, a ubiquitously expressed isoform of plectin, we have identified the first cytolinker protein capable of physically interlinking mitochondria with the IF network system of cells. By generating a mouse line selectively lacking this isoform, combined with the phenotypic analysis of primary fibroblasts derived from these animals, we were able to establish a direct link between P1b expression and mitochondrial shape. Mitochondrial morphology might be regulated by anchoring mitochondrial membranes or membrane-associated molecules to the IF cytoskeleton. IF-based P1b could either provide a scaffolding platform for signaling proteins – which establish and/or maintain mitochondrial morphology on the surface of the organelle, such as proteins involved in fusion/fission events – or the interlinkage between mitochondria and IFs mediated by P1b could play a mechanical role in shaping the organelle. Future studies will be aimed at elucidating the precise molecular mechanism underlying this phenomenon and its applicability to other cell types expressing different IF networks systems. It will also be of interest to investigate whether other cytolinker proteins play a similar role and whether P1b-mediated cross talk between mitochondria and the cytoskeleton is affected in plectin-related epidermolysis bullosa simplex and other human diseases involving mitochondrial dysfunctions.
Materials and Methods
Cell culture and transfection of fibroblasts
Primary mouse fibroblasts derived from wt, P1b−/−, and P0 mice (Andrä et al., 1997), and immortalized cell lines from wt/p53−/− and P0/p53−/− double knockout mice (Andrä et al., 2003) were isolated and cultivated as described previously (Andrä et al., 1998). Primary myoblasts derived from wt and P1b−/− mice were isolated and cultivated according to Rando and Blau (1994).
For generating cell lines stably expressing fusion proteins of EGFP and N-terminal fragments of P1b, P1, or EGFP alone, we used immortalized P0 fibroblasts and the Phoenix retroviral expression system as described previously (Gregor et al., 2006).
Expression plasmids encoding full-length P1b and P1 have been described previously (Rezniczek et al., 2003). For generating plasmids harboring only the exon 1b sequence, the corresponding cDNA (Rezniczek et al., 2003) was cloned into expression vectors pEGFP-N1 and pEGFP-C1 (Clontech Laboratories, Inc.). For transfections, FuGENE 6 transfection reagent (Roche) was used.
Cellular and mitochondrial subfractionation
Cells were scraped off in 250 mM sucrose, 10 mM MOPS-KOH, pH 7.2, and 1 mM EDTA (SME); homogenized using a Dounce tissue grinder (Wheaton), and centrifuged at 700 g for 10 min. The supernatant was collected and the pellet was resuspended in SME and recentrifuged (nuclear pellet). Pooled supernatants were centrifuged at 15,000 g for 15 min to obtain a cytosolic fraction and a mitochondrial pellet, which was dissolved in SME.
Mitochondria were subjected to extraction in 100 mM Na2CO3, pH 11.5, for 30 min on ice. Membranes were isolated by centrifugation at 100,000 g for 30 min at 4°C, and proteins in the supernatant were precipitated using trichloroacetic acid. Alternatively, mitochondria were solubilized with 1% Triton X-100 or left untreated and then incubated with increasing concentrations of proteinase K (Roche) for 30 min on ice.
Immunoblotting
For immunoblotting (Rezniczek et al., 2004), antiserum No. 9 recognizing all plectin isoforms (Andrä et al., 2003) and antibodies to P1b, P1 (Abrahamsberg et al., 2005), ATPS (Invitrogen), cyt c (BD Biosciences), porin (Invitrogen), actin (Sigma-Aldrich), vim (from P. Traub, University of Bonn, Bonn, Germany), phospho-PKC (kit 9921), AMPKβ1/2 (both from Cell Signaling Technology Inc.), and RACK1 (BD Biosciences) were used in combination with HRP-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories).
Immunofluorescence and time-lapse video microscopy
Cells were fixed with 4% PFA, permeabilized with 0.1% Triton X-100, and immunostained using antibodies to GFP (Invitrogen), cyt c, Drp-1 (both from BD Biosciences), and vim (P. Traub) in combination with donkey anti–rabbit IgG Alexa Fluor 488 (Invitrogen), donkey anti–mouse IgG Rhodamine red, donkey anti–mouse IgG Cy5, and donkey anti–goat IgG Texas red (all from Jackson ImmunoResearch Laboratories). Microscopy was performed at room temperature using a confocal microscope (LSM 510; Carl Zeiss, Inc.) equipped with a Plan-Apochromat 63× 1.4 NA objective lens (Carl Zeiss Inc.). Images were recorded using the LSM510 module and the LSM software (Carl Zeiss Inc.) and processed using the Photoshop 7.0 (Adobe) software package.
Mitochondrial morphologies were classified as fragmented (individual round- or rod-shaped organelles, >80% displaying an axial length of <5 μm), intermediate (majority ∼5 μm), or tubular (often interconnected in branched networks, >80% displaying a length of >5 μm). Because of their smaller size, myoblast mitochondria were classified as fragmented (round, <2 μm) or elongated (tubular, >2 μm).
Cells expressing mito-PAGFP (from R.J. Youle, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD) were kept in a closed POCmini cultivation system (Carl Zeiss, Inc.). Photoactivation of mito-PAGFP was performed as described previously (Karbowski et al., 2004), using a confocal microscope (LSM Live DuoScan) and a Plan-Apochromat 63× 1.4 NA objective lens (Carl Zeiss, Inc.).
Fibroblasts were transfected with cDNA encoding ATPS subunit 9 presequence (provided by N. Pfanner, Institute of Biochemistry and Molecular Biology, University of Freiburg, Freiburg, Germany) fused to EGFP (mito-GFP), and live-cell imaging was performed using an inverted microscope (Axiovert S100TV; Carl Zeiss, Inc.) as described previously (Osmanagic-Myers et al., 2006). Frames were collected with a Plan-Apochromat 100× 1.4 NA objective lens (Carl Zeiss, Inc.) and individual mitochondria were tracked using Metamorph 6.3 software (MDS Analytical Technologies).
Flow cytometry
Cells were resuspended in medium containing 1 μM NAO or 0.1 μM MitoTracker red CMXRos, incubated for 15 min at 37°C in the dark, and analyzed using a cytometer (LSR1; BD Biosciences).
Online supplemental material
Fig. S1 shows the subcellular localization of isoform 1b. Fig. S2 depicts the targeted disruption of P1b for generating knockout mice, Southern blot analysis, and PCR to identify mutant ES cell clones and mouse lines, as well as an RNase protection assay and the analyses of adhesion, migration, and proliferation properties of P1b−/− fibroblasts. Details about the generation of P1b−/− mice are provided in the legend. Fig. S3 presents a subcellular fractionation after PMA treatment. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200710151/DC1.
Supplementary Material
Acknowledgments
We thank R.J. Youle and N. Pfanner for cDNAs, P. Traub for antibodies, and T. Sauer for excellent support with flow cytometry.
This work was supported by grants F 006-11 and P17862-B09 from the Austrian Science Research Fund.
Abbreviations used in this paper: ATPS, ATP synthase; cyt c, cytochrome c; IF, intermediate filament; mito-PAGFP, mitochondrial matrix–targeted photoactivatable GFP; NAO, 10-n-nonyl-acridine orange; P1b, plectin 1b; RACK1, receptor for activated C kinase 1; ROI, region of interest; vim, vimentin; wt, wild type.
References
- Abrahamsberg, C., P. Fuchs, S. Osmanagic-Myers, I. Fischer, F. Propst, A. Elbe-Bürger, and G. Wiche. 2005. Targeted ablation of plectin isoform 1 uncovers role of cytolinker proteins in leukocyte recruitment. Proc. Natl. Acad. Sci. USA. 102:18449–18454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrä, K., H. Lassmann, R. Bittner, S. Shorny, R. Fässler, F. Propst, and G. Wiche. 1997. Targeted inactivation of plectin reveals essential function in maintaining the integrity of skin, muscle, and heart cytoarchitecture. Genes Dev. 11:3143–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrä, K., B. Nikolic, M. Stöcher, D. Drenckhahn, and G. Wiche. 1998. Not just scaffolding: plectin regulates actin dynamics in cultured cells. Genes Dev. 12:3442–3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrä, K., I. Kornacker, A. Jörgl, M. Zörer, D. Spazierer, P. Fuchs, I. Fischer, and G. Wiche. 2003. Plectin-isoform-specific rescue of hemidesmosomal defects in plectin (−/−) keratinocytes. J. Invest. Dermatol. 120:189–197. [DOI] [PubMed] [Google Scholar]
- Anesti, V., and L. Scorrano. 2006. The relationship between mitochondrial shape and function and the cytoskeleton. Biochim. Biophys. Acta. 1757:692–699. [DOI] [PubMed] [Google Scholar]
- Capetanaki, Y., R.J. Bloch, A. Kouloumenta, M. Mavroidis, and S. Psarras. 2007. Muscle intermediate filaments and their links to membranes and membranous organelles. Exp. Cell Res. 313:2063–2076. [DOI] [PubMed] [Google Scholar]
- Cubitt, A.B., R. Heim, S.R. Adams, A.E. Boyd, L.A. Gross, and R.Y. Tsien. 1995. Understanding, improving and using green fluorescent proteins. Trends Biochem. Sci. 20:448–455. [DOI] [PubMed] [Google Scholar]
- Elliott, C.E., B. Becker, S. Oehler, M.J. Castañón, R. Hauptmann, and G. Wiche. 1997. Plectin transcript diversity: identification and tissue distribution of variants with distinct first coding exons and rodless isoforms. Genomics. 42:115–125. [DOI] [PubMed] [Google Scholar]
- Fuchs, P., M. Zörer, G.A. Rezniczek, D. Spazierer, S. Oehler, M.J. Castañón, R. Hauptmann, and G. Wiche. 1999. Unusual 5′ transcript complexity of plectin isoforms: novel tissue-specific exons modulate actin binding activity. Hum. Mol. Genet. 8:2461–2472. [DOI] [PubMed] [Google Scholar]
- Gregor, M., A. Zeöld, S. Oehler, K.A. Marobela, P. Fuchs, G. Weigel, D.G. Hardie, and G. Wiche. 2006. Plectin scaffolds recruit energy-controlling AMP-activated protein kinase (AMPK) in differentiated myofibres. J. Cell Sci. 119:1864–1875. [DOI] [PubMed] [Google Scholar]
- Karbowski, M., D. Arnoult, H. Chen, D.C. Chan, C.L. Smith, and R.J. Youle. 2004. Quantitation of mitochondrial dynamics by photolabeling of individual organelles shows that mitochondrial fusion is blocked during the Bax activation phase of apoptosis. J. Cell Biol. 164:493–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh, A., B. Larsson, G. von Heijne, and E.L. Sonnhammer. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305:567–580. [DOI] [PubMed] [Google Scholar]
- Osmanagic-Myers, S., and G. Wiche. 2004. Plectin-RACK1 (receptor for activated C kinase 1) scaffolding: a novel mechanism to regulate protein kinase C activity. J. Biol. Chem. 279:18701–18710. [DOI] [PubMed] [Google Scholar]
- Osmanagic-Myers, S., M. Gregor, G. Walko, G. Burgstaller, S. Reipert, and G. Wiche. 2006. Plectin-controlled keratin cytoarchitecture affects MAP kinases involved in cellular stress response and migration. J. Cell Biol. 174:557–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Olle, R., M.A. Lopez-Toledano, D. Goryunov, N. Cabrera-Poch, L. Stefanis, K. Brown, and R.K. Liem. 2005. Mutations in the neurofilament light gene linked to Charcot-Marie-Tooth disease cause defects in transport. J. Neurochem. 93:861–874. [DOI] [PubMed] [Google Scholar]
- Rando, T.A., and H.M. Blau. 1994. Primary mouse myoblast purification, characterization, and transplantation for cell-mediated gene therapy. J. Cell Biol. 125:1275–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapaport, D. 2003. Finding the right organelle. Targeting signals in mitochondrial outer-membrane proteins. EMBO Rep. 4:948–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezniczek, G.A., C. Abrahamsberg, P. Fuchs, D. Spazierer, and G. Wiche. 2003. Plectin 5′-transcript diversity: short alternative sequences determine stability of gene products, initiation of translation and subcellular localization of isoforms. Hum. Mol. Genet. 12:3181–3194. [DOI] [PubMed] [Google Scholar]
- Rezniczek, G.A., L. Janda, and G. Wiche. 2004. Plectin. Methods Cell Biol. 78:721–755. [PubMed] [Google Scholar]
- Ryan, M.T., W. Voos, and N. Pfanner. 2001. Assaying protein import into mitochondria. Methods Cell Biol. 65:189–215. [DOI] [PubMed] [Google Scholar]
- Shore, G.C., H.M. McBride, D.G. Millar, N.A. Steenaart, and M. Nguyen. 1995. Import and insertion of proteins into the mitochondrial outer membrane. Eur. J. Biochem. 227:9–18. [DOI] [PubMed] [Google Scholar]
- Toh, B.H., S.J. Lolait, J.P. Mathy, and R. Baum. 1980. Association of mitochondria with intermediate filaments and of polyribosomes with cytoplasmic actin. Cell Tissue Res. 211:163–169. [DOI] [PubMed] [Google Scholar]
- Toivola, D.M., G.Z. Tao, A. Habtezion, J. Liao, and M.B. Omary. 2005. Cellular integrity plus: organelle-related and protein-targeting functions of intermediate filaments. Trends Cell Biol. 15:608–617. [DOI] [PubMed] [Google Scholar]
- Uttam, J., E. Hutton, P.A. Coulombe, I. Anton-Lamprecht, Q.C. Yu, T. Gedde-Dahl Jr., J.D. Fine, and E. Fuchs. 1996. The genetic basis of epidermolysis bullosa simplex with mottled pigmentation. Proc. Natl. Acad. Sci. USA. 93:9079–9084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waizenegger, T., T. Stan, W. Neupert, and D. Rapaport. 2003. Signal-anchor domains of proteins of the outer membrane of mitochondria: structural and functional characteristics. J. Biol. Chem. 278:42064–42071. [DOI] [PubMed] [Google Scholar]
- Wiche, G. 1998. Role of plectin in cytoskeleton organization and dynamics. J. Cell Sci. 111:2477–2486. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}