

Abstract
Bisphosphonates are a widely used class of drugs that have been proven to be extremely useful in the prevention and treatment of osteoporosis, hypercalcemia of malignancy, and bone metastases associated with multiple myeloma, breast cancer, and other solid tumors. In the past several years there have been numerous reports describing the occurrence of Osteonecrosis of the Jaws (ONJ) associated with these drugs. In the great majority of cases this condition develops after surgical manipulation of the oral tissues. The natural history and pathophysiology of ONJ however, remains unknown at the present time. Furthermore, whether the ONJ lesion initiates in the oral mucosa or derives from the underlying bone has not been determined. In this report we describe the effect of pamidronate, a second generation bisphosphonate, on oral mucosal cells. Our results show that bisphosphonate pre-treatment of oral mucosal cells inhibits proliferation and wound healing at clinically relevant doses and that this inhibition is not due to cellular apoptosis.
Background
Bisphosphonates are a widely used class of drugs with known efficacy in the prevention and treatment of postmenopausal and steroid-induced osteoporosis; Paget’s disease of bone; hypercalcemia of malignancy; osteolytic lesions of multiple myeloma; and bone metastases associated with breast, prostate, lung, and other soft tissue tumors (1–4). Beginning in 2003, numerous reports of Osteonecrosis of the Jaws (ONJ) associated with bisphosphonate treatment have appeared in the literature (5). Overwhelmingly, ONJ cases are seen in the oncologic population, where patients are often receiving concomitant chemotherapeutic agents and are consequently immunosuppressed. To date, our knowledge regarding ONJ has been derived from case report studies (6–11). The largest and most comprehensive study by Hoff et al. reported on 4,000 cancer patients where 33 developed ONJ. This retrospective study found the overall occurrence rate of ONJ was 0.8 %, with 2.8 % occurring in multiple myeloma patients and 1.2% in breast cancer patients. While this and numerous other studies strongly suggest an association between bisphosphonates and ONJ, the true incidence, etiology, pathogenesis, and natural history of this condition have yet to be elucidated (12).
The development of ONJ lesions appear to be associated with previous traumatic injury in the vast majority of cases. Spontaneous cases occur in about 30 % of patients; however, such cases are typically localized to areas that are easily injured with very thin overlying mucosa, such as in the mylohyoid ridge region (5–12). Several theories have been proposed regarding the pathoetiology of ONJ with most speculation focusing on origin from the bone. For instance, many authors have hypothesized that ONJ is related to an over-suppression of bone turnover by bisphosphonates (6, 7). This theory, however, fails to explain why ONJ occurs almost exclusively in the maxillofacial region. Another hypothesis is that ONJ is a result of the inhibition of angiogenesis by the bisphosphonates. While several reports have shown that bisphosphonates decrease angiogenesis, almost all of these studies have only been performed in vitro and in transgenic animal models (13–18).
Several definitions for ONJ have recently been proposed and all include exposure of maxillary or mandibular bone. A breech in the oral mucosa is an absolute requirement for such a definition to apply (19–22). At the present time, it is not definitively known whether the ONJ lesion, in fact, initiates in the bone, or whether it may originate in the mucosa. Toxicity of bisphosphonates to gastric mucosal cells has been previously documented. Several authors (23–26) have shown that a number of bisphosphonates, including pamidronate, are cytotoxic to human intestinal epithelial cells. The effect of bisphosphonates on other epithelial cells such as those derived from the oral mucosa, have not been investigated. Interestingly, a recent report describing a patient who developed a palatal ulceration after holding aldendronate tablets under a denture suggests a potentially toxic effect of bisphosphonates on oral mucosa similar to that seen in the gastrointestinal studies (27). We propose that oral epithelial cells are subjected to local increases in BP concentration following a traumatic event, and that presence of such BPs may inhibit normal epithelial wound healing, thus contributing to persistent exposure of underlying bone and development of ONJ. In this study, we aim to examine the effects of pamidronate, a nitrogen-containing bisphosphonate, on oral mucosal cells using an in vitro wound healing model.
Materials and Methods
Isolation of Oral Keratinocytes
Mouse oral keratinocytes were obtained from Satrajit Sinha and LeeAnn Sinha at the State University of New York at Buffalo. All protocols were approved by the Institutional Animal Care and Use Committee. Adult 129Sv mice were euthanized; their heads were removed, rinsed in 70% ethanol and then cut open to expose the oral cavity. Blocks of tissue containing oral epithelium (primarily buccal and palatal regions) were removed and rinsed again briefly in 70% ethanol and washed with phosphate-buffered saline (PBS). After a brief incubation in 1% penicillin/streptomycin the tissue was placed in a solution of 1% gentamicin sulfate for an additional five minutes. Following these decontaminating steps, the tissue was rinsed several times with PBS and transferred into a tube containing a solution of dispase (neutral protease grade II from Bacillus polymixa in PBS, Roche). The tissues were incubated at 4ºC overnight in sufficient dispase solution to cover the samples. After dispase treatment, the tissues were rinsed again and the oral epithelium was carefully removed from the underlying submucosal layer. The isolated oral epithelium was then transferred to a tissue culture plate containing 0.5 ml of TrypLE Express cell dissociation reagent (Invitrogen) and cut into small pieces with a razor blade. The epithelial sheets were then incubated for five minutes at 37ºC and then filtered through a 100 micron Cell-Strainer (BD Biosciences). The dissociated keratinocytes were pelleted, washed, and resuspended in 5 ml of low calcium DMEM supplemented with 15% chelated fetal bovine serum. The media was changed to remove non-adherent cells after 24 hours. The keratinocytes were cultured in low calcium media until colonies formed. When cells reached confluence, they were subcultured using TrypLE Express to dissociate the cells and replated. The proliferating cells expressed basal keratinocyte marker K14 and underwent visible signs of differentiation upon switching to medium containing high concentration of calcium (LGS and SS unpublished data).
Cell Proliferation Assay
Murine oral keratinocytes were grown on 10 cm tissue-culture treated plates (BD Falcon) in low calcium (0.1mM) keratinocyte growth media supplemented with 15% fetal bovine serum (FBS, Hyclone, Logan, UT). The plates were maintained at 37°C and 5% CO2. For proliferation experiments, 5 × 103 cells were seeded per well in 96 well plates in 100μl of media and the plates were incubated with media overnight. The following day pamidronate was added to the wells at 0, 0.003, 0.01, 0.03 and 0.1 mM and proliferation was measured at 24, 48, 72, and 168 hours. A MTS/PMS reagent based cell proliferation assay kit from Promega (Promega, Madison, WI) was then used to quantify proliferation. Reagents were added directly in the incubation media and incubated at 37°C for an hour. Absorbance at 490 nm was then measured with a plate reader.
Apoptosis Assays
To determine whether oral keratinocytes exposed to pamidronate undergo apoptosis, cultures were assayed by three different methods. Caspases (Cysteine-requiring Aspartate proteases) belong to a family of degradative enzymes that are critical in the process of apoptosis and mediate programmed cell death. Caspase-3 is a key enzyme in the apoptotic cascade as it cleaves and activates procaspases 2, 6, 7, and 9 as well as mediating DNA condensation, DNA fragmentation, and cell blebbing (27–29). Cells were also assayed by TUNEL (Terminal transferase deoxy-UTP Nick End Labeling) the most commonly used in situ test for apoptosis where DNA strand breaks are detected by the binding of terminal deoxynuclotidyl transferase to the 3′ ends of DNA fragments. DAPI (4′, 6-Diamidino-2-phenylindole dihydrochloride) is a blue fluorescent stain that binds to double stranded DNA resulting in a 20-fold enhancement of fluorescence. Cells that have undergone apoptosis show pyknotic nuclei that can be visualized following DAPI staining.
Caspase-3 Assay
In order to assess the mechanism of BP toxicity on oral keratinocytes, the upregulation of Caspase-3 was examined in oral keratinocytes. Oral keratinocytes were grown to confluence in 6-well plates containing low Ca++ incubation media. Cells were treated with pamidronate for 24 and 48 hrs at 0, 0.03, 0.1 and 0.3 mM. Positive controls were generated by incubation of cells with 200nM staurosporine and 10μM cycloheximide (Sigma, St. Louis, MO) for 24 hours. The incubation media was aspirated off and centrifuged at 1,200 rpm for 5 minutes in order to collect the nonadherent cells. Adherent cells were removed from the plate using the lysis buffer provided in the kit and a cell scraper. The lysate was added to the nonadherent cell pellet and incubated on ice for 10 minutes followed by centrifugation at 12,000 rpm for 10 minutes at 4°C. Total protein concentrations in samples were determined using a Bradford reagent based protein assay (Sigma, St. Louis, MO). A standard curve was developed using known concentrations of bovine serum albumen (Bio-Rad, Hercules, CA). The standard curve equation was used to normalize sample protein concentrations. The Caspase-3 assay was performed using a colorimetric kit from Sigma. Appropriate concentrations of sample and control lysates were added to assay buffer in a 96-well plate. Caspase-3 substrate (Acetyl-Asp-Glu-Val-Asp p-nitroanilide) was then added to the plate and incubated at 37°C for 90 minutes. Absorbance was read at 405nm.
TUNEL Assay
Permanox chamber slides (Nunc, Rochester, NY) were seeded with 1×104 cells in growth media. Once wells were confluent, appropriate pamidronate (Sigma, St. Louis, MO) concentrations were added with fresh media and allowed to incubate. Positive controls were obtained by incubating cells with 200nm staurosporine and 10μM cycloheximide (Sigma, St. Louis, MO). Slides were fixed with 4% paraformaldehyde in PBS for 1 hour at room temperature. Following fixation, slides were washed with PBS and permeabilized with a 0.1% sodium citrate, 0.1% Triton X-100 solution for 2 minutes on ice. Samples were washed with PBS and then incubated with enzyme and labeling solution (Roche, Indianapolis, IN) for 1 hour at 37°C in a humidified chamber. The enzyme and labeling solutions catalyze the polymerization of labeled nucleotides to free 3′-OH DNA ends. Slides were then washed three times with PBS and mounted with Vectashield mounting solution (Vector Labs, Burlingame, CA). Detection of fragmented DNA was then observed with fluorescent microscopy.
DAPI staining
Permanox chamber slides (Nunc, Rochester, NY) were seeded with 1×105 cells in growth media. When confluence was reached, appropriate pamidronate (Sigma, St. Louis, MO) concentrations were added with fresh media and allowed to incubate. Positive controls were obtained by incubating cells with 200nM staurosporine and 10μM cycloheximide (Sigma, St. Louis, MO). Samples were fixed with 4% paraformaldehyde in PBS for 10 mins then washed three times with PBS. Slides were mounted with Vectashield mounting solution with DAPI (Vector Labs, Burlingame, CA) and observed under fluorescent microscopy.
Wounding Assay
The in vitro model of wound healing was used to examine the effects of pamidronate treatment on the ability of oral keratinocytes to migrate and proliferate in a monolayer culture. Oral keratinocytes were seeded into 6 well plates and incubated overnight. One group was preincubated with 0, 0.003, 0.01, 0.06, and 0.1 mM of pamidronate for 3 days prior to wounding. Wounds were created with a standardized protocol using a trimmed comb containing 7 tines spaced 2.5 mm apart (Goody Products Inc., Peachtree City, GA) that had been washed with 70% ethanol and dried before use. Immediately following wounding the culture media was returned to the cells following a brief wash. In a second plate bisphosphonate was added in identical concentrations as the preincubated group immediately after wounding. Migration of cells into wounded areas of the plate was observed over several days.
Statistical Analysis
Statistical analysis was performed by ANOVA and Tukey-Kramer poc-hoc test for all pairwise comparisons (JMP IN® 5.1 for Windows, SAS Instititute Inc.). Results are presented as a representation from a series of three separate experiments.
Results
Pamidronate inhibits cell proliferation
Oral keratinocyte proliferation was assayed at 24, 48, 72, and 168 hours with 0, 0.03, 0.1, and 0.3 mM pamidronate concentrations. The results, shown in Figures 1 and 2 demonstrate that pamidronate inhibits cellular proliferation at a concentration of 0.1 mM. When the cells were exposed to concentrations below 0.1 mM there was no significant difference in cell growth however, exposure to pamidronate at higher doses caused the cells be released from the plate within 2 hrs (data not shown). The relative number of viable cells in the 0.1 mM bisphosphonate group did remain constant over the course of 7 days (Figures 1 and 2).
Figure 1. Cellular proliferation of oral keratinocytes with pamidronate.
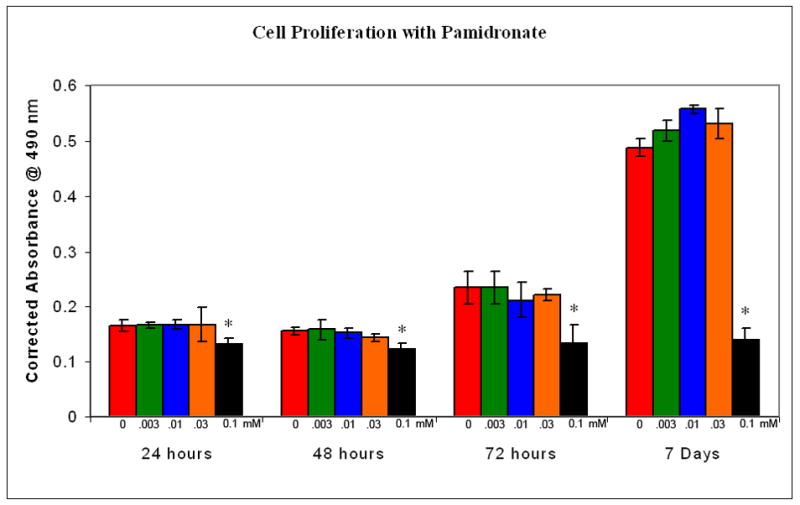
Low concentrations of pamidronate (0.003 - 0.03 mM) did not effect cell proliferation over the course of 7 days. A higher dose (0.1mM) significantly inhibited proliferation. * - p<0.05 when compared to controls.
Figure 2. Cellular proliferation of oral keratinocytes with pamidronate.
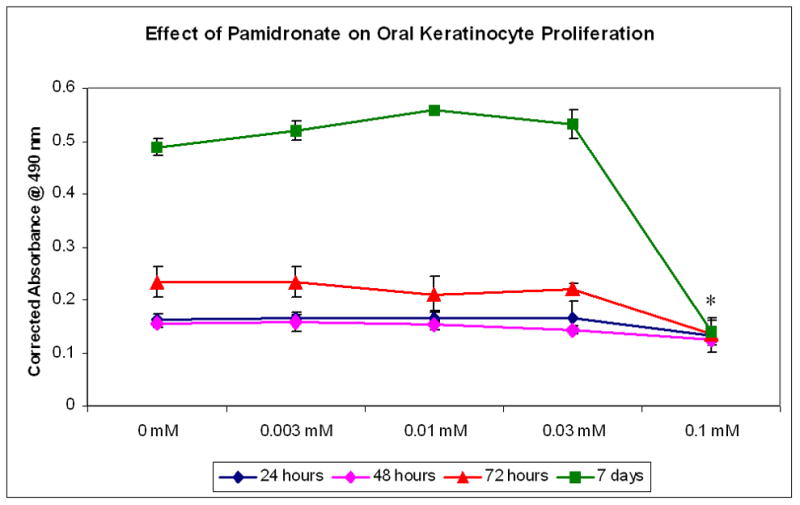
Cells exposed to 0.1mM pamidronate did not proliferate from 24 hours to 7 days. * - p<0.05 when compared to controls.
Pamidronate does not cause apoptosis of oral keratinocytes
Apoptosis or programmed cell death is an organized process where a cascade of events ultimately results in activation of several different caspase enzymes that result in DNA fragmentation. The ability of pamidronate to induce apoptosis in oral keratinocytes was evaluated using three different methods; the upregulation of caspase-3, fragmentation of DNA by TUNEL assay, and analysis of apoptotic nuclei visualized with DAPI staining.
The results of the caspase-3 assays are shown in Figure 3. Enzyme levels in the wells treated with 0.03 and 0.1 mM pamidronate showed a significant increase at 24 and 48 hrs when compared to the 6 hr sample however caspase-3 levels were not significantly different from the control levels. Cells that were treated with 0.3 mM of pamidronate showed a decrease in caspase-3 levels at 24 and 48 hrs coincident with the release of cells from the plate. The cells treated with staurosporine and cycloheximide, agents known to cause significant apoptosis, showed an upregulation of caspase-3 at all time points that was between 4 and 6 fold higher levels than that seen in any of the cells that received pamidronate.
Figure 3. Caspase-3 activity in oral keratinocytes incubated with pamidronate.
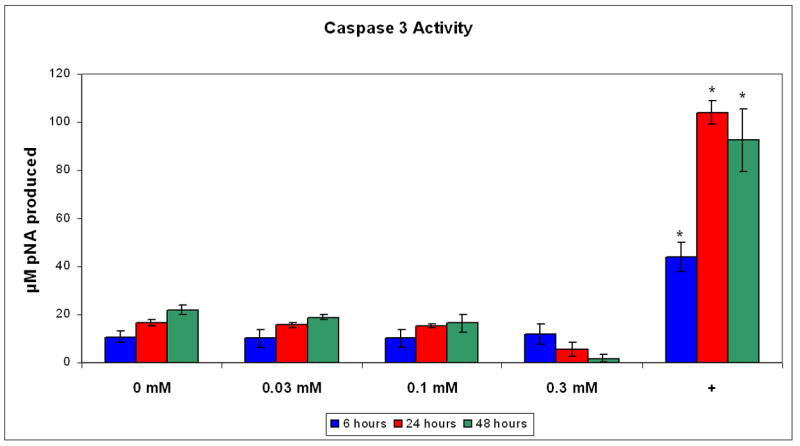
Pamidronate did not significantly increase caspase-3 activity in cells over a 48 hour period when compared with controls. The positive control (+) was obtained by incubating cells with staurosporine and cycloheximide. * - Significant increase compared with controls, p<0.05.
Analysis of the percentage of apoptotic cells was performed by counting the number of TUNEL positive cells in five high power fields of approximately 100 cells. The results for the 24 hr time point are shown in Figure 4. Approximately 2% of cells treated with 0.03, 0.1, and 0.3 mM pamidronate for 24h were TUNEL positive, similar to untreated control cells. In contrast, the positive control, cells treated with 200nM staurosporine and 10um cycloheximide, had nearly 100% TUNEL positive cells. At 48 hrs the findings were similar to that seen at 24 hrs however many of the cells in the 0.3 mM pamidronate samples had floated off of the slide. The six hour time point showed minimal effects in the pamidronate treated samples with only about 50 % TUNEL positive cells seen in the positive control (data not shown). These results are consistent with the caspase-3 assay results (discussed above) as well as quantitation of DAPI stained pyknotic nuclei in cells treated with 0, 0.03, 0.1, and 0.3 mM pamidronate respectively (data not shown).
Figure 4. Apoptosis in oral keratinocytes incubated with pamidronate.

TUNEL assay performed on cells incubated with 0.03, 0.1, and 0.3 mM pamidronate did not increase the percentage of TUNEL positive cells when compared to nontreated cells. Staurosporine and cycloheximide treated positive control cells were all apoptotic by 24 hours.
Wound healing of Oral keratinocytes is inhibited by Pamidronate
The in vitro model of wound healing was used to examine the effects of Pamidronate treatment on the ability of oral keratinocytes to migrate and proliferate in a monolayer culture. The results are shown in Figure 5. In the preincubated samples, the control and 0.003, 0.01, and 0.06 mM pamidronate groups cellular migration was appreciated beginning at 6 hrs and the wound was completely closed at 72 hrs. The 0.1 mM pre-incubated group had a significantly delayed wound closure at all time points but did close when incubated for 5 days (data not shown). The plates that had bisphosphonate added immediately after wounding (no pre-incubation) had a response that was identical to the control samples.
Figure 5. Wound healing in oral keratinocytes with pamidronate.

Cells were observed for 96 hours after wounding in cell culture plates. Control wells exhibited significant migration into the wounded area from time of injury (A) to 96 hours (B). Cells pre-incubated for 72 hrs with 0.1mM pamidronate prior to wounding had greatly reduced migration from 0 (C) to 96 hours (D). Data shown is representative of 3 independent experiments.
Discussion
We show here for the first time that pamidronate, a representative nitrogen-containing bisphosphonate, suppresses cellular proliferation and delays wound healing of oral mucosal cells. Furthermore, the effects of pamidronate are observed at doses that would theoretically be expected in the maxillofacial bone and soft tissues of patients receiving the drug by the intravenous route. These effects however, were not the result of apoptotic cell death.
In late 2003 a preliminary report described 36 patients with avascular necrosis of the jaws in patients receiving intravenous bisphosphonates. With the exception of one subject being treated for osteoporosis, all patients were receiving BPs as an adjunct to cancer therapy (5). Subsequently, Ruggiero et al. reported on 63 cases of ONJ. In this series fifty-five patients were under treatment for cancer (28 multiple myeloma, 20 breast cancer) and 7 patients were being treated for osteoporosis. Since these initial reports, numerous case series have been reported in the literature (6). As previously mentioned the great majority of information regarding ONJ has been from case series (5–11). There are a limited number of retrospective studies available; the largest is a series of 4,000 cancer patients reported by Hoff et al. This study, similar to other reports showed an association of ONJ with previous dental trauma (most commonly extraction). Lesions were more commonly seen in the mandible and multiple myeloma patients followed by breast cancer patients were most often affected (12). An association with bisphosphonate treatment, especially IV administration appears to have been established however the incidence, etiology, pathogenesis, and natural history of ONJ are not known (5–12).
It has been postulated that the ONJ bone lesion is a result of an over-suppression of bone turnover by the BPs (6, 7). This theory however, fails to explain why exposed necrotic lesions are rarely seen in bones other than the jaws. Several authors have suggested that a compromised blood supply secondary to BPs is the initiating event in the development of the ONJ lesion (7). While a significant number of studies have shown that BPs inhibit angiogenesis, the overwhelming majority of these investigations were performed in vitro (most commonly using human umbilical vein endothelial cells) or in vivo in animal models (13–18). Apoptosis or programmed cell death is a process where living cells participate in a cascade of events that ends with the cell dying. In contrast to necrosis, the sequence of events that take place during apoptosis are organized with several unique features that can be studied in the laboratory.
Apoptosis is important in development as well as in many disease processes and cancer. Two different pathways of apoptosis have been identified; the extrinsic or death receptor pathway and the intrinsic or mitochondrial pathway both resulting in upregulation of proteolytic enzymes called caspases (cysteinyl asprtate-specific proteases) with subsequent DNA fragmentation, DNA budding, and chromatin condensation. Stimulation of the apoptotic pathways depend on the cell type and the mechanism of insult. In order to demonstrate that a cell has undergone apoptosis it is necessary to utilize several different but complementary techniques including observation of cell morphological changes, DNA fragmentation assays, and biochemical assays for caspases (28–30). We have found that pamidronate was not able to promote apoptosis of oral mucosal cells at any tested concentration however, at BP doses greater than 0.1 mM there appeared a toxic effect that resulted in the detachment of the adherent cells. Thus, it is possible that pamidronate may lead to cell necrosis. This possibility will be further explored in the future.
Bisphosphonate toxicity to epithelial cells has been well documented. The development of gastric erosions and ulcers is a well known side effect associated with oral administration of nitrogen-containing BPs. Several studies have demonstrated the adverse effects of exposing gastrointestinal cells to BPs (23–26). Wallace et al (25) used an ex vivo gastric chamber model to show concentration-dependent epithelial injury and loss of integrity after exposure to pamidronate and alendronate. Exposure to the drug caused necrosis of the gastric epithelium and the mechanism of damage was independent of microvascular injury, possibly due to disruption of surface active phospholipids within the mucosal layer. Twiss et al (23) confirmed the cytotoxic effects of pamidronate on Caco-2 intestinal cells by demonstrating increased cell permeability after BP exposure in vitro. In a subsequent investigation, this same group (24) incubated Caco-2 epithelial cell cultures with various concentrations of two nitrogen-containing BPs, pamidronate and alendronate. They observed that in the presence of calcium, both BPs were toxic to the cells, however, only pamidronate exhibited toxicity when calcium was absent from the media. Furthermore, they demonstrated that pamidronate forms insoluble complexes with calcium that deposit on the cell surface, exposing them to locally increased and cytotoxic concentrations of the drug. They concluded that neither of the BPs induced apoptosis, however, cellular morphometry measured by microscopic observation was the only assay method used to assess programmed cell death. Suri et al (26) exposed Caco-2 cells to various BPs in vitro and evaluated apoptosis by both nuclear morphometry and TUNEL assays. In contrast to the results of Twiss et al., they found a dose-dependent increase in apoptotic cells, reduction in cell viability, and inhibition of cell proliferation with pamidronate, among other BPs. Apoptosis and loss of viability was prevented when BP-induced inhibition of farnyldiphosphate (FPP) synthase—an enzyme in the mevalonate pathway—was bypassed by addition of Geranylgeraniol, a downstream protein. They postulated that the toxic effects that BPs had on intestinal cells were likely related to disruption of the mevalonate pathway, which thereby reduces production of cholesterol precursors and interferes with cell membrane biosynthesis. In the present study we evaluated whether oral keratinocytes undergo apoptosis following exposure to pamidronate by three different methods. DAPI (4′, 6-diamidino-2-phenylindole), a dye that incorporates into DNA in viable cells and becomes highly fluorescent was used to visualize chromatin condensation. TUNEL assays were used to evaluate nuclear fragmentation and caspase-3 activity was measured with a biochemical assay. All assays failed to show any significant increase in oral keratinocyte apoptosis secondary to pamidronate exposure. While these findings differ from those seen by Suri et al. it is important to note that Caco-2 cells, although they are considered a good model for gastric epithelium, are a cell line that is derived from a gastric adenocarcinoma. The effect of using a tumor cell line as model for normal gastric epithelium as in the Twiss and Suri studies is not known, however, apoptosis is known to be altered in many cancers (28–30).
To our knowledge, there is no data on actual bisphosphonate concentrations in bone from treated patients, and at best such concentrations can be extrapolated from other clinical pharmacokinetic data and preclinical data on bisphosphonate concentrations in bone tissue. In patients with metastatic bone disease the mean amount of pamidronate delivered to the skeleton has been estimated to be 65% of an intravenous dose, a percentage stable during subsequent administrations (31, 32). Extrapolating this data and assuming no substantial elimination, 4 years of treatment with 90 mg of monthly intravenous pamidronate will lead to a mean bone concentration of around 200 ng/mg (854 nmol/g). Such calculations, which also have been made by Reid for zoledronate (33), result in an estimated pamidronate bone concentration remarkably similar to those extrapolated from data on actual bone concentrations in animals such as dogs (240 ng/mg) and mice (155 ng/mg) assessed using 14C-labeled pamidronate (34, 35). A range of 50 to 500 ng/mg, or 214 to 2140 nmol/g, therefore seems a reasonable estimate of the mean pamidronate concentration in bone tissue in humans treated for a period of 4 years with monthly 90 mg infusions. Little is known about the concentration of BP in various parts of the skeleton, as well as concentrations in bone sites with increased bone turnover such as bone metastases or alveolar bone following a surgical procedure, although 99mTc-labeled BP data show preferential uptake in these sites. There is also no data on BP concentrations in gingival crevicular fluid, but it has been speculated that these concentrations are similar to bone concentrations (33). Nevertheless, we hypothesize that exposure of the oral epithelium to toxic concentrations of BP derived from the underlying bone results in an inhibition of wound healing. Our results show that proliferation and wound healing of murine oral keratinocytes is inhibited at pamidronate concentrations above 0.1 mM, concentrations that may be reached in the maxillofacial bones and crevicular fluid of cancer patients on intravenous therapy.
To our knowledge, the effect of nitrogen-containing bisphosphonates on oral mucosal cells has not been previously investigated. Such studies are of interest as they may shed light on the pathophysiological mechanisms underlying the newly described phenomenon ONJ. Although the focus thus far in ONJ research has involved BPs and bone—an “inside-out” theory—it is intriguing to speculate that the adverse effects of BP on oral epithelium may play a critical role in the initiation of ONJ—an “outside-in” hypothesis. The results of this study suggest that at clinically relevant concentrations pamidronate inhibits cell proliferation and the capacity for wound healing in murine oral keratinocytes. These effects however are not due to cell apoptosis. Future studies will investigate the effects of bisphosphonates on human oral keratinocytes in vitro as well as the development of an in vivo wound healing model. At the present time there are no available therapeutic regimens for the treatment of ONJ, hence a major focus has been on prevention of this condition. While many of the professional societies have published recommendations for preventive strategies (19–22), these protocols are not based on knowledge about the pathobiology of ONJ nor are they evidence based. The in vitro studies and the wounding model described herein will allow future investigations to dissect the specific molecular mechanisms of oral mucosal cell injury by bisphosphonates and the development of rational treatment protocols.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rosen LS, et al. Zoledronic acid versus pamidronate in the treatment of skeletal metastases in patients with breast cancer or osteolytic lesions of multiple myeloma: a phase III, double-blind, comparative trial. Cancer J. 2001;7:377–87. [PubMed] [Google Scholar]
- 2.Saad F, et al. A randomized, placebo-controlled trial of zoledronic acid in patients with hormone-refractory metastatic prostate carcinoma. J Natl Cancer Inst. 2002;94:1458–68. doi: 10.1093/jnci/94.19.1458. [DOI] [PubMed] [Google Scholar]
- 3.Major P. The use of zoledronic acid, a novel, highly potent bisphosphonate, for the treatment of hypercalcemia of malignancy. Oncologist. 2002;7:481–91. doi: 10.1634/theoncologist.7-6-481. [DOI] [PubMed] [Google Scholar]
- 4.Van Poznak CH. The use of bisphosphonates in patients with breast cancer. Cancer Control. 2002;9:480–9. doi: 10.1177/107327480200900605. [DOI] [PubMed] [Google Scholar]
- 5.Marx RE. Pamidronate (Aredia) and zoledronate (Zometa) induced avascular necrosis of the jaws: a growing epidemic. J Oral Maxillofac Surg. 2003;61:1115–7. doi: 10.1016/s0278-2391(03)00720-1. [DOI] [PubMed] [Google Scholar]
- 6.Ruggiero SL, Mehrotra B, Rosenberg TJ, Engroff SL. Osteonecrosis of the jaws associated with the use of bisphosphonates: a review of 63 cases. J Oral Maxillofac Surg. 2004;62(5):527–34. doi: 10.1016/j.joms.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 7.Marx RE, Fortin M, Broumand V. Bisphosphonate-induced exposed bone (Osteonecrosis/Osteopetrosis) of the Jaws: Risk factors, recognition, prevention and treatment. J Oral Maxillofacial Surg. 2005;63:1567–1575. doi: 10.1016/j.joms.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 8.Migliorati CA. Bisphosphonates and oral cavity avascular bone necrosis. J Clin Oncol. 2003;21(22):4253–4. doi: 10.1200/JCO.2003.99.132. [DOI] [PubMed] [Google Scholar]
- 9.Wang J, Goodger NM, Pogrel MA. Osteonecrosis of the jaws associated with cancer chemotherapy. J Oral Maxillofac Surg. 2003;61:1104–7. doi: 10.1016/s0278-2391(03)00328-8. [DOI] [PubMed] [Google Scholar]
- 10.Pogrel MA. Bisphosphonates and bone necrosis. J Oral Maxillofac Surg. 2004;62:391–2. doi: 10.1016/j.joms.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 11.Lugassy G, et al. Severe osteomyelitis of the jaw in long-term survivors of multiple myeloma: a new clinical entity. Am J Med. 2004;117:440–1. doi: 10.1016/j.amjmed.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 12.Hoff AO, et al. Proc Am Soc Clin Oncol. 2006;24:475s. Abstract 8528. [Google Scholar]
- 13.Wood J, et al. Novel antiangiogenic effects of the bisphosphonate compound zoledronic acid. J Pharmacol Exp Ther. 2002;302:1055–61. doi: 10.1124/jpet.102.035295. [DOI] [PubMed] [Google Scholar]
- 14.Bezzi M, et al. Zoledronate sensitizes endothelial cells to tumor necrosis factor-induced programmed cell death: evidence for the suppression of sustained activation of focal adhesion kinase and protein kinase B/Akt. J Biol Chem. 2003;31(278):43603–14. doi: 10.1074/jbc.M308114200. [DOI] [PubMed] [Google Scholar]
- 15.Hamma-Kourbali Y, et al. A novel non-containing-nitrogen bisphosphonate inhibits both in vitro and in vivo angiogenesis. Biochem Biophys Res Commun. 2003;310:816–23. doi: 10.1016/j.bbrc.2003.09.083. [DOI] [PubMed] [Google Scholar]
- 16.Giraudo E, Inoue M, Hanahan D. An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. J Clin Invest. 2004;114:623–33. doi: 10.1172/JCI22087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Okamoto T, et al. Incadronate disodium inhibits advanced glycation end products-induced angiogenesis in vitro. Biochem Biophys Res Commun. 2002;297:419–24. doi: 10.1016/s0006-291x(02)02218-0. [DOI] [PubMed] [Google Scholar]
- 18.Santini D, et al. Pamidronate induces modifications of circulating angiogenetic factors in cancer patients. Clin Cancer Res. 2002;8:1080–1084. [PubMed] [Google Scholar]
- 19.Advisory Task Force on Bisphosphonate-Related Ostenonecrosis of the Jaws, American Association of Oral and Maxillofacial Surgeons. American Association of Oral and Maxillofacial Surgeons position paper on bisphosphonate-related osteonecrosis of the jaws. J Oral Maxillofac Surg. 2007;65:369–76. doi: 10.1016/j.joms.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 20.Kyle RA, Yee GC, Somerfield MR, Flynn PJ, Halabi S, Jagannath S, Orlowski RZ, Roodman DG, Twilde P, Anderson K American Society of Clinical Oncology. American Society of Clinical Oncology 2007 clinical practice guideline update on the role of bisphosphonates in multiple myeloma. J Clin Oncol. 2007;25:2464–72. doi: 10.1200/JCO.2007.12.1269. [DOI] [PubMed] [Google Scholar]
- 21.Salvatore Ruggiero, Gralow Julie, Marx Robert E, Hoff Ana O, Schubert Mark M, Huryn Joseph M, Toth Bela, Damato Kathryn. Practical Guidelines for the Prevention, Diagnosis, and Treatment of Osteonecrosis of the Jaw in Patients With Cancer. Vicente Valero J Oncol Prac. 2006;1:7–14. doi: 10.1200/jop.2006.2.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khosla S, et al. Bisphosphonate-Associated Osteonecrosis of the Jaw: Report of a Task Force of the American Society for Bone and Mineral Research. J Bone Miner Res. 2007;22:1479–1489. doi: 10.1359/jbmr.0707onj. [DOI] [PubMed] [Google Scholar]
- 23.Twiss IM, et al. Cytotoxic effects of pamidronate on monolayers of human intestinal epithelial (Caco-2) cells and its epithelial transport. J Pharm Sci. 1994;83:699–703. doi: 10.1002/jps.2600830521. [DOI] [PubMed] [Google Scholar]
- 24.Twiss IM, et al. The effects of nitrogen-containing bisphosphonates on human epithelial (Caco-2) cells, an in vitro model for intestinal epithelium. J Bone Miner Res. 1999;14:784–91. doi: 10.1359/jbmr.1999.14.5.784. [DOI] [PubMed] [Google Scholar]
- 25.Wallace JL, Dicay M, McKnight W, Bastaki S, Blank MA. N-bisphosphonates cause gastric epithelial injury independent of effects on the microcirculation. Alimen Pharmacol Ther. 1999;13:1675–82. doi: 10.1046/j.1365-2036.1999.00658.x. [DOI] [PubMed] [Google Scholar]
- 26.Suri S, et al. Nitrogen-containing bisphosphonates induce apoptosis of Caco-2 cells in vitro by inhibiting the mevalonate pathway: a model of bisphosphonate-induced gastrointestinal toxicity. Bone. 2001;29:336–43. doi: 10.1016/s8756-3282(01)00589-0. [DOI] [PubMed] [Google Scholar]
- 27.de Groen PC, et al. Esophagitis associated with the use of alendronate. N Engl J Med. 1996;335:1016–21. doi: 10.1056/NEJM199610033351403. [DOI] [PubMed] [Google Scholar]
- 28.Loro L, Vintermyr OK, Johannessen AC. Apoptosis in normal and diseased oral tissues. Oral Dis. 2005;11:274–87. doi: 10.1111/j.1601-0825.2005.01117.x. [DOI] [PubMed] [Google Scholar]
- 29.Huerta S, Goulet EJ, Huerta-Yepez S, Livingston EH. Screening and detection of apoptosis. J Surg Res. 2007;139:143–56. doi: 10.1016/j.jss.2006.07.034. [DOI] [PubMed] [Google Scholar]
- 30.Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411:342–8. doi: 10.1038/35077213. [DOI] [PubMed] [Google Scholar]
- 31.Leyvraz S, et al. Pharmacokinetics of pamidronate in patients with bone metastases. J Natl Cancer Inst. 1992;84:788–92. doi: 10.1093/jnci/84.10.788. [DOI] [PubMed] [Google Scholar]
- 32.Cremers, et al. Skeletal retention of bisphosphonate (palmidronate) and its relation to the rate of bone resorption in patients with breast cancer and bone metstasis. JBMR. 2005;20:1543–154. doi: 10.1359/JBMR.050522. [DOI] [PubMed] [Google Scholar]
- 33.Reid IR, Bolland MJ, Grey AB. Is bisphosphonate-associated osteonecrosis of the jaw caused by soft tissue toxicity? Bone. 2007;41:318–20. doi: 10.1016/j.bone.2007.04.196. [DOI] [PubMed] [Google Scholar]
- 34.King LE, Vieth R. Extraction and measurement of pamidronate from bone samples using automated pre-column derivatization, high-performance liquid chromatography and fluorescence detection. J Chromatogr B Biomed Appl. 1996;678:325–30. doi: 10.1016/0378-4347(95)00531-5. [DOI] [PubMed] [Google Scholar]
- 35.Hoggarth CR, Bennett R, Daley-Yates PT. The Pharmacokinetics and distribution of pamidronate for a range of doses in the mouse. Calcif Tissue Int. 1991;49:416–420. doi: 10.1007/BF02555853. [DOI] [PubMed] [Google Scholar]