

Abstract
The TGF-β pathway has tumor suppressor activity in many epithelial tissues. Since TGF-β is a potent inhibitor of epithelial cell proliferation, it has been widely assumed that this property underlies the tumor suppressor effect. Here we have used a xenograft model of breast cancer to show that endogenous TGF-β has the potential to suppress tumorigenesis through a novel mechanism, involving effects at two distinct levels in the hierarchy of cellular progeny that make up the epithelial component of the tumor. Firstly TGF-β reduces the size of the putative cancer stem or early progenitor cell population, and secondly it promotes differentiation of a more committed, but highly proliferative, progenitor cell population to an intrinsically less proliferative state. We further show that reduced expression of the type II TGF-β receptor correlates with loss of luminal differentiation in a clinical breast cancer cohort, suggesting that this mechanism may be clinically relevant. At a molecular level, the induction of differentiation by TGF-β involves down-regulation of Id1, and forced overexpression of Id1 can promote tumorigenesis despite persistence of the anti-proliferative effect of TGF-β. These data suggest new roles for the TGF-β pathway in regulating tumor cell dynamics that are independent of direct effects on proliferation.
Introduction
TGF-βs are important mediators of cell-cell and cell-environmental sensing in the dynamic processes that are responsible for tissue development and homeostasis in many organ systems. Perturbations in this normal transactional process can promote tumorigenesis, and many lines of evidence suggest that an impaired TGF-β response in epithelial tissues is associated with enhanced tumorigenesis, at least in the early stages of the process (1). Since epithelial cells are exquisitely sensitive to growth inhibition by TGF-β, the antiproliferative effect of TGF-β is widely assumed to be critical for its tumor suppressor activity. However, loss of TGF-β can promote tumorigenesis in the absence of overt effects on cell proliferation (2), and conversely some transformed cells can form tumors despite persistence of a growth inhibitory response to TGF-β (3). These findings suggest that other mechanisms may be important. As a result, we undertook a detailed analysis of the mechanism of tumor suppression by TGF-β in an experimentally tractable breast cancer model system.
Most human breast cancer cell lines are derived from pleural effusions or ascites (4), and therefore represent a very late stage in the disease process, when the tumor suppressor effects of TGF-β have largely been abrogated (1). Miller and co-workers have developed a xenograft model of human breast cancer progression based on a spontaneously immortalized human breast epithelial cell line, MCF10A, which has a defective Ink4 locus (5–7). The MCF10ACa1h subline (“Ca1h”) that we have used here is a tumorigenic, but non-metastatic derivative of MCF10A, transformed with the activated Ha-ras oncogene (6). Ca1h xenografts exhibit well-differentiated glandular areas, which contain estrogen receptor positive cells (6;8). Although ras mutations per se are relatively rare in breast cancer (<5% of cases), hyperactivation of the ras pathway is a common feature of many breast tumors (9). Furthermore, epigenetic inactivation of the Ink4A locus is a frequent early event in human breast cancer (10). The model therefore has several key molecular features of low grade human breast cancers. Thus, this cell line permits analysis of control processes that may regulate a transformed breast epithelial cell relatively early in the carcinogenic process, before they are lost on further progression.
Many tumors are now thought to consist of a hierarchy of cells with different proliferative and developmental potential. A very small number of “cancer stem cells” or “tumor-initiating cells” are hypothesized to give rise to a much larger population of highly proliferative, but committed progenitor cells, which may then undergo limited differentiation in a caricature of normal histogenesis (11). The key features of the tumor-initiating cells that allow them to be termed “stem” cells are the ability to self-renew and to give rise to diverse offspring. Cell populations enriched for cells with putative cancer stem cell properties have now been prospectively identified for a number of different tumor types, including breast cancer (reviewed in (12;13)). Since stem cells divide infrequently and are relatively drug resistant, the efficacy of most conventional therapies is likely to be limited by the fact that they are targeted primarily against the more committed, proliferative progeny and not against the cancer stem cells themselves (12;13). Thus the issue of how cancer stem cells are regulated becomes central to the design of successful preventive and therapeutic agents. Here we show that TGF-β has the potential to function as a tumor suppressor in breast cancer by depleting the putative cancer stem or early progenitor cell population, and by promoting differentiation of the more committed progeny. These effects of TGF-β appear to be independent of its ability to directly inhibit cell proliferation.
Materials and Methods
Cell culture and retroviral infections
The MCF10CA1h cell line (Barbara Ann Karmanos Cancer Institute, Cell Line Resource, Detroit MI) was cultured as previously described (14). Wnt-1 cells derived from a mammary tumor in an MMTV-Wnt mouse (gift of Yi Li, Baylor College of Medicine, TX) and MDA MB231 cells (American Type Culture Collection, Rockville MD) were cultured in DMEM, 10% FBS. Retroviral transduction with the Myc-tagged dominant negative type II TGF-β receptor (“DNR”) construct in pLPCX was done as previously described (14). The pBABE retrovirus expressing Id1 was as described (15). Following infection, cells were maintained under positive selection for 7 days and in vivo experiments were performed within 2–3 weeks of transduction, using pools of transduced cells.
Growth inhibition assays and Western blot analysis
Growth inhibition and Western blot analyses were performed as previously described (14). Antibodies were obtained from the following sources: c-Myc, Active Motif LLC (Carlsbad, California, USA); p27, cycD1, p21, cdk2, cdk4, cdc25a and Id1 were all from Santa Cruz Biotechnology Inc. (Santa Cruz, California, USA);
Tumorigenesis
Animal studies were performed under protocols approved by the NCI, in accordance with AAALAC guidelines and policies established by the NIH. MCF10CA1h cells were suspended in DMEM/F-12 at varying densities from 5×103 cells/ml to 2.5×106cells/ml for titration experiments, and five-week-old female athymic NCr nu/nu mice were inoculated subcutaneously on the hind flank with 0.2 ml of the cell suspension. Mice were palpated up to 3 times weekly, palpable lesions were measured with calipers, and tumor volumes were calculated as previously described (14). All tumors were examined by a board-certified veterinary pathologist (M.R. Anver), and the fraction of tumor area that comprised each of the three predominant histological types in the tumor (cribriform, clear cell or pleoimorphic regions) was assessed.
Immunohistochemistry, tissue arrays and in vivo proliferation indices
Formalin-fixed, paraffin-embedded tumor sections from xenografted tumors or a human breast cancer tissue array (IMH-364 from Imgenex, CA) were stained with antibodies recognizing CK8 (Hybridoma Bank, IA), CK14 (Covance, CA), CK6 (Research Diagnostics, Inc. MA), TGF-β RII (Santa Cruz, CA), phospho-Histone H3 (Upstate Biotechnology Inc., VA). The tissue microarray was scored blinded for staining intensity in the epithelial compartment by a board-certified pathologist (A. Ooshima) on a scale of 0 to 3+. For CK8, a score of 1–3 was considered positive. For TβRII, a score of 0 or 1 was classified as low TβRII expression, and a score of 2 or 3 was classified as high expression. Overall proliferation indices in xenografted tumors was assessed by bromodeoxyuridine labeling as previously described (14). To determine proliferation indices in well-differentiated luminal regions of the xenografted tumors compared with the less differentiated regions containing progenitor cells, tumor sections were double immunostained for the luminal marker CK8 (brown DAPI stain) and phospho-histone H3 (blue acid phosphatase stain), and staining was quantitated as detailed in Suppl. Fig. 1.
Real-time RT-PCR quantitation of mRNA
Total tumor RNA was extracted using Trizol® according to manufacturer’s instructions (Invitrogen, Carlsbad, CA). Real-time PCR was performed in a two-step reaction using the SuperScript™III First Strand Synthesis System (Invitrogen, CA) to generate cDNAs from tumor RNA samples. The second step was performed in a fluorescent temperature cycler (Bio-Rad, CA). Brilliant SyBR Green QPCR Master Mix (Stratagene, CA) and specific primers for each of the target genes (SuperArray Bioscience Corp., MD) were used. 18S ribosomal RNA was used as a reference transcript for normalization.
Flow cytometry analyses for SP fractions and differentiation markers
SP fractions
Tumor xenografts were minced with scalpels and then digested in DMEM/F12 medium containing 300U/ml collagenase (Stem Cell Technologies, Vancouver, Canada) and 100U/ml hyaluronidase (Stem Cell Technologies) for 16h at 37°C. Cells were then washed and resuspended to a final concentration of 106cells/ml in DMEM/2% FBS. Preliminary studies were performed to determine optimal Hoechst dye concentrations and exposure time for quantitation of the Hoechst dye effluxing side population (SP) (16). Cell suspensions from the tumor samples or cultured cell lines were stained with Hoechst-33342 (5µg/ml, Sigma) at 37°C for 90 minutes. Verapamil (5µM, Sigma) was added to parallel control tubes to inhibit Hoechst efflux. FACS analysis was performed on an LSR II (BD Biosciences, CA). The SP fraction was then determined using FlowJo software (Tree Star Inc, San Carlos, CA) to be the region of the dot plot where the majority of Hoechst-effluxing cells were shifted to the higher fluorescence intensity by verapamil inhibition.
Differentiation markers
Subconfluent cells in DMEM/F12 containing 1% calf serum were treated with TGF-β1 (5 ng/ml) or vehicle control (4mM HCl, 0.1% BSA) for 4 days. Cells were trypsinized and single cells were fixed and permeabilized with BD Cytofix/Cytoperm™ (Becton-Dickinson, San Jose, CA, USA). FITC-conjugated MUC1 (BD Pharmingen) and Cytokeratin 8 (Hybridoma Bank, IA) antibodies were incubated with cells at 4°C overnight. Isotype-matched control antibodies were used in each experiment. Stained cells were analyzed using a FACS Caliber (Becton-Dickinson, San Jose, CA, USA) and FlowJo software.
Tumorsphere Formation
Single cells were plated in ultra low attachment 24-well plates (Corning, NY) at a density of 1500 cells/ml in regular growth medium (17). After 7–10 days, wells were examined under an inverted microscope at 40X magnification, and the number of spheres of ≥100µm in diameter were counted for a total of 20–25 independent fields/well, and 3 replicate wells/condition. To determine the effect of TGF-β treatment on tumorsphere-forming efficiency, cells were allowed to grow to ~75% confluence for 3 days in standard tissue culture dishes in standard growth medium, and then were switched to medium containing 1% calf serum for 5 hours and then treated with 5 ng/ml TGF-β for 24h prior to trypsinization, washing and seeding into low attachment dishes as above. The Alk5 kinase inhibitor SB431542 (5µM final conc; Sigma-Aldrich, St. Louis, MO) was included in the growth medium in the low attachment dishes to ensure that there was no persistence of the TGF-β effect during growth in the low attachment conditions.
In silico datamining of published array data
The publicly available primary data from Sorlie et al. (18) were downloaded from the Stanford Microarray Database. For each spot, channel signals were calculated as the mean foreground minus median background intensity. Flagged spots or spots with the intensity lower than 50 in any channel were filtered out. Log base 2 ratios of red and green signals were calculated for each qualified cDNA probe (mammary tumor/normal tissue vs. the reference of human cell lines mixture) and normalized with the Lowess smoother. The values plotted represent mean differences among the cancer subtypes relative to median level of TGFB1 expression across all samples. Differential expression of TGFB1 was tested using one-way ANOVA (overall F-test and specific comparison between the Luminal Subtype A and Basal-like groups). The calculation was performed using R 2.4.0 and Bioconductor gregmisc package.
Statistical analyses
Statistical analysis on data other than microarray data was performed using analysis tools on the VassarStats Statistical Computation site10. Data were analyzed using the parametric unpaired Student t-test, or the non-parametric Mann-Whitney test unless otherwise stated. Cross-categorized frequency data were analyzed using the Fisher Exact test. All p-values are two-sided.
Results
TGF-β-dependent tumor suppression in Ca1h tumors does not involve the expected molecular mechanisms of growth inhibition
We have previously shown that reduction or loss of TGF-β response by introduction of a dominant negative type II TGF-β receptor (DNR) enhances tumorigenesis in the Ca1h cell line (see Ref.(14) and an independent data set in Fig. 1a). TGF-β inhibited the proliferation of Ca1h cells in vitro, and the effect was largely blocked by the DNR (Fig. 1b). As expected, loss of TGF-β response was associated with increased proliferation in Ca1h tumors in vivo (Fig. 1c). For many epithelial cells, inhibition of proliferation by TGF-β correlates with effects on expression of a constellation of cell cycle regulators (19). However, our original assumption that TGF-β was functioning as a tumor suppressor in vivo through direct effects on cell proliferation was challenged by finding that the expression of canonical TGF-β target genes involved in cell cycle regulation was unchanged or changed in the opposite to the expected direction in the Ca1h-DNR tumors (Fig. 1d). Thus, Ca1h-DNR tumors paradoxically expressed lower c-myc and higher p27 levels than the Ca1h tumors, while p21 levels were unchanged. These observations led us to hypothesize either that TGF-β inhibits proliferation by different molecular mechanisms in vitro and in vivo, or that TGF-β may function as a tumor suppressor by a novel mechanism that only indirectly affects cell proliferation.
Fig. 1. Effects of loss of TGF-β response on tumorigenesis and proliferation in Ca1h cells.

(a) Tumor growth kinetics for Ca1h parental or retrovirally transduced cells growing as subcutaneous xenografts in nude mice. 7.5 × 105 cells were injected/site. Results are the mean +/− S.D. for 4 (Ca1h) or 10 (Ca1h-CON and Ca1h-DNR) tumors/group. (b) Effect of 200pM TGF-β on proliferation of Ca1h cells in vitro determined by 3H-Thymidine incorporation. Results are mean +/− S.D. for 3 determinations. * indicates p<0.05. (c) Effect of loss of TGF-β response on proliferation of Ca1h tumors in vivo, determined by quantitation of BrdU-labelled tumor cells (6 tumors/genotype group; ≥25 high power fields quantitated/tumor). (d) Western blot analysis of expression of cell cycle regulators in protein extracts from Ca1h tumors. Data are shown for two representative tumors of each genotype group.
Loss of TGF-β-dependent tumor suppression is associated with acquisition of a “basal” gene expression profile
To gain insight into such mechanisms, we compared gene expression patterns between Ca1h and Ca1h-DNR tumors, and identified a 26-gene signature that was associated with loss of TGF-β-mediated tumor suppression in this model. The gene list included several genes already known to be directly regulated by TGF-β, including Id1, CTGF, and thrombospondin (Suppl. Table 1). In trying to make biological sense of this gene expression signature, we noticed that loss of TGF-β response changed the relative expression of a number of genes that had recently been shown to be differentially expressed between type I (luminal) and type II (basal) human mammary epithelial cells in culture (20). Overall, the CA1h-DNR tumors had a more basal and less luminal gene expression profile (Suppl. Table 1). This finding was of interest, as a number of studies have shown that human breast cancers with a “basal” gene expression profile have a worse prognosis than those with a “luminal” profile (18;21). Using RTQ-PCR, we confirmed that loss of TGF-β mediated tumor suppression in Ca1h tumors was also associated with a downregulation of the canonical luminal markers cytokeratin 18 (CK18), estrogen receptor-alpha (ESR1) and GATA binding protein 2 (GATA2), and an upregulation of the “basal” markers cytokeratin 5 (CK5), and frizzled7 (FZD7) and p63 (TP73L) (Fig. 2a).
Fig. 2. Loss of TGF-β response is associated with loss of differentiation in the Ca1h model.
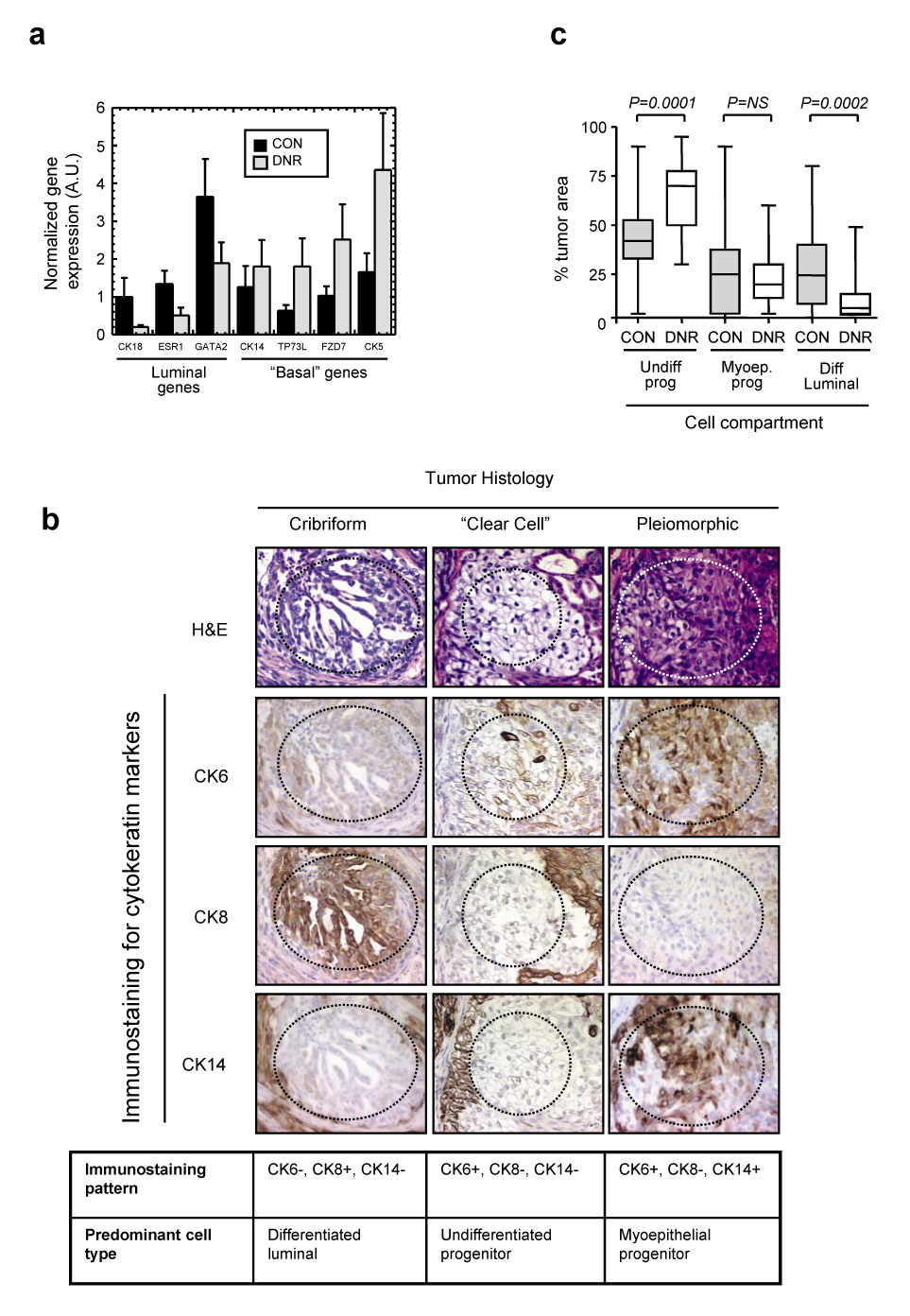
(a) RTQ-PCR analysis of relative expression of basal and luminal marker genes in Ca1h-CON and Ca1h-DNR tumors. Results are the mean +/−SEM for 3 tumors of each genotype and are normalized to expression of the 18S rRNA transcript. CON, tumors from Ca1h cells transduced with empty retrovirus; DNR, tumors from Ca1h cells transduced with the dominant negative TβRII. (b) Representative H&E stained sections of a Ca1h tumor, showing the three predominant histologies that make up the tumor, and immunohistochemical analysis of cyokeratin expression within a given histologic region. In each case, the histology of interest lies within the dotted lines. The table below summarizes cytokeratin marker staining patterns and the deduced identity of the dominant cell type for each histology. (c) Quantitation of the % tumor area occupied by each of the three major cell compartments, identified as above. Prog, progenitor; Diff, differentiated; Undiff, undifferentiated; CON, tumors from Ca1h cells transduced with empty retrovirus; DNR, tumors from Ca1h cells transduced with the dominant negative TβRII. The boxes indicated the median and quartile values, while the whiskers show the 95% confidence interval. Data represent the combined results of 3 independent experiments, for a total of 32 (CON) and 30 (DNR) tumors analyzed.
Loss of TGF-β-dependent tumor suppression induces a less differentiated, intrinsically more proliferative phenotype in the Ca1h tumors
Several markers that are referred to as “basal” on the basis of their expression patterns in stratified epithelia (eg. cytokeratins 5,6), are expressed in the putative progenitor compartment in the breast, rather than the basally-located myoepithelial compartment (22;23). Thus our array data led us to hypothesize that TGF-β might function as a tumor suppressor in part by promoting differentiation of the proliferative progenitor cell population, to form intrinsically less proliferative glandular structures expressing luminal markers.
Ca1h cells form tumors with a mixed histology, having regions of well-organized cribriform structures, “clear cell” areas, and sheets or nests of highly pleiomorphic cells (Suppl. Fig. 2 and Fig 2b). We assessed the cell lineage and differentiation state of the different histologies by immunostaining for cytokeratin markers (Fig. 2b). The cribriform regions were positive for the luminal marker CK8, and were often surrounded by a layer of cells that were positive for the myoepithelial marker CK14, suggesting that these areas represented relatively well-differentiated glandular structures containing predominantly luminal cells. CK8 is found exclusively in luminal cells (22). Both pleiomorphic and clear cell regions, but not the cribriform regions, stained for the progenitor marker CK6. However, the pleiomorphic areas were also positive for CK14. Based on the schema of Boecker and Buerger (24), the clear cells probably represent uncommitted progenitor cells, while the pleiomorphic areas may represent early myoepithelial progenitors.
In agreement with our original hypothesis, we found that loss of TGF-β response in Ca1h tumors was associated with a highly significant decrease in the area of tumor occupied by differentiated luminal structures, and a corresponding increase in uncommitted progenitor structures (Fig. 2c). By double immunostaining for CK8 and the proliferation marker phospho-histone H3, we showed that the differentiated luminal structures in Ca1h tumors had a ~2x lower proliferation rate than the progenitor structures (3.2 +/− 0.5 vs 6.5 +/− 0.6 mitotic cells/hpf; n=8 tumors/group; P = 0.0009; see Suppl. Fig. 1). Loss of TGF-β response did cause a mild increase in proliferation within the progenitor compartment (from 6.4 +/− 1.6 to 8.3 +/− 2.5 labelled cells/hpf, P = 0.05), but not the luminal compartment. Similar results were obtained by counting mitoses in the different compartments (data not shown). Thus TGF-β may suppress tumor growth rate more profoundly by promoting differentiation of the large progenitor cell population to an intrinsically less proliferative state, than through direct inhibition of tumor cell proliferation. Treatment of Ca1h cells with TGF-β in vitro caused an upregulation of the differentiated luminal markers MUC1 and CK8, as determined by FACS analysis (Fig. 3a), suggesting that the tumor cell is the direct target for this differentiating effect of TGF-β.
Fig. 3. TGF-β promotes differentiation of Ca1h cells in vitro, and TGF-β pathway status correlates with differentiation in clinical breast cancer samples.
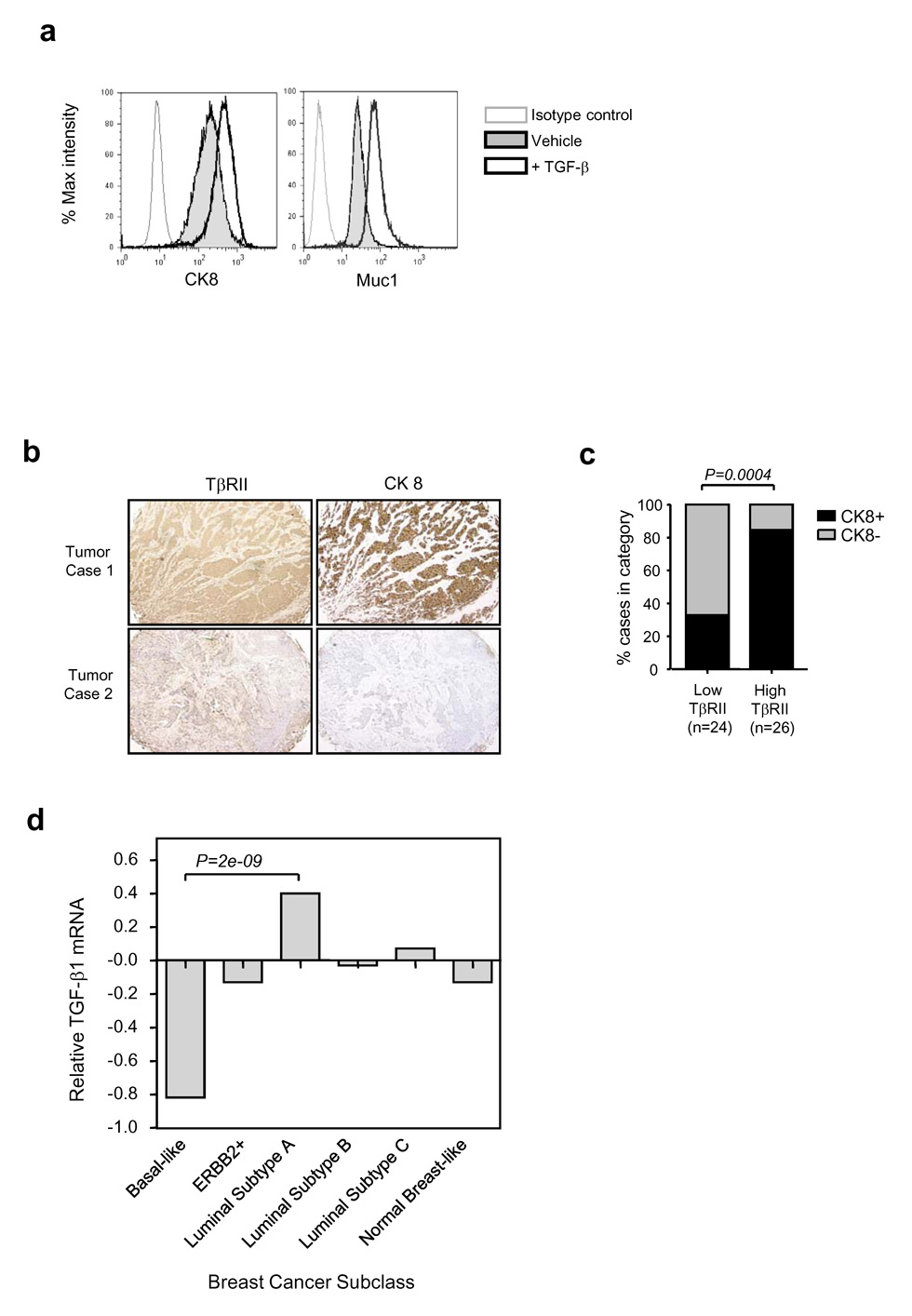
(a) TGF-β effects on expression of luminal markers by Ca1h cells in vitro. Expression of the differentiated luminal markers cytokeratin 8 (CK8) or MUC1 was determined by FACS analysis following treatment with 5ng/ml TGF-β (bold line, no shading) or vehicle control (bold line, gray shading) for 3 days. The thin line indicates staining with the isotype control antibody. (b) Correlation between TβRII and differentiation in clinical breast cancer samples. Adjacent sections of the Imgenex human breast cancer array were immunostained for TβRII or cytokeratin 8 (CK8). Brown indicates positive staining. Two representative tumors are shown. (c) Individual tumors were scored as high or low for TβRII expression, and positive or negative for CK8. The % of cases in each of the two TβRII categories that were CK8 positive (black bars) or CK8 negative (grey bars) is indicated. n gives the number of tumors analyzed. (d) Relative expression of TGF-β1 mRNA in different subclasses of breast cancer. The publicly available cDNA gene expression data (18) were downloaded from the Stanford Microarray Database and normalized. Classification of breast carcinomas was adopted from Sorlie et al. (18). The values represent mean differences among the cancer subtypes relative to median level of TGFB1 expression across all samples. Differential expression in TGFB1 was tested using one-way ANOVA (overall F-test p = 4.73E-07) and specific comparison between the Luminal Subtype A and Basal-like groups (p= 2.24E-09).
If TGF-β does act as a tumor suppressor in part by promoting differentiation, we would predict that a reduction in TGF-β ligand or receptor should be associated with loss of luminal differentiation in human breast cancer samples. To address this question, we immunostained adjacent sections of a human breast cancer tissue array for the type II TGF-β receptor (TβRII) and the luminal marker CK8 (Fig. 3b). For the 50 samples that were evaluable, the results showed that TβRII expression was indeed significantly correlated with CK8 expression (Fig. 3c). Thus >80% of the individual tumors with high TβRII expression, were also positive for CK8. Furthermore, in comparing TGF-β1 mRNA expression across different breast cancer subtypes from a large clinical microarray dataset (18), we found that TGF-β1 was significantly underexpressed in the “basal-like” and overexpressed in the “luminal A” subclasses of tumors (Fig. 3d), again consistent with a role for the TGF-β pathway in inducing or maintaining the luminally differentiated state.
TGF-βreduces the size of the putative cancer stem cell population
Since TGF-β family members are enriched in the stem cell compartments of many tissues (25;26), we further hypothesized that TGF-β might also suppress tumorigenesis through more upstream effects on the cancer stem cells themselves. For many tissues, normal stem cells are highly enriched within a minor subpopulation, the side population or “SP” fraction, that can be visualized by FACS analysis by virtue of their ability to efflux the fluorescent dye Hoechst 33342 (27). As predicted by the cancer stem cell model, freshly isolated human tumors also have an SP fraction (28). More surprisingly, cancer cell lines seem to retain intrinsic stem cell hierarchies in vitro (29), and have SP fractions that are enriched for cancer stem cell activity (28;30). We found that the Ca1h cells in vitro have a small SP fraction (0.03–0.08% of total cells), that was dramatically reduced following treatment with TGF-β (Fig. 4a,b). TGF-β also reduced the SP fraction in the human breast cancer cell line MDA MB231, and in the mouse tumor cell line Wnt1 (Fig.4b). Furthermore, FACS analysis of cells freshly recovered from Ca1h and Ca1h-DNR tumors showed that the Ca1h-DNR tumors have a considerably higher proportion of cells in the SP gate, consistent with the presence of a larger equilibrium stem cell population in vivo (Fig. 4c).
Fig. 4. TGF-β treatment reduces the size of the SP fraction and the ability to form tumorspheres.

(a) FACS analysis of Hoechst dye staining patterns for Ca1h cells in vitro. The SP fraction, which actively pumps out the Hoechst dye, is identified as the poorly staining cell population (indicated by red triangle) that largely disappears when the ATP binding cassette (ABC) transporters are inhibited with verapamil. (b) Effect of TGF-β treatment on the SP fraction for 3 different mammary/breast cancer cell lines. The size of the SP population was determined by FACS analysis following treatment with TGF-β (5ng/ml) for 24 hours. Results for Ca1h cells are the mean +/− S.E.M. for 3 independent experiments. (c) Blocking TGF-β response with the DNR increases the equilibrium SP fraction in Ca1h tumors. Ca1h tumors were digested and FACS analysis was performed on the resulting single cell suspension. Results represent the mean +/− SEM for 3 individual tumors/genotype. CON, control; DNR, dominant negative TβRII. (d) Effect of TGF-β on tumorsphere formation by Ca1h cells. The ability of Ca1h cells cultured in low attachment dishes to form tumorspheres was determined as described in Methods. Where indicated, cells were pre-treated with 5ng/ml TGF-β for 24 hours prior to trypsinization and replating in low attachment dishes in the absence of TGF-β. Results represent the means +/− SEM of three replicate wells for each condition. Inset: phase contrast image of tumorsphere formed by Ca1h cells.
Normal mammary stem cells can undergo anchorage-independent growth in vitro to form “mammospheres” (31), and human breast tumors contain cells with the same property, suggesting that the ability to form such spheres may also be a feature of breast cancer stem cells (17). Pre-treatment of Ca1h cells with TGF-β for 24 hours prior to assay reduced the efficiency of tumorsphere formation by ~2-fold, consistent with TGF-β reducing the size of the stem/progenitor cell population, while expression of the DNR increased the efficiency of tumorsphere formation by ~3-fold (Fig. 4d).
The most definitive test for a cancer stem cell is the ability to initiate and sustain tumorigenesis. If TGF-β does indeed decrease the cancer stem cell population, then the CA1h-DNR cells, which cannot respond to TGF-β, should be able to form tumors at lower initial cell inocula. In agreement with this prediction, we found that CA1h-DNR cells were 10–20x more efficient at forming tumors than are the parental Ca1h cells (Table 1). From our data, we calculate that the Ca1h-DNR cultures contain ~1 tumor initiating cell/10,000 cells. However, this calculation assumes a 100% efficiency of tumor formation by the tumor initiating cells, and hence is likely to underestimate the true representation of these cells within the population.
Table 1. Effect of loss of TGF-β response on tumor take rate of Ca1h cells.
Ca1h cells were transduced with control (CON), dominant negative TβRII (DNR) or Id1 expressing retroviruses and then injected into the flanks of nude mice at varying initial cell inocula. Mice were monitored over 3 months for the appearance of tumors. The relative number of tumor initiating cells in the population was determined by calculating the minimum cell inoculum required to generate a tumor.
| Experimental tumor take rate | ||||||
|---|---|---|---|---|---|---|
| Initial cell inoculum | 5 × 105 | 105 | 104 | 103 | Calculated # of tumor initiating cells/million cells | |
| Expt. 1 | ||||||
| Cell genotype | Ca1h-CON | 5/5 | 5/6 | 0/6 | 0/6 | 8 |
| Ca1h-DNR | 5/5 | 5/6 | 4/6 | 1/6 | 70–170 | |
| Expt. 2 | ||||||
| Cell genotype | Ca1h-CON | ND | 5/5 | 0/8 | 0/7 | <12 |
| Ca1h-DNR | ND | 5/5 | 3/4 | 1/5 | 75–200 | |
| Ca1h-Id1 | ND | 5/5 | 0/4 | 0/5 | <24 | |
TGF-βpromotes differentiation of progenitor cells in part through down-regulation of Id1
Scrutiny of our array data suggested Id1 as a potential mediator of the effects of TGF-β on tumorigenesis. Id1 is a dominant inhibitor of basic helix-loop-helix transcription factors, that has been shown to inhibit lineage commitment and differentiation in many cell types (32), and it is a direct TGF-β target (33). Blocking TGF-β response with the DNR increased the expression of endogenous Id1 in Ca1h cells in vitro (Suppl. Fig. 3). Overexpression of Id1 in the Ca1h cells had no impact on the growth inhibitory effect of TGF-β (Fig. 5a), but did block the ability of TGF-β to induce luminal differentiation in vitro (Fig. 5b). Overexpression of Id1 enhanced tumor growth in vivo (Fig. 5c), with a corresponding reduction in luminal differentiation in the tumors (Fig. 5d), though to a slightly lesser extent than that seen when TGF-β response was blocked with the DNR. Thus overexpression of Id1 selectively uncouples the differentiation-promoting response to TGF-β from the growth inhibitory effect, permitting enhanced tumorigenesis despite the persistence of the anti-proliferative response. Induction of Id1 by BMP has been shown to sustain self-renewal in embryonic stem cells (34). However, forced expression of Id1 had no effect on the efficiency of tumorsphere formation, the size of the SP fraction (Suppl. Fig. 4), or on the efficiency of tumor formation in vivo (Table 1), suggesting that down-regulation of Id1 by TGF-β may only mediate the differentiating effect of TGF-β on the proliferative progenitor cells, and not its effects on reducing the putative tumor stem cell population.
Fig. 5. Id1 blocks promotion of differentiation and partially blocks suppression of tumorigenesis by TGF-β, while not affecting growth inhibition.

(a) Antiproliferative effect of TGF-β on genetically modified Ca1h cells, as assessed by 3H-Thymidine incorporation. All data are normalized to the no TGF-β condition for the particular cell line. Results are the means +/− SD for 3 replicate wells. (b) Ability of TGF-β to promote luminal differentiation in genetically modified Ca1h cells. Expression of the differentiated luminal marker Muc1 in genetically modified Ca1h cells was determined by FACS analysis following treatment with 5ng/ml TGF-β (bold line, no shading) or vehicle control (bold line, gray shading) for 3 days. The thin line indicates staining with the isotype control antibody. (c) Effect of Id1 overexpression on growth of Ca1h tumors in vivo. Results are the mean +/− S.D. for 4 (Ca1h) or 12 (Ca1h-CON, Ca1h-DNR and Ca1h-Id1) tumors/group. (d) Effect of Id1 overexpression on Ca1h tumor differentiation. The % of tumor area occupied by differentiated luminal structures as determined histologically is shown for the three tumor genotypes. The boxes indicate the median and quartile values, while the whiskers show the 95% confidence interval. 12 tumors were analyzed/genotype group.
Discussion
Here we have used a xenograft model of early stage breast cancer to show that TGF-β has the potential to suppress tumorigenesis through effects at two distinct levels in the developmental hierarchy of cell types that make up the tumor parenchyma (see model in Suppl. Fig. 5). Firstly endogenous TGF-β appears to restrict the size of the putative cancer stem cell compartment. This effect decreases the efficiency of tumor establishment, and could underlie the earlier clinical observation that reduction in TβRII expression in early hyperplastic breast lesions is associated with increased probability of subsequent development of invasive breast cancer (35). Secondly, TGF-β acts on the proliferative progenitor cell compartment to promote differentiation to a more organized, intrinsically less proliferative state, characterized by enhanced expression of luminal markers. This effect impacts on the growth rate, bulk and histological appearance of the tumor, and could underlie the previous clinical observation that reduction in TβRII staining in invasive breast cancer, is associated with a higher mitotic index and higher tumor grade (36). Here, we demonstrated a significant association between TβRII protein expression and luminal differentiation in 50 primary breast cancers, and in silico datamining of a large-scale breast cancer microarray study showed TGF-β1 mRNA to be significantly upregulated in the good prognosis “luminal A” subclass and downregulated in the poor prognosis “basal-like” subclass of tumors. While direct anti-proliferative effects of TGF-β are still seen in the breast cancer model that we used here, they appear to be less important than the effects on differentiation, as overexpression of Id1 blocks the differentiating effect of TGF-β and enhances tumorigenesis, while not affecting the ability of TGF-β to inhibit cell proliferation.
TGF-βs are plausible candidates for critical regulators of cancer stem cell dynamics. TGF-β family members are enriched in all populations of normal stem cells that have been isolated so far, and likely contribute to the complex molecular network that specifies “stemness” (25;26). Depending on the specific TGF-β family member and the target cell involved, TGF-β family members may either maintain pluripotency or induce commitment in germinal, embryonal and somatic stem cells (37). In the mammary gland, putative stem cells have been identified by ultrastructural and functional criteria (reviewed in (31)), and recently, mammary glands have been reconstituted from single mammary epithelial cells, providing definitive evidence for the existence of a multipotent mammary stem cell (38;39). Although a role for endogenous TGF-β in normal mammary stem cell dynamics has not yet been demonstrated, transgenic overexpression of active TGF-β in the mouse mammary epithelium resulted in diminished regenerative capacity of the mammary gland in serial transplantation experiments, suggesting that excess TGF-β can induce premature senescence of the mammary stem cell compartment (40). Mice overexpressing TGF-β1 in the mammary gland were relatively resistant to induction of mammary tumors (41;42), as would be expected if TGF-β reduced either the normal or the cancer stem cell compartments. Conversely, mice in which the TGF-β response was compromised showed increased rates of spontaneous or chemically-induced tumorigenesis (43;44). Since experimental inactivation of Bmpr1a in the mouse intestine was recently shown to cause an expansion of the intestinal stem and progenitor cell populations, leading to intestinal polyposis (45), other TGF-β superfamily members may suppress the early stages of tumorigenesis by similar mechanisms in other epithelial tissues.
Stem cells can undergo three different types of division. Self-renewal by symmetric division gives rise to two daughter stem cells, and occurs during allometric growth. Self-renewal by asymmetric division gives one stem cell and one committed daughter cell and is the mechanism used for tissue maintenance. Finally, the third option is symmetric division to give two committed daughter cells, which depletes the stem cell population. Since loss of TGF-β response increases the apparent size of the putative cancer stem/early progenitor cell fraction, our data suggest that local TGF-β signaling might normally reduce the probability of symmetric self-renewal of the stem cell in favor of either asymmetric self-renewal or symmetric division to two committed daughters. Alternatively, although we do not see a pro-apoptotic effect of TGF-β on the bulk cell population, conceivably TGF-β might induce apoptosis specifically of the stem cell population. These various possibilities are under active investigation. The mechanisms underlying the apparently paradoxical downregulation of Myc and upregulation of p27 in the more rapidly proliferating, poorly differentiated Ca1h tumors following TGF-β pathway blockade are also still obscure, though Myc has been shown to deplete epidermal stem cells and promote their terminal differentiation (46).
The ability of TGF-β to induce differentiation of the committed progenitors, but not its ability to deplete the cancer stem/early progenitor cell population, was dependent on the down-regulation of Id1, a known TGF-β target gene (33). Id proteins are highly expressed during embyrogenesis and have been implicated in the regulation of self-renewal and differentiation in many tissues (47). Forced overexpression of Id1 in the SCp2 mammary epithelial cell line blocks functional differentiation in response to lactogenic hormones (48). In breast cancer, Id1 is expressed more frequently in infiltrating ductal carcinomas than ductal carcinoma in situ (15), and Id1 is a negative predictor of survival (49). A number of oncogenes relevant to breast cancer can upregulate Id1, including ErbB2, Myc, and Ras (47). Since oncogene overexpression frequently also upregulates TGF-β expression (50), down-regulation of Id1 by TGF-β may be an important homeostatic mechanism to oppose these oncogenic insults early in the carcinogenic process. Once the TGF-β pathway is compromised however, the oncogene-induced increase in Id1 could block differentiation and cause expansion of the progenitor compartment, leading to a rapidly growing tumor with an aggressive histology. Overexpression of Id1 results in tumors that are 30–50% smaller than those seen with TGF-β pathway blockade (Fig. 5c), suggesting that the differentiating effect of TGF-β could contribute at least half of its effective tumor suppressor activity in the Ca1h model.
In summary, we have used a breast cancer model system to show that loss of TGF-β response can increase the size of the putative cancer stem cell/early progenitor compartment and block further differentiation of the lineage-restricted progeny, thus promoting tumorigenesis by a mechanism that is independent of direct effects on proliferation. The data suggest that strategies to restore or enhance TGF-β response in early carcinogenesis might constitute a novel form of differentiation therapy for prevention or treatment of epithelial tumors.
Supplementary Material
Acknowledgements
We thank Barbara Taylor (National Cancer Institute FACS Core Facility) for expert assistance with the FACS analysis, and Glenn Merlino, Fred Miller, Gil Smith, Jeffrey Rosen and Yi Li for many helpful discussions.
Footnotes
Financial support: This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (Lalage Wakefield), and with Federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. N01-C0-12400 (Miriam Anver).
References
- 1.Derynck R, Akhurst RJ, Balmain A. TGF-β signaling in tumor suppression and cancer progression. Nat Genet. 2001;29:117–129. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- 2.Engle SJ, Hoying JB, Boivin GP, Ormsby I, Gartside PS, Doetschman T. Transforming growth factor β1 suppresses nonmetastatic colon cancer at an early stage of tumorigenesis. Cancer Res. 1999;59:3379–3386. [PubMed] [Google Scholar]
- 3.Tobin SW, Douville K, Benbow U, Brinckerhoff CE, Memoli VA, Arrick BA. Consequences of altered TGF-β expression and responsiveness in breast cancer: evidence for autocrine and paracrine effects. Oncogene. 2002;21:108–118. doi: 10.1038/sj.onc.1205026. [DOI] [PubMed] [Google Scholar]
- 4.Neve RM, Chin K, Fridlyand J, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dawson PJ, Wolman SR, Tait L, Heppner GH, Miller FR. MCF10AT: a model for the evolution of cancer from proliferative breast disease. Am J Pathol. 1996;148:313–319. [PMC free article] [PubMed] [Google Scholar]
- 6.Santner SJ, Dawson PJ, Tait L, et al. Malignant MCF10CA1 cell lines derived from premalignant human breast epithelial MCF10AT cells. Breast Cancer Res Treat. 2001;65:101–110. doi: 10.1023/a:1006461422273. [DOI] [PubMed] [Google Scholar]
- 7.Iavarone A, Massague J. Repression of the CDK activator Cdc25A and cell-cycle arrest by cytokine TGF- β in cells lacking the CDK inhibitor p15. Nature. 1997;387:417–422. doi: 10.1038/387417a0. [DOI] [PubMed] [Google Scholar]
- 8.Strickland LB, Dawson PJ, Santner SJ, Miller FR. Progression of premalignant MCF10AT generates heterogeneous malignant variants with characteristic histologic types and immunohistochemical markers. Breast Cancer Res Treat. 2000;64:235–240. doi: 10.1023/a:1026562720218. [DOI] [PubMed] [Google Scholar]
- 9.Clark GJ, Der CJ. Aberrant function of the Ras signal transduction pathway in human breast cancer. Breast Cancer Res Treat. 1995;35:133–144. doi: 10.1007/BF00694753. [DOI] [PubMed] [Google Scholar]
- 10.Crawford YG, Gauthier ML, Joubel A, et al. Histologically normal human mammary epithelia with silenced p16(INK4a) overexpress COX-2, promoting a premalignant program. Cancer Cell. 2004;5:263–273. doi: 10.1016/s1535-6108(04)00023-6. [DOI] [PubMed] [Google Scholar]
- 11.Dalerba P, Cho RW, Clarke MF. Cancer stem cells: models and concepts. Annu Rev Med. 2007;58:267–284. doi: 10.1146/annurev.med.58.062105.204854. [DOI] [PubMed] [Google Scholar]
- 12.Wicha MS, Liu S, Dontu G. Cancer stem cells: an old idea--a paradigm shift. Cancer Res. 2006;66:1883–1890. doi: 10.1158/0008-5472.CAN-05-3153. [DOI] [PubMed] [Google Scholar]
- 13.Zhang M, Rosen JM. Stem cells in the etiology and treatment of cancer. Curr Opin Genet Dev. 2006;16:60–64. doi: 10.1016/j.gde.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 14.Tang B, Vu M, Booker T, et al. TGF- β switches from tumor suppressor to prometastatic factor in a model of breast cancer progression. J Clin Invest. 2003;112:1116–1124. doi: 10.1172/JCI18899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin CQ, Singh J, Murata K, et al. A role for Id-1 in the aggressive phenotype and steroid hormone response of human breast cancer cells. Cancer Res. 2000;60:1332–1340. [PubMed] [Google Scholar]
- 16.Smalley MJ, Clarke RB. The mammary gland "side population": a putative stem/progenitor cell marker? J Mammary Gland Biol Neoplasia. 2005;10:37–47. doi: 10.1007/s10911-005-2539-0. [DOI] [PubMed] [Google Scholar]
- 17.Ponti D, Costa A, Zaffaroni N, et al. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005;65:5506–5511. doi: 10.1158/0008-5472.CAN-05-0626. [DOI] [PubMed] [Google Scholar]
- 18.Sorlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Massague J. G1 cell-cycle control and cancer. Nature. 2004;432:298–306. doi: 10.1038/nature03094. [DOI] [PubMed] [Google Scholar]
- 20.Park JS, Noh DY, Kim SH, et al. Gene expression analysis in SV40-immortalized human breast luminal epithelial cells with stem cell characteristics using a cDNA microarray. Int J Oncol. 2004;24:1545–1558. [PubMed] [Google Scholar]
- 21.Sorlie T, Tibshirani R, Parker J, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci U S A. 2003;100:8418–8423. doi: 10.1073/pnas.0932692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gusterson BA, Ross DT, Heath VJ, Stein T. Basal cytokeratins and their relationship to the cellular origin and functional classification of breast cancer. Breast Cancer Res. 2005;7:143–148. doi: 10.1186/bcr1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith GH, Mehrel T, Roop DR. Differential keratin gene expression in developing, differentiating, preneoplastic, and neoplastic mouse mammary epithelium. Cell Growth Differ. 1990;1:161–170. [PubMed] [Google Scholar]
- 24.Boecker W, Buerger H. Evidence of progenitor cells of glandular and myoepithelial cell lineages in the human adult female breast epithelium: a new progenitor (adult stem) cell concept. Cell Prolif. 2003;36 Suppl 1:73–84. doi: 10.1046/j.1365-2184.36.s.1.7.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tumbar T, Guasch G, Greco V, et al. Defining the epithelial stem cell niche in skin. Science. 2004;303:359–363. doi: 10.1126/science.1092436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramalho-Santos M, Yoon S, Matsuzaki Y, Mulligan RC, Melton DA. "Stemness": transcriptional profiling of embryonic and adult stem cells. Science. 2002;298:597–600. doi: 10.1126/science.1072530. [DOI] [PubMed] [Google Scholar]
- 27.Zhou S, Schuetz JD, Bunting KD, et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med. 2001;7:1028–1034. doi: 10.1038/nm0901-1028. [DOI] [PubMed] [Google Scholar]
- 28.Hirschmann-Jax C, Foster AE, Wulf GG, et al. A distinct "side population" of cells with high drug efflux capacity in human tumor cells. Proc Natl Acad Sci U S A. 2004;101:14228–14233. doi: 10.1073/pnas.0400067101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Locke M, Heywood M, Fawell S, Mackenzie IC. Retention of intrinsic stem cell hierarchies in carcinoma-derived cell lines. Cancer Res. 2005;65:8944–8950. doi: 10.1158/0008-5472.CAN-05-0931. [DOI] [PubMed] [Google Scholar]
- 30.Patrawala L, Calhoun T, Schneider-Broussard R, Zhou J, Claypool K, Tang DG. Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2+ and ABCG− cells are similarly tumorigenic. Cancer Res. 2005;65:6207–6219. doi: 10.1158/0008-5472.CAN-05-0592. [DOI] [PubMed] [Google Scholar]
- 31.Dontu G, Abdallah WM, Foley JM, et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003;17:1253–1270. doi: 10.1101/gad.1061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Norton JD. ID helix-loop-helix proteins in cell growth, differentiation and tumorigenesis. J Cell Sci. 2000;113:3897–3905. doi: 10.1242/jcs.113.22.3897. [DOI] [PubMed] [Google Scholar]
- 33.Kang Y, Chen CR, Massague J. A self-enabling TGF β response coupled to stress signaling: Smad engages stress response factor ATF3 for Id1 repression in epithelial cells. Mol Cell. 2003;11:915–926. doi: 10.1016/s1097-2765(03)00109-6. [DOI] [PubMed] [Google Scholar]
- 34.Ying QL, Nichols J, Chambers I, Smith A. BMP induction of Id proteins suppresses differentiation and sustains embryonic stem cell self-renewal in collaboration with STAT3. Cell. 2003;115:281–292. doi: 10.1016/s0092-8674(03)00847-x. [DOI] [PubMed] [Google Scholar]
- 35.Gobbi H, Dupont WD, Simpson JF, et al. Transforming growth factor- β and breast cancer risk in women with mammary epithelial hyperplasia. J Natl Cancer Inst. 1999;91:2096–2101. doi: 10.1093/jnci/91.24.2096. [DOI] [PubMed] [Google Scholar]
- 36.Gobbi H, Arteaga CL, Jensen RA, et al. Loss of expression of transforming growth factor β type II receptor correlates with high tumour grade in human breast in-situ and invasive carcinomas. Histopathology. 2000;36:168–177. doi: 10.1046/j.1365-2559.2000.00841.x. [DOI] [PubMed] [Google Scholar]
- 37.Mishra L, Derynck R, Mishra B. Transforming growth factor- β signaling in stem cells and cancer. Science. 2005;310:68–71. doi: 10.1126/science.1118389. [DOI] [PubMed] [Google Scholar]
- 38.Shackleton M, Vaillant F, Simpson KJ, et al. Generation of a functional mammary gland from a single stem cell. Nature. 2006;439:84–88. doi: 10.1038/nature04372. [DOI] [PubMed] [Google Scholar]
- 39.Stingl J, Eirew P, Ricketson I, et al. Purification and unique properties of mammary epithelial stem cells. Nature. 2006;439:993–997. doi: 10.1038/nature04496. [DOI] [PubMed] [Google Scholar]
- 40.Kordon EC, McKnight RA, Jhappan C, Hennighausen L, Merlino G, Smith GH. Ectopic TGF β 1 expression in the secretory mammary epithelium induces early senescence of the epithelial stem cell population. Dev Biol. 1995;168:47–61. doi: 10.1006/dbio.1995.1060. [DOI] [PubMed] [Google Scholar]
- 41.Boulanger CA, Smith GH. Reducing mammary cancer risk through premature stem cell senescence. Oncogene. 2001;20:2264–2272. doi: 10.1038/sj.onc.1204312. [DOI] [PubMed] [Google Scholar]
- 42.Pierce DF, Jr, Gorska AE, Chytil A, et al. Mammary tumor suppression by transforming growth factor β1 transgene expression. Proc Natl Acad Sci U S A. 1995;92:4254–4258. doi: 10.1073/pnas.92.10.4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gorska AE, Jensen RA, Shyr Y, Aakre ME, Bhowmick NA, Moses HL. Transgenic mice expressing a dominant-negative mutant type II transforming growth factor-β receptor exhibit impaired mammary development and enhanced mammary tumor formation. Am J Pathol. 2003;163:1539–1549. doi: 10.1016/s0002-9440(10)63510-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bottinger EP, Jakubczak JL, Haines DC, Bagnall K, Wakefield LM. Transgenic mice overexpressing a dominant-negative mutant type II transforming growth factor β receptor show enhanced tumorigenesis in the mammary gland and lung in response to the carcinogen 7,12-dimethylbenz-[a]-anthracene. Cancer Res. 1997;57:5564–5570. [PubMed] [Google Scholar]
- 45.He XC, Zhang J, Tong WG, et al. BMP signaling inhibits intestinal stem cell self-renewal through suppression of Wnt-β-catenin signaling. Nat Genet. 2004;36:1117–1121. doi: 10.1038/ng1430. [DOI] [PubMed] [Google Scholar]
- 46.Arnold I, Watt FM. c-Myc activation in transgenic mouse epidermis results in mobilization of stem cells and differentiation of their progeny. Curr Biol. 2001;11:558–568. doi: 10.1016/s0960-9822(01)00154-3. [DOI] [PubMed] [Google Scholar]
- 47.Perk J, Iavarone A, Benezra R. Id family of helix-loop-helix proteins in cancer. Nat Rev Cancer. 2005;5:603–614. doi: 10.1038/nrc1673. [DOI] [PubMed] [Google Scholar]
- 48.Desprez PY, Hara E, Bissell MJ, Campisi J. Suppression of mammary epithelial cell differentiation by the helix-loop-helix protein Id-1. Mol Cell Biol. 1995;15:3398–3404. doi: 10.1128/mcb.15.6.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schoppmann SF, Schindl M, Bayer G, et al. Overexpression of Id-1 is associated with poor clinical outcome in node negative breast cancer. Int J Cancer. 2003;104:677–682. doi: 10.1002/ijc.11009. [DOI] [PubMed] [Google Scholar]
- 50.Anzano MA, Roberts AB, De Larco JE, et al. Increased secretion of type β transforming growth factor accompanies viral transformation of cells. Mol Cell Biol. 1985;5:242–247. doi: 10.1128/mcb.5.1.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.