

Abstract
NHEJ (non-homologous end joining) is the predominant mechanism for repairing DNA double-stranded breaks in human cells. One essential NHEJ factor is the Ku heterodimer, which is composed of Ku70 and Ku86. Here we have generated heterozygous loss-of-function mutations for each of these genes in two different human somatic cell lines, HCT116 and NALM-6 using gene targeting. Previous work had suggested that phenotypic differences might exist between the genes and/or between the cell lines. By providing a side-by-each comparison of the four cell lines, we demonstrate that there are indeed subtle differences between loss-of-function mutations for Ku70 versus Ku86, which is accentuated by whether the mutations were derived in the HCT116 or NALM-6 genetic background. Overall, however, the phenotypes of the four lines are quite similar and they provide a compelling argument for the hypothesis that Ku loss-of-function mutations in human somatic cells result in demonstrable haploinsufficiencies. Collectively, these studies demonstrate the importance of proper biallelic expression of these genes for NHEJ and telomere maintenance and they provide insights into why these genes are uniquely essential for primates.
1. Introduction
The integrity of chromosomes must be maintained in order to ensure survival. This requirement is difficult for cells to sustain since chromosomal DNA is damaged continuously by both exogenous and endogenous agents. Among the many forms of DNA damage that can occur, DNA DSBs (double-stranded breaks) are the most dangerous (reviewed by [1, 2]). DSBs can occur in response to external stimuli like IR (ionizing radiation) and also by exposure to clinical chemotherapeutic agents like bleomycin and etoposide. Moreover, DSBs also arise as a result of natural processes, such as V(D)J and switch recombination, lymphoid-specific processes needed for the maturation of T and B cells (reviewed by [3, 4]). Consequently, to ensure their survival, mammals have evolved intricate and efficient mechanisms for the repair of DSBs.
In eukaryotic cells, two major processes are responsible for repair of DSBs, namely HR (homologous recombination; reviewed by [5, 6]) and NHEJ (reviewed by [2]). HR carries out accurate repair by utilizing a homologous chromosome or an undamaged sister chromatid as a template. In contrast, NHEJ uses no, or very little, sequence homology for repair events that can occur in an error-prone manner. Both pathways are conserved throughout eukaryotic evolution but their relative importance varies between organisms. Simpler organisms like S. cerevisiae rely mainly on HR to repair damaged DNA while in higher eukaryotes, and particularly in humans, NHEJ is the predominant repair mechanism. This bias is not well understood although it is possible that in the complex human genome where a very small percentage of the DNA actually codes for any protein, the errors made by NHEJ are better tolerated. There are at least seven proteins that play important roles in NHEJ: Ku70, Ku86, DNA-PKCS (DNA-dependent protein kinase complex catalytic subunit), XRCC4 (X-ray cross complementing 4), LIGIV (DNA ligase IV), Artemis and Cernunnos/XLF (XRCC4-like factor) (reviewed by [2, 7]). In humans, mutations have been described for LIGIV [8, 9], Artemis [10] and XLF [11] that cause IRS (IR sensitivity), immune deficiency and/or cancer predisposition. The deleterious phenotypes associated with these mutations substantiate the importance of NHEJ in humans.
Mutations for the other NHEJ factors (Ku70, Ku86, DNA-PKcs and XRCC-4), however, have yet to be described in humans. In particular, even heterozygous mutations for either Ku70 or Ku86 have yet to be documented. Ku is a heterodimeric protein composed of 70 and 86 kDa subunits (Ku70 and Ku86, respectively) and this protein binds to all forms of ds (double-stranded) DNA ends in a sequence non-specific manner [7]. The ability to bind virtually all broken dsDNA ends can be explained by the crystal structure of Ku. Ku forms an open, ring-type structure that can be threaded onto a dsDNA end [12]. One side of the ring cradles one face of the DNA while the other side is more open, presumably to allow other NHEJ factors to access the broken DNA end [13, 14]. In vertebrates, Ku recruits DNA-PKCS to the sites of DNA damage during DNA DSB repair. The interaction of DNA-PKCS with the DNA-bound Ku heterodimer leads to the formation of the DNA-PK holoenzyme and this complex exhibits a DNA-dependent protein kinase activity that is essential for NHEJ (reviewed by [4]). Cells that lack Ku are IRS (IR sensitive), immune deficient and defective for DNA DSB repair (reviewed in [7]). Mice containing targeted disruption in either the Ku70 [15] or Ku86 gene [16, 17] show increased sensitivity to IR and a failure to carry out V(D)J recombination. Moreover, the inactivation of the Ku86 gene in the mouse is known to cause growth retardation in cells [18], induce a marked increase in chromosomal aberrations [19-21] and to also cause premature senescence [22]. In summary, mouse models of Ku loss-of-function mutations unequivocally validate the importance of Ku for NHEJ, but a direct demonstration of this is still lacking in human patients.
A description of mutations is also lacking for DNA-PKCS. DNA-PKCS is an ∼460 kDa polypeptide and its C-terminus contains sequence homology to the catalytic domains of the proteins of the PIKK (phosphatidyl inositol 3-kinase-like kinase) family (reviewed by [4]). DNA-PKCS is the product of the scid (severe combined immune deficiency; prkdc/XRCC7) gene and the loss of this gene results in defects in DNA DSB repair, immune deficiency and IRS in mice, Arabian horses and Jack Russell terriers. The only human cell line known to lack DNA-PKCS is M059J [23]. This cell line, which was isolated from a malignant glioma, lacks DNA-PKCS activity due to defective mRNA turnover associated with a frameshift mutation in the prkdc gene [24]. Importantly, this mutation appears to have been generated during propagation of the glioma during cell culture and was absent in the patient and from the tumor from which it was derived [23], emphasizing again the complete lack of human patients with DNA-PK mutations.
Besides NHEJ, an additional important role for the DNA-PK complex is in the protection of telomeres, the end structures of chromosomes (reviewed by [25]). Interestingly, all three components of the DNA-PK complex — Ku70, Ku86 and DNA-PKCS — play some role in the protection of telomeres [21, 26-29]. Moreover, MEFs (mouse embryo fibroblasts) from DNA-PKCS-/- mice display a significant increase in chromosome fusions, even though the actual length of telomeres is not altered [29]. Ku70-/- and Ku86-/- MEFs also show elevated chromosome end fusions [26-28]. These observations suggest that Ku is directly involved in a telomere capping function and indeed Ku has been physically located at telomeres by biochemical studies [21, 30]. The role of Ku in actual telomere length maintenance is, however, less clear. There are conflicting reports of Ku-defective mice showing telomere shortening [21] and also telomere elongation [28]. Also, deletion of both alleles of Ku in Arabidopsis thaliana causes telomere elongation with no apparent telomere fusions [31]. The effect of Ku or DNA-PKCS mutations on telomere function in human patients is not known.
In order to better understand the roles Ku and DNA-PK play in NHEJ and telomere maintenance in humans, our laboratory used gene targeting to functionally inactivate the Ku86 locus in the human adenocarcinoma somatic tissue culture cell line, HCT116 [32]. The null cell lines were not viable, a finding that was unexpected given the existence of many other Ku86-null organisms. Moreover, the derivative human cell lines heterozygous for Ku86 showed significant haploinsufficient phenotypes, with defects in cell proliferation, IRs, elevated levels of p53, polyploidy, shortened telomeres and elevated levels of GCRs (gross chromosomal rearrangements) [33]. All of these phenotypes suggested that, if anything, Ku86 played an even more critical role in NHEJ and telomere maintenance in humans than in other mammals. Importantly, most of these phenotypes have been independently confirmed by laboratories utilizing Ku70 antisense DNA [34], Ku86 antisense DNA [35], Ku86 cRNA [36], or Ku86 RNAi [37] approaches to reduce Ku expression in a variety of human somatic cell lines. Recently, however, Uegaki et al. have reported that heterozygous inactivation of Ku70 and Ku86 resulted in human cell lines that did not show haploinsufficient phenotypes [38]. These studies were carried out in the pre-B leukemic cell line, NALM-6 [39, 40]. The reasons for the discrepancies between these two studies are not known. A likely possibility was that the discrepancies might be due to the different cell lines utilized, especially since NALM-6 has been reported to be greatly up-regulated for HR ([41, 42]; reviewed by [43]), and thus, presumably, less sensitive to mutations in NHEJ genes. To address this issue, we have generated Ku70 and Ku86 heterozygous mutations in both HCT116 and NALM-6 cell lines and have compared the resulting lines side-by-each. These studies have revealed that while there are some important differences, especially related to radiation sensitivity, the overall effects of these mutations are quite similar for all cell lines.
2. Materials and methods
2.1. Cell culture
HCT116 cells were cultured in McCoy’s 5A media containing 10% fetal calf serum, 100 U/ml penicillin and 50 U/ml streptomycin. The media was also supplemented with L-glutamine. NALM-6 cells were grown in RPMI media containing 10% fetal calf serum, 100 U/ml penicillin and 50 U/ml streptomycin. The cells were incubated at 37°C in a humidified incubator with 5% CO2. All cell lines derived from correct targeting events were grown in the presence of 1 mg/ml G418.
2.2. Targeting vector construction
The targeting vectors were constructed utilizing the system described by Kohli et al. [44]. Briefly, the right and left homology arms of the Ku70 targeting were constructed by PCR from HCT116 genomic DNA. The primers used to construct the left homology arm for Ku70 were BR5, 5′-ATACATACGCGGCCGCCTATACTTTAAGTCATCTATAGGTTACTC-3′ and BR6, 5′-GCTCCAGCTTTTGTTCCCTTTAGCTTACCACTAAATGGAAGCTCC-3′. The right homology arm was constructed using the primers BR7, 5′-CGCCCTATAGTGAGTCGTATTACTGTGTACCTGACTTCAGGCATGTG-3′ and BR8, 5′-ATACATACGCGGCCGCGCCCAGCCTGCTTCTGATTTAAGGGCAG-3′. The arms were used in a fusion PCR reaction, together with a 4 kb PvuI restriction enzyme fragment containing the drug selection marker. The fusion PCR product was gel purified and ligated to the pAAV backbone using NotI restriction enzyme sites to construct the final targeting vector. For the Ku86 targeting vector, the primers used to construct the left homology arm were BR9, 5′-ATACATACGCGGCCGCAGGGAGACAAGGACCACTGACAAG-3′ and BR10, 5′-GCTCCAGCTTTTGTTCCCTTTAGCTTGGAAGGGAGGAGTCAAGGT-3′. For the right homology arms, the primers used were BR11, 5′-CGCCCTATAGTGAGTCGTATTACCCGGACTGGGGATCCGGAGAGGTG-3′ and BR12, 5′-ATACATACGCGGCCGCCAATGAGGAGTTGAGGGAACTAGGGATC-3′. Fusion PCR and the following ligation were performed as described above for the Ku70 vector.
2.3. Packaging and isolating virus
The targeting vector (8 μg), together with pAAV-RC and pHelper plasmids (8 μg of each) from the AAV Helper-Free System (Stratagene) was transfected into AAV 293 cells (Invitrogen) using Lipofectamine 2000 (Invitrogen). Virus was isolated from the AAV 293 cells 48 hr after transfection using a freeze-thaw method [44]. Identical methods were used for the Ku70 and Ku86 vectors.
2.4. Infections
HCT116 and NALM-6 cells were grown to ∼70-80% confluence in 6-well tissue culture plates. Fresh media (1.5 ml) was added to the cells 3 hr prior to addition of the virus. The required volume of the virus was added drop-wise to the plates. After a 2 hr incubation at 37°C, another 1.5 ml of media was added to the plates. After a further 48 hr incubation, the cells were transferred to 96-well plates at the appropriate density and placed under selection (1 mg/ml G418) to obtain single colonies.
2.5. Isolation of genomic DNA and Southern hybridizations
Chromosomal DNA was prepared, digested, subjected to electrophoresis and then transferred to a nitrocellulose membrane as described [45]. The membrane was hybridized with probe c (Fig. 1C) to detect correct targeting of the Ku70 targeting vector. The probe corresponds to ∼400 bp of the selection cassette and was made by PCR with the primers KU70PR7, 5′-GGCGCGCATGCCCGACGGCG-3′ and KU70PR8, 5′-GTGGGAGTGGCACCTTCCAAG-3′. The PCR product was electrophoresed on a 1% agarose gel and gel purified prior to use. The Prime-It ®II kit (Stratagene) was used to radiolabel the Southern probe with {32P}-α-dATP. Three different probes, a’, b’ and c’ (Fig. 1D), were used to confirm the targeting events generated with the Ku86 targeting vector. Probe a’ lies 5′ to the targeted locus and is a ∼400 bp PCR product made using primers 86-1F1, 5′-CACGCATCACCACCCTGACATTTTG-3′ and 86-1R1, 5′-CCTCTGTGCAGCTAGGCATCTG-3′. Probe b’, also ∼400 bp in length, lies 3′ to the targeted locus and is the PCR product of the primers 86-1F2, 5′-CATGACTGAGATTTTGTCTAGGGCAAG-3′ and 86-1R2, 5′-GGATTTACAGCATAAGACAATGGG-3′. Probe c’ is an internal probe 416bp in length and was obtained by digesting the selection cassette with the restriction enzymes AflIII and ApaLI.
Fig. 1.

Scheme for functional inactivation of the human Ku70 and Ku86 loci. (A) Partial Ku70 and Ku86 genomic loci. Open rectangles with numbers represent exons. (B) The cartoon of the targeting vector. In the targeting vector open boxes designate the left and right inverted terminal repeats; triangles, loxP sites; PGK, phosphoglycerate kinase eukaryotic promoter; Neo, neomycin resistance gene; EM7, EM7 prokaryotic promoter; Zeo, zeomycin resistance gene; dotted rectangles, left and right homology arms to facilitate targeting by homologous recombination. (C) In the targeted allele of Ku70, exons 3 & 4 have been replaced by the targeting vector. a and b are external probes while c is an internal probe. HindIII (Hi) restriction enzyme sites and an unique FspI (Fs) restriction site in the targeting construct are shown. Also shown are the primers used for the diagnostic PCR screening. (D) In the targeted allele of Ku86, exon 1 has been replaced by the targeting vector. a’ and b’ are 5′ and 3′ external probes respectively while c’ is an internal probe. Ap is a ApaLI restriction site in the targeting construct. Also shown are the primers used for the diagnostic PCR screening.
2.6. Isolation of genomic DNA and genomic PCR
Genomic DNA for PCR screening was isolated using the PUREGENE® DNA Purification Kit (Gentra Systems). DNA was dissolved in a final volume of 30 μl, 2 μl of which was used in each PCR reaction. For Ku70 targeting events, PCR was carried out at both the 5′- and 3′-sides of the targeted locus. For the 5′-end, a control PCR was performed using the primer set LArmR, 5′-GCTCCAGCTTTTGTTCCCTTTAG-3′ and BR5.1, 5′-ATACATACGCGGCCGCCTATACTTTAAGTCATCTATAGGTTACTC-3′ (Fig. 1C). Correct targeting was determined using LArmR and KU703,4F1, 5′-CAGTGACTAGCTATAGTACATGTTCTGC-3′ (Fig. 1C). For the 3′-end, the control PCR was carried out using the primer set RArmF, 5′-CGCCCTATAGTGAGTCGTATTAC-3′ and BR8, 5-ATACATACGCGGCCGCGCCCAGCCTGCTTCTGATTTAAGGGCAG-3′ (Fig. 1C). PCR to screen for correctly targeted clones was performed using RArmF and KU703,4R1, 5′-GTCTACCCCCACGAAATGGAGGGCTGTG-3′ (Fig. 1C). For the Ku86 targeted clones, 3′-end control PCR was performed using RArmF and BR12, 5′-ATACATACGCGGCCGCCAATGAGGAGTTGAGGGAACTAGGGATC-3′. Correct targeting was determined at the 3′-end using RArmF and KU861R, 5′-CATGAATTAGGTATCCAGTAAGTGCCATC-3′. At the 5′-end, control PCR was performed using LArmR and BR9, 5′-ATACATACGCGGCCGCAGGGAGACAAGGACCACTGACAAG-3′. Correct targeting was confirmed at the 5′-end using the primer set LArmR and KU861F, 5′-TATTCCTCTGAGGCCAAGTGCAGTTG-3′.
2.7. Whole cell extract preparation
Cells were trypsinized and washed twice with PBS. For whole cell extraction, cells were boiled in DNase buffer (Gibco) for 10 min. The samples were then digested with DNaseI (0.1 U/μl; Gibco) for 10 min at 37°C. The samples were finally boiled in 5X SDS buffer (0.225 M Tris-HCl, pH 6.8, 50% {v/v} glycerol, 5% SDS, 0.05% bromophenol blue, 0.14 M β-mercaptoethanol).
2.8. Nuclear extract preparation
Nuclear extracts were prepared according to the protocol supplied with the CelLytic™ NuCLEAR™ Extraction kit (Sigma). Briefly, cells were collected and washed with cold PBS. The cells were then resuspended in lysis buffer (50 mM TrisHCl, pH 7.5, 10 mM MgCl2, 15 mM CaCl2, 1.5 M sucrose) and kept on ice for 15 min, followed by mixing by inversion. Centrifugation was carried out to obtain a pellet that was then resuspended in extraction buffer (20 mM HEPES, pH 7.9, 1.5 mM MgCl2, 0.42 M NaCl, 0.2 M EDTA, 25% {v/v} glycerol). The suspension was agitated for 30 min at 4°C and centrifuged. The supernatant from the final centrifugation step was collected as the nuclear extract.
2.9. Immunoblotting
For immunoblot detection, proteins were subjected to electrophoresis on a 4-20% gradient gel (Bio-Rad), electroblotted onto a nitrocellulose membrane and detected as described [46].
2.10. DNA end-binding assay
Ku DNA end-binding activity was measured by a gel mobility shift assay as described [46] with some modifications. Briefly, a 43 bp dsDNA obtained by annealing two single-stranded oligonucleotides was radiolabeled with {32P}-γ-dATP using the Klenow fragment polymerase. Radiolabeled DNA (∼4 ng) was incubated with 1 μg of nuclear protein in 15 μl of binding buffer (10 mM TrisHCl, pH 8.0, 1 mM EDTA, 10% glycerol, 200 mM NaCl), with 1 μg of circular TA plasmid (Invitrogen) as competitor on ice for 5 min. Electrophoresis was then carried out using a 5% native polyacrylamide gel for 3 hr at 130V. The gel was dried and exposed to X-ray film for ∼15-30 min at -80°C.
2.11. DNA-PK kinase assay
The DNA-PK kinase assays were performed as follows. Nuclear extracts were incubated on ice for 15 min with pre-swollen dsDNA-cellulose (Sigma). 100 μg of nuclear extract was used with each sample. Following incubation on ice, the samples were washed twice in Z’ 0.05 buffer (25 mM HEPES/KOH, 50 mM KCl, 10 mM MgCl2, 20% glycerol, 0.1% IGEPAL™, 1 mM DTT). After the washing steps, the samples were centrifuged and the precipitate was resuspended in 100 μl Z’ 0.05 buffer. The sample was then incubated at 30°C for 15 min with either a functional DNA-PK substrate peptide (EPPLSQEAFADLLKK) or a mutant peptide (EPPLSEQAFADLLKK) and {32P}-γ-ATP. The functional peptide can be phosphorylated by DNA-PK at the underlined serine residue while the mutant peptide is not recognized by DNA-PK. Following incubation, polyacrylamide gel electrophoresis was carried out. The gel was vacuum dried and exposed to X-ray film. The amount of phosphorylated peptide was quantitated by using a phosphoimager.
2.12. Cell proliferation assay
To obtain a growth curve, 3 × 104 or 1 × 105 HCT116 or NALM-6 cells, respectively, were plated out in each well of 6-well plates or 25 cm2 flasks, respectively, in triplicate. Cell numbers were determined using a hemacytometer everyday thereafter starting at day 4 utilizing growth media without selection.
2.13. X-ray survival assay
For HCT116 cell lines, 300 cells were seeded into each well of a 6-well tissue culture plate about 10-12 hr before irradiation. For NALM-6 clones, three 96-well plates were used for each clone at each dosage (0, 2.5 and 5.0 Gy). For the 0 Gy control, each well of the 96-well plate contained either 2.5, 5 or 10 cells. For the irradiated samples, each well contained 10, 100 or 1000 cells. After irradiation, HCT116 cells were allowed to grow for 10-14 days before the colonies were fixed, stained, counted and a cell survival percentage was calculated [45]. NALM-6 cells were allowed to grow for ∼3 weeks before the colonies were counted.
2.14. Telomeric TRF analysis
TRF (terminal restriction fragment) analysis was performed as described [33, 47], utilizing the restriction enzymes AluI and MboI (New England Biolabs). The probes were constructed by PCR using telomere-specific oligonucleotides (Operon).
2.15. Cytogenetic analysis
G-banding cytogenetic analyses were performed in the Cytogenetics Core Laboratory at the University of Minnesota.
3. Results
3.1. Generation of heterozygous Ku70+/- HCT116 and NALM-6 cells lines and a Ku86+/- NALM-6 cell line
A rAAV (recombinant adeno-associated virus) was used for the delivery of the targeting vector into the desired cell line [44]. The targeting vectors contained ∼900 bp-long left and right homology arms (LHA and RHA, respectively) flanking a neomycin-resistance selection cassette (Fig. 1B). The LHA for the Ku70 targeting vector contained sequences 5′ of exon 3 while the RHA contained sequences 3′ of exon 4. For the Ku86 targeting vector, the LHA contained sequences upstream of exon 1 and the RHA contained sequences downstream of exon 1. The selection cassette was a ∼2.8 kb restriction fragment which contained the PGK promoter, the neomycin resistance gene, the EM7 promoter and the zeomycin resistance gene. The selection cassette was also flanked on either end by loxP sites. Correct targeting of the endogenous genomic Ku70 locus (Fig. 1A) removes exons 3 and 4, resulting in the generation of a G418-resistant cell line (Fig. 1C). The removal of exons 3 and 4 should also generate a null mutation, since hypothetical splicing of exon 2 to exon 5 results in an out-of-frame mRNA. Gene targeting of the Ku70 locus was carried out in two different human cells lines, HCT116 and NALM-6. For the targeting of the Ku86 locus only NALM-6 cells were used since we have previously described the generation of HCT116 cell lines deficient for Ku86 [33]. HCT116 is an immortalized colon cancer cell line [48] with a stable diploid karyotype that responds normally to DNA DSBs [49-51]. Over 20 different loci — including Ku86 — have been inactivated via gene targeting in this cell line (reviewed by [43]). NALM-6 is a human pre-B cell line [39] that also contains a stable diploid karyotype and responds normally to DNA DSBs. NALM-6 has also been successfully used for gene targeting, albeit at frequencies 20-fold higher than similar events in HCT116 (reviewed by [43]), prompting speculation that HR is up-regulated in this cell line [42].
Targeting events resulted in the generation of neomycin/G418-resistant colonies. Genomic DNA was isolated from these colonies and a diagnostic PCR analysis was carried out to screen for correctly targeted clones. For the Ku70 clones, a control PCR was performed using the primers RArmF and BR8 (Fig. 1C). This was done to check for the integrity of the isolated DNA and to optimize PCR conditions. The experimental PCR was carried out using the primers RArmF and Ku703,4R1 (Fig. 1C). RArmF corresponded to sequences in the targeting vector while Ku703,4R1 lay in the 3′-flanking region. A total of 437 HCT116 clones were screened in this manner and correct targeting events were seen in three independent clones (#53, #106 and #441) for a targeting frequency of 0.69%. Correct targeting events resulted in the production of a 1.3 kb band using RArmF and Ku703,4R1 (3′ PCR, Fig. 2A, upper panel). The correct targeting event was confirmed using a 5′-flanking PCR strategy and two of the clones, #53 and #106, showed the expected 2 kb PCR band (Fig. 2A, lower panel). The third clone, #441 showed a slightly smaller band using 5′-PCR (data not shown) suggesting that it had undergone an accompanying deletional event and thus this clone was not characterized further. The PCR bands resulting from correct targeting events were absent from both a randomly targeted clone (#108) and the parental HCT116 cell line (Fig. 2A). An identical PCR analysis was carried out to screen the clones obtained from targeting events using NALM-6 cells. Two independent correctly targeted clones, N31 and N35, were obtained from a total of 121 clones screened for a targeting frequency of 1.65%. This targeting frequency was 2.4-fold higher than the frequency observed with the same vector in HCT116 cells, consistent with a higher rate of HR in this cell line ([41, 42]; reviewed by [43]). As expected, both N31 and N35 genomic DNA produced the diagnostic 1.3 kb (3′-PCR) and 2.0 kb (5′-PCR) bands (Fig. 2B). Again, genomic DNA obtained from a randomly targeted clone (N50) and the parental NALM-6 cell line did not generate either band (Fig. 2B). To further confirm that correct targeting events had taken place, the 1.3 kb PCR fragment was cloned and sequenced and this confirmed that the fragment corresponded to the region of the Ku70 locus that was being targeted (data not shown).
Fig. 2.

Identification of Ku70 and Ku86 heterozygous cell lines. (A) Diagnostic genomic PCR carried out with HCT116 Ku70+/- cells. The 3′-PCR was carried out using the primer set RArmF and Ku703,4R1 (Fig. 1C) while the 5′-PCR was performed using the primers LArmR and Ku703,4F1 (Fig. 1C). An ethidium-bromide stained agarose gel picture is shown. Genomic DNA was isolated from two heterozygous clones (#53 and #106), a randomly targeted clone (#108) and from WT HCT116 cell line. (B) Diagnostic genomic PCR performed with the NALM-6 clones. The same primer sets were used as described above in (A). The gel shows the PCR performed with DNA from WT, two Ku70+/- clones (N31 and N35) and a randomly targeted clones (N50). (C) Southern blot analysis of WT (HCT116 and NALM-6) and Ku70+/- cell lines (#53, #106, N31 and N35). Genomic DNA was doubly digested with HindIII and FspI and hybridized with probe c (Fig. 1C). (D) Genomic PCR carried out on WT NALM-6, Ku86+/- clones (N197 and N397) and two randomly targeted clones (N205 and N310). The upper panel shows the 3′-PCR using the primers RArmF and KU861R while the lower panel shows the 5′-PCR using the primers LArmR and KU861F. M indicates the DNA ladder lane. (E) Southern hybridization of Ku86+/- NALM-6 clones using 5′- and 3′-flanking probes, a’ and b’, respectively. Approximate molecular markers are shown on the left. The 5′-probe and 3′ probe are shown in Fig. 1D. (F) Southern hybridization of Ku86+/- NALM-6 clones using an internal probe, c’ (Fig. 1D).
To screen for the targeting events in the Ku86 locus, the control PCR was done using the primer set RArmF and BR12 (Fig. 1D). The experimental PCR was carried out at the 3′-end using the primers RArmF and KU861R. A total of 379 clones were screened in this manner and two independent clones, N197 and N397, showed the expected PCR band (Fig. 2D, upper panel) for a targeting frequency of 0.53%. To confirm the PCR results, N197 and N397 were also subjected to an additional diagnostic PCR at the 5′-end using the primers LArmR and KU861F (Fig. 1D) and these two Ku86+/- clones displayed the expected PCR band of ∼1.3kb (Fig. 2D, lower panel). The diagnostic PCR bands at both the 3′- and 5′-ends were absent from the wild-type NALM-6 and two randomly targeted clones, N205 and N310 (Fig. 2D).
As an additional confirmation, Southern hybridization was carried out for all of the heterozygous clones. Genomic DNA was isolated and digested with the restriction enzymes HindIII and FspI (for the Ku70 clones) and with ApaLI (for the Ku86 clones). A Southern blot analysis was then performed using an internal probe (probe c, Fig. 1C) for the Ku70 clones. A 4.8 kb band, caused by the presence of an FspI site introduced on the targeting vector (Fig. 1C), confirmed that a single, correctly targeted event had occurred in each clone (Fig. 2C). This unique 4.8 kb band was present in all four clones (#53, #106, N31 and N35), which were deemed by PCR screening as being correctly targeted. The band was absent, as expected, from the parental HCT116 and NALM-6 cell lines since these cells did not contain the targeting vector. For the Ku86 clones three different probes were utilized (Fig. 1D) after digesting the genomic DNA with the restriction enzyme ApaLI. Using probe a’ (5′ flanking region), the genomic locus was identified by the presence of a ∼11 kb band. This band was present in the wild-type, the heterozygous clone and also in two incorrectly targeted clones (Fig. 2E, upper panel). An additional expected 6.3 kb band was also observed in the lanes corresponding to clones N197 and N397. This band results from the introduction of an ApaLI site present in the neomycin selection cassette (Fig. 1D) and it should only be seen in the event of correct targeting. This band was absent from the lanes corresponding to wild-type and the randomly targeted clones. Confirmation of this was obtained using probe b’, derived from the 3′ flanking region. A ∼7 kb band, predicted for correct targeting events, was seen only in the heterozygous clones, N197 and N397 (Fig. 2E, lower panel). Finally we used an internal probe, c’, corresponding to the sequences in the selection cassette. Correct targeting events should result in a ∼6.3 kb band when the ApaLI restriction enzyme is used and this was indeed observed for clones N197 and N397 (Fig. 2F). This band was absent from the wild-type cell line since these cells do not contain the selection cassette. The randomly targeted clones displayed bands that were of different sizes resulting from the random integration of the selection cassette in the genome. In summary, the PCR and Southern analyses confirmed the isolation of four independent Ku70+/- cell lines; two each with HCT116 and NALM-6 genetic backgrounds, respectively, and two independent Ku86+/- cell lines in the NALM-6 background.
3.2. Primary characterizations
Immunoblot analysis was carried out on the Ku70+/- and Ku86+/- cell lines. All six cell lines (#53, #106, N31, N35, N197 and N397) showed the expected reductions in expression of both Ku subunits anticipated from the inactivation of one of the Ku subunits (Supplemental Fig. 1). The reduction of expression in one of the subunits was expected perforce due to the gene targeting and the reduction in the other subunit was similarly expected because it is known that in the absence of one subunit, the other Ku subunit is unstable [7]. Thus, the levels of both Ku subunits were diminished in the different cell lines, although the effect seemed more pronounced in HCT116 cells (Supplemental Fig. 1).
The reduction in Ku levels was confirmed by quantitating the amount of non-specific, dsDNA end-binding activity (an activity that can be solely ascribed to Ku; [7]) within the cell lines. An EMSA (electrophoretic mobility shift assay) [52] was carried out using radioactive dsDNA probes and nuclear extracts to detect this activity of the Ku heterodimer. All six cell lines (#53, #106, N31, N35, N197 and N397) showed the expected reductions in DNA end-binding activity (Supplemental Fig. 2). As a control, two independent, randomly targeted clones, N6 and N401, and both of the parental cell lines were also included in this assay. These cell lines did not show any appreciable decrease in Ku binding, confirming that the effect was Ku-specific (Supplemental Fig. 2).
Once bound to the end of a dsDNA, Ku recruits DNA-PKCS to generate the DNA-PK holoenzyme complex. When Ku expression is lowered in cells by either gene targeting [32] or by anti-sense RNA or cDNA expression [35, 36], there is a down regulation of DNA-PK kinase activity. Thus, DNA-PK kinase assays were performed to further validate the DEB results. In this assay, a peptide (+) derived from p53 with a consensus phosphorylation site for DNA-PK is used along with a control peptide (-) in which that site has been mutated [53]. Also included as controls in the assay were extracts from two human glioma cell lines, M059K and M059J, which are known to be positive and negative, respectively, for DNA-PK kinase activity [23]. All six cell lines (#53, #106, N31, N35, N197 and N397) showed the expected reductions in DNA-PK kinase activity, although the reduction observed for N397 was not statistically significant (Supplemental Fig. 3). Again, control randomly targeted cell lines, N6 and N401, and the parental cell lines displayed wild-type levels of kinase activity (Supplemental Fig. 3).
Collectively, the data presented above demonstrated that all six of the cell lines generated by gene targeting displayed the expected biochemical characteristics of mammalian cell lines deficient for Ku.
3.3. Biological endpoints
Human Ku86+/- HCT116 cells display a haploinsufficient growth defect [32], which has not been observed for any other species. We therefore carried out a growth assay on both HCT116 and NALM-6 Ku70+/- cell lines to investigate whether they had growth defects. Both HCT116 Ku70+/- clones, #53 and #106 showed a significantly reduced rate of growth compared to the parental HCT116 cells (Fig. 3A). The growth rate of clone #53 was very similar to the HCT116 Ku86+/- cell line, 70-32, while clone #106 grew even slower. A randomly targeted clone, #98, did not show any significant growth defect when compared to the WT HCT116 cells (Fig. 3A). The NALM-6 Ku70+/- clones, N31 and N35, grew somewhat differently. While N35 grew appreciably slower than the wild-type clone, N31 showed no significant growth defect (Fig. 3B). A randomly targeted clone, N48, also had no growth defect. These experiments were performed three independent times, each time being done in triplicate. Since one of the Ku70+/- NALM-6 clones grew slower while the other grew like wild-type, no conclusion concerning a correlation between Ku70 levels in NALM-6 cells with growth defects could be deduced.
Fig. 3.

Growth defects of human somatic cells with reduced Ku expression. (A) Ku70+/- HCT116 cells show defects in their proliferation rate. 3 × 103 cells from the indicated clones were seeded on tissue culture plates and the increase in cell number was determined using trypan blue staining and a hemocytometer in daily intervals. The average of three experiments, each done in triplicate is shown. (B) Ku70+/- NALM-6 clones. The proliferation rate was determined as described above. (C) Ku86+/- NALM-6 cells. The proliferation rate was determined as in (A).
To pursue this issue, growth assays were performed with the Ku86+/- NALM-6 cell lines. Both Ku86 heterozygous clones, N197 and N397, showed a significant decrease in their ability to grow when compared to the parental wild-type cell line (Fig. 3C) although clone N397 cells grew reproducibly slower than the clone N197 cells. A randomly targeted clone (N401) had a growth profile identical to that of the wild-type cell line (Fig. 3C). Thus, lowering of Ku86 levels in NALM-6 cells caused a growth defect phenotype that was similar to the growth defect phenotype in Ku86+/- and Ku70+/- HCT116 cells.
3.4. Ionizing radiation sensitivity
An important role of the Ku heterodimer in human cells is in the repair of damaged DNA. A lowered level of Ku would thus be expected to sensitize cells to DNA damaging agents. Not surprisingly, HCT116 Ku70+/- cells were slightly more sensitive to IR when compared to the parental line (Fig. 4A). Their IR sensitivity, which was most obvious at higher doses (e.g., 5.0 Gy), was similar to that of a HCT116 Ku86+/- cell line, 70-32 (Fig. 4A). In marked contrast to HCT116 Ku70+/- cell lines, NALM-6 cells that were heterozygous for Ku70, N31 and N35, or for Ku86, N197 and N397, displayed no (N31, N35 and N397) or only a slightly increased (N197) sensitivity to IR when compared to the parental (WT) NALM-6 cells or to randomly targeted clones, N50 (Fig. 4B) or N401 (Fig. 4C). These results indicated that there are cell line-specific differences in the repair of DNA damage caused by IR.
Fig. 4.
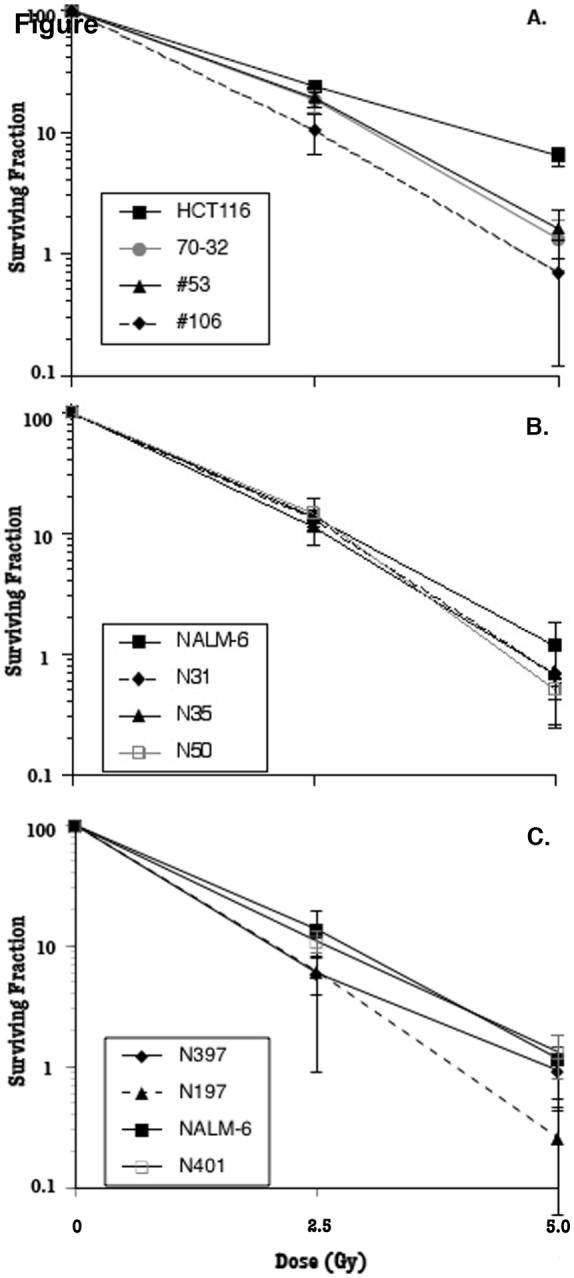
The IR sensitivity profiles of human somatic cells expressing reduced levels of Ku. HCT116-derived, but not NALM-6-derived, cell lines are IRs. For HCT116 (A), 300 cells of the indicated cell lines were seeded on tissue culture plates and X-irradiated at the indicated doses. Cells surviving to form colonies between 10-14 days later were scored. For NALM-6 Ku70+/- clones (B), control plates (no irradiation) contained 10, 5 and 2.5 cells per well of a 96-well plate. Plates that were used for irradiation had 10, 100 or 1000 cells per well of a 96-well plate. For NALM-6 Ku86+/- clones (C), control plates (no irradiation) contained 5, 10 and 20 cells per well while the plates used for irradiation had 100, 500 and 1000 cells per well of a 96-well tissue culture plate. Following irradiation, all the plates were allowed to recover for 20 days before wells containing colonies were scored.
3.5. Telomere length assays (TRFs)
Telomeres are the terminal structures of linear chromosomes and are theoretically a naturally occurring dsDNA end. Not surprisingly, Ku physically associates with telomeres in a variety of organisms [54]. Confusingly, there are reports of both shortening [21] and lengthening [28, 31] of telomeres in cells devoid of Ku. We have shown that functional inactivation of even a single allele of Ku86 in HCT116 cells caused profound telomere shortening [33]. To investigate whether this observation was also true for the Ku70 allele and to also determine if this phenotype varies between different human somatic cell lines, we carried out the well-established TRF assay [55]. TRF analysis takes advantage of the fact that telomeric DNA, which is comprised of only a T2AG3 repetitive sequence, is devoid of recognition sequences for any restriction enzymes. Thus, when genomic DNA is digested with very frequent-cutting restriction enzymes like AluI and MboI, the telomeric DNA remains largely intact. The telomeric DNA can then be detected with a radioactive d(C3TA2)3 probe in a Southern blotting procedure.
There was a severe effect of reduced Ku70 expression on the telomere lengths of HCT116 cells. While wild-type HCT116 cells showed telomeres ranging from 2 to 10 kb in length, the Ku70 heterozygous clones #53 and #106 had TRFs ranging from only 1 to 4 kb (Fig. 5A). This severe telomere shortening was identical to that described for HCT116 Ku86+/- cell lines [33]. The effect of lowered Ku70 expression on the telomere lengths in NALM-6 cells also resulted in shorter telomeres, but the effect was much less pronounced. The telomere lengths of NALM-6 parental cell line ranged from 2 to 12 kb while both Ku70 heterozygous clones, N31 and N35, ranged only from 2 to 10 kb (Fig. 5B). Inactivation of a single allele of Ku86 in NALM-6 cells resulted in disparate TRFs. N197 exhibited severely reduced telomeres (Fig. 5C), comparable to the reduction observed in the HCT116 cell lines (Fig. 5A and [33]). Clone N397 cells, in contrast, displayed only marginally shorter telomeres when compared to the wild-type NALM-6 cells (Fig. 5C) and resembled much more the Ku70+/- clones, N31 and N35 (Fig. 5B). From these results we concluded that telomere shortening accompanies a reduction in Ku expression in both cell types, but that the effect is much more pronounced in HCT116, than in NALM-6 cells.
Fig. 5.
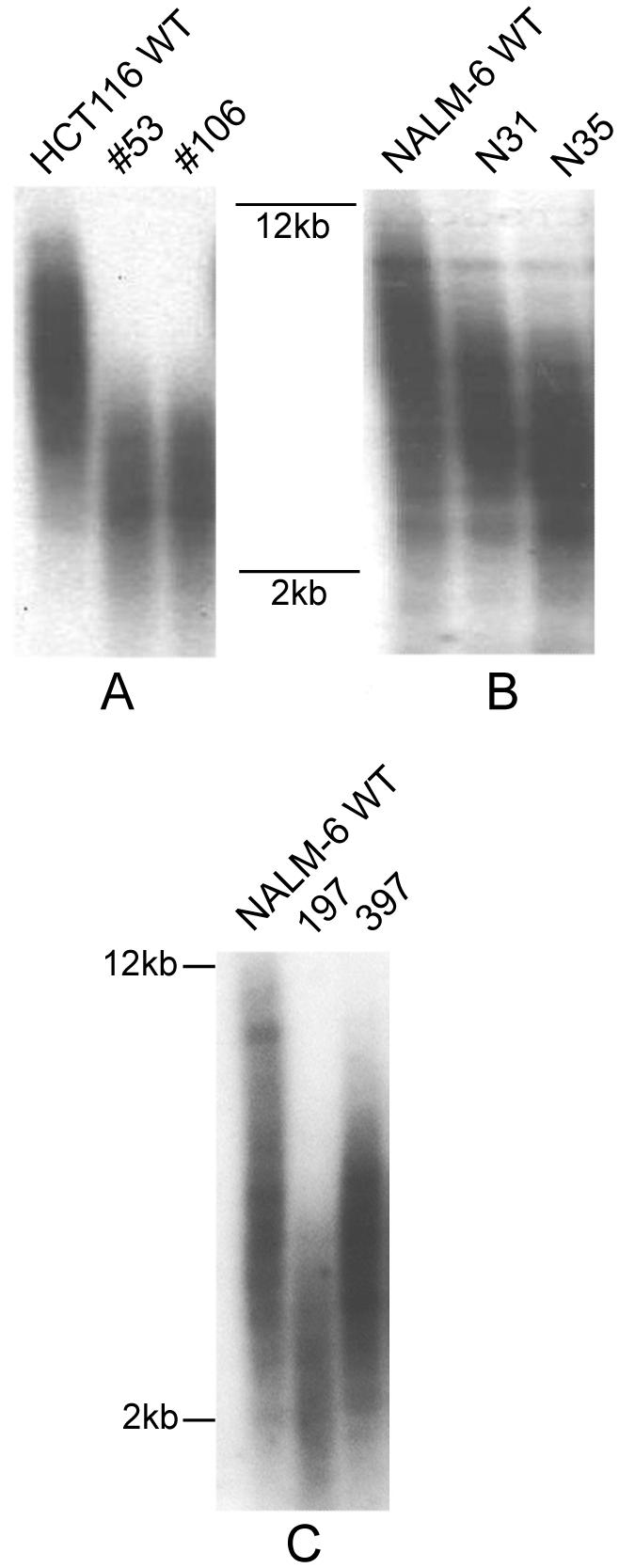
Telomere shortening in human Ku70 and Ku80-deficient cell lines. Genomic DNA was purified from the indicated cell lines, digested to completion with AluI and MboI, and then subjected to terminal restriction fragment Southern blot analysis under denaturing conditions with a (C3TA2)3 5′-end-radiolabeled oligonucleotide probe. (A) Telomere lengths of Ku70+/- HCT116 clones. (B) Telomere lengths of Ku70+/- NALM-6 clones. (C) Telomere lengths of Ku86+/- NALM-6 clones. Approximate molecular size markers are shown.
3.6. Genomic (in)stability
Defects in telomere length maintenance have been repeatedly linked to defects in genomic stability (reviewed by [1]) although most of these studies have been carried out with non-human systems. Consistent with these observations, however, human HCT116 Ku86+/- cell lines have very short telomeres and a high level of GCRs (gross chromosomal rearrangements), i.e., genomic instability [33]. To see if this phenotype could be extended to other human cell lines, the six Ku heterozygous cell lines generated for this study were cytogenetically analyzed by G-banding. The parental HCT116 cell line had a fairly high level of genetic instability, with 11.8% of all metaphases examined containing some aberrant chromosomal feature (usually a translocation, but also duplications and deletions — data not shown) (Table 1). Surprisingly, the HCT116 Ku70+/- cell lines did not show any increase in this frequency (Table 1). The NALM-6 parental cell showed only 5.4% aberrant metaphases. Again, a reduction of Ku70 expression resulted in no increase in GCRs and potentially even a reduction (Table 1). The NALM-6 Ku86+/- cell lines, in contrast, were 5-fold more unstable than the corresponding parental cell line (Table 1). This result, however, was somewhat misleading, as the N197 cell line was only slightly unstable (2 GCRs in 20 metaphases) whereas the N397 cell line was quite unstable (9 GCRs in 20 metaphases). From these studies we concluded that while human somatic cell lines with reduced Ku expression can be genetically unstable, there is no direct correlation between the reduction in Ku and the frequency of instability.
Table 1.
Genomic Stability of Ku Heterozygous Cells
| Cell Clone | Aberrant Metaphases | Parental Metaphases | GCR Frequency |
|---|---|---|---|
| HCT116 (WT) | 13 | 97 | 11.8% |
| HCT116 (Ku70+/-) | 5 | 35 | 12.5% |
| NALM-6 (WT) | 2 | 35 | 5.4% |
| NALM-6 (Ku70+/-) | 0 | 40 | 0.0% |
| NALM-6 (Ku86+/-) | 11 | 29 | 27.5% |
4. Discussion
We have demonstrated that HCT116 cell lines that are heterozygous for Ku86 are haploinsufficient [32, 33] and that the null cell lines are not viable [32]. In contrast, Uegaki et al., recently demonstrated that inactivation of either Ku70 or Ku86 in the human leukemic B-cell line, NALM-6, resulted in subclones that were essentially aphenotypic [38]. In an attempt to understand these discrepancies, we created 6 new cell lines heterozygous for either Ku70 or Ku86 in either the HCT116 or NALM-6 genetic backgrounds using gene targeting and characterized side-by-each their phenotypes. Inactivation of either gene in either cell type resulted in the biochemical deficits that are classically associated with Ku loss-of-function mutations: mutation of one gene resulted in concomitant lowering of protein expression for both subunits, and there was reduced Ku DEB and DNA-PK kinase activities (Supplemental Figs. 1 & 2). Thus, all of the cell lines behaved genetically and biochemically as expected for Ku heterozygous cell lines. Deletion of a Ku70 allele in HCT116 cells generated two independent cell lines (#53 and #106) that were virtually indistinguishable from the two independent cell lines we have described following disruption of Ku86 (clones #44 and #70) [32, 33]: the cell lines grew slowly, were slightly IRs and they contained extremely shortened telomeres. Because all four independent cell lines behaved identically, we conclude that the resultant phenotypes are directly related to the reduction in Ku expression. This hypothesis is strengthened by our demonstration that the re-expression of a Ku86 cDNA in the HCT116 Ku86+/- cell lines rescued the proliferation and radiation sensitivity defects and partially rescue the short telomere phenotype [56]. There is, however, one critical difference between the Ku86+/- and Ku70+/- HCT116 cell lines and that is whereas the Ku86+/- lines are genetically unstable [33], the Ku70 lines are not (Table 1). We had previously inferred that the short telomeres and the genetic instability in HCT116 Ku86+/- cell lines were directly linked because this connection has been rigorously documented in numerous other systems [1]. That conclusion now seems less tenable in the face of the stability of the Ku70+/- cell lines and even more so since the genetic instability of Ku86+/- cell lines could not be rescued by the re-expression of Ku86 [56].
Although the generation of Ku70+/- HCT116 cell lines was important, the main impetus for carrying out these studies was to confirm the lack of phenotypes for the corresponding Ku mutations in NALM-6 cells and to understand mechanistically why this might be so. Indeed, we could confirm some of the phenotypes reported by Uegaki et al. [38]. Specifically, our Ku-deficient NALM-6 cell lines were also not IRs (Fig. 4). In this instance, the difference is likely related to the basal inherent enhanced radiosensitivity of the NALM-6 line, which is typical for cells of lymphoid origin [57]. In this already radiosensitive background, it may be difficult to detect the effect of the loss of a single allele of Ku on IRS. Consistent with this view, the inactivation of a single allele of LIGIV was essentially equally as radioresistant as the parental NALM-6 population and it was only upon the subsequent removal of the remaining LIGIV allele that profound IRS was observed [41]. In contrast, the inactivation of one LIGIV allele in HCT116 cells results in cells with a demonstrable sensitivity, which is intermediate between the parental and the null cell lines, to etoposide (Sehyun Oh & EAH, unpublished data). Thus, there are clearly cell line-specific differences between HCT116 and NALM-6 (and, by extension, presumably between other human cell lines).
Similarly, we also did not observe, with one exception, a significant telomere length defect. Thus, Uegaki et al. have reported that none of their Ku heterozygous NALM-6 clones showed telomere shortening and indeed, in the data they presented, even slight or significant (clone H2) telomere lengthening could be observed [38]. This was unexpected since our HCT116 Ku heterozygous cell lines showed severe telomere shortening ([33] and Fig. 5) and other groups using complementary RNAi approaches have also observed telomere shortening [37]. Nonetheless, we confirm that our NALM-6 Ku heterozygous clones also have only slight telomere shortening (Fig. 5). Why might NALM-6 cells be less sensitive in comparison to HCT116 cells to Ku-induced telomere defects? One explanation may be the apparent up-regulation of HR in NALM-6 cells ([41, 42]; reviewed by [43]). The conclusion that HR is up-regulated in NALM-6 cells is based upon the increased frequency of gene targeting reported for this cell line [41, 42]. It is perhaps important to note, however, that we have not observed this phenomenon. Although the frequency of correctly targeting the Ku70 locus was a modest 2.4-fold higher in NALM-6 versus HCT116 cells (1.65% versus 0.69%) the Ku86 targeting frequency was virtually identical (0.53% versus 0.56% [32]) and the frequency of DNA-PKCS targeting is actually reduced (0.66% versus 3.64%; BR and EAH, unpublished data). Nonetheless, there exists an additional mechanism of telomere maintenance (ALT; alternative lengthening of telomeres) that is known to be HR-dependent (reviewed by [58]). Thus, although ALT is not thought to be operational in cell lines that express telomerase — which NALM-6 does [38] — it may be that up-regulated HR activity frequently compensates for the telomere defects that Ku loss-of-function normally causes [33, 37]. This compensation may not be absolute, however, as the N197 cell line does contain severely shortened telomeres. Why N197 telomeres should be so short when N397, N31 and N35 were only marginally shortened is not clear. We have noted, however, that the telomere length of random NALM-6 subclones can be quite variable and that clones derived from single cells (as these lines were) can stochastically and frequently have either very short or very long telomeres (data not shown). Thus, clonal variability is currently the best “explanation” for this result.
Significantly, however, we report here that our Ku-defective NALM-6 cell lines — again with a single exception — have growth defects, a trait that was not observed earlier [38]. Of the four cell lines that we generated, two of them (N35 and N397) grew quite slowly, one (N197) grew modestly slowly and one (N31) grew indistinguishably from the parental line (Fig. 3). Since all four HCT116 cell lines grow slowly (one of them, #106, extremely so; Fig. 3A) we conclude that slow growth/proliferation defects are a common feature of Ku-deficient cell lines. The existence of clones #106 (Fig. 3A) and N31 (Fig. 3B) suggests that the slow growth can occasionally either be augmented (clone #106) or corrected (clone N31) by additional clonal genetic variations.
How might the differences between our results and those of Uegaki et al. be explained? One possibility is that real differences in the cell lines might exist because of the different targeting strategies employed. For example, Uegaki et al. deleted exon 2 for their Ku86 knockout [38]. This is a small exon and most importantly it results in an in-frame deletion if splicing should occur between exons 1 and 3. In our strategy, we have removed exon 1 (Fig. 1D), which disrupts the initiator ATG and thus only potentially allows for protein expression from cryptic downstream methionines. This genetic difference, subtle differences in cell culture methodology, or clonal variability are thus the most likely sources of the discrepancies observed.
Lastly, it should be noted that we do not see any coherent correlation between telomere length and genetic stability. Thus, while HCT116 Ku70+/- cell lines have short telomeres (Fig. 5A) that are indistinguishable in length from the telomeres in HCT116 Ku86+/- cell lines [33], the Ku70+/- cells are as genetically stable as the parental population (Table 1), whereas the Ku86+/- cells are not [33]. The NALM-6 Ku86+/- cell lines were also unstable, perhaps suggesting a gene-specific effect, but this conclusion is weakened by the observation that the N197 cell line, which had very short telomeres (Fig. 5C), was only modestly unstable (Table 1) whereas N397, which has near wild-type length telomeres (Fig. 5C), was more unstable (Table 1). Moreover, the demonstration that we cannot complement the genetic instability of the HCT116 Ku86+/- cell lines by re-expression of a Ku86 cDNA [56] also suggests that the genetic instability is not linked to the Ku expression levels of a cell.
In conclusion, we have generated a series of new human somatic cell lines whose phenotypes reaffirm the haploinsufficient nature of the Ku70 and Ku86 loci and reaffirm the necessity for their proper biallelic expression. Moreover, our data suggest that either cell proliferation and/or telomeric functions/defects are likely responsible for the essential nature of these genes. Secondly, we do observe subtle cell line-specific differences. These differences suggest that one must take care in extrapolating any given result to other human cell lines and, most obviously, to human patients. Lastly, in a additional cautionary note to other researchers, we report that clonal variability was observed for almost every phenotype analyzed. Thus, only by analyzing multiple, independent clones and/or by the reciprocal approach of functional complementation can one be certain that a particular phenotype is ascribable to the activity — or lack thereof — of the gene of interest.
Supplementary Material
Acknowledgements
The G-banding cytogenetic analyses were performed in the Cytogenetics Core Laboratory at the University of Minnesota with support from the Comprehensive Cancer Center NIH Grant no. P30 CA077598-09. This work has been supported in part by grants HL079559 and GM069576 from the NIH to EAH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Maser RS, DePinho RA. Connecting chromosomes, crisis, and cancer. Science. 2002;297:565–569. doi: 10.1126/science.297.5581.565. [DOI] [PubMed] [Google Scholar]
- [2].Hefferin ML, Tomkinson AE. Mechanism of DNA double-strand break repair by non-homologous end joining. DNA Repair. 2005;4:639–648. doi: 10.1016/j.dnarep.2004.12.005. [DOI] [PubMed] [Google Scholar]
- [3].Jung D, Alt FW. Unraveling V(D)J recombination. Insights into gene regulation. Cell. 2004;116:299–311. doi: 10.1016/s0092-8674(04)00039-x. [DOI] [PubMed] [Google Scholar]
- [4].Meek K, Gupta S, Ramsden DA, Lees-Miller SP. The DNA-dependent protein kinase: the director at the end. Immunol. Rev. 2004;200:132–141. doi: 10.1111/j.0105-2896.2004.00162.x. [DOI] [PubMed] [Google Scholar]
- [5].Cahill D, Connor B, Carney JP. Mechanisms of eukaryotic DNA double strand break repair. Front Biosci. 2006;11:1958–1976. doi: 10.2741/1938. [DOI] [PubMed] [Google Scholar]
- [6].van Veelen L, Wesoly J, Kanaar R. In: DNA Damage Recognition. Seide W, Kow YW, Doetsch P, editors. Taylor and Francis Group; New York: 2006. pp. 581–607. [Google Scholar]
- [7].Hendrickson EA, Huffman JL, Tainer JA. In: DNA Damage Recognition. Seide W, Kow YW, Doetsch P, editors. Taylor and Francis Group; New York: 2006. pp. 629–684. [Google Scholar]
- [8].Riballo E, Critchlow SE, Teo S-H, Doherty AJ, Priestley A, Broughton B, Kysela B, Beamish H, Plowman N, Arlett CF, Lehmann AR, Jackson SJ, Jeggo PA. Identification of a defect in DNA ligase IV in a radiosensitive leukemia patient. Current Biol. 1999;9:699–702. doi: 10.1016/s0960-9822(99)80311-x. [DOI] [PubMed] [Google Scholar]
- [9].O’Driscoll M, Cerosaletti KM, Girard PM, Dai Y, Stumm M, Kysela B, Hirsch B, Gennery A, Palmer SE, Seidel J, Gatti RA, Varon R, Oettinger MA, Neitzel H, Jeggo PA, Concannon P. DNA ligase IV mutations identified in patients exhibiting developmental delay and immunodeficiency. Mol. Cell. 2001;8:1175–1185. doi: 10.1016/s1097-2765(01)00408-7. [DOI] [PubMed] [Google Scholar]
- [10].Moshous D, Callebaut I, de Chasseval R, Corneo B, Cavazzana-Calvo M, Le Deist F, Tezcan I, Sanal O, Bertrand Y, Philippe N, Fischer A, de Villartay JP. Artemis, a novel DNA double-strand break repair/V(D)J recombination protein, is mutated in human severe combined immune deficiency. Cell. 2001;105:177–186. doi: 10.1016/s0092-8674(01)00309-9. [DOI] [PubMed] [Google Scholar]
- [11].Buck D, Malivert L, de Chasseval R, Barraud A, Fondaneche MC, Sanal O, Plebani A, Stephan JL, Hufnagel M, le Deist F, Fischer A, Durandy A, de Villartay JP, Revy P. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell. 2006;124:287–299. doi: 10.1016/j.cell.2005.12.030. [DOI] [PubMed] [Google Scholar]
- [12].Walker JR, Corpina RA, Goldberg J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature. 2001;412:607–614. doi: 10.1038/35088000. [DOI] [PubMed] [Google Scholar]
- [13].Yoo S, Kimzey A, Dynan WS. Photocross-linking of an oriented DNA repair complex. Ku bound at a single DNA end. J. Biol. Chem. 1999;274:20034–20039. doi: 10.1074/jbc.274.28.20034. [DOI] [PubMed] [Google Scholar]
- [14].Ribes-Zamora A, Mihalek I, Lichtarge O, Bertuch AA. Distinct faces of the Ku heterodimer mediate DNA repair and telomeric functions. Nat. Struct. Mol. Biol. 2007;14:301–307. doi: 10.1038/nsmb1214. [DOI] [PubMed] [Google Scholar]
- [15].Ouyang H, Nussenzweig A, Kurimasa A, Soares VC, Li X, Cordon-Cardo C, Li WH, Cheong N, Nussenzweig M, Iliakis G, Chen D, Li G. Ku70 is required for DNA repair but not for T cell antigen receptor gene recombination In vivo. J. Exp. Med. 1997;15:921–929. doi: 10.1084/jem.186.6.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Nussenzweig A, Chen C, da Costa Soares V, Sanchez M, Sokol K, Nussenzweig MC, Li GC. Requirement for Ku80 in growth and immunoglobulin V(D)J recombination. Nature. 1996;382:551–555. doi: 10.1038/382551a0. [DOI] [PubMed] [Google Scholar]
- [17].Zhu C, Bogue MA, Lim D-S, Hasty P, Roth DB. Ku86-deficient mice exhibit severe combined immunodeficiency and defective processing of V(D)J recombination intermediates. Cell. 1996;86:379–389. doi: 10.1016/s0092-8674(00)80111-7. [DOI] [PubMed] [Google Scholar]
- [18].Nussenzweig A, Sokol K, Burgman P, Li L, Li GC. Hypersensitivity of Ku80-deficient cell lines and mice to DNA damage: the effects of ionizing radiation on growth, survivial and development. Proc. Natl. Acad. Sci. USA. 1997;94:13588–13593. doi: 10.1073/pnas.94.25.13588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Karanjawala ZE, Grawunder U, Hsieh CL, Lieber MR. The nonhomologous DNA end joining pathway is important for chromosome stability in primary fibroblasts. Curr. Biol. 1999;9:1501–1504. doi: 10.1016/s0960-9822(00)80123-2. [DOI] [PubMed] [Google Scholar]
- [20].Difilippantonio MJ, Zhu J, Chen HT, Meffre E, Nussenzweig MC, Max EE, Ried T, Nussenzweig A. DNA repair protein Ku80 suppresses chromosomal aberrations and malignant transformation. Nature. 2000;404:510–514. doi: 10.1038/35006670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].d’Adda di Fagagna F, Hande MP, Tong W-M, Roth DB, Lansdorp PM, Wang Z-Q, Jackson SP. Effects of DNA nonhomologous end-joining factors on telomere length and chromosomal stability in mammalian cells. Current Biol. 2001;11:1192–1196. doi: 10.1016/s0960-9822(01)00328-1. [DOI] [PubMed] [Google Scholar]
- [22].Vogel H, Lim D-S, Karsenty G, Finegold M, Hasty P. Deletion of Ku86 causes early onset of senescence in mice. Proc. Natl. Acad. Sci. USA. 1999;96:10770–10775. doi: 10.1073/pnas.96.19.10770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lees-Miller SP, Godbout R, Chan DW, Weinfeld M, Day RS, 3rd, Barron GM, Allalunis-Turner J. Absence of p350 subunit of DNA-activated protein kinase from a radiosensitive human cell line. Science. 1995;267:1183–1185. doi: 10.1126/science.7855602. [DOI] [PubMed] [Google Scholar]
- [24].Anderson CW, Dunn JJ, Freimuth PI, Galloway AM, Allalunis-Turner MJ. Frameshift mutation in PRKDC, the gene for DNA-PKcs, in the DNA repair- defective, human, glioma-derived cell line M059J. Radiat. Res. 2001;156:2–9. doi: 10.1667/0033-7587(2001)156[0002:fmiptg]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- [25].de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19:2100–2110. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- [26].Bailey SM, Meyne J, Chen DJ, Kurimasa A, Li GC, Lehnert BE, Goodwin EH. DNA double-strand break repair proteins are required to cap the ends of mammalian chromosomes. Proc. Natl. Acad. Sci. U S A. 1999;96:14899–14904. doi: 10.1073/pnas.96.26.14899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hsu HL, Gilley D, Galande SA, Hande MP, Allen B, Kim SH, Li GC, Campisi J, Kohwi-Shigematsu T, Chen DJ. Ku acts in a unique way at the mammalian telomere to prevent end joining. Genes Dev. 2000;14:2807–2812. doi: 10.1101/gad.844000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Samper E, Goytisolo FA, Slijepcevic P, van Buul PPW, Blasco MA. Mammalian Ku86 protein prevents telomeric fusions independently of the length of TTAGGG repeats and the G-strand overhang. EMBO Reports. 2000;1:244–252. doi: 10.1093/embo-reports/kvd051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Goytisolo FA, Samper E, Edmonson S, Taccioli GE, Blasco MA. The absence of the DNA-dependent protein kinase catalytic subunit in mice results in anaphase bridges and in increased telomeric fusions with normal telomere length and G-strand overhang. Mol. Cell. Biol. 2001;21:3642–3651. doi: 10.1128/MCB.21.11.3642-3651.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hsu HL, Gilley D, Blackburn EH, Chen DJ. Ku is associated with the telomere in mammals. Proc. Natl. Acad. Sci. U S A. 1999;96:12454–12458. doi: 10.1073/pnas.96.22.12454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Riha K, Watson JM, Parkey J, Shippen DE. Telomere length deregulation and enhanced sensitivity to genotoxic stress in Arabidopsis mutants deficient in Ku70. EMBO J. 2002;21:2819–2826. doi: 10.1093/emboj/21.11.2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Li G, Nelsen C, Hendrickson EA. Ku86 is essential in human somatic cells. Proc. Natl. Acad. Sci. U S A. 2002;99:832–837. doi: 10.1073/pnas.022649699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Myung K, Ghosh G, Fattah FJ, Li G, Kim H, Dutia A, Pak E, Smith S, Hendrickson EA. Regulation of telomere length and suppression of genomic instability in human somatic cells by Ku86. Mol. Cell. Biol. 2004;24:5050–5059. doi: 10.1128/MCB.24.11.5050-5059.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Omori S, Takiguchi Y, Suda A, Sugimoto T, Miyazawa H, Tanabe N, Tatsumi K, Kimura H, Pardington PE, Chen F, Chen DJ, Kuriyama T. Suppression of a DNA double-strand break repair gene, Ku70, increases radio- and chemosensitivity in a human lung carcinoma cell line. DNA Repair. 2002;1:299–310. doi: 10.1016/s1568-7864(02)00006-x. [DOI] [PubMed] [Google Scholar]
- [35].Sadji Z, Le Romancer M, Lewin MJ, Reyl-Desmars F. Human colon carcinoma cell-line HCT116 transfected by antisense cDNA as a tool to study the Ku86 involvement in cell proliferation. Cell. Signal. 2000;12:745–750. doi: 10.1016/s0898-6568(00)00126-1. [DOI] [PubMed] [Google Scholar]
- [36].Marangoni E, Le Romancer M, Foray N, Muller C, Douc-Rasy S, Vaganay S, Abdulkarim B, Barrois M, Calsou P, Bernier J, Salles B, Bourhis J. Transfer of Ku86 RNA antisense decreases the radioresistance of human fibroblasts. Cancer Gene Ther. 2000;7:339–346. doi: 10.1038/sj.cgt.7700111. [DOI] [PubMed] [Google Scholar]
- [37].Jaco I, Munoz P, Blasco MA. Role of human Ku86 in telomere length maintenance and telomere capping. Cancer Res. 2004;64:7271–7278. doi: 10.1158/0008-5472.CAN-04-1381. [DOI] [PubMed] [Google Scholar]
- [38].Uegaki K, Adachi N, So S, Iiizumi S, Koyama H. Heterozygous inactivation of human Ku70/Ku86 heterodimer does not affect cell growth, double-strand break repair, or genome integrity. DNA Repair. 2006;5:303–311. doi: 10.1016/j.dnarep.2005.10.008. [DOI] [PubMed] [Google Scholar]
- [39].Hurwitz R, Hozier J, LeBien T, Minowada J, Gajl-Peczalska K, Kubonishi I, Kersey J. Characterization of a leukemic cell line of the pre-B phenotype. Int. J. Cancer. 1979;23:174–180. doi: 10.1002/ijc.2910230206. [DOI] [PubMed] [Google Scholar]
- [40].Drexler HG, MacLeod RA, Borkhardt A, Janssen JW. Recurrent chromosomal translocations and fusion genes in leukemia-lymphoma cell lines. Leukemia. 1995;9:480–500. [PubMed] [Google Scholar]
- [41].Grawunder U, Zimmer D, Fugmann S, Schwarz K, Lieber MR. DNA ligase IV is essential for V(D)J recombination and DNA double-strand break repair in human precursor lymphocytes. Mol. Cell. 1998;2:477–484. doi: 10.1016/s1097-2765(00)80147-1. [DOI] [PubMed] [Google Scholar]
- [42].Adachi N, So S, Iiizumi S, Nomura Y, Murai K, Yamakawa C, Miyagawa K, Koyama H. The human pre-B cell line Nalm-6 is highly proficient in gene targeting by homologous recombination. DNA Cell Biol. 2006;25:19–24. doi: 10.1089/dna.2006.25.19. [DOI] [PubMed] [Google Scholar]
- [43].Hendrickson EA. In: Sourcebook of models for biomedical research. Conn M, editor. The Humana Press Inc.; Totowa, NJ: 2007. in press. [Google Scholar]
- [44].Kohli M, Rago C, Lengauer C, Kinzler KW, Vogelstein B. Facile methods for generating human somatic cell gene knockouts using recombinant adeno-associated viruses. Nucl. Acids Res. 2004;32:e3. doi: 10.1093/nar/gnh009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lee SE, Pulaski CR, He DM, Benjamin DM, Voss MJ, Um J, Hendrickson EA. Isolation of mammalian cell mutants that are X-ray sensitive, impaired in DNA double-strand break repair and defective for V(D)J recombination. Mutat. Res. 1995;336:279–291. doi: 10.1016/0921-8777(95)00002-2. [DOI] [PubMed] [Google Scholar]
- [46].Han Z, Johnston C, Reeves WH, Carter T, Wyche JH, Hendrickson EA. Characterization of a Ku86 variant protein that results in altered DNA binding and diminished DNA-dependent protein kinase activity. J. Biol. Chem. 1996;271:14098–14104. doi: 10.1074/jbc.271.24.14098. [DOI] [PubMed] [Google Scholar]
- [47].van Steensel B, Smogorzewska A, de Lange T. TRF2 protects human telomeres from end-to-end fusions. Cell. 1998;92:401–413. doi: 10.1016/s0092-8674(00)80932-0. [DOI] [PubMed] [Google Scholar]
- [48].Shirasawa S, Furuse M, Yokoyama N, Sasazuki T. Altered growth of human colon cancer cell lines disrupted at activated Ki-ras. Science. 1993;260:85–88. doi: 10.1126/science.8465203. [DOI] [PubMed] [Google Scholar]
- [49].Waldman T, Kinzler KW, Vogelstein B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res. 1995;55:5187–5190. [PubMed] [Google Scholar]
- [50].Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- [51].Bunz F, Fauth C, Speicher MR, Dutriaux A, Sedivy JM, Kinzler KW, Vogelstein B, Lengauer C. Targeted inactivation of p53 in human cells does not result in aneuploidy. Cancer Res. 2002;62:1129–1133. [PubMed] [Google Scholar]
- [52].Rathmell WK, Chu G. Involvement of the Ku autoantigen in the cellular response to DNA double-strand breaks. Proc. Natl. Acad. Sci. USA. 1994;91:7623–7627. doi: 10.1073/pnas.91.16.7623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Finnie NJ, Gottlieb TM, Blunt T, Jeggo PA, Jackson SP. DNA-dependent protein kinase activity is absent in xrs-6 cells: implications for site-specific recombination and DNA double-strand break repair. Proc. Natl. Acad. Sci. USA. 1995;92:320–324. doi: 10.1073/pnas.92.1.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Gullo C, Au M, Feng G, Teoh G. The biology of Ku and its potential oncogenic role in cancer. Biochim. Biophys. Acta. 2006;1765:223–234. doi: 10.1016/j.bbcan.2006.01.001. [DOI] [PubMed] [Google Scholar]
- [55].McElligott R, Wellinger RJ. The terminal DNA structure of mammalian chromosomes. EMBO J. 1997;16:3705–3714. doi: 10.1093/emboj/16.12.3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Ghosh G, Li G, Myung K, Hendrickson EA. The lethality of Ku86 (XRCC5) loss-of-function mutations in human cells is independent of p53 (TP53) Radiat. Res. 2007;167:66–79. doi: 10.1667/RR0692.1. [DOI] [PubMed] [Google Scholar]
- [57].Hendry JH. Survival of cells in mammalian tissues after low doses of irradiation: a short review. Int. J. Radiat. Biol. 1988;53:89–94. doi: 10.1080/09553008814550451. [DOI] [PubMed] [Google Scholar]
- [58].Reddel RR, Bryan TM, Colgin LM, Perrem KT, Yeager TR. Alternative lengthening of telomeres in human cells. Radiat. Res. 2001;155:194–200. doi: 10.1667/0033-7587(2001)155[0194:alotih]2.0.co;2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.